

ABSTRACT
Enveloped viruses typically encode their own fusion machinery to enter cells. Herpesviruses are unusual, as they fuse with a number of cellular compartments throughout their life cycles. As uncontrolled fusion of the host membranes should be avoided in these events, tight regulation of the viral fusion machinery is critical. While studying herpes simplex virus 1 (HSV-1) glycoprotein gM, we identified the cellular protein E-Syt1 (extended synaptotagmin 1) as an interaction partner. The interaction took place in both infected and transfected cells, suggesting other viral proteins were not required for the interaction. Most interestingly, E-Syt1 is a member of the synaptotagmin family of membrane fusion regulators. However, the protein is known to promote the tethering of the endoplasmic reticulum (ER) to the plasma membrane. We now show that E-Syt1, along with the related E-Syt3, negatively modulates viral release into the extracellular milieu, cell-to-cell viral spread, and viral entry, all processes that implicate membrane fusion events. Similarly, these E-Syt proteins impacted the formation of virus-induced syncytia. Altogether, these findings hint at the modulation of the viral fusion machinery by the E-Syt family of proteins.
IMPORTANCE Viruses typically encode their own fusion apparatus to enable them to enter cells. For many viruses, this means a single fusogenic protein. However, herpesviruses are large entities that express several accessory viral proteins to regulate their fusogenic activity. The present study hints at the additional participation of cellular proteins in this process, suggesting the host can also modulate viral fusion to some extent. Hence E-Syt proteins 1 and 3 seem to negatively modulate the different viral fusion events that take place during the HSV-1 life cycle. This could represent yet another innate immunity response to the virus.
KEYWORDS: E-Syt1, E-Syt2, E-Syt3, gM, UL10, HSV, syncytia, fusion, synaptotagmin
INTRODUCTION
Synaptotagmins are members of a membrane-trafficking protein family that were originally defined as containing an N-terminal transmembrane region, a variable linker, and two so-called C-terminal C2 domains that bind phospholipids and calcium (1). There are 17 known mammalian synaptotagmins found in the brain and located in distinct subcellular compartments (2, 3). They participate in the maturation of secretory vesicles, exocytosis, and endocytosis, including protein and membrane recycling at nerve terminals (4–6). Most of these proteins engage and regulate SNARE proteins, the core cellular fusion machinery (7), and many act as Ca2+ sensors and form homo- or heterodimers (1, 8–11). Moreover, they finely control membrane blending by acting on fusion pore opening and expansion (12). However, the functions of several synaptotagmins remain to be determined.
The extended synaptotagmins (E-Syts) belong to a recently discovered family of transmembrane proteins that are ubiquitously expressed. The three mammalian homologs (E-Syt1, E-Syt2, and E-Syt3) of the Saccharomyces cerevisiae tricalbins (tricalbins 1, 2, and 3) have a domain structure similar to that of the classical synaptotagmins. They also contain an SMP (synaptotagmin-like, mitochondrial-lipid-binding protein) domain and multiple C2 domains (five in E-Syt1 and three in E-Syt2 and E-Syt3) (13). Despite this structural homology, they do not seem to interact with SNARE proteins to modulate fusion events as synaptotagmins do. Rather, they mediate the tethering of the endoplasmic reticulum (ER) to the plasma membrane (PM) (14), specific lipid transfer between the two membranes (15), and receptor signaling (16). At the intracellular level, E-Syt1 broadly associates with the ER, while E-Syt2 and E-Syt3 are at ER-PM contact sites (14, 17, 18). These surface contacts are mediated by C2 domain-dependent binding of phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] in the PM and, in the case of E-Syt1, by elevated levels of cytosolic Ca2+ (19). As for synaptotagmins, E-Syt proteins can form homo- or heterodimers with each other, which promotes the above-mentioned ER-PM interactions (14). However, E-Syts are not essential, and cells that lack all three isoforms do not exhibit obvious major defects aside from growing more slowly (15). Despite these findings, the precise roles played by E-Syts are mainly unknown, and their functional link with the SNARE cellular fusion machinery, if any, remains to be found.
Enveloped viruses enter cells by fusion with either the plasma membrane or an internal compartment, such as endosomes. The process is driven by virus-encoded proteins that are activated downstream of their initial interactions with specific host receptors. Although fusion is often mediated by a unique viral protein, herpesviruses exhibit a complex fusion apparatus composed of a core of four distinct viral proteins. For herpes simplex virus 1 (HSV-1), these are glycoprotein B (gB), gD, and the gH/gL complex, with the first exhibiting the fusogenic activity (20). While all four proteins are present on the surfaces of virions, they are also detected on various cellular compartments during infection, including the nuclear envelopes, trans-Golgi network (TGN), endosomes, and plasma membrane (21–27). This implies that gB is tightly regulated to avoid the unforeseen and uncontrolled fusion of various cellular compartments. This is particularly critical, as the virus undergoes 3 or 4 distinct fusion events during its replication cycle (28). As discussed above, the virus initially enters cells by fusion (1st fusion event) and subsequently releases its genome into the nucleus. There, it assembles new viral particles that are too large to escape via the nuclear pores and rather bud through the inner nuclear envelope, producing transient enveloped viruses that also contain the above-mentioned core fusion machinery (29–31). This is followed by the rapid fusion of these primary enveloped virions with the outer nuclear envelope, thereby releasing naked viral capsids into the cytoplasm (2nd fusion event). Following their reenvelopment within the cell and subsequent budding from that compartment to yield double-enveloped viruses (32), the viruses fuse with the plasma membrane to release singly enveloped mature virions (3rd fusion event). Finally, HSV-1 can use an alternative exit pathway and infect neighboring cells via direct cell-cell fusion induced by the virus, representing a potential fourth fusion event (33).
A large body of literature shows that HSV-1 gB is regulated by the viral proteins gD and gH/gL and by cellular receptors (nectin 1 or 2, HVEM, and 3-O-sulfated heparin sulfate) (20). There is evidence that other molecules also participate in this process, including the viral proteins UL20, gK, US3, TK, gM, and gN (34–43). Many of these activities were detected in the context of mutants that promote syncytia, cell-cell fusion events that can arise when cells expressing the core HSV-1 fusion machinery at their surfaces contact adjacent cells. The viral gM protein is an interesting case, as it is present on most of the cellular compartments where viral fusion takes place, as well as on the above-mentioned primary enveloped virions and on mature extracellular virions (27, 44, 45). It also reduces the surface expression of gB and the gH/gL complex and negatively impacts syncytium formation in transfected cells (46–48). In contrast, gM instead promotes viral entry at the cell surface and stimulates gH/gL incorporation into mature virions (41, 49, 50). Interestingly, it is among the first viral glycoproteins found at the inner nuclear membrane, well before the core fusion machinery is targeted there (45). This raises the questions of whether and how gM may modulate viral fusion and is itself regulated, both positively and negatively. So far, the HSV-1 proteins UL46 and gN have been identified as gM partners (43, 51–53), with gN directly binding to gM and stimulating virus-induced syncytium formation (43). Another open question is whether cellular proteins, aside from the known HSV-1 receptors, could participate in this process. Thus, little is known about the role and modulation of gM during infection.
To understand how gM might regulate viral fusion, a search for novel binding partners was performed, with an emphasis on host-derived proteins using HSV-1 gM-transfected cells. We found by proteomics and confirmed in coimmunoprecipitation (co-IP) studies that HSV-1 gM binds to the cellular E-Syt1 protein. Knocking E-Syt1 down stimulated the release of the virus into the extracellular medium at the expense of cell-associated infectious particles. Conversely, overexpressing E-Syt1 led to reduced levels of mature virions in the medium, hinting at a negative regulation. E-Syt1 did not act alone but in combination with the related E-Syt3, which exhibited a similar phenotype. Most interestingly, these E-Syt proteins impacted viral entry, as well as cell-cell fusion (syncytia) and viral plaque size (cell-to-cell spread), suggesting they somehow acted on the viral fusion machinery. These findings highlight functional links between the viral and cellular fusion machineries and the membrane-tethering E-Syt family.
RESULTS
E-Syt1 interacts with HSV-1 gM.
The reported ability of the HSV-1 gM protein to modulate fusion both negatively and positively points to the likely regulation of gM itself. To identify new gM partners, particularly cellular ones, they were probed in hemagglutinin (HA)-gM-transfected cells. Whole-cell lysates were used to immunoprecipitate gM using an HA monoclonal antibody for greater specificity (available gM antibodies are polyclonal). As controls, preimmune sera or cells treated only with the transfection reagent were tested in parallel. Analysis of the entire band tracks by silver staining (Fig. 1) and mass spectrometry (MS) (Table 1) led to the identification of only a few proteins present in two independent experiments, aside from gM. They were specific to the immunoprecipitate and absent from the lipofectamine or preimmune controls. Of particular interest was E-Syt1, a 1,104-amino-acid protein with a predicted molecular mass of 122.9 kDa that is a tethering factor related to synaptotagmins. To confirm this interaction, reciprocal immunoprecipitations with either gM or E-Syt1 antibodies were performed, using mock-treated or gM-transfected cells. As expected, the data corroborated the above-mentioned findings, with gM antibodies bringing down E-Syt1 (Fig. 2B) and vice versa (Fig. 2C). Given their interaction in transfected cells, it appeared that the two proteins formed a complex in the absence of other viral proteins. To probe whether such a complex is relevant in the context of infection, the experiments were repeated in cells infected for 18 h, a time when structural proteins such as viral glycoproteins are typically expressed in large amounts. Once again, co-IP experiments confirmed that the two proteins did bind one another (Fig. 3). As expected, this was dependent on the use of gM or E-Syt1 antibodies and the presence of the virus.
FIG 1.

Silver staining of samples analyzed by mass spectrometry. Cells were transiently transfected with a plasmid expressing HA-gM or mock transfected (no DNA). Twenty-four hours posttransfection, the cells were harvested and lysed in RIPA buffer. The lysates were immunoprecipitated using a preimmune serum or an anti-HA monoclonal antibody, and the bead-bound proteins were analyzed on precast 4 to 12% SDS-PAGE gradient gels. The proteins were subsequently silver stained, and entire lane tracks were processed for mass spectrometry as detailed in Materials and Methods. Bands specifically detected by silver staining in the gM immunoprecipitation are marked by stars. The numbers on the left indicate molecular mass in kilodaltons.
TABLE 1.
Proteins identified by proteomicsa
| Accession no. | UniProt ID | GeneID | Description | Mass (kDa) | Total no. of spectra (% coverage) |
|
|---|---|---|---|---|---|---|
| 1st expt | 2nd expt | |||||
| A8K8U1_HUMAN | Q86VP6 | CAND1 | Cullin-associated and neddylation-dissociated 1 | 136.3 | 8 (9) | 12 (11) |
| B3KMV5_HUMAN | Q9BSJ8 | ESYT1 | Extended synaptotagmin 1 | 122.9 | 3 (3) | 3 (3) |
| B4DDH8_HUMAN | Q96N66 | MBOAT7 | Lysophospholipid acyltransferase 7 | 50.6 | 2 (6) | 3 (10) |
| GM_HHV11 | P04288.1 | 2703379 | HSV-1 gM | 51.4 | 26 (27) | 23 (25) |
HeLa cells transfected with HA-gM were immunoprecipitated with an HA antibody, resolved on gradient gels, and processed for mass spectrometry. Peptides were analyzed with Mascot and Scaffold using a combined HSV-1 and human in-house trypsin database. The thresholds for detection were 99% for the proteins and 0.1% FDR for the peptides with at least 2 peptides.
FIG 2.

HSV-1 gM and E-Syt1 are reciprocally coimmunoprecipitated in transfected cells. HeLa cells were transfected with plasmids expressing HA-tagged gM. After 24 h of transfection, the cells were lysed as described in Materials and Methods. The lysates were immunoprecipitated using a monoclonal anti-E-Syt1 antibody or with a monoclonal anti-HA antibody and loaded onto a 10% SDS-PAGE gel. (A) A Western blot of the total lysates (2% loaded) served as a positive antibody control. (B and C) In parallel, the immunoprecipitates were analyzed by Western blotting with anti-gM (4C10) (B) or anti-E-Syt1 (C) antibodies. The numbers on the left indicate molecular mass in kilodaltons. −, transfection control; +, cells transfected with an HA-gM plasmid.
FIG 3.

E-Syt1 interacts with HSV-1 gM in infected cells. HeLa cells were infected with wild-type virus at an MOI of 5, and the cells were harvested at 18 hours postinfection (hpi) and then lysed. Total cell lysates were loaded on gels (A) or immunoprecipitated with the polyclonal (4C10) anti-gM (B) or anti-E-Syt1 (C) antibody (Ab). Uninfected cells and immunoprecipitations performed using the preimmune serum served as negative controls. The bead-bound proteins were resolved in a 10% gel and immunoblotted with either E-Syt1 or the PAS980 gM antibodies, as indicated. The numbers on the left indicate molecular mass in kilodaltons. −, preimmune serum as a negative control; +, immunoprecipitation with the antibody of interest.
E-Syt1 modulates viral egress.
E-Syt1 is a tethering molecule that enables contacts between the ER and the cell surface. It is also a member of the synaptotagmin family, which typically interacts with the SNARE machinery, the core cellular element that drives membrane fusion. To determine if E-Syt1 has any functional relevance for infection, extracellular viral yields were measured in the presence or absence of E-Syt1 using commercially available small interfering RNA (siRNA) pools. The data revealed that knocking down E-Syt1 (80% efficacy of the siRNA) (Fig. 4A and B) led to a highly significant increase of infectious virions in the supernatant (Fig. 4C). To evaluate if E-Syt1 modulates viral-particle production, intracellular yields were monitored next. Figure 4D shows that E-Syt1 depletion reduced the number of cell-associated viral particles, suggesting that E-Syt1 may hinder their release across the plasma membrane. In contrast, the virus did not impact the level of E-Syt1 expression, since levels were similar in mock-treated and infected cells (Fig. 5).
FIG 4.

Inhibition of E-Syt1 enhances HSV-1 infection. HeLa cells were seeded in 6-well plates and transfected with siRNAs targeting E-Syt1. Forty-eight hours later, the cells were infected with wild-type HSV-1 at an MOI of 5 for 20 h. (A and B) Cell lysates were analyzed by Western blotting against E-Syt1 (A), and the efficacy of the siRNA was measured with a ChemiDoc instrument and Image Lab software, which give a linear response over several log units (B). (C and D) The extracellular medium (C) and cell lysates (D) were then collected and titrated on Vero cells. Viral plaques were then quantified and normalized to 100%, using the no-siRNA control (transfection reagent only). The error bars show the standard errors of the mean (SEM) of four independent experiments, each done in duplicate. Bilateral Student's t tests were performed to detect significant hits compared to the control (Ctrl). The exact P values were 0.0003 (B), 0.0004 (C), and 0.020 (D) (*, P < 0.05; ***, P < 0.001).
FIG 5.
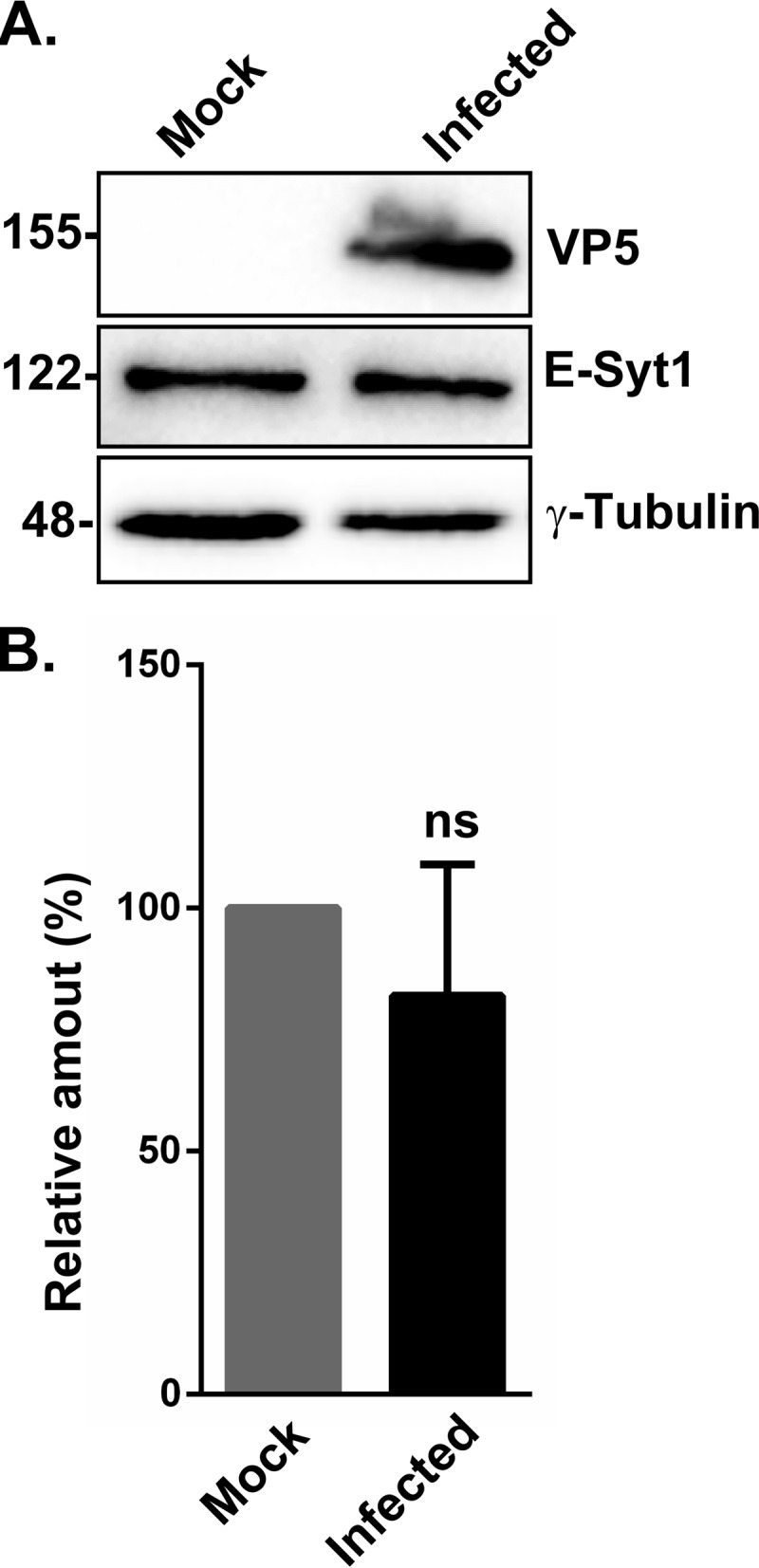
Impact of the virus on E-Syt1 expression. HeLa cells were mock treated or infected with wild-type HSV-1 at an MOI of 5 for 20 h. (A) Cell lysates were prepared as described above, and VP5, E-Syt1, and γ-tubulin expression was determined by Western blotting. The numbers on the left indicate molecular mass in kilodaltons. (B) Quantification was performed with a ChemiDoc instrument and Image Lab software. The values (means and SEM) were derived from six independent experiments. Bilateral Student's t tests were performed to detect significant hits compared to the mock treatment (P = 0.541). ns, not significant.
The E-Syt3 isoform also takes part in HSV-1 egress.
E-Syt1 is one of three related human isoforms. To determine whether other E-Syt proteins also influence the HSV-1 life cycle, we first evaluated their expression in HeLa cells by reverse transcription (RT)-PCR. The results indicated that all three isoforms were indeed present in these cells (Fig. 6A). This was further confirmed by Western blotting using E-Syt1-, -2-, or -3-specific antibodies (data not shown). To see if E-Syt2 and -3 also physically interact with gM, co-IP experiments were done in infected cells using gM antibodies. Most interestingly, both of them were also coimmunoprecipitated with gM while they were absent from the control uninfected cell lysates (Fig. 6B). Although undetected by mass spectrometry, this was not surprising, given the ability of E-Syts to form heterodimers (14). We finally probed whether the E-Syt proteins colocalized with gM in infected cells. Unfortunately, we could not find antibodies that detect endogenous E-Syt proteins by immunofluorescence and consequently resorted to E-Syt overexpression. Oddly, only minimal colocalization was noted between gM and the E-Syt proteins (Fig. 7), hinting at the presence of large pools of gM and E-Syts not binding one another in this context.
FIG 6.

E-Syt2 and E-Syt3 are expressed and interact with HSV-1 gM in infected cells. (A) Expression of E-Syt1, E-Syt2, and E-Syt3 was monitored in HeLa cells by RT-PCR. Total RNA was isolated from the cells, reverse transcribed, and amplified with E-Syt1-, -2-, or -3-specific primers by PCR. Plasmids coding for each isoform served as PCR and specificity controls. A representative experiment is shown among the three performed. (B) HeLa cells were mock infected or infected with wild-type virus at an MOI of 5. At 18 hpi, the cells were harvested and then lysed with RIPA buffer. The immunoprecipitation was performed using the polyclonal (4C10) anti-gM for infected cells and with uninfected cells as a negative control. +, presence, and −, absence of the antibody used for immunoprecipitation. Recovered proteins (IP) and aliquots of the lysates (Input) were separated by SDS-PAGE and detected by Western blotting with either E-Syt2, E-Syt3, or the PAS980 gM antibodies as indicated. One representative experiment out of four independent experiments is shown.
FIG 7.

HSV-1 gM only minimally colocalizes with overexpressed E-Syt proteins. HeLa cells grown on coverslips were transfected for 12 h with plasmids coding for myc E-Syt1 (A), myc E-Syt2 (B), or myc E-Syt3 (C). They were then infected with wild-type HSV-1 for 12 h, fixed, permeabilized, and immunostained with an antibody against myc to detect E-Syt1, E-Syt2, or E-Syt3 (red signals) and with an anti-gM (4C10) antibody to detect infected cells (green signals), while nuclei were stained with Hoechst (blue signals). Scale bar, 10 μm.
We next examined if knocking down the E-Syt isoforms individually affected viral output. As they can dimerize, we also depleted them in pairs. The efficacies of the commercial siRNA pools were monitored by Western blotting and digitally quantified. All the interference reagents proved efficient, with reductions reaching 78% (E-Syt1), 86% (E-Syt2), and 91% (E-Syt3) (Fig. 8A). Given their efficacy, we next proceeded to test the impacts of the E-Syt proteins on extracellular viral yields by plaque assays. These results indicated that E-Syt2 depletion had a slight but not statistically significant effect on HSV-1 production, while depletion of E-Syt1 alone or in combination with E-Syt3 led to significant and larger increases in mature infectious particles in the extracellular milieu (Fig. 8B). To examine if the E-Syt proteins impaired only HSV-1 release into the extracellular medium or if they also impacted cell-to-cell spread, plaque sizes were also measured. The data were in agreement with the above-mentioned findings in that depletion of E-Syt1, E-Syt3, or both increased the propagation of the virus to neighboring cells, while siRNAs against E-Syt2 seem to hamper the depletion of E-Syt1 or -3 (Fig. 9).
FIG 8.

Efficient knockdown of E-Syt proteins and impact on viral release. (A to C) Efficacy of siRNAs. (A) HeLa cells were transfected for 48 h with the transfection reagent only (Ctrl) or siRNAs against E-Syt1, E-Syt2, or E-Syt3, as indicated. The cells were then harvested and lysed, and the levels of E-Syt proteins were detected by Western blotting, as shown. Immunoblots of cell lysates were also probed with β-calnexin or γ-tubulin as an internal loading control. The results were quantified with a ChemiDoc station and Image Lab. The histograms under the gels indicate the mean values from three experiments and the standard errors of the mean. The asterisks indicate the results of bilateral Student's t tests, which were as follows: siE-Syt1, 0.00035; siE-Syt2, 0.0002; siE-Syt3, 0.0002. (B) Virus production in E-Syt-depleted cells. The supernatants from the above-described infections were collected, and their viral contents were titrated on Vero cells by plaque assays to monitor viral output. The titers were normalized to the mean values obtained with samples treated with transfection reagent only. The error bars show the SEM of three independent experiments, each done in duplicate. Bilateral Student's t tests were performed to detect significant hits compared to the control. Exact P values were as follows: NC1, 0.63; siE-Syt1, 0.0004; siE-Syt2, 0.286; siE-Syt3, 0.341; siE-Syt1 and -2, 0.659; siE-Syt1 and -3, 0.006; siE-Syt2 and -3, 0.0.38 (**, P < 0.01; ***, P < 0.001; ns, not significant).
FIG 9.

E-Syt1 and E-Syt3 downregulation increases cell-to-cell spread. An experiment was performed essentially as for Fig. 7, and plaque size was determined in square pixels using ImageJ. The graph shows the mean size ± SEM of 100 plaques under each condition from a representative experiment. They were also statistically analyzed with bilateral Student's t tests (n = 3 independent experiments). P values were as follows: NC1, 0.955; siE-Syt1, 0.0006; siE-Syt2, 0.80; siE-Syt3, 0.00004; siE-Syt1 and -2, 0.368; siE-Syt1 and -3, 7.3 × 10−11; siE-Syt2 and -3, 0.038 (***, P < 0.001; ns, not significant).
To better define how E-Syts modulated the virus, infected cells were monitored by transmission electron microscopy under E-Syt depletion conditions. Figure 10 shows representative images and quantitation of these observations from two independent experiments. With wild-type levels of E-Syts, the virus was readily detected within the nucleus (nuclear capsids) or occasionally between the two nuclear envelopes (primary enveloped virions). The virus was also prevalent in the cytoplasm (with or without an envelope) and at the cell surface (extracellular virions). Under these conditions, the viral particles were distributed roughly equally among the nucleus (38%), cytoplasm (32%), and cell surface (30%). Upon the depletion of E-Syt1, E-Syt3, or both, no obvious impact on the nuclear pool of the virus was noted. A modest and statistically significant decrease of nuclear particles was noted in siE-Syt1-treated cells, but such an impact was not confirmed in the double knockdowns. In contrast, the proportions of virions at the cell surface strongly and significantly jumped to 53%, 48%, and 55% (siE-Syt1, siE-Syt3, and both, respectively) at the expense of reduced levels of viral particles in the cytoplasm (15%, 16%, and 13%, respectively). The E-Syt proteins thus seemed to favor the exit of the virus from the cytoplasm into the extracellular milieu without much affecting the nuclear levels of the virus. Most importantly, these data showed that there were no gross virion morphogenesis defects.
FIG 10.

HeLa cells were mock transfected or treated with E-Syt1 or E-Syt3 siRNA pools and then infected with wild-type HSV-1 at an MOI of 5 for 18 h. The cells were then fixed and processed for thin-section electron microscopy as described in Materials and Methods. (A) Representative cells for each condition. The boxed areas are enlarged on the right. (B) Values representing the average numbers and percentages of viral particles counted from two independent experiments, each using six randomly selected cells per condition (up to 2,418 particles per condition). The results were compared to those for the HSV-1 samples using bilateral Student's t tests with HSV-1. *, E-Syt 1, P = 0.0311 (nuclear capsids), P = 0.0276 (cytoplasmic capsids), P = 0.0194 (extracellular virions); E-Syt 3, P = 0.042 (cytoplasmic capsids), P = 0.0227 (extracellular virions); E-Syt 1 and -3, P = 0.0194 (cytoplasmic capsids), P = 0.0262 (extracellular virions); *, P < 0.05.
Given that reduced levels of E-Syt1 and E-Syt3 promoted the release of virions from the cells, we sought to determine if, conversely, overexpressing them would reduce viral yields. In full agreement with the above-mentioned findings, overexpression of E-Syt proteins caused a significant reduction, roughly 40%, for E-Syt1 and E-Syt3, while the reduction for E-Syt2 was not statistically significant (Fig. 11). Thus, E-Syt1 and E-Syt3 under both reduced and overexpressed conditions influenced virus production, while E-Syt2 did not seem to play a significant role.
FIG 11.

Overexpression of E-Syt1 or E-Syt-3 reduces viral yields. HeLa cells were transfected with plasmids expressing myc E-Syt1, myc E-Syt2, or myc E-Syt3. Twenty-four hours later, the cells were infected with wild-type HSV-1 at an MOI of 5 for 18 h. (A) Cell lysates were prepared, and exogenous protein expression was tested by Western blotting using anti-myc antibodies. (B) Viusl production under the conditions in panel A was evaluated by plaque assays. The bars represent the means of three independent experiments, along with the SEM. Bilateral Student's t tests were performed to detect significant hits compared to the control. P values were as follows: myc E-Syt1, 0.030; myc E-Syt2, 0.061; myc E-Syt3, 0.016 (*, P < 0.05; ns, not significant).
E-Syt1 and E-Syt3 also modulate viral entry.
While exiting cells, HSV-1 must fuse with the plasma membrane to be released in the extracellular environment. Our data so far supported the involvement of E-Syt1 and E-Syt3 in this step. As mentioned above, this is not the only fusion event the virus undertakes at the plasma membrane, as it also fuses with the plasma membrane upon entry at the beginning of the replication cycle. This is particularly relevant, as HSV-1 transiently induces the release of Ca2+ from ER stores (54–56) and E-Syt1 promotes membrane contacts in a Ca2+-dependent fashion (17, 18). Consequently, a viral entry assay was used to directly test this hypothesis. The assay relies on a recombinant HSV coding for a luciferase (57) and a short, 1-h infection time to monitor viral entry before viral genome replication kicks in. In this assay, no luminescence was detectable upon infection with wild-type virus or at 4°C, a temperature that prohibits viral entry (Fig. 12). In contrast, infections with HSV-1 Lox-Luc gave a strong signal, which we normalized to 100%. Most interestingly, knocking down E-Syt1, E-Syt3, or both promoted viral entry into the cells, with the strongest effect seen with the double knockdowns (Fig. 12). Thus, as for viral egress, E-Syt1 and E-Syt3 modulated the passage of the virus during viral entry.
FIG 12.

E-Syt1 and E-Syt3 impair HSV-1 entry. HeLa cells seeded in 6-well plates were transfected with siRNAs targeting E-Syt1, E-Syt3, or both for 48 h. Control nontargeting siRNAs (NC1) were also used. The cells were subsequently mock infected or infected at an MOI of 30 for 1 h at 4°C (viral adsorption step) with wild-type HSV-1 (luciferase negative) or HSV-1 Lox-Luc (luciferase positive). The cells were then incubated at 37°C for another hour to enable viral entry and subsequently lysed at room temperature for 30 min in the presence of luciferin and energy. As an additional control, one sample was left at 4°C throughout the experiment to prevent viral entry. All the samples were next transferred to 96-well plates and analyzed with a luminometer. The values represent the mean relative light units (RLU) from three independent experiments, and the error bars indicate the standard errors of the mean. The asterisks indicate the results of bilateral Student's t tests upon comparison to the HSV-1 Lox-Luc positive control. The following P values were obtained: mock treated, 8 × 10−9; HSV-1 17+, 1 × 10−9; 4°C, 0.0004; NC1, 0.86; siE-Syt1, 0.001; siE-Syt3, 0.028; siE-Syt1 and -3, 0.024 (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant).
E-Syt1 and E-Syt3 limit syncytium formation.
As mentioned above, a number of viral mutants deregulate the viral fusion machinery and lead to a cell-cell fusion phenotype (syncytia). We recently demonstrated that this deregulation can also occur by overexpressing regulators of the machinery (43). The present findings somehow position E-Syt proteins as negative regulators of fusion in the context of an HSV-1 infection, since deleting them stimulates viral entry and egress through the plasma membrane, and vice versa if overexpressed. To test if E-Syt1 and E-Syt3 also modulate virus-induced syncytium formation, they were overexpressed, and cell-cell fusion was monitored. As expected, no syncytia were recorded in the absence of infection (data not shown). Similarly, only rare and small syncytia were found in the presence of the wild-type virus (Fig. 13A). Knocking down E-Syt1, E-Syt3, or both all resulted in increased detection of syncytia, including large ones that reached up to 14 nuclei per cell (Fig. 13B to D). Once again, the combined effect of E-Syt1 and E-Syt3 gave the strongest phenotype, with half the cells being multinuclear. This indicated that the E-Syts do indeed regulate virus-induced membrane fusion.
FIG 13.

E-Syt1 and E-Syt3 knockdown increases cell-to-cell fusion in infected cells. HeLa cells grown on coverslips were mock transfected (A) or treated for 48 h with each of the different siRNA pools targeting E-Syt1 (B), E-Syt3 (C), or E-Syt1 and E-Syt3 (D). They were subsequently infected at an MOI of 2 with wild-type HSV-1 for 14 h. To visualize cell-to-cell fusion, the cells were fixed and incubated with the membrane marker Alexa Fluor 647-labeled WGA, a lectin that binds to N-acetyl-d-glucosamine and sialic acid present on the cell surface and internal glycoproteins. The stained cells were processed for fluorescence microscopy and randomly (blindly) photographed in order to determine the presence of syncytia, defined as single cells containing two or more nuclei. For clarity, the outlines of some of the cells are indicated by dashed lines. Quantification was done by counting 200 cells per condition in each of 2 independent experiments. The reported values represent averages from these independent experiments, and the error bars indicate the standard errors of the mean. The fluorescence images show typical examples for each condition. Scale bars, 10 μm.
DISCUSSION
Enveloped viruses are typically fully autonomous and encode their own fusion machinery, whether a single protein, such as the vesicular stomatitis virus G glycoprotein, or multiple viral proteins, as for herpesviruses (20, 58). It is most interesting that HSV-1 depends on the E-Syt1 and E-Syt3 host proteins, in addition to its cellular receptors, to regulate its fusion activities across cellular membranes. These findings were corroborated in multiple ways, including the interaction of E-Syt1, -2, and -3 with the HSV-1 glycoprotein M, itself a regulator of viral fusion. In addition, E-Syt1 and the related E-Syt3 curbed viral egress, viral entry, and cell-to-cell viral spread, as well as HSV-1-induced cell-cell fusion. E-Syts thus appear to impact many of the possible fusion steps occurring during the HSV-1 life cycle. It now remains to be seen if E-Syts also affect the fusion of the primary enveloped virions with the outer nuclear membrane. This is a distinct possibility, given the presence of E-Syt1 in the ER and its continuity with the outer nuclear envelope. Another critical aspect is whether the E-Syt proteins and gN compete for gM, as they seemingly have opposite effects (43). Further work is required to elucidate the fine-tuning of the viral fusion machinery, but the lack of gN antibodies complicates this endeavor (43).
E-Syt proteins promote the close apposition of the ER and the plasma membrane and the transfer of lipids between the two compartments and can form both homo- and heterodimers (14, 15). While only E-Syt1 was initially identified by mass spectrometry as an HSV-1 gM binding partner, E-Syt3 was also found to modulate virus-dependent fusion steps, supporting the view that they act cooperatively. The minimal colocalization of gM with exogenously expressed E-Syt1 suggests that most of these molecules are not associated with one another, and E-Syt3 may consequently have eluded detection in our original co-IP/MS assays. It may also be that E-Syt3 is bound to E-Syt1 instead of gM, though we cannot exclude indirect binding of E-Syt1 with gM. In the latter scenario, the molecule bridging gM and E-Syt1 would be cellular in nature, since E-Syt1 does bind to gM in transfected cells in the absence of other viral proteins. The fact that E-Syt2 did not exhibit similar phenotypes in these events suggests that the three E-Syts are not merely redundant cellular isoforms. In this respect, it is worth nothing that while E-Syt2 did not statistically impact viral spread or plaque size, it did seem to have an effect opposite that of E-Syt1 and -3 in siRNA-depleted cells. Moreover, depleting cells of E-Syt2 in conjunction with E-Syt1 or -3 nullified their phenotypes, an aspect that remains to be clarified and may be linked to the ability of E-Syt2 to form heterodimers with other E-Syt isoforms and perhaps shield the virus from E-Syt1 or -3.
In all the above-described experiments, E-Syt1 or E-Syt3 downregulation stimulated the production of infectious viral particles, while E-Syt1 and/or E-Syt3 overexpression had the opposite effect. While the present data do not exclude the possibility that E-Syt overexpression inhibited viral yields by triggering the unfolded-protein response, this is perhaps not a major issue, as HSV-1 is known to efficiently dampen such a response via ICP0 (59). Thus, our results seem most consistent with a negative role for E-Syts on virus-induced fusion events. This is somewhat in contrast with the contribution of the related synaptotagmins, which promote exocytosis in neurons (60), and the current lack of evidence that E-Syt proteins modulate cellular fusion events. However, not all members of the synaptotagmin family act in the same way. For instance, in contrast to other synaptotagmins, synaptotagmin V11 limits pore expansion during lysosomal exocytosis (61), while synaptotagmin 11 minimizes vesicle formation along the clathrin-coated-vesicle pathway (62). It is therefore plausible that synaptotagmins modulate fusion by various means, both positively and negatively. One important question is whether the E-Syts somehow cooperate with or antagonize the SNARE fusion machinery, as do synaptotagmins in neurons (63). While the existing literature points to a tethering function for E-Syts, the membrane proximity they promote is nonetheless an essential step toward and prerequisite for membrane fusion. Whatever the case might be, the present findings suggest a previously unforeseen link between the HSV-1 fusion apparatus and the cellular E-Syt protein family.
MATERIALS AND METHODS
Cells and viruses.
Vero (African green monkey kidney) and HeLa (human cervical cancer) cells were purchased from the ATCC, and all the cell lines were tested yearly for the absence of mycoplasma contaminants. They were grown at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (Sigma-Aldrich) supplemented with 5% bovine growth serum (BGS) (HyClone) and 2 mM l-glutamine (Invitrogen). Wild-type HSV-1 strain 17+ and the HSV-1 (17+) Lox-Luc viral strain (both obtained from Beate Sodeik) were propagated on BHK cells, and their titers were determined by plaque assay on Vero cells. The latter virus is derived from strain 17+ and encodes a Gaussian luciferase gene under the constitutively expressed immediate-early cytomegalovirus (CMV) promoter positioned between the UL55 and UL56 HSV-1 genes (57).
Antibodies.
The primary antibodies and dilutions used in this study were as follows: anti-myc rabbit polyclonal (1:1,000; 2272; Cell Signaling Technology), anti-myc mouse polyclonal 9B11 (1:100; 2276; Cell Signaling Technology), anti-HA mouse monoclonal (1:1,000; SC-7392; Santa Cruz), anti E-Syt1 rabbit polyclonal (1:1,000; A303-362A; Bethyl Laboratories), anti E-Syt2 rabbit polyclonal (1:1,000; NBP1-59988; Novus Biologicals), anti E-Syt3 rabbit polyclonal (1:1,000; NBP1-91354; Novus Biologicals), anti-β-actin mouse monoclonal (1:2,500; ab6276; Abcam), anti γ-tubulin mouse monoclonal (1:5,000; T6557; Sigma-Aldrich), anti-HSV VP5 mouse monoclonal (1:2,000; Virusys), anti-HSV gM rabbit polyclonal PAS980 (1:1,000; courtesy of Lynn Enquist), anti-HSV gM rabbit polyclonal 4c10 (1:1,000; courtesy of Joel Baines), and anti-human DDX3 rabbit R648 polyclonal (1:4,000; courtesy of A. Patel). Horseradish peroxidase-coupled secondary antibodies (goat anti-mouse or -rabbit) and light-chain-specific antibody (mouse anti-rabbit) were all used at 1:10,000 and were purchased from Jackson ImmunoResearch.
Plasmids.
Plasmids encoding myc E-Syt1, myc E-Syt2, and myc E-Syt3 were a gift from Pietro de Camilli (14). The empty vector pcDNA3.1 myc was from Invitrogen. The plasmid encoding pHA-UL10 (expressing HA-tagged HSV-1 gM) was generated by Mutagenex (Suwanee, GA, USA).
Mass spectrometry.
HeLa cells were transiently transfected with plasmid expressing HA-gM or mock transfected (no DNA). Twenty-four hours posttransfection, the cells were harvested and lysed in radioimmunoprecipitation assay (RIPA) buffer (0.15 M NaCl, 10 mM Tris-HCl, pH 7.4, 1% deoxycholic acid, 1% NP-40, 0.1% SDS). The lysates were then immunoprecipitated using a preimmune serum or an anti-HA monoclonal antibody, and the bead-bound proteins were analyzed on precast 4 to 12% SDS-PAGE gradient gels and by silver staining. For each lane, four similarly sized parts were excised, taking care to exclude the heavy chain of the precipitating antibodies, and destained with 15 mM potassium ferricyanide (Sigma-Aldrich) and 50 mM sodium thiosulfate (Sigma-Aldrich). Each part was shrunk in 50% acetonitrile (ACN), reconstituted in 50 mM ammonium bicarbonate with 10 mM TCEP [Tris(2-carboxyethyl)phosphine hydrochloride; Thermo Fisher Scientific], and vortexed for 1 h at 37°C. Chloroacetamide (Sigma-Aldrich) was added for alkylation to a final concentration of 55 mM. Samples were vortexed for another hour at 37°C. One microgram of trypsin was added, and digestion was performed for 8 h at 37°C. Peptide extraction was conducted with 90% ACN. The extracted peptide samples were dried down and solubilized in 5% ACN-0.2% formic acid (FA). The samples were loaded on a home-made C18 precolumn (0.3-mm inside diameter [i.d.] by 5 mm) connected directly to the switching valve. They were separated on a home-made reversed-phase column (150-μm i.d. by 150 mm) with a 56-min gradient from 10 to 30% ACN-0.2% FA and a 600-nl/min flow rate on a Nano-LC-Ultra-2D (Eksigent, Dublin, CA) connected to an LTQ-Orbitrap Elite (Thermo Fisher Scientific, San Jose, CA). Each full MS spectrum acquired at a resolution of 60,000 was followed by 12 tandem-MS (MS-MS) spectra on the most abundant multiply charged precursor ions. Tandem-MS experiments were performed using collision-induced dissociation (CID) at a collision energy of 25%. The data were processed using Mascot 2.1 (Matrix Science, London, United Kingdom) and a concatenated UniProt human and herpesvirus database. Mass tolerances on precursor and fragment ions were 10 ppm and 0.01 Da, respectively. Variable selected posttranslational modifications were carbamidomethyl (C), oxidation (M), deamidation (NQ), and phosphorylation (STY). The data were visualized with Scaffold 4.3.0 (protein threshold, 99%, with at least 2 peptides identified and a false-discovery rate [FDR] of 0.1% for peptides).
Coimmunoprecipitation.
For immunoprecipitation studies, HeLa cells were grown on 10-cm dishes and transfected at 80% confluence with the indicated plasmids (24 μg/well) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Twenty-four hours posttransfection, the cells were harvested, washed with phosphate-buffered saline (PBS), and lysed for 30 min at 4°C with gentle agitation in 1 ml of RIPA buffer supplemented with a protease inhibitor cocktail (Roche). For immunoprecipitation studies in the context of infections, cells were instead infected with wild-type HSV-1 at a multiplicity of infection (MOI) of 5 for 18 h. They were then lysed as described above. In both cases, the supernatants obtained after centrifugation were precleared by incubation with preimmune serum for 1 h. Protein A agarose beads were then added and allowed to react for an additional 1 h at 4°C with rotation. After centrifugation at 10,000 × g for 5 min to remove the beads, the precleared lysates were incubated with appropriate antibodies (anti-HA for proteomics; anti-gM or anti-E-Syt1 for co-IP) overnight at 4°C. Fresh protein A agarose beads were added for 1 h, and the immune complexes were washed three times with RIPA buffer and once with Tris-HCl, pH 7.4, to remove unbound proteins. The immunoprecipitates were analyzed by Western blotting as detailed below.
SDS-PAGE and Western immunoblotting assay.
Immunoblotting was carried out essentially as described previously (43). Briefly, protein concentrations in each sample were measured using a Bradford protein essay (Bio-Rad, Mississauga, Ontario, Canada), and 25 μg of proteins was loaded onto 10% acrylamide SDS-PAGE gels. Afterward, the proteins were transferred to polyvinylidene difluoride membranes and then blocked for 1 h at room temperature with blocking buffer (10% nonfat dry milk in 13.7 mM NaCl, 0.27 mM KCl, 0.2 mM KH2PO4, 1 mM Na2HPO4, 0.1% Tween 20). The membranes were ultimately reacted overnight with the primary antibodies as indicated in each figure legend, followed by an appropriate secondary antibody conjugated to horseradish peroxidase for 1 h at room temperature. Detection was done using Clarity Western ECL substrate (Bio-Rad) and a ChemiDoc MP system (Bio-Rad), which, unlike films, enables the reliable and linear quantification of proteins over a wide range (57). The images were acquired and analyzed with Image Lab software version 5.0 (Bio-Rad).
RNA isolation and RT-PCR amplification.
The relative levels of the mRNA transcripts for E-Syt1, E-Syt2, and E-Syt3 were detected by RT-PCR as follows. First, total RNA was extracted from HeLa cells using the SV Total RNA isolation system (Promega Corporation, Madison, WI, USA) according to the manufacturer's instructions. cDNAs were next synthesized using 2 μg of RNA with a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA) following the manufacture's protocol. The resulting cDNAs were subjected to PCR assay using Taq DNA polymerase and ThermoPol buffer (M0267; New England BioLabs, Ipswich, MA, USA) in a total volume of 50 μl. The following primers were used: E-Syt1 forward primer, 5′-GGGGCTGTGTCAATGTTCTT-3′; E-Syt1 reverse primer, 5′-TCCTTCACTTGCACATCGAG-3′; E-Syt2 forward primer, 5′-GAACCTGTGTGGGAGGAAAA-3′; E-Syt2 reverse primer, 5′-GCCTTTGGCAAGTTCTTCAG-3′; E-Syt3 forward primer, 5′-AGACCTGTCCCCACAACAAG-3′; and E-Syt3 reverse primer, 5′-TAGGGATCAGCTCCACTGCT-3′. The PCR products were subsequently separated on a 1% agarose gel, stained with ethidium bromide, and visualized and photographed using the above-mentioned ChemiDoc MP system (Bio-Rad). The expected size of the PCR E-Syt1, E-Syt2, and E-Syt3 fragments were 869 bp, 869 bp, and 874 bp, respectively.
siRNA transfection.
HeLa cells grown in 6-well plates were transfected with siRNA pools targeting each E-Syt (see below) using PepMute (SignaGen Laboratories, Rockville, MD, USA). The siRNA transfection mixture was composed of 100 μl of PepMute transfection buffer, 50 nM pooled siRNAs, and 1.5 μl of PepMute reagent. After 48 h of transfection, the cells were directly harvested or, where indicated, infected as described above. The siRNAs used were as follows: E-Syt1, On-Targetplus Smart pools (L-010652-00-0005; Dharmacon, Mississauga, ON, Canada); E-Syt2, human ESYT2 HSC.RNAI.N020728.12.5 and -12.1 (Integrated DNA Technologies, Inc. [IDT], Coralville, IA, USA); and E-Syt3, human ESYT3 HSC.RNAI.N031913.12.6 (IDT). The nontargeting control (NC1) was from IDT.
Purification of extracellular virus.
HeLa cells were grown on 500-cm2 dishes until 80% confluent. The cells were mock treated or infected with wild-type HSV-1 at an MOI of 5. Eighteen hours later, the extracellular medium was harvested and then clarified by centrifugation at 300 × g for 5 min at 4°C. The samples were filtered through a 0.45-μm filter to eliminate any remaining intact cells and large cellular debris. Extracellular virions were subsequently pelleted by centrifugation at 20,000 × g for 1 h at 4°C in a Beckman SW32-Ti rotor. The viral pellets were finally resuspended in MNT buffer (30 mM MES [morpholineethanesulfonic acid], 100 mM NaCl, 20 mM Tris HCl, pH 7.4) prior to analysis by Western blotting (see above).
Viral titers.
Virus titers were measured by plaque assay with Vero cells as described previously, using serial dilutions of the viral preparations (26). Once plaques appeared, the cells were fixed with cold methanol and stained with 0.1% crystal violet.
Entry assay.
The entry assay was performed essentially as described previously (57). For instance, 6 × 105 HeLa cells/well were plated in 6-well plates and transfected 16 h later with E-Syt-specific siRNAs using PepMute. As a negative control, cells were transfected with the nontargeting NC1 siRNA. Forty-eight hours posttransfection, the cells were inoculated at an MOI of 30 at 4°C for 1 h with HSV-1 (17+) Lox-Luc or wild-type HSV-1 as a negative control. To allow virus penetration, the cells were then incubated at 37°C for another hour, washed with PBS, and lysed for 30 min at room temperature with 100 μl/well of lysis buffer from a luciferase assay kit (Biotium, Burlington, Ontario, Canada). The viral strain used carries a luciferase gene under the constitutively expressed immediate-early CMV promoter positioned between the UL55 and UL56 HSV-1 genes. Samples were then transferred to 96-well plates and analyzed with a Lumistar Galaxy luminometer and software version 4.30-0 (BMG Labtech). In each experiment, a control sample was left at 4°C to prevent viral entry into the cells.
Electron microscopy.
HeLa cells were transfected with E-Syt-specific siRNAs and then infected 2 days later with wild-type HSV-1 at an MOI of 5 for 18 h. They were next fixed for an hour with 2.5% glutaraldehyde-2% paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.2). The cells were washed and scraped off the plates in cacodylate buffer and pelleted by low-speed centrifugation. The pellets were postfixed with 1% OsO4 in cacodylate buffer for an hour at 4°C and then dehydrated in a graded series of ethanol and embedded in Epon (64). Ultrathin sections were obtained using a Reichert Ultracut ultramicrotome and mounted on naked nickel grids. These sections were stained with 3% aqueous uranyl acetate and examined with a Philips CM100 transmission electron microscope. Electron micrographs were captured using an AMT XR80 digital camera. Quantification of the capsids found in the samples was done from randomly selected fields from two independent experiments.
Syncytial assays.
HeLa cells were grown on glass coverslips in 24-well plates prior to transfection with siRNA oligonucleotides as described above. Forty-eight hours posttransfection, the cells were infected with wild-type HSV-1 at an MOI of 2 for 1 h at 37°C and then grown for 14 h at 37°C. The cells were fixed at 4°C for 30 min with 4% paraformaldehyde made in PBS and washed with PBS. To visualize syncytium formation, the plasma membrane was labeled with 5 μg/ml of wheat germ agglutinin (WGA) conjugated to Alexa Fluor 647 (Molecular Probes) and stained with 0.1 g/ml Hoechst 33,342 to identify the nuclei. Fluorescence images were captured with a Zeiss Axio-Imager Z2 epifluorescence microscope and assembled with Adobe Photoshop CS6. Cells with more than one nucleus were defined as a syncytium and were scored from representative microscopic fields of view (100 cells per condition per independent experiment).
Statistical analysis.
Virus titers, protein abundance, and syncytium formation were normalized to the values obtained for the controls as mentioned in each figure legend and analyzed with bilateral Student's t tests using GraphPad Prism version 5 (GraphPad Software).
ACKNOWLEDGMENTS
We are indebted to Pietro de Camilli, A. Patel, Joel Baines, and Lynn Enquist for antibodies and plasmids.
This study was funded by the Canadian Institutes of Health Research (grant MOP258030), the Natural Sciences and Engineering Research Council of Canada (grant RGPIN-2016-04277), and the Canadian Foundation for Innovation (to R.L.). The Institute for Research in Immunology and Cancer (IRIC) receives funds from IRICoR, the Canadian Foundation for Innovation, and the Fonds de Recherche du Québec-Santé (FRQS). The IRIC proteomics facility is a node of the Canadian Genomic Innovation Network and is supported by the Canadian government through Genome Canada. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We all actively participated in the present study. I.E.K. performed the bulk of the experiments, analyzed the data, and contributed to the writing of the manuscript. M.L. made the initial discovery that E-Syt1 modulates HSV-1, and B.K. performed some of the later experiments. J.D. performed all the plaque assays. E.B. and P.T. did the mass spectrometry of the samples. R.L. designed the experiments and wrote the manuscript.
We have no conflict of interest to declare.
REFERENCES
- 1.Sudhof TC. 2002. Synaptotagmins: why so many? J Biol Chem 277:7629–7632. doi: 10.1074/jbc.R100052200. [DOI] [PubMed] [Google Scholar]
- 2.Dean C, Dunning FM, Liu H, Bomba-Warczak E, Martens H, Bharat V, Ahmed S, Chapman ER. 2012. Axonal and dendritic synaptotagmin isoforms revealed by a pHluorin-syt functional screen. Mol Biol Cell 23:1715–1727. doi: 10.1091/mbc.E11-08-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adolfsen B, Saraswati S, Yoshihara M, Littleton JT. 2004. Synaptotagmins are trafficked to distinct subcellular domains including the postsynaptic compartment. J Cell Biol 166:249–260. doi: 10.1083/jcb.200312054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li C, Ullrich B, Zhang JZ, Anderson RG, Brose N, Sudhof TC. 1995. Ca(2+)-dependent and -independent activities of neural and non-neural synaptotagmins. Nature 375:594–599. doi: 10.1038/375594a0. [DOI] [PubMed] [Google Scholar]
- 5.Ahras M, Otto GP, Tooze SA. 2006. Synaptotagmin IV is necessary for the maturation of secretory granules in PC12 cells. J Cell Biol 173:241–251. doi: 10.1083/jcb.200506163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poskanzer KE, Marek KW, Sweeney ST, Davis GW. 2003. Synaptotagmin I is necessary for compensatory synaptic vesicle endocytosis in vivo. Nature 426:559–563. doi: 10.1038/nature02184. [DOI] [PubMed] [Google Scholar]
- 7.Hong W, Lev S. 2014. Tethering the assembly of SNARE complexes. Trends Cell Biol 24:35–43. doi: 10.1016/j.tcb.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Nagy G, Milosevic I, Mohrmann R, Wiederhold K, Walter AM, Sorensen JB. 2008. The SNAP-25 linker as an adaptation toward fast exocytosis. Mol Biol Cell 19:3769–3781. doi: 10.1091/mbc.E07-12-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hui E, Bai J, Wang P, Sugimori M, Llinas RR, Chapman ER. 2005. Three distinct kinetic groupings of the synaptotagmin family: candidate sensors for rapid and delayed exocytosis. Proc Natl Acad Sci U S A 102:5210–5214. doi: 10.1073/pnas.0500941102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugita S, Shin OH, Han W, Lao Y, Sudhof TC. 2002. Synaptotagmins form a hierarchy of exocytotic Ca(2+) sensors with distinct Ca(2+) affinities. EMBO J 21:270–280. doi: 10.1093/emboj/21.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brose N, Petrenko AG, Sudhof TC, Jahn R. 1992. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science 256:1021–1025. doi: 10.1126/science.1589771. [DOI] [PubMed] [Google Scholar]
- 12.Wang CT, Bai J, Chang PY, Chapman ER, Jackson MB. 2006. Synaptotagmin-Ca2+ triggers two sequential steps in regulated exocytosis in rat PC12 cells: fusion pore opening and fusion pore dilation. J Physiol 570:295–307. doi: 10.1113/jphysiol.2005.097378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Creutz CE, Snyder SL, Schulz TA. 2004. Characterization of the yeast tricalbins: membrane-bound multi-C2-domain proteins that form complexes involved in membrane trafficking. Cell Mol Life Sci 61:1208–1220. doi: 10.1007/s00018-004-4029-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giordano F, Saheki Y, Idevall-Hagren O, Colombo SF, Pirruccello M, Milosevic I, Gracheva EO, Bagriantsev SN, Borgese N, De Camilli P. 2013. PI(4,5)P(2)-dependent and Ca(2+)-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell 153:1494–1509. doi: 10.1016/j.cell.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saheki Y, Bian X, Schauder CM, Sawaki Y, Surma MA, Klose C, Pincet F, Reinisch KM, De Camilli P. 2016. Control of plasma membrane lipid homeostasis by the extended synaptotagmins. Nat Cell Biol 18:504–515. doi: 10.1038/ncb3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herdman C, Moss T. 2016. Extended-synaptotagmins (E-Syts); the extended story. Pharmacol Res 107:48–56. doi: 10.1016/j.phrs.2016.01.034. [DOI] [PubMed] [Google Scholar]
- 17.Min SW, Chang WP, Sudhof TC. 2007. E-Syts, a family of membranous Ca2+-sensor proteins with multiple C2 domains. Proc Natl Acad Sci U S A 104:3823–3828. doi: 10.1073/pnas.0611725104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernandez-Busnadiego R, Saheki Y, De Camilli P. 2015. Three-dimensional architecture of extended synaptotagmin-mediated endoplasmic reticulum-plasma membrane contact sites. Proc Natl Acad Sci U S A 112:E2004–E2013. doi: 10.1073/pnas.1503191112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Idevall-Hagren O, Lu A, Xie B, De Camilli P, Fernandez-Busnadiego R, Saheki Y, De Camilli P. 2015. Triggered Ca2+ influx is required for extended synaptotagmin 1-induced ER-plasma membrane tethering. EMBO J 34:2291–2305. doi: 10.15252/embj.201591565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Norrild B, Virtanen I, Pedersen B, Pereira L. 1983. Requirements for transport of HSV-1 glycoproteins to the cell surface membrane of human fibroblasts and Vero cells. Arch Virol 77:155–166. doi: 10.1007/BF01309264. [DOI] [PubMed] [Google Scholar]
- 22.Compton T, Courtney RJ. 1984. Virus-specific glycoproteins associated with the nuclear fraction of herpes simplex virus type 1-infected cells. J Virol 49:594–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilbert R, Ghosh HP. 1993. Immunoelectron microscopic localization of herpes simplex virus glycoprotein gB in the nuclear envelope of infected cells. Virus Res 28:217–231. doi: 10.1016/0168-1702(93)90023-G. [DOI] [PubMed] [Google Scholar]
- 24.Miranda-Saksena M, Boadle RA, Armati P, Cunningham AL. 2002. In rat dorsal root ganglion neurons, herpes simplex virus type 1 tegument forms in the cytoplasm of the cell body. J Virol 76:9934–9951. doi: 10.1128/JVI.76.19.9934-9951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beitia Ortiz de Zarate I, Kaelin K, Rozenberg F. 2004. Effects of mutations in the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B on intracellular transport and infectivity. J Virol 78:1540–1551. doi: 10.1128/JVI.78.3.1540-1551.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turcotte S, Letellier J, Lippé R. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J Virol 79:8847–8860. doi: 10.1128/JVI.79.14.8847-8860.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loret S, Guay G, Lippé R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 29.Wright CC, Wisner TW, Hannah BP, Eisenberg RJ, Cohen GH, Johnson DC. 2009. Fusion between perinuclear virions and the outer nuclear membrane requires the fusogenic activity of herpes simplex virus gB. J Virol 83:11847–11856. doi: 10.1128/JVI.01397-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farnsworth A, Wisner TW, Webb M, Roller R, Cohen G, Eisenberg R, Johnson DC. 2007. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc Natl Acad Sci U S A 104:10187–10192. doi: 10.1073/pnas.0703790104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Padula ME, Sydnor ML, Wilson DW. 2009. Isolation and preliminary characterization of herpes simplex virus 1 primary enveloped virions from the perinuclear space. J Virol 83:4757–4765. doi: 10.1128/JVI.01927-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henaff D, Radtke K, Lippé R. 2012. Herpesviruses exploit several host compartments for envelopment. Traffic 13:1443–1449. doi: 10.1111/j.1600-0854.2012.01399.x. [DOI] [PubMed] [Google Scholar]
- 33.Johnson DC, Webb M, Wisner TW, Brunetti C. 2001. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J Virol 75:821–833. doi: 10.1128/JVI.75.2.821-833.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melancon JM, Foster TP, Kousoulas KG. 2004. Genetic analysis of the herpes simplex virus type 1 UL20 protein domains involved in cytoplasmic virion envelopment and virus-induced cell fusion. J Virol 78:7329–7343. doi: 10.1128/JVI.78.14.7329-7343.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanders PG, Wilkie NM, Davison AJ. 1982. Thymidine kinase deletion mutants of herpes simplex virus type 1. J Gen Virol 63:277–295. doi: 10.1099/0022-1317-63-2-277. [DOI] [PubMed] [Google Scholar]
- 36.Bond VC, Person S. 1984. Fine-structure physical map locations of alterations that affect cell-fusion in herpes-simplex virus type-1. Virology 132:368–376. doi: 10.1016/0042-6822(84)90042-4. [DOI] [PubMed] [Google Scholar]
- 37.Debroy C, Pederson N, Person S. 1985. Nucleotide sequence of a herpes simplex virus type 1 gene that causes cell fusion. Virology 145:36–48. doi: 10.1016/0042-6822(85)90199-0. [DOI] [PubMed] [Google Scholar]
- 38.Hutchinson L, Goldsmith K, Snoddy D, Ghosh H, Graham FL, Johnson DC. 1992. Identification and characterization of a novel herpes simplex virus glycoprotein, gK, involved in cell fusion. J Virol 66:5603–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pogue-Geile KL, Lee GT, Shapira SK, Spear PG. 1984. Fine mapping of mutations in the fusion-inducing MP strain of herpes simplex virus type 1. Virology 136:100–109. doi: 10.1016/0042-6822(84)90251-4. [DOI] [PubMed] [Google Scholar]
- 40.Ruyechan WT, Morse LS, Knipe DM, Roizman B. 1979. Molecular genetics of herpes simplex virus. II. Mapping of the major viral glycoproteins and of the genetic loci specifying the social behavior of infected cells. J Virol 29:677–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim IJ, Chouljenko VN, Walker JD, Kousoulas KG. 2013. Herpes simplex virus 1 glycoprotein M and the membrane-associated protein UL11 are required for virus-induced cell fusion and efficient virus entry. J Virol 87:8029–8037. doi: 10.1128/JVI.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wisner TW, Wright CC, Kato A, Kawaguchi Y, Mou F, Baines JD, Roller RJ, Johnson DC. 2009. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J Virol 83:3115–3126. doi: 10.1128/JVI.01462-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.El Kasmi I, Lippé R. 2015. Herpes simplex virus 1 gN partners with gM to modulate the viral fusion machinery. J Virol 89:2313–2323. doi: 10.1128/JVI.03041-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baines JD, Wills E, Jacob RJ, Pennington J, Roizman B. 2007. Glycoprotein M of herpes simplex virus 1 is incorporated into virions during budding at the inner nuclear membrane. J Virol 81:800–812. doi: 10.1128/JVI.01756-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J, Nagel CH, Sodeik B, Lippé R. 2009. Early, active, and specific localization of herpes simplex virus type 1 gM to nuclear membranes. J Virol 83:12984–12997. doi: 10.1128/JVI.01180-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klupp BG, Nixdorf R, Mettenleiter TC. 2000. Pseudorabies virus glycoprotein M inhibits membrane fusion. J Virol 74:6760–6768. doi: 10.1128/JVI.74.15.6760-6768.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koyano S, Mar EC, Stamey FR, Inoue N. 2003. Glycoproteins M and N of human herpesvirus 8 form a complex and inhibit cell fusion. J Gen Virol 84:1485–1491. doi: 10.1099/vir.0.18941-0. [DOI] [PubMed] [Google Scholar]
- 48.Crump CM, Bruun B, Bell S, Pomeranz LE, Minson T, Browne HM. 2004. Alphaherpesvirus glycoprotein M causes the relocalization of plasma membrane proteins. J Gen Virol 85:3517–3527. doi: 10.1099/vir.0.80361-0. [DOI] [PubMed] [Google Scholar]
- 49.Ren Y, Bell S, Zenner HL, Lau SY, Crump CM. 2012. Glycoprotein M is important for the efficient incorporation of glycoprotein H-L into herpes simplex virus type 1 particles. J Gen Virol 93:319–329. doi: 10.1099/vir.0.035444-0. [DOI] [PubMed] [Google Scholar]
- 50.Lau SY, Crump CM. 2015. HSV-1 gM and the gK/pUL20 complex are important for the localization of gD and gH/L to viral assembly sites. Viruses 7:915–938. doi: 10.3390/v7030915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347–354. doi: 10.1016/j.virol.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 52.Fossum E, Friedel CC, Rajagopala SV, Titz B, Baiker A, Schmidt T, Kraus T, Stellberger T, Rutenberg C, Suthram S, Bandyopadhyay S, Rose D, von Brunn A, Uhlmann M, Zeretzke C, Dong YA, Boulet H, Koegl M, Bailer SM, Koszinowski U, Ideker T, Uetz P, Zimmer R, Haas J. 2009. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog 5:e1000570. doi: 10.1371/journal.ppat.1000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Striebinger H, Funk C, Raschbichler V, Bailer SM. 2016. Subcellular trafficking and functional relationship of the HSV-1 glycoproteins N and M. Viruses 8:83. doi: 10.3390/v8030083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheshenko N, Del Rosario B, Woda C, Marcellino D, Satlin LM, Herold BC. 2003. Herpes simplex virus triggers activation of calcium-signaling pathways. J Cell Biol 163:283–293. doi: 10.1083/jcb.200301084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheshenko N, Liu W, Satlin LM, Herold BC. 2007. Multiple receptor interactions trigger release of membrane and intracellular calcium stores critical for herpes simplex virus entry. Mol Biol Cell 18:3119–3130. doi: 10.1091/mbc.E07-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheshenko N, Trepanier JB, Stefanidou M, Buckley N, Gonzalez P, Jacobs W, Herold BC. 2013. HSV activates Akt to trigger calcium release and promote viral entry: novel candidate target for treatment and suppression. FASEB J 27:2584–2599. doi: 10.1096/fj.12-220285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khadivjam B, Stegen C, Hogue-Racine MA, El Bilali N, Dohner K, Sodeik B, Lippe R. 2017. The ATP-dependent RNA helicase DDX3X modulates herpes simplex virus type 1 gene expression. J Virol 91:e02411-. doi: 10.1128/JVI.02411-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim IS, Jenni S, Stanifer ML, Roth E, Whelan SP, van Oijen AM, Harrison SC. 2017. Mechanism of membrane fusion induced by vesicular stomatitis virus G protein. Proc Natl Acad Sci U S A 114:E28–E36. doi: 10.1073/pnas.1618883114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burnett HF, Audas TE, Liang G, Lu RR. 2012. Herpes simplex virus-1 disarms the unfolded protein response in the early stages of infection. Cell Stress Chaperones 17:473–483. doi: 10.1007/s12192-012-0324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Sudhof TC. 1994. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- 61.Jaiswal JK, Chakrabarti S, Andrews NW, Simon SM. 2004. Synaptotagmin VII restricts fusion pore expansion during lysosomal exocytosis. PLoS Biol 2:E233. doi: 10.1371/journal.pbio.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang C, Wang Y, Hu M, Chai Z, Wu Q, Huang R, Han W, Zhang CX, Zhou Z. 2016. Synaptotagmin-11 inhibits clathrin-mediated and bulk endocytosis. EMBO Rep 17:47–63. doi: 10.15252/embr.201540689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang S, Li Y, Ma C. 2016. Synaptotagmin-1 C2B domain interacts simultaneously with SNAREs and membranes to promote membrane fusion. eLife 5:e14211. doi: 10.7554/eLife.14211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luft JH. 1961. Improvements in epoxy resin embedding methods. J Biophys Biochem Cytol 9:409–414. doi: 10.1083/jcb.9.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]