

Abstract
Key points
NMDA receptor (NMDAR)‐mediated Ca2+ signalling plays a critical role in modulating hypothalamic neurosecretory function. However, whether an altered NMDAR‐evoked changes in Ca2+ (NMDAR‐ΔCa2+) signalling in magnocellular neurosecretory cells (MNCs) may contribute to neurohumoral activation during disease states is unknown.
We show that activation of NMDARs evoked similar inward currents in MNCs of sham and renovascular hypertensive (RVH) rats. Despite this, a prolonged and larger NMDAR‐ΔCa2+ response was observed in the latter.
The exacerbated NMDAR‐ΔCa2+ responses in MNCs of RVH rats affected both somatic and dendritic compartments.
Inhibition of the endoplasmic reticulum sarcoendoplasmic reticulum calcium trasport ATPase (SERCA) pump prolonged NMDAR‐ΔCa2+ responses in sham rats, but not in RVH rats.
Our study supports an altered spatiotemporal dynamic of NMDAR‐ΔCa2+ signalling in MNCs from RVH rats, partly due to blunted endoplasmic reticulum Ca2+ buffering capacity.
Abstract
A growing body of evidence supports an elevated NMDA receptor (NMDAR)‐mediated glutamate excitatory function in the supraoptic nucleus and paraventricular nucleus of hypertensive rats that contributes to neurohumoral activation in this disease. However, the precise mechanisms underlying altered NMDAR signalling in hypertension remain to be elucidated. In this study, we performed simultaneous electrophysiology and fast confocal Ca2+ imaging to determine whether altered NMDAR‐mediated changes in intracellular Ca2+ levels (NMDAR‐ΔCa2+) occurred in hypothalamic magnocellular neurosecretory cells (MNCs) in renovascular hypertensive (RVH) rats. We found that despite evoking a similar excitatory inward current, activation of NMDARs resulted in a larger and prolonged ΔCa2+ in MNCs from RVH rats. Changes in NMDAR‐ΔCa2+ dynamics were observed both in somatic and dendritic compartments. Inhibition of the sarcoendoplasmic reticulum calcium trasport ATPase (SERCA) pump activity with thapsigargin prolonged NMDAR‐ΔCa2+ responses in MNCs of sham rats, but this effect was occluded in RVH rats, thus equalizing the magnitude and time course of the NMDA‐ΔCa2+ responses between the two experimental groups. Taken together, our results support (1) an exacerbated NMDAR‐ΔCa2+ response in somatodendritic compartments of MNCs of RVH rats, and (2) that a blunted ER Ca2+ buffering capacity contributes to the altered NMDAR‐ΔCa2+ dynamics in this condition. Thus, altered spatiotemporal dynamics of the NMDAR‐ΔCa2+ response stands as an underlying mechanism contributing to neurohumoral activation in neurogenic hypertension.
Keywords: hypothalamus, glutamate, NMDA, endoplasmic reticulum
Key points
NMDA receptor (NMDAR)‐mediated Ca2+ signalling plays a critical role in modulating hypothalamic neurosecretory function. However, whether an altered NMDAR‐evoked changes in Ca2+ (NMDAR‐ΔCa2+) signalling in magnocellular neurosecretory cells (MNCs) may contribute to neurohumoral activation during disease states is unknown.
We show that activation of NMDARs evoked similar inward currents in MNCs of sham and renovascular hypertensive (RVH) rats. Despite this, a prolonged and larger NMDAR‐ΔCa2+ response was observed in the latter.
The exacerbated NMDAR‐ΔCa2+ responses in MNCs of RVH rats affected both somatic and dendritic compartments.
Inhibition of the endoplasmic reticulum sarcoendoplasmic reticulum calcium trasport ATPase (SERCA) pump prolonged NMDAR‐ΔCa2+ responses in sham rats, but not in RVH rats.
Our study supports an altered spatiotemporal dynamic of NMDAR‐ΔCa2+ signalling in MNCs from RVH rats, partly due to blunted endoplasmic reticulum Ca2+ buffering capacity.
Abbreviations
- eNMDAR
extrasynaptic NMDAR
- ER
endoplasmic reticulum
- MNC
magnocellular neurosecretory cell
- NMDAR
NMDA receptor
- OT
oxytocin
- PVN
paraventricular nucleus
- RVH rat
renovascular hypertensive rat
- SERCA
sarco/endoplasmic reticulum Ca2+‐ATPase
- SON
supraoptic nucleus
- TG
thapsigargin
- VP
vasopressin
Introduction
Vasopressin and oxytocin magnocellular neurosecretory cells (MNCs) of the hypothalamic supraoptic (SON) and paraventricular (PVN) nuclei play critical roles in fluid balance, and cardiovascular and reproductive homeostasis (Silverman & Zimmerman, 1983). Moreover, exacerbated activity of MNCs contributes to neurohumoral activation during cardiovascular diseases such as hypertension and heart failure (Riegger et al. 1985; Packer et al. 1987; Packer, 1988; Yemane et al. 2010; Littlejohn et al. 2013). Neurohumoral activation has a direct impact into morbidity/mortality in these diseases (Cohn et al. 1984; Yemane et al. 2010). Thus, elucidating the precise cellular mechanisms contributing to altered MNC activity in hypertension is of critical importance.
The amino acid glutamate, acting primarily on ionotropic NMDA receptors (NMDARs) is the major excitatory neurotransmitter within the SON and PVN (van den Pol et al. 1990). Binding of glutamate to NMDARs results in an influx of Ca2+ that evokes a direct membrane depolarization and the adoption of burst firing activity (Hu & Bourque, 1992; Nissen et al. 1995), which optimizes neuropeptide release from the posterior pituitary (Cazalis et al. 1985). Importantly, a growing body of evidence supports an elevated glutamate tone (Zhang et al. 2017), an increased glutamatergic innervation density (Biancardi et al. 2010) and increased expression of NMDARs (Li et al. 2014; Glass et al. 2015) in the SON and PVN of hypertensive rats.
In addition to evoking membrane depolarization, the NMDAR‐mediated Ca2+ influx results in an increase in intracellular free Ca2+ levels (ΔCa2+) (McBain & Mayer, 1994) affecting in turn a variety of downstream signalling mechanisms, including activation of Ca2+‐sensitive channels (Petersen, 2002; Sah & Faber, 2002), changes in the balance of kinase/phosphatase activities (Colbran & Brown, 2004) and stimulation of nitric oxide production (Bredt & Snyder, 1989), all of which could further alter MNC activity.
We recently showed that, in addition to acting on synaptically located NMDARs (sNMDARs) to mediate transient excitatory postsynaptic currents (EPSCs), glutamate also binds to extrasynaptic NMDARs (eNMDARs), activation of which mediates a tonic, sustained excitatory current that strongly stimulates firing activity in MNCs (Fleming et al. 2011). Moreover, we showed that eNMDARs (but not sNMDARs) are functionally coupled, in a Ca2+‐dependent manner, to the A‐type K+ channel‐mediated current I A (Naskar & Stern, 2014) and that an augmented eNMDAR–I A coupling is a critical mechanism contributing to exacerbated MNC activity in renovascular hypertension (RVH) (Zhang et al. 2017). However, whether a change in NMDAR‐ΔCa2+ contributed to the altered NMDAR–I A coupling and increased MNC activity in RVH rats remains unknown.
The functional consequences of the NMDAR‐ΔCa2+ are largely dependent on the magnitude and time course of the Ca2+ signal, which in turn are determined not only by the amount of Ca2+ influx following NMDAR activation, but also by intracellular Ca2+ buffering mechanisms, including the endoplasmic reticulum via the sarco/endoplasmic reticulum Ca2+‐ATPase (SERCA) pump (Toescu & Verkhratsky, 1998). Surprisingly, the precise spatiotemporal Ca2+ dynamics in MNCs following NMDAR activation have not been investigated in detail, and more importantly, whether an altered NMDAR‐ΔCa2+ dynamic in MNCs occurs during hypertension, contributing in turn to its exacerbated actions, remains unknown. To this end, we performed in this study simultaneous patch‐clamp electrophysiological recordings and fast confocal Ca2+ imaging to characterize NMDAR‐ΔCa2+ spatiotemporal dynamics in MNCs in sham and RVH rats.
Methods
Ethical approval
All experimental procedures were in strict compliance with NIH guidelines, and were approved by the Augusta University Institutional Animal Care and Use Committee, and conform to the principles and regulations as described in Grundy (2015).
Animals
Male Wistar rats purchased from Harlan (Indianapolis, IN, USA, 4–5 weeks old) were housed under standardized conditions (12 h:12 h light–dark cycle) with food and water available ad libitum. Rats weighing between 150 and 180 g (approximately 5–6 weeks old) were used to induce the renovascular 2K1C Goldblatt hypertension model (RVH), a well‐characterized and widely used model. Rats were anaesthetized with isoflurane (3%) throughout the surgery. Following an abdominal incision, the left kidney was exposed, and a 0.2 mm clip was placed over the left renal artery, partially occluding it. Sham rats were subjected to the same surgical procedure, although the artery was not occluded. Post‐operative care included proper management of associated pain (buprenorphine, 0.25 mg kg−1, subcutaneous, as needed). Systolic blood pressure was measured at the beginning of the sixth week post‐surgery, using a tail‐cuff method. Mean systolic blood pressure values for sham and RVH were 139.9 ± 2.7 (n = 14) and 200.0 ± 3.1 mmHg (n = 16), respectively (P < 0.0001, Student's unpaired t test). All rats were used for experiments during the sixth to seventh week post‐surgery.
Hypothalamic slices
Rats were anaesthetized with pentobarbital (50 mg kg−1 i.p.), quickly decapitated and brains dissected out. Coronal slices were cut (250 μm thick) utilizing a vibroslicer (Leica VT1200s, Leica Microsystems (Buffalo Grove, IL, USA)) as previously described (Fleming et al. 2011). An oxygenated ice cold artificial cerebrospinal fluid (ACSF) was used during slicing (containing in mm): 119 NaCl, 2.5 KCl, 1 MgSO4, 26 NaHCO3, 1.25 NaH2PO4, 20 d‐glucose, 0.4 ascorbic acid, 2.0 CaCl2 and 2.0 pyruvic acid; pH 7.4; 295–305 mosmol l−1. After sectioning, slices were placed in a holding chamber containing ACSF and kept at room temperature (22°C) until used.
Electrophysiological recordings
Slices were placed in a submersion‐style recording chamber, and bathed with solutions (3.0 ml min−1) that were bubbled continuously with a gas mix of 95% O2–5% CO2, and maintained at near physiological temperature (32°C). Thin‐walled (1.5 mm o.d., 1.17 mm i.d.) borosilicate glass (G150TF‐3, Warner Instruments, Sarasota, FL, USA) was used to pull patch pipettes (3–5 MΩ) on a horizontal Flaming/Brown micropipette puller (P‐97, Sutter Instruments, Novato, CA, USA). Whole‐cell patch‐clamp recordings from SON neurons were visually made using differential interference contrast (DIC) videomicroscopy as previously described (Fleming et al. 2011). Recordings were obtained with a Multiclamp 700A amplifier (Axon Instruments, Union City, CA, USA). The voltage output was digitized at 16‐bit resolution, 10 kHz (Digidata 1440, Axon Instruments), and saved on a computer to be analysed offline using pCLAMP10 software (Axon Instruments). Mean series resistance was 12.5 ± 0.4 MΩ and experiments were discarded in cases in which the series resistance was unstable or changed >20%. The internal solution contained (in mm): 140 potassium gluconate, 0.2 EGTA, 10 HEPES, 10 KCl, 0.9 MgCl2, 4 MgATP, 0.3 NaGTP and 20 phosphocreatine (Na+); pH was adjusted to 7.2–7.3 with 1 mm KOH and the osmolarity was 280–290 mosmol l−1. The ACSF contained (in mm): 85.09 NaCl, 30 TEA, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 20 d‐glucose, 0.4 ascorbic acid, 2.0 CaCl2 and 2.0 pyruvic acid. Tetrodotoxin (0.5 μm) was added to prevent generation of action potentials. All recordings were obtained in low Mg2+ (10 μm MgSO4) and in the presence of glycine (10 μm) in order to facilitate measurements of NMDA currents (I NMDA), as we previously reported (Fleming et al. 2011). I NMDA was activated by focal, transient application of NMDA via a picospritzer. I NMDA current densities were determined by dividing the current amplitude by the cell capacitance, obtained by integrating the area under the transient capacitive phase of a 5 mV depolarizing step pulse, in voltage clamp mode.
Confocal Ca2+ imaging
SON neurons were loaded through the patch pipette with fluo‐5F pentapotassium salt (100 μm; Thermo Fisher Scientific, Waltham, MA, USA). Once the whole‐cell configuration was established, the dye was allowed to dialyse into the cell for at least 15 min before the initiation of the recordings. Calcium imaging was conducted using the Yokogawa real time live cell laser confocal system (CSU‐10) combined with a highly sensitive EMCCD camera (iXon+ 885, Andor Technology, South Windsor, CT, USA) (Son et al. 2013). Fluorescence images were obtained using a diode‐pumped solid‐state laser (Melles Griot, Carlsbad, CA, USA), and fluorescence emission was collected at >495 nm. Images were acquired at a rate of 4 frames s−1. The fractional fluorescence (F/F 0) was determined by dividing the fluorescence intensity (F) within a region of interest (ROI; 10 × 10 pixels) by a baseline fluorescence value (F 0) determined from 30 images before brief application of NMDA (a period showing no change in intracellular calcium levels), as previously described (Son et al. 2013). The ROI was positioned within the cell soma, and care was taken to avoid the cell nucleus. Data were analysed using Andor IQ software (Andor Technology) and Image J (NIH).
Statistical analysis
All values are expressed as means ± SEM. Drug effects in sham and RVH rats were assessed using two‐way analysis of variance repeated measures (ANOVA‐RM), as indicated. Where the F ratio was significant, post hoc comparisons were completed using the Bonferroni post hoc test. When needed, and as indicated, Student's unpaired t test was used to compare basal differences between sham and RVH rats. Pearson's correlation test was used to determine if correlations existed between two parameters. Differences were considered statistically significant at P < 0.05 and n refers to the number of cells. Statistical significance was tested at the 95% (P < 0.05) confidence level. All statistical analyses were conducted using GraphPad Prism (GraphPad Software, San Diego, CA, USA).
Results
NMDA‐evoked currents do not differ between sham and RVH rats
To analyse and compare the properties of NMDA currents in MNCs from sham and RVH rats, patched MNCs were held at various membrane potentials and subjected to repetitive focal applications of NMDA (100 μm, 500 ms duration). Mean cell capacitance (sham: 38.5 ± 5.8 pF, RVH: 37.7 ± 1.0 pF) and input resistance (sham: 796.5 ± 58.3 MΩ, RVH: 868.3 ± 32.9 MΩ) did not differ between experimental groups. As shown in the representative examples of Fig. 1 A NMDA evoked fast inward currents from holding potentials of −50 mV to 0 mV, reversing to outward currents at positive membrane potentials. Figure 1 B shows plots of the mean current amplitude and density, respectively, as a function of the holding potential. Results from a two‐way ANOVA indicated an overall small though significantly diminished I NMDA peak amplitude in RVH compared to sham rats (F = 1.6, P < 0.05, n = 6 in each group). However, post hoc analysis indicated that no individual differences were observed for any of the voltages measured. When data were expressed as current density (i.e. normalized by cell capacitance), similar results were observed (F = 1.6, P < 0.05). Conversely, no differences in I NMDA area were observed between groups (F = 0.0001, P = 0.9, not shown). As we recently reported, I NMDA reversal potential was not different between MNCs in sham and RVH rats (Zhang et al. 2017).
Figure 1. NMDAR activation evokes similar currents in MNCs from sham and RVH rats.
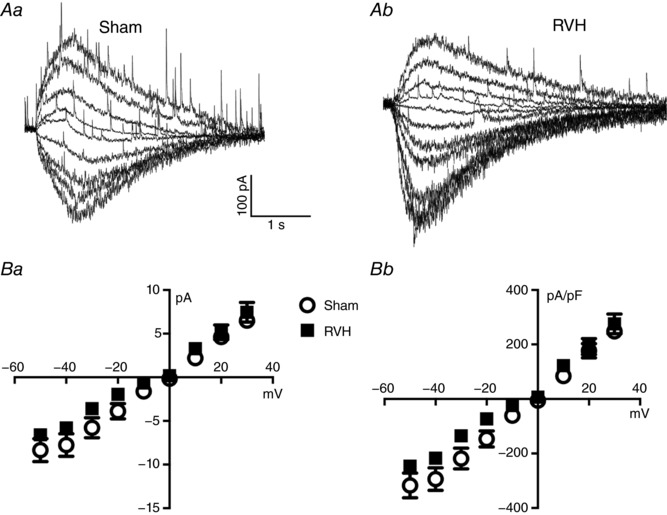
A, representative examples of evoked NMDAR‐mediated currents (I NMDA) in a MNC from a sham (Aa) and an RVH (Ab) rat, following a puff of NMDA (picospritzer, 100 μm, 4 p.s.i., 500 ms), while holding the neurons at varying holding potentials (from −50 to +30 mV in 10 mV increments). B, mean plots of I NMDA peak amplitude (Ba) and current density (Bb) as a function of the holding Vm in sham and RVH rats (n = 6 each).
The time course of NMDAR‐evoked changes in intracellular somatic Ca2+ levels is prolonged in RVH rats
In addition to evoking direct membrane depolarization, activation of NMDARs leads to an increase in intracellular levels of Ca2+. Thus, to compare the NMDAR‐evoked changes in intracellular Ca2+ (NMDAR‐ΔCa2+) between MNCs in sham and RVH rats, we obtained simultaneous patch‐clamp and confocal Ca2+ imaging recordings in MNCs loaded with fluo‐5 (Methods). In order to better detect and assess changes in intracellular Ca2+ dynamics, longer puffs of NMDA, but at a lower concentration, were used (20 μm, 5 s). As shown in Fig. 2, focal application of NMDA onto the somata of the recorded neurons evoked a robust increase in somatic [Ca2+]i whose time course surpassed the duration of the evoked I NMDA. We found in this set of studies, similar to what we reported in Fig. 1, that the peak amplitude, area and decay kinetics of the focally evoked I NMDA was not different between MNCs in sham and RVH rats (n = 24 in each group) (Fig. 3 A–C).
Figure 2. Representative examples of NMDAR‐evoked increase in intracellular Ca2+ levels in sham and RVH rats.
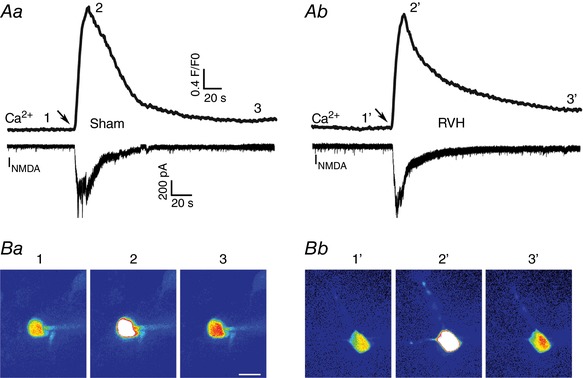
A, simultaneous I NMDA (voltage clamp, lower traces) and intracellular Ca2+ (upper traces) measurements in response to a focal brief application of NMDA (50 μm, 4 p.s.i., 5 s) in a sham (Aa) and RVH (Ab) rat. B, representative confocal Ca2+ images (pseudocolour) of the patched neurons at the time points (1–3/1′–3′) indicated in the ΔCa2+ traces are shown below. Scale bar in Ba: 15 μm. Arrows in A point to the time of the NMDA puffs. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 3. NMDAR‐evoked increase in intracellular Ca2+ is prolonged in MNCs from RVH rats.
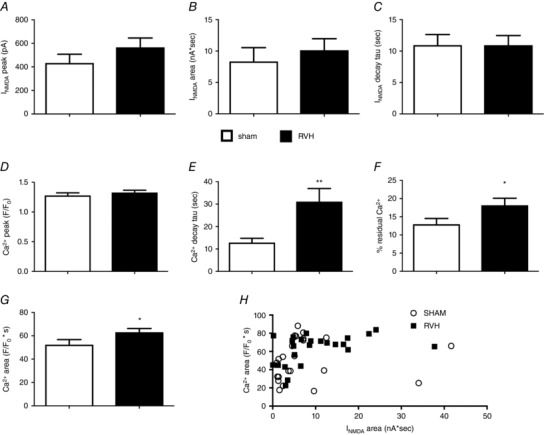
Summary of the mean I NMDA peak amplitude (A), area (B) and decay time constant (τ) (C) in MNCs from sham and RVH rats. Summary of the mean ΔCa2+ peak (D), decay time constant (τ) (E), % residual Ca2+ (F) and area (G) in MNCs from sham and RVH rats. H, plot of ΔCa2+ area as a function of I NMDA area in MNCs from sham and RVH rats. n = 24 in each group. * P < 0.05 and ** P < 0.01 vs. Sham, unpaired t test.
Measurements of the NMDAR‐ΔCa2+ signal revealed no significant differences in the [Ca2+]i signal peak amplitude between groups (Fig. 3 D). However, the decay time course of the NMDAR‐ΔCa2+ signal following the NMDA stimulus was notably slower in MNCs from RVH rats. Thus, the decay time constant (τ) in the latter was significantly longer compared to MNCs in sham rats (P < 0.01, Fig. 3 E). In many cases, as shown in the representative trace in Fig. 2 B, [Ca2+]i did not return completely to the baseline level before NMDAR stimulation, at least within the time period of the recording. Thus, when this phenomenon was quantified as the percentage NMDAR‐ΔCa2+ residual at the end of the recording, relative to the [Ca2+]i peak, a significantly larger residual Δ[Ca2+]i was observed in MNCs from RVH compared to sham rats (P < 0.02, Fig. 3 F). The slower decay time course of the NMDAR‐ΔCa2+ signal in MNCs from RVH rats resulted consequently in a significantly larger NMDAR‐ΔCa2+ signal area compared to those in sham rats (P < 0.05, Fig. 3 G). We found no significant correlations between the magnitude of I NMDA and the magnitude of the evoked [Ca2+]i (R 2 = 0.05 and 0.18 for sham and RVH, respectively (Fig. 3 H).
In a few instances, we observed a more complex response to the NMDA stimulation, consisting of an initial fast inward component, reflecting the rapid summation of excitatory postsynaptic currents (EPSCs). This response was then followed by a slower, and more persistent I NMDA component. These two phases of the I NMDA response were also clearly reflected in the evoked NMDAR‐ΔCa2+ signal, in which concomitant fast and slow components were also observed (Fig. 4). These responses were equally observed both in sham and RVH rats (n = 6/group).
Figure 4. Complex I NMDA and ΔCa2+ responses following NMDAR activation in a subset of MNCs.
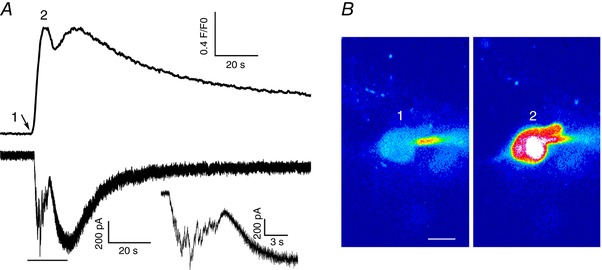
A, representative example of simultaneous I NMDA (voltage clamp, lower trace) and intracellular Ca2+ (upper trace) measurements in response to a focal brief application of NMDA, in which an initial fast inward component, likely representing summation of fast EPSPs (arrow, see also expanded inset corresponding to the underlined part of the main trace) is followed by a slower, longer lasting response. These two clearly separate components in I NMDA were also reflected as two separate peaks in the intracellular ΔCa2+ response. B, representative confocal Ca2+ images (pseudocolour) of the patched neuron at the time points (1 and 2) indicated in the ΔCa2+ traces are shown. Scale bar = 10 μm. The arrow in A points to the time of the NMDA puff. [Color figure can be viewed at wileyonlinelibrary.com]
Changes in NMDAR‐evoked increases in dendritic intracellular Ca2+ levels in RVH rats
In a limited number of cases (n = 5 and 7 in sham and RVH rats, respectively), intracellular loading with fluo5‐F was efficient enough to load and visualize neuronal dendrites, allowing us to monitor also dendritic changes in NMDAR‐ΔCa2+ signal. A representative image is shown in Fig. 5. For these recordings, we quantified changes at the most distal segment of the visible dendrite. This length ranged from 12.6 to 160.8 μm. Similar to what we observed for the soma, a significantly prolonged decay time course and a larger NMDAR‐ΔCa2+ area were observed (P < 0.02 for both τ and area) (Fig. 5 C). As shown in Fig. 5 D, no significant correlations between the dendritic length and ΔCa2+ peak (R 2 = 0.25 sham and 0.15 RVH), area (R 2 = 0.50 sham and 0.01 RVH) or decay τ (R 2 = 0.001 sham and 0.13 RVH) were observed (P > 0.2 in all cases).
Figure 5. NMDAR‐evoked increase in dendritic intracellular Ca2+ is prolonged in MNCs from RVH rats.
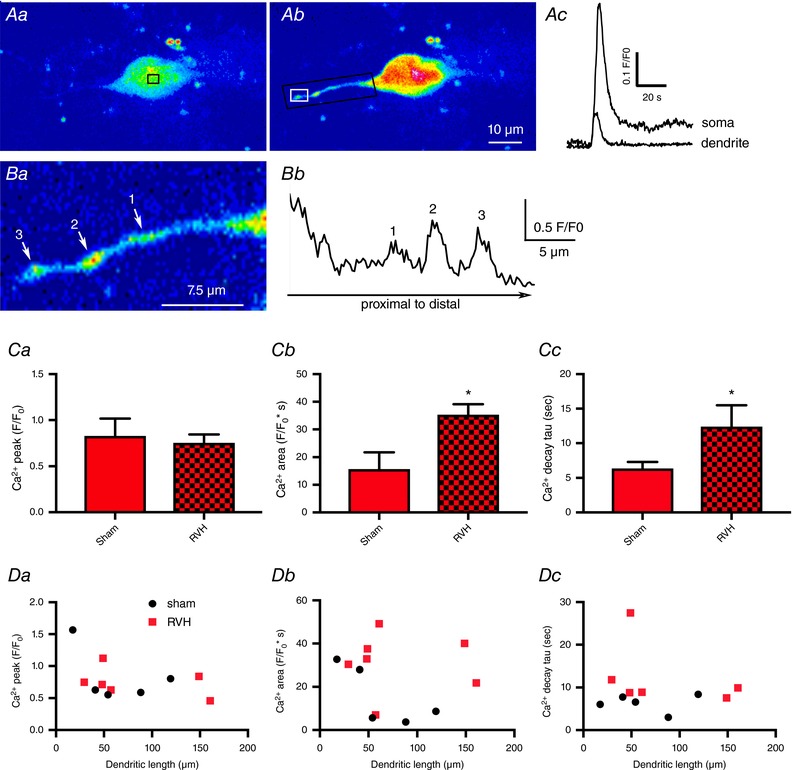
Aa and b, representative confocal Ca2+ images (pseudocolour) of a fluo‐5‐loaded MNC before (Aa) and after (Ab) a puff of NMDA (50 μm, 4 p.s.i., 5 s). Ac, somatic and dendritic ΔCa2+ measurements (F/F 0) obtained from the small squared areas indicated in Aa and Ab, respectively. Ba, an expanded dendritic segment (black rectangular area in Ab) obtained at the peak of the ΔCa2+ response is shown. Note the three dendritic varicosities (1–3), which showed the largest ΔCa2+ response, as shown quantitatively in Bb, which represents a line scan plot (F/F 0 as a function of distance) along that dendritic segment. Ca–c, summary of the mean dendritic ΔCa2+ peak (Ca), area (Cb) and decay time constant (τ) (Cc) for MNCs in sham (n = 5) and RVH (n = 7) rats. Da–c, plots of ΔCa2+ peak (Da), area (Db) and decay time constant (τ) (Dc) as a function of the dendritic length from soma. * P < 0.02 compared to Sham, unpaired t test. [Color figure can be viewed at wileyonlinelibrary.com]
Interestingly, line scans obtained from a subset of dendrites in these recordings indicated that the most predominant dendritic ΔCa2+ signals following NMDAR activation occurred within defined dendritic “hot spots”, which likely represent dendritic varicosities (see representative example in Fig. 5 B).
A blunted endoplasmic reticulum buffering capacity contributes to the prolonged NMDAR‐ΔCa2+ in MNCs from RVH rats
A major factor influencing the shape and time course of intracellular Ca2+ in MNCs is buffering by the endoplasmic reticulum (ER) via the sarco/endoplasmic reticulum Ca2+‐ATPase (SERCA) pump (Toescu & Verkhratsky, 1998). Thus to determine whether the prolonged NMDAR‐ΔCa2+ in MNCs from RVH rats was due to a diminished ER Ca2+ buffering capacity, we repeated a set of experiments in slices preincubated with thapsigargin (TG) (3 μm, 45 min), an endoplasmic reticulum SERCA pump blocker (Lytton et al. 1991) that leads to depletion of ER Ca2+ stores.
To first test the efficacy of TG in our experimental conditions, MNCs from slices incubated in the presence or absence of TG were exposed to a focal application of caffeine (50 μm, 5 s), known to mobilize Ca2+ from ER (Komori et al. 2010). As shown in Fig. 6, the magnitude of the caffeine‐evoked ΔCa2+ signal was significantly blunted in slices preincubated in TG (P < 0.0001, n = 7 and 10 in control and TG, respectively), supporting that TG significantly blocked SERCA pump activity and ER Ca2+ uptake.
Figure 6. Thapsigargin (TG) blunts caffeine‐induced increases in intracellular Ca2+ levels.
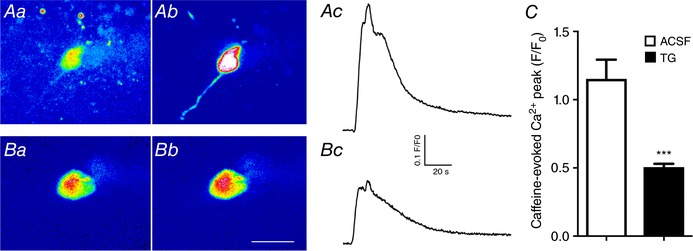
Aa and b, representative confocal Ca2+ images (pseudocolour) of a fluo‐5‐loaded MNC before (Aa) and after (Ab) a puff of caffeine (50 μm, 5 s) in control ACSF. Ac, somatic ΔCa2+ measurements (F/F 0) in response to caffeine obtained from the same MNC shown in Aa and b. Ba and b, representative confocal Ca2+ images (pseudocolour) of a different fluo‐5‐loaded MNC before (Ba) and after (Bb) a puff of caffeine (50 μm, 5 s) in a slice preincubated in TG (3 μm, 45 min). Bc, somatic ΔCa2+ measurements (F/F 0) in response to caffeine obtained from the same MNC shown in Ba and b. C, summary data of mean ΔCa2+ peak amplitude in response to caffeine in slices incubated in normal ACSF (n = 7) or in TG (n = 10). *** P < 0.0001 vs. ACSF unpaired t test. Scale bar in Bb = 20 μm. [Color figure can be viewed at wileyonlinelibrary.com]
In separate sets of studies, we then repeated focal applications of NMDA to MNCs from sham (n = 19) and RVH (n = 26) rats in slices preincubated in TG as above. Representative examples of these recordings are shown in Fig. 7 A and B. Importantly, we found that in slices preincubated in TG, the decay time course of the NMDAR‐ΔCa2+ in sham rats was slowed down, to the same level as we previously found in RVH rats. Moreover, no further increases in the decay τ were observed in RVH rats in the TG group (Fig. 7 Ca). Similar results were observed when the NMDAR‐ΔCa2+ magnitude (area) was compared among groups (Fig. 7 Cb). Thus, the prolonged and enhanced NMDAR‐ΔCa2+ observed in RVH rats in control conditions was absent in slices preincubated in TG. Unfortunately, we were unable to obtain in this set of studies sufficient MNCs with dendritic labelling, which precluded us from obtaining a similar analysis to that shown in Fig. 5.
Figure 7. Thapsigargin (TG) blunts caffeine‐induced increases in intracellular Ca2+ levels.
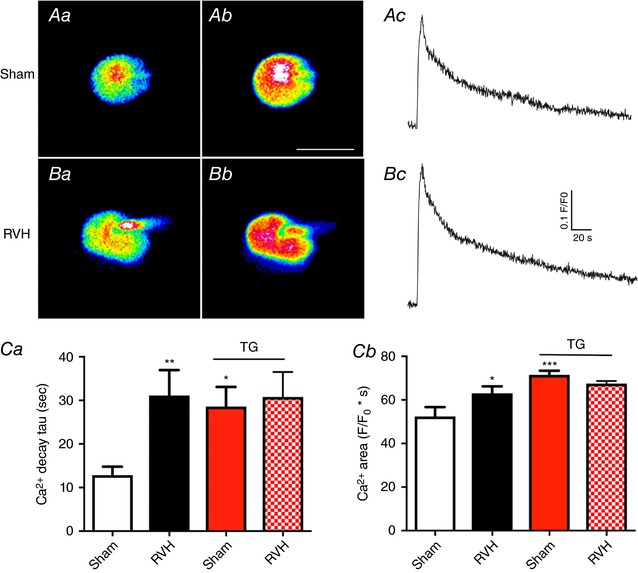
Aa and b, representative confocal Ca2+ images (pseudocolour) of a fluo‐5‐loaded MNC in a sham rat before (Aa) and after (Ab) a puff of NMDA (50 μm, 5 s) in slices preincubated in TG. Ac, somatic ΔCa2+ measurements (F/F 0) in response to NMDA obtained from the same MNC shown in Aa and b. Ba and b, representative confocal Ca2+ images (pseudocolour) of a fluo‐5‐loaded MNC in an RVH rat before (Aa) and after (Ab) a puff of NMDA (50 μm, 5 s) in slices preincubated in TG. Bc, somatic ΔCa2+ measurements (F/F 0) in response to NMDA obtained from the same MNC shown in Ba and b. Ca and b, combined summary data of mean ΔCa2+ decay time constant (τ) and area, respectively, in MNCs from sham and RVH rats in control ACSF (n = 24 and 24, respectively, for sham and RVH) and in slices preincubated in TG (n = 19 and 26, respectively, for sham and RVH, red columns). Please note that the data for control ACSF is the same as shown in Fig. 3; it was included here for better comparison with data obtained in TG. * P < 0.05, ** P < 0.01 and *** P < 0.001 vs. Sham, Bonferroni's post hoc test. Scale bar in Ab = 20 μm. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
Using a combination of patch‐clamp electrophysiology with simultaneous fast confocal Ca2+ imaging in a renovascular hypertensive rat model (RVH), we aimed in this study to determine whether NMDAR‐evoked changes in intracellular Ca2+ dynamics (NMDAR‐ΔCa2+) in MNCs was altered during hypertension, and if so, to identify potential underlying mechanisms. Our main findings indicate that (1) despite a similar magnitude of NMDA receptor‐mediated currents (I NMDA), an altered spatiotemporal profile in the ΔCa2+ response was evoked in MNCs of hypertensive rats; (2) the NMDAR‐ΔCa2+ response was significantly slower and larger over time, resulting in an overall exacerbated ΔCa2+ response per unit of I NMDA; (3) the prolonged and larger NMDA‐ΔCa2+ responses in MNCs of RVH rats affected both somatic and dendritic compartments; (4) inhibition of the ER SERCA pump activity with thapsigargin prolonged NMDAR‐ΔCa2+ responses in MNCs of sham rats, but this effect was occluded in RVH rats, equalizing the responses observed in sham and RVH rats in this condition. Taken together, these results support an exacerbated NMDAR‐ΔCa2+ signalling in MNCs of hypertensive rats, and suggest that a blunted ER Ca2+ uptake capacity is a contributing underlying mechanism to this effect.
Exacerbated NMDAR‐ΔCa2+ signalling in RVH rats: role of altered ER Ca2+ buffering capacity
Several studies from our laboratory and others support an exacerbated glutamatergic function in the SON/PVN of hypertensive rats (Li & Pan, 2007; Zheng et al. 2011; Gabor & Leenen, 2012; Glass et al. 2015). However, the precise underlying mechanisms and pathways ultimately leading to increased neuronal activity and increased neurohumoral outflow from the SON/PVN during this condition remain to be fully elucidated. Here, we focused on addressing the contribution of changes in the spatiotemporal pattern of intracellular Ca2+ dynamics, a critical process in determining the overall efficacy and downstream actions on membrane excitability that follows the activation of glutamate NMDARs.
One of the key findings in this study is that despite a similar I NMDA magnitude between MNCs from sham and RVH rats, a more prolonged and larger NMDAR‐ΔCa2+ was observed in the latter. The spatiotemporal pattern and magnitude of the changes in intracellular Ca2+ levels that follow the activation of NMDARs is influenced by a number of different factors, including the expression density of NMDA receptors, the single‐channel NMDAR Ca2+ permeability, as well as intracellular Ca2+ buffering mechanisms. Since Ca2+ influx is a major component mediating I NMDA (McBain & Mayer, 1994), and since the magnitude of I NMDA was not different between sham and RVH rats, it is reasonable to argue that an increase in NMDA receptor numbers and/or in Ca2+ permeability were not factors contributing to the larger NMDAR‐ΔCa2+ observed in MNCs from RVH rats.
Following an initial rise in intracellular Ca2+ levels, Ca2+ buffering and cytosolic clearance mechanisms gradually decrease cytosolic free Ca2+ levels, resulting in a slowly decaying Ca2+ time course, Thus, a diminished efficacy of Ca2+ buffering mechanisms is expected to mostly affect the decay phase of the Ca2+ transient. In MNCs, the endoplasmic reticulum Ca2+ transport ATPase (ER‐SERCA) has been shown to efficiently shape somatic Ca2+ transients (Kim et al. 2003; Komori et al. 2010; Dayanithi et al. 2012). Thus, to determine whether a blunted ER‐SERCA buffering mechanism contributed to the prolonged NMDA‐ΔCa2+ in RVH rats, we compared the effects of thapsigargin (TG), a SERCA blocker, on the NMDAR‐ΔCa2+ signalling both in sham and RVH rats. We found that in MNCs from sham rats, TG significantly prolonged the decay time course and increased the overall magnitude of the NMDAR‐ΔCa2+ signal to a similar extent to that observed under control conditions in MNCs of RVH rats. Conversely, TG failed to affect the NMDAR‐ΔCa2+ signal in RVH rats. Taken together, these results are in line with the notion that (1) ER‐SERCA efficiently restricts the magnitude and time course of NMDAR‐ΔCa2+ signalling in MNCs, and (2) that a blunted ER‐SERCA buffering capacity contributes to the prolonged NMDAR‐ΔCa2+ signal observed in MNCs in RVH rats. Other alternative mechanisms, however, including changes in NMDAR desensitization properties, known to affect I NMDA decay kinetic properties (Jonas & Spruston, 1994) could not be completely ruled out.
It is important to acknowledge that the ER could also act as a source of intracellular free Ca2+ levels by releasing stored Ca2+, in most instances, in a Ca2+‐dependent manner (i.e. Ca2+‐induced Ca2+ release) (Hongpaisan et al. 2001). In fact, NMDAR activation has been shown to evoke ER‐Ca2+ release in hippocampal neurons, contributing to the overall NMDAR‐ΔCa2+ waveform (Emptage et al. 1999). In our study, however, TG treatment never resulted in a reduction of the NMDAR‐ΔCa2+ signal. Taken together, our study indicates that in MNCs, the ER takes up (but does not release) Ca2+ in response to NMDAR activation.
The precise mechanism ultimately leading to altered ER Ca2+ buffering and enhanced NMDAR‐ΔCa2+ signalling in RVH rats is at present unknown. ER stress, a process that involves the accumulation of unfolded or misfolded proteins in the ER lumen, has been proposed to be a critical mechanism in the development and maintenance of neurogenic hypertension (Young et al. 2012; Chao et al. 2013). ER stress could result from various pathological processes, including angiotensin II‐mediated oxidative stress (Chao et al. 2013), or subsequent to a cytoplasmic Ca2+ overload (Richter et al. 2016). In fact, sustained NMDAR activation in hippocampal neurons was recently shown to contribute to ER stress via Ca2+ overload (Dong et al. 2017). Conversely, once established, ER stress affects ER Ca2+ homeostasis, furthering in turn cytosolic Ca2+ accumulation (Richter et al. 2016). Nevertheless, whether the enhanced tonic NMDAR activation that occurs in MNCs during hypertension (Zhang et al. 2017) is a critical factor itself contributing to ER stress and disrupted intracellular Ca2+ homeostasis in this condition, remains to be determined.
As reported, we observed in some instances a multimodal response to NMDA consisting of an initial fast inward component that reflected the rapid summation of excitatory postsynaptic currents (EPSCs) followed by the most typical slower, and more persistent I NMDA component. While this phenomenon was not further explored in this work, the initial fast component could reflect activation of presynaptic NMDARs resulting in enhanced endogenous neurotransmitter release efficacy, as reported elsewhere (Bouvier et al. 2015).
In relation to this, one limitation of our study is that a puff of NMDA, due to the relatively slow and diffuse spatiotemporal profile of agonist delivery, results most likely in the activation of extrasynaptic over synaptic NMDARs, largely because the latter are contained within a spatially restricted structure, and desensitize much more rapidly than eNMDARs, which can remain activated in a sustained manner in the presence of the agonist (Sah et al. 1989; Okamoto et al. 2009). We recently showed that eNMDARs carry the majority of the charge transfer mediated by glutamate excitatory actions in MNCs, strongly stimulating activity in these neurons (Fleming et al. 2011). Moreover, we recently showed that eNMDARs (but not synaptic NMDARs) are functionally coupled, in a Ca2+‐dependent manner, to the A‐type K+ channel‐mediated current I A (Naskar & Stern, 2014) and that an augmented eNMDAR–I A coupling is a critical mechanism contributing to exacerbated MNC activity in RVH rats (Zhang et al. 2017). Thus, the fact that we activated eNMDARs in this study is functionally relevant in the context of these previous studies. However, it remains unknown whether glutamate changes in intracellular Ca2+ following activation of synaptic NMDARs would be similarly affected in RVH rats.
Another limitation of our study is that we did not distinguish between vasopressin (VP) and oxytocin (OT) MNC phenotypes. Nevertheless, the NMDAR‐ΔCa2+ signals in sham and RVH rats were relatively consistent across all MNCs tested in each group, suggesting that both cell types were likely affected in hypertensive rats. Future studies will be needed, however, to determine if NMDAR‐ΔCa2+ signals in VP and OT MNCs are differentially affected in RVH rats.
Functional consequences of altered NMDAR‐ΔCa2+ signalling in RVH rats
Prolonged and larger NMDAR‐ΔCa2+ signalling could have numerous downstream consequences on MNC excitability in RVH rats. Critical ion channels and neurotransmitter pathways that influence MNC activity and neurosecretory outflow are modulated in a Ca2+‐dependent manner. Thus, a rise in intracellular Ca2+ levels following NMDAR activation could lead to activation of inhibitory channels, such as the small conductance Ca2+‐dependent K+ channels (SK), or to excitatory channels such as Ca2+‐activated cation (CAN) channels, including TRPM5/6 (Teruyama et al. 2011), which would act in a negative or positive feedback, respectively, to the NMDAR‐mediated excitatory effect. We recently showed that NMDARs in MNCs are functionally coupled to A‐type K+ channels (Naskar & Stern, 2014), which efficiently affect MNC firing by regulating spike onset and interspike interval during repetitive firing (Bourque, 1988; Luther & Tasker, 2000). Specifically, we showed that NMDAR activation leads to a Ca2+‐dependent inhibition of A‐type K+ channels, an effect that potentiates the excitatory effect of NMDAR activation (Naskar & Stern, 2014). Moreover, we also reported that an augmented NMDAR tone contributed to blunted A‐type K+ channel activity, and thus firing discharge in MNCs in RVH rats (Zhang et al. 2017). Thus, the results from the present work support the notion that a blunted ER Ca2+ buffering capacity could be a contributing mechanism leading to the enhanced NMDAR‐I A coupling and exacerbated firing activity of MNCs during hypertension. Clearly, future studies are warranted to further assess how the various downstream Ca2+‐dependent signalling mechanisms are affected in MNCs during hypertension, as a consequence of the enhanced NMDAR‐ΔCa2+ signalling in this condition.
We found that the increased NMDAR‐ΔCa2+ signal observed in MNCs from RVH rats was not only restricted to the soma, but also involved dendritic compartments. In MNCs, this phenomenon is expected to have important physiological consequences. VP and OT are not only released from neurohypophysial terminals into the circulation, but also intranuclearly from the dendrites of MNCs (Ludwig & Leng, 2006). Dendritic release of these neuropeptides is activity‐ and Ca2+‐dependent (Ludwig & Leng, 2006), and activation of NMDARs, via the subsequent increase in intracellular Ca2+, serves as a powerful mechanism to trigger dendritic release (de Kock et al. 2004; Son et al. 2013). Intranuclear release of VP and OT acts as a powerful autoregulatory mechanism to regulate MNC firing activity and to optimize systemic release (Ludwig & Leng, 1997; Gouzenes et al. 1998). Moreover, we recently showed that dendritic release of VP, via diffusion in the extracellular space, stimulates the activity of neighbouring presympathetic neurons, increasing sympathoexcitatory outflow from the PVN (Son et al. 2013). Thus, the enhanced NMDAR‐ΔCa2+ in RVH rats may lead to an augmented dendritic VP release, contributing in turn to enhanced neurohumoral outflow during hypertension.
In summary, results from the present study show that NMDAR‐ΔCa2+ signalling in MNCs is exacerbated in MNCs from RVH rats, and that a blunted ER Ca2+ buffering capacity contributes as an underlying mechanism. Future studies are warranted to further assess the precise mechanisms leading to altered ER function in RVH rats as well as the overall contribution of this mechanism to neurohumoral activation in neurogenic hypertension.
Additional information
Competing interests
None declared.
Author contributions
M.Z. carried out the experiments, analysed, interpreted data and contributed to manuscript writing. J.E.S. advised on the experiments and the analysis, and wrote the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship and all those who qualify for authorship are listed.
Funding
This work was supported by an NIH National Heart, Lung, and Blood Institute grant (R01 HL112225) to J.E.S.
Edited by: Harold Schultz & Julie Chan
References
- Biancardi VC, Campos RR & Stern JE (2010). Altered balance of gamma‐aminobutyric acidergic and glutamatergic afferent inputs in rostral ventrolateral medulla‐projecting neurons in the paraventricular nucleus of the hypothalamus of renovascular hypertensive rats. J Comp Neurol 518, 567–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque CW (1988). Transient calcium‐dependent potassium current in magnocellular neurosecretory cells of the rat supraoptic nucleus. J Physiol 397, 331–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier G, Bidoret C, Casado M & Paoletti P (2015). Presynaptic NMDA receptors: Roles and rules. Neuroscience 311, 322–340. [DOI] [PubMed] [Google Scholar]
- Bredt DS & Snyder SH (1989). Nitric oxide mediates glutamate‐linked enhancement of cGMP levels in the cerebellum. Proc Natl Acad Sci USA 86, 9030–9033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalis M, Dayanithi G & Nordmann JJ (1985). The role of patterned burst and interburst interval on the excitation‐coupling mechanism in the isolated rat neural lobe. J Physiol 369, 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao YM, Lai MD & Chan JY (2013). Redox‐sensitive endoplasmic reticulum stress and autophagy at rostral ventrolateral medulla contribute to hypertension in spontaneously hypertensive rats. Hypertension 61, 1270–1280. [DOI] [PubMed] [Google Scholar]
- Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, Simon AB & Rector T (1984). Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med 311, 819–823. [DOI] [PubMed] [Google Scholar]
- Colbran RJ & Brown AM (2004). Calcium/calmodulin‐dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol 14, 318–327. [DOI] [PubMed] [Google Scholar]
- Dayanithi G, Forostyak O, Ueta Y, Verkhratsky A & Toescu EC (2012). Segregation of calcium signalling mechanisms in magnocellular neurones and terminals. Cell Calcium 51, 293–299. [DOI] [PubMed] [Google Scholar]
- de Kock CP, Burnashev N, Lodder JC, Mansvelder HD & Brussaard AB (2004). NMDA receptors induce somatodendritic secretion in hypothalamic neurones of lactating female rats. J Physiol 561, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Kalueff AV & Song C (2017). N‐methyl‐d‐aspartate receptor‐mediated calcium overload and endoplasmic reticulum stress are involved in interleukin‐1beta‐induced neuronal apoptosis in rat hippocampus. J Neuroimmunol 307, 7–13. [DOI] [PubMed] [Google Scholar]
- Emptage N, Bliss TV & Fine A (1999). Single synaptic events evoke NMDA receptor‐mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron 22, 115–124. [DOI] [PubMed] [Google Scholar]
- Fleming TM, Scott V, Joe N, Naskar K, Brown CH & Stern JE (2011). State‐dependent changes in astrocyte regulation of extrasynaptic NMDA receptor signalling in neurosecretory neurons. J Physiol 589, 3929–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabor A & Leenen FH (2012). Central neuromodulatory pathways regulating sympathetic activity in hypertension. J Appl Physiol (1985) 113, 1294–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass MJ, Wang G, Coleman CG, Chan J, Ogorodnik E, Van Kempen TA, Milner TA, Butler SD, Young CN, Davisson RL, Iadecola C & Pickel VM (2015). NMDA receptor plasticity in the hypothalamic paraventricular nucleus contributes to the elevated blood pressure produced by angiotensin II. J Neurosci 35, 9558–9567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouzenes L, Desarmenien MG, Hussy N, Richard P & Moos FC (1998). Vasopressin regularizes the phasic firing pattern of rat hypothalamic magnocellular vasopressin neurons. J Neurosci 18, 1879–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongpaisan J, Pivovarova NB, Colegrove SL, Leapman RD, Friel DD & Andrews SB (2001). Multiple modes of calcium‐induced calcium release in sympathetic neurons II: a [Ca2+]i‐ and location‐dependent transition from endoplasmic reticulum Ca accumulation to net Ca release. J Gen Physiol 118, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B & Bourque CW (1992). NMDA receptor‐mediated rhythmic bursting activity in rat supraoptic nucleus neurones in vitro . J Physiol 458, 667–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P & Spruston N (1994). Mechanisms shaping glutamate‐mediated excitatory postsynaptic currents in the CNS. Curr Opin Neurobiol 4, 366–372. [DOI] [PubMed] [Google Scholar]
- Kim MH, Lee SH, Park KH & Ho WK (2003). Distribution of K+‐dependent Na+/Ca2+ exchangers in the rat supraoptic magnocellular neuron is polarized to axon terminals. J Neurosci 23, 11673–11680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori Y, Tanaka M, Kuba M, Ishii M, Abe M, Kitamura N, Verkhratsky A, Shibuya I & Dayanithi G (2010). Ca2+ homeostasis, Ca2+ signalling and somatodendritic vasopressin release in adult rat supraoptic nucleus neurones. Cell Calcium 48, 324–332. [DOI] [PubMed] [Google Scholar]
- Li DP & Pan HL (2007). Glutamatergic inputs in the hypothalamic paraventricular nucleus maintain sympathetic vasomotor tone in hypertension. Hypertension 49, 916–925. [DOI] [PubMed] [Google Scholar]
- Li DP, Zhu LH, Pachuau J, Lee HA & Pan HL (2014). mGluR5 upregulation increases excitability of hypothalamic presympathetic neurons through NMDA receptor trafficking in spontaneously hypertensive rats. J Neurosci 34, 4309–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlejohn NK, Siel RB Jr, Ketsawatsomkron P, Pelham CJ, Pearson NA, Hilzendeger AM, Buehrer BA, Weidemann BJ, Li H, Davis DR, Thompson AP, Liu X, Cassell MD, Sigmund CD & Grobe JL (2013). Hypertension in mice with transgenic activation of the brain renin‐angiotensin system is vasopressin dependent. Am J Physiol Regul Integr Comp Physiol 304, R818–R828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig M & Leng G (1997). Autoinhibition of supraoptic nucleus vasopressin neurons in vivo: a combined retrodialysis/electrophysiological study in rats. Eur J Neurosci 9, 2532–2540. [DOI] [PubMed] [Google Scholar]
- Ludwig M & Leng G (2006). Dendritic peptide release and peptide‐dependent behaviours. Nat Rev Neurosci 7, 126–136. [DOI] [PubMed] [Google Scholar]
- Luther JA & Tasker JG (2000). Voltage‐gated currents distinguish parvocellular from magnocellular neurones in the rat hypothalamic paraventricular nucleus. J Physiol 523, 193–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytton J, Westlin M & Hanley MR (1991). Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca‐ATPase family of calcium pumps. J Biol Chem 266, 17067–17071. [PubMed] [Google Scholar]
- McBain CJ & Mayer ML (1994). N‐Methyl‐d‐aspartic acid receptor structure and function. Physiol Rev 74, 723–760. [DOI] [PubMed] [Google Scholar]
- Naskar K & Stern JE (2014). A functional coupling between extrasynaptic NMDA receptors and A‐type K+ channels under astrocyte control regulates hypothalamic neurosecretory neuronal activity. J Physiol 592, 2813–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen R, Hu B & Renaud LP (1995). Regulation of spontaneous phasic firing of rat supraoptic vasopressin neurones in vivo by glutamate receptors. J Physiol 484, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto S, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK, Zhang D, Vincent Chen HS, Tong G, Hayden MR & Lipton SA (2009). Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med 15, 1407–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer M (1988). Neurohormonal interactions and adaptations in congestive heart failure. Circulation 77, 721–730. [DOI] [PubMed] [Google Scholar]
- Packer M, Lee WH, Kessler PD, Gottlieb SS, Bernstein JL & Kukin ML (1987). Role of neurohormonal mechanisms in determining survival in patients with severe chronic heart failure. Circulation 75, IV80–92. [PubMed] [Google Scholar]
- Petersen OH (2002). Cation channels: homing in on the elusive CAN channels. Curr Biol 12, R520–522. [DOI] [PubMed] [Google Scholar]
- Richter M, Vidovic N, Honrath B, Mahavadi P, Dodel R, Dolga AM & Culmsee C (2016). Activation of SK2 channels preserves ER Ca2+ homeostasis and protects against ER stress‐induced cell death. Cell Death Differ 23, 814–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegger GA, Liebau G, Bauer E & Kochsiek K (1985). Vasopressin and renin in high output heart failure of rats: hemodynamic effects of elevated plasma hormone levels. J Cardiovasc Pharmacol 7, 1–5. [DOI] [PubMed] [Google Scholar]
- Sah P & Faber ES (2002). Channels underlying neuronal calcium‐activated potassium currents. Prog Neurobiol 66, 345–353. [DOI] [PubMed] [Google Scholar]
- Sah P, Hestrin S & Nicoll R (1989). Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science 246, 815–818. [DOI] [PubMed] [Google Scholar]
- Silverman AJ & Zimmerman EA (1983). Magnocellular neurosecretory system. Annu Rev Neurosci 6, 357–380. [DOI] [PubMed] [Google Scholar]
- Son SJ, Filosa JA, Potapenko ES, Biancardi VC, Zheng H, Patel KP, Tobin VA, Ludwig M & Stern JE (2013). Dendritic peptide release mediates interpopulation crosstalk between neurosecretory and preautonomic networks. Neuron 78, 1036–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teruyama R, Sakuraba M, Kurotaki H & Armstrong WE (2011). Transient receptor potential channel M4 and M5 in magnocellular cells in rat supraoptic and paraventricular nuclei. J Neuroendocrinol 23, 1204–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toescu EC & Verkhratsky A (1998). Principles of Calcium Signalling. Plenum Press, New York. [Google Scholar]
- van den Pol A, Wuarin J & Dudek F (1990). Glutamate, the dominant excitatory transmitter in neuroendocrine regulation. Science 250, 1276–1278. [DOI] [PubMed] [Google Scholar]
- Yemane H, Busauskas M, Burris SK & Knuepfer MM (2010). Neurohumoral mechanisms in deoxycorticosterone acetate (DOCA)‐salt hypertension in rats. Exp Physiol 95, 51–55. [DOI] [PubMed] [Google Scholar]
- Young CN, Cao X, Guruju MR, Pierce JP, Morgan DA, Wang G, Iadecola C, Mark AL & Davisson RL (2012). ER stress in the brain subfornical organ mediates angiotensin‐dependent hypertension. J Clin Invest 122, 3960–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Biancardi VC & Stern JE (2017). An increased extrasynaptic NMDA tone inhibits A‐type K+ current and increases excitability of hypothalamic neurosecretory neurons in hypertensive rats. J Physiol 595, 4647–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Liu X, Li Y, Sharma NM & Patel KP (2011). Gene transfer of neuronal nitric oxide synthase to the paraventricular nucleus reduces the enhanced glutamatergic tone in rats with chronic heart failure. Hypertension 58, 966–973. [DOI] [PMC free article] [PubMed] [Google Scholar]