

Abstract
Key points
Endothelial cell function in resistance arteries integrates Ca2+ signalling with hyperpolarization to promote relaxation of smooth muscle cells and increase tissue blood flow. Whether complementary signalling occurs in lymphatic endothelium is unknown.
Intracellular calcium and membrane potential were evaluated in endothelial cell tubes freshly isolated from mouse collecting lymphatic vessels of the popliteal fossa. Resting membrane potential measured using intracellular microelectrodes averaged ∼−70 mV.
Stimulation of lymphatic endothelium by acetylcholine or a TRPV4 channel agonist increased intracellular Ca2+ with robust depolarization. Findings from Trpv4 −/− mice and with computational modelling suggest that the initial mobilization of intracellular Ca2+ leads to influx of Ca2+ and Na+ through TRPV4 channels to evoke depolarization.
Lymphatic endothelial cells lack the Ca2+‐activated K+ channels present in arterial endothelium to generate endothelium‐derived hyperpolarization. Absence of this signalling pathway with effective depolarization may promote rapid conduction of contraction along lymphatic muscle during lymph propulsion.
Abstract
Subsequent to a rise in intracellular Ca2+ ([Ca2+]i), hyperpolarization of the endothelium coordinates vascular smooth muscle relaxation along resistance arteries during blood flow control. In the lymphatic vasculature, collecting vessels generate rapid contractions coordinated along lymphangions to propel lymph, but the underlying signalling pathways are unknown. We tested the hypothesis that lymphatic endothelial cells (LECs) exhibit Ca2+ and electrical signalling properties that facilitate lymph propulsion. To study electrical and intracellular Ca2+ signalling dynamics in lymphatic endothelium, we excised collecting lymphatic vessels from the popliteal fossa of mice and removed their muscle cells to isolate intact LEC tubes (LECTs). Intracellular recording revealed a resting membrane potential of ∼−70 mV. Acetylcholine (ACh) increased [Ca2+]i with a time course similar to that observed in endothelium of resistance arteries (i.e. rapid initial peak with a sustained ‘plateau’). In striking contrast to the endothelium‐derived hyperpolarization (EDH) characteristic of arteries, LECs depolarized (>15 mV) to either ACh or TRPV4 channel activation. This depolarization was facilitated by the absence of Ca2+‐activated K+ (KCa) channels as confirmed with PCR, persisted in the absence of extracellular Ca2+, was abolished by LaCl3 and was attenuated ∼70% in LECTs from Trpv4 −/− mice. Computational modelling of ion fluxes in LECs indicated that omitting K+ channels supports our experimental results. These findings reveal novel signalling events in LECs, which are devoid of the KCa activity abundant in arterial endothelium. Absence of EDH with effective depolarization of LECs may promote the rapid conduction of contraction waves along lymphatic muscle during lymph propulsion.
Keywords: endothelium‐derived hyperpolarizing factor, TRP channels, calcium‐activated K+ channel, mathematical model
Key points
Endothelial cell function in resistance arteries integrates Ca2+ signalling with hyperpolarization to promote relaxation of smooth muscle cells and increase tissue blood flow. Whether complementary signalling occurs in lymphatic endothelium is unknown.
Intracellular calcium and membrane potential were evaluated in endothelial cell tubes freshly isolated from mouse collecting lymphatic vessels of the popliteal fossa. Resting membrane potential measured using intracellular microelectrodes averaged ∼−70 mV.
Stimulation of lymphatic endothelium by acetylcholine or a TRPV4 channel agonist increased intracellular Ca2+ with robust depolarization. Findings from Trpv4 −/− mice and with computational modelling suggest that the initial mobilization of intracellular Ca2+ leads to influx of Ca2+ and Na+ through TRPV4 channels to evoke depolarization.
Lymphatic endothelial cells lack the Ca2+‐activated K+ channels present in arterial endothelium to generate endothelium‐derived hyperpolarization. Absence of this signalling pathway with effective depolarization may promote rapid conduction of contraction along lymphatic muscle during lymph propulsion.
Abbreviations
- ACh
acetylcholine
- AECT
arterial endothelial cell tube (popliteal)
- [Ca2+]i
intracellular Ca2+ concentration
- [Ca2+]o
extracellular Ca2+ concentration
- [Ca2+]IS
Ca2+ concentration in IP3‐sensitive store
- CaCC
Ca2+‐activated Cl− channel
- BKCa
large‐conductance Ca2+‐activated K+ channel
- EC
endothelial cell
- EDH
endothelial derived hyperpolarization
- eNOS
endothelial nitric oxide synthase
- ER
endoplasmic reticulum
- GSK101
GSK1016790A, TRPV4 channel activator
- KATP
ATP‐sensitive K+
- KCa
Ca2+‐activated K+
- KIR
inward‐rectifying K+
- [K+]o
extracellular K+ concentration
- Ing‐Ax
inguinal efferent‐axillary afferent
- IP3
inositol 1,4,5‐trisphosphate
- IP3R
inositol trisphosphate receptor
- LEC
lymphatic endothelial cell
- LECT
lymphatic endothelial cell tube (popliteal)
- LMC
lymphatic muscle cell
- NaK
Na+–K+‐ATPase
- NKCC
Na+–K+–Cl− cotransporter
- NO
nitric oxide
- PMCA
plasma membrane Ca2+‐ATPase
- PSS
physiological salt solution
- SKCa/IKCa
small‐ and intermediate‐conductance Ca2+‐activated K+ channels
- SERCA
sarco(endo)plasmic reticulum Ca2+‐ATPase
- SMC
smooth muscle cell (vascular)
- TRP
transient receptor potential
- TRPV4
transient receptor potential vanilloid type 4
- Trpv4−/−
Trpv4 knockout
- Vm
membrane potential
- VRAC
volume‐regulated anion channel
- WT
wild‐type
Introduction
Endothelium‐derived hyperpolarization (EDH) is integral to the control of vascular resistance, tissue blood flow and oxygen delivery (Busse et al. 2002; Garland & Dora, 2017). In contrast to endothelial cell (EC)‐dependent signalling mediated through the release and diffusion of autacoids (e.g. nitric oxide (NO) and prostaglandins), the EDH pathway serves as a direct electrical signal that spreads through gap junctions from EC to EC and from ECs to smooth muscle cells (SMCs) to promote vasodilatation (Bagher & Segal, 2011). The longitudinal spread of hyperpolarization conforms to passive cable properties demonstrated in both intact resistance arteries containing SMCs (Emerson et al. 2002) and their freshly isolated endothelial cell tubes (Behringer & Segal, 2012). EDH entails an increase in cytosolic Ca2+ concentration ([Ca2+]i) that activates small‐ and intermediate‐conductance Ca2+‐activated K+ channels (SKCa/IKCa) in the plasma membrane (Félétou, 2016; Garland & Dora, 2017). As such, the initiation of EDH is a classic example of the intimate interaction between Ca2+ homeostasis and alterations in V m (Busse et al. 1988; Behringer & Segal, 2015) and this relationship is integral to blood flow regulation throughout the body (Félétou, 2016).
Collecting lymphatic vessels perform multiple distinct physiological roles (Aspelund et al. 2016), including the rapid coordination of contraction and relaxation driven by lymphatic muscle cells (LMCs) to pump fluid centrally from peripheral tissues (von der Weid et al. 1996; Scallan & Davis, 2013; Scallan et al. 2013). Whereas lymphatic endothelial cell function has focused largely on the NO pathway (Chakraborty et al. 2015), the potential for EDH signalling in isolated endothelium of collecting lymphatics is unknown. Resolving the determinants of electrical signalling in lymphatic vessels is integral to understanding how they maintain the ability to contract rapidly to move lymph against a pressure gradient. Thus, if the EDH pathway is activated in lymphatic vessels, relaxation of LMCs may interfere with effective pumping. Indeed, lymphatic vessels isolated from mice that lack endothelial nitric oxide synthase (i.e. eNOS−/−) do not dilate in response to the muscarinic receptor type 3 (M3) agonist acetylcholine (ACh). Instead, they display enhanced constriction and increased contraction frequency (Scallan & Davis, 2013). The absence of residual dilatation to ACh in lymphatics from eNOS−/− mice suggests that LECs lack EDH signalling characteristic of blood vessels in response to SKCa/IKCa activation. If lymphatic endothelium indeed lacks signalling via the EDH pathway, it would be markedly different from endothelium studied in the context of blood flow control.
In the present study, we tested the hypothesis that LECs exhibit Ca2+ and electrical signalling properties that permit lymph propulsion. To investigate simultaneous [Ca2+]i and electrical responses of native intact lymphatic ECs in the absence of LMC influences, LEC tubes (LECTs) were isolated from popliteal collecting lymphatic vessels of C57BL/6 mice (1–2 months old) using mild enzymatic digestion and gentle trituration. Arterial endothelial cell tubes (AECTs) isolated from popliteal arteries were studied under identical conditions as a positive control for EDH signalling in blood vessels. Sharp microelectrodes and Fura‐2 photometry were used to measure membrane potential (V m) and [Ca2+]i, respectively, in response to ACh, which involves the internal release of Ca2+ followed by Ca2+ influx, or in response to the transient receptor potential vanilloid type 4 (TRPV4) agonist GSK1016790A (GSK101), which mediates Ca2+ influx. We found that despite the similar time course of intracellular [Ca2+]i dynamics in response to ACh, LECTs depolarize instead of hyperpolarizing in the manner of AECTs, indicating the lack of an EDH pathway. Using intact pressurized lymphatic vessels, we verified that lymphatic endothelium depolarized to ACh. This finding was supported by RT‐PCR demonstrating the absence of mRNA for Kcnn3 and Kcnn4 (which encode SKCa and IKCa proteins, respectively) in LECTs, which contrasts with their known expression in AECTs. Despite the absence of Ca2+‐activated K+ (KCa) channels and their association with TRPV4 channels in blood vessels (Sonkusare et al. 2012, 2014), LECTs of Trpv4 −/− mice exhibited significantly reduced responses to ACh with both [Ca2+]i (∼50%) and membrane depolarization (∼70%) versus C57BL/6 controls. To gain further insight into which ion channel(s) enable depolarization in response to the rise in [Ca2+]i, a computational model of lymphatic endothelium was applied.
Methods
Animal care and use
All animal care and experimental procedures were approved by the Animal Care and Use Committee of the University of Missouri and performed in accord with the National Research Council's Guide for the Care and Use of Laboratory Animals (8th edn, 2011). Mice were housed in an enriched environment maintained on a 12:12 h light–dark cycle at ∼23°C with fresh tap water and standard chow available ad libitum. Experiments were performed on male C57BL/6 mice (1–2 months old; n = 33) purchased from Charles River Laboratories (Wilmington, MA, USA). In complementary experiments, male Trpv4 −/− mice (C57BL/6 background, n = 4) bred at the University of Missouri (breeders obtained from GlaxoSmithKline (King of Prussia, PA, USA)) were used to investigate the role of TRPV4 (Thorneloe et al. 2008) in mediating [Ca2+]i influx. Each mouse was anaesthetized using pentobarbital sodium (60 mg kg−1, i.p. injection). Upon completion of tissue removal, the anaesthetized mouse was killed by exsanguination.
Solutions
Popliteal collecting lymphatic vessels were dissected while immersed in a Krebs buffer containing (in mm): 146.9 NaCl, 4.7 KCl, 2 CaCl2·2H2O, 1.2 MgSO4, 1.2 NaH2PO4·H2O, 1.2 NaHCO3, 1.5 Hepes, 5 d‐glucose. Control physiological salt solution (PSS) was used for the dissociation procedure and all electrical and Ca2+ recordings, and contained the following (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 Hepes, 10 d‐glucose. The pH of all solutions was adjusted to 7.4 before use. During dissociation of lymphatic or arteriolar muscle cells to obtain endothelial cell tubes, PSS contained 0.62 mg ml−1 papain (≥6 units), 1.5 mg ml−1 collagenase (≥15 units), 1.0 mg ml−1 dithioerythritol, 0.1% bovine serum albumin (USB Corp., Cleveland, OH, USA) and 0.1 mm CaCl2. For experiments with nominally zero [Ca2+]o, CaCl2 was isosmotically replaced with NaCl. Reagents were purchased from Sigma‐Aldrich (St Louis, MO, USA) unless indicated otherwise.
Surgery and microdissection
Once a mouse was anaesthetized, the fur covering each hindlimb was removed by shaving. With the mouse in the prone position, a ∼1 cm L‐shaped incision was made from the ankle towards the knee to expose the saphenous vein. While viewing through a stereomicroscope, connective tissue surrounding the saphenous vein was carefully cut to expose a superficial popliteal lymphatic vessel intimately associated with the vein. The collecting lymphatic vessel and associated adipose tissue were then carefully removed from the vein using microdissection and placed in Krebs buffer. Two popliteal collecting lymphatic vessels were removed per mouse (one per leg). In separate experiments, the popliteal artery was removed from each leg in similar fashion. Vessels were then pinned to the bottom of a chamber coated with transparent Sylgard® rubber (Dow Corning, Midland, MI, USA) before adipose and connective tissue were removed using microdissection, then vessels were transferred to a glass tube containing the dissociation buffer and enzymes. We found it unnecessary to clean lymphatic vessels completely with microdissection as performed previously (Scallan & Davis, 2013) since enzymatic digestion facilitated the removal of connective tissue.
For endothelial V m measurements of isolated and pressurized lymphatic vessels, inguinal efferent‐axillary afferent (Ing‐Ax) internodal collecting vessels were used due to their amenability for removal of connective tissue, which facilitated access of the microelectrode to the endothelial layer while limiting trauma to smooth muscle and ECs. In brief, a midline dorsal incision and retraction of the skin fold revealed the large Ing‐Ax paralleling the superficial epigastric vein. The Ing‐Ax lymphatic vessel and surrounding connective tissue was removed by dissection and further cleaned in a Sylgard‐coated dish in Krebs buffer supplemented with 0.5% BSA.
Endothelial cell tube isolation and superfusion
Popliteal AECTs and LECTs were prepared as recently described (Socha & Segal, 2013). Briefly, vessel segments were then rinsed with PSS lacking enzymes and transferred to a tissue chamber (RC‐27N, Warner Instruments, Hamden, CT, USA) containing PSS at room temperature. To dissociate muscle cells from the endothelial layer, a vessel segment was incubated with an enzyme cocktail (see ‘Solutions’) at 34°C for either 30 min (arteries) or 20 min (lymphatic vessels). The vessel segment was then gently triturated using aspiration and ejection from a micropipette during visual inspection at ×200. Dissociation pipettes were prepared from borosilicate glass capillary tubes (1.0 mm outer diameter (o.d.)/ 0.58 mm internal diameter (i.d.); World Precision Instruments (WPI), Sarasota, FL, USA) pulled (P‐97; Sutter Instruments, Novato, CA, USA) and heat‐polished at one end (tip i.d.: ∼50–70 μm). The tissue chamber containing the freshly isolated EC tube (width: ∼40 μm, length: 0.3–1 mm) was secured to an aluminium platform (width: 14.5 cm, length: 24 cm, thickness: 0.4 cm). A micromanipulator (DT3‐100, Siskiyou Corp., Grants Pass, OR, USA) mounted at each end of the platform held a blunt‐ended, heat‐polished micropipette (o.d., 60–100 μm) used to position and secure the EC tube against the bottom (coverslip) of the tissue chamber. The aluminium platform was mounted on an inverted microscope (Eclipse TS100, Nikon) located on a vibration‐isolated table (Technical Manufacturing Corp., Peabody, MA, USA) and superfused with control PSS at ∼4 ml min−1 along the axis of the EC tube. Throughout the experiments, temperature was maintained at 28 °C using an in‐line heater (SH‐27B, Warner) and heating platform (PH6, Warner) coupled to a temperature controller (TC‐344B, Warner). Preparations were stable for the duration of experiments (1–2 h) under these conditions.
Ca2+ photometry
Ca2+ photometry was performed using an IonOptix system (Milford, MA, USA) as described (Socha et al. 2011; Behringer & Segal, 2015). Briefly, prior to loading Fura‐2 dye, the preparation was maintained at room temperature for 10 min while autofluorescence was recorded at 510 nm during alternate excitation at 340 and 380 nm (10 Hz). Fura‐2 AM dye (5 μm; F14185, Thermo Fisher Scientific, Waltham, MA, USA) was loaded for 20 min followed by 20 min washout to allow time for intracellular de‐esterification and removal of excess dye. Temperature was raised to 28°C during the final 10 min. Autofluorescence during excitation at 340 and 380 nm (average values over 30 s acquisition) was subtracted from the respective recordings at 510 nm. Using a ×40 objective (Nikon S Fluor; numerical aperture, 0.90), the imaging window was 140 μm (length) by ∼40 μm (width) and contained ∼40–50 ECs (Behringer & Segal, 2015).
Intracellular recording of V m
Endothelial cell tubes
Microelectrodes were pulled (P‐97; Sutter) from glass capillary tubes (GC100F‐10, Warner) and backfilled with 2 m KCl (tip resistance, ∼150 MΩ). Membrane potential was measured using Axoclamp 2B (Molecular Devices, Sunnyvale, CA, USA) and Warner IE‐210 electrometers as described (Behringer & Segal, 2012; Behringer et al. 2012). An Ag/AgCl pellet placed in effluent PSS served as a reference electrode. Amplifier outputs were connected to an analog‐to‐digital converter (Digidata 1322A, Molecular Devices) and data were recorded at 1000 Hz on a Dell personal computer using Axoscope 10.1 software (Molecular Devices). Individual cells were impaled along the midline of the EC tube while viewing at ×400 magnification. For lymphatic endothelium, successful impalements were indicated by a sharp negative deflection of V m and typically required ∼5 min for stabilization to resting V m (approx. range: −50 to −80 mV) before applying pharmacological agents. For AECTs, V m typically stabilized within 1 min (resting V m range: −25 to −40 mV) following impalement (Behringer & Segal, 2012, 2015 Behringer et al. 2012). For simultaneous Ca2+ photometry and electrophysiology, the photometric window for acquiring Ca2+ measurements was positioned adjacent to the electrode recording V m (Behringer & Segal, 2015).
Intact Ing‐Ax lymphatic vessels
The vessel was cannulated on glass pipettes (∼80 μm) in an observation chamber with a Burg‐style V‐track mounting system and transferred to an inverted microscope (Zeiss, model IM35). Both pipettes were attached to a single pressure reservoir with an output line that was split with a T‐connector. Pressure was set to 8 cmH2O and the vessel was lengthened to remove axial slack. Afterward, pressure was set to 3 cmH2O for the duration of the experiment. A peristaltic pump constantly exchanged the Krebs buffer in the observation chamber with fresh buffer at 0.5 ml min−1. A heat exchanger connected to the water‐jacketed observation chamber warmed the vessel to 37°C over 30–60 min. Spontaneous contractions typically began within 10 min and stabilized by 30 min. Wortmannin (2 μm) was then added to minimize, but not completely abolish, contraction amplitude to prevent dislodging the recording electrode. Membrane potential in the endothelium was recorded using intracellular microelectrodes (250–300 MΩ) filled with 1 m KCl and advanced through the smooth muscle layer. Impalements of LECs were confirmed by the lack of action potentials (an indicator of the electrical activity of adjacent smooth muscle cells) and resting V m ∼ −70 mV (Von der Weid & Van Helden, 1997). Membrane potential was sampled at 1 kHz using a NPI SEC‐05X amplifier (ALA instruments, Farmingdale, NY, USA) and acquired on a personal computer using a custom (Davis et al. 2012) LabVIEW program (National Instruments, Austin, TX, USA). Once impaled, membrane potential was allowed to stabilize over a period of a 2 min before ACh was applied. Upon completion of recording, the microelectrode was retracted and V m values were adjusted, if appropriate, based on the offset potential (typically ≤ 5 mV).
Pharmacology
Acetylcholine (ACh; 1 μm) was used to activate Gq protein‐coupled signalling through muscarinic receptors, which, in arteriolar ECs, activates Ca2+ release through IP3 receptors and Ca2+ influx via transient receptor potential (TRP) channels (Busse et al. 2002). The TRPV4 agonist GSK1016790A (GSK101; 10–100 nm) was used to stimulate Ca2+ influx and thereby elevate [Ca2+]i. GSK101 was applied only once to each experimental preparation. Complementary experiments were performed using LECTs isolated from Trpv4 −/− mice in which the gene encoding TRPV4 was deleted (Thorneloe et al. 2008).
Other pharmacological agents included LaCl3 (100 μm; non‐selective cation channel blocker), BaCl2 (100 μm; KIR channel blocker), NS309 (10 μm; SKCa/IKCa activator; Tocris Bioscience, Bristol, UK), apamin (300 nm; SKCa blocker; Anaspec, Fremont, CA, USA) + charbydotoxin (100 nm; IKCa and BKCa blocker; Anaspec), paxilline (1 μm; BKCa blocker; Tocris), levcromakalim (10 μm; KATP channel opener; Tocris) and glyburide (10 μm; KATP channel blocker; Tocris). A maximal concentration of 0.1% DMSO was applied to any given preparation and vehicle controls were performed to confirm that [Ca2+]i and V m were unaffected by this concentration of DMSO.
Endpoint RT‐PCR
After validating the primers (Table 1), EC tubes were isolated from an artery and from a lymphatic vessel of the popliteal fossa as described above. RNA expression for the Kcnn3 and Kcnn4 genes was then determined using RT‐PCR, and the message levels were compared against those found in respective intact vessels. Briefly, total RNA was extracted from tissues by using the Arcturus PicoPure RNA Isolation Kit (Thermo Fisher Scientific, Waltham, MA, USA) with on‐column DNase I treatment (Qiagen, Valencia, CA, USA) according to the manufacturer's protocol. Due to the low volume of tissue in a single EC tube, RNA was eluted with 24 μl of nuclease‐free water. Purified RNA (8 μl) was then used for cDNA synthesis via reverse transcription using random hexamer and oligo (dT) primers, and SuperScript III First‐Strand Synthesis System (Thermo Fisher Scientific). PCR was performed in a reaction mixture containing first‐strand cDNA as the template (2 μl), 2 mm MgCl2, 0.25 μm primers, 0.2 mm deoxynucleotide triphosphates and GoTaq Flexi DNA polymerase (Promega, Madison, WI, USA). The PCR programme comprised an initial denaturation step at 95°C for 4 min, followed by 35 cycles of denaturation for 30 s at 95°C, annealing 30 s at 56°C and elongation for 30 s at 72°C, and a final extension step at 72°C for 5 min. PCR amplification products were separated on a 2% agarose gel by electrophoresis, stained with SYBR‐Safe (Thermo Fisher Scientific) and visualized by UV transillumination. Forward and reverse primers used for this study and the expected size of amplicons are listed in Table 1.
Table 1.
Primer sets used for RT‐PCR
| Protein | Gene | Strand | Sequence | Accession no. | Amplicon |
|---|---|---|---|---|---|
| SK3 | Kcnn3 | S | GAC CGA ACT GTC TTG GGG TT | NM_080466 | 294 |
| AS | GCC CGA GAT GGG GTA TAG GA | ||||
| IK | Kcnn4 | S | GAC ATC AGC GCA AGA TGC TG | NM_008433 | 290 |
| AS | CTT CTG TGA GTT CAT GTG GAG C | ||||
| α‐Actin | Acta2 | S | GTG AAG AGG AAG ACA GCA CAG | NM_007392 | 146 |
| AS | GCC CAT TCC AAC CAT TAC TCC | ||||
| VE‐cadherin | Cdh5 | S | CTT CCT TAC TGC CCT CAT TGT | NM_009868 | 313 |
| AS | CTG TTT CTC TCG GTC CAA GTT |
Mathematical model: Ca2+ and electrical signalling of lymphatic endothelium
The mathematical model of LECT Ca2+ signalling and electrophysiology was adapted from a model developed for mesenteric vascular endothelial cells (Silva et al. 2007). The plasma membrane is modelled based on the Hodgkin–Huxley model, which has been used for a variety of cell types (e.g. neurons and cardiomyocytes) (Lindblad et al. 1996; Yang et al. 2003; Koenigsberger et al. 2005; Silva et al. 2007). In addition to Ca2+, the dynamics of all major ions (Na+, K+ and Cl−) are included (see Fig. 7) to provide a comprehensive analysis of ion fluxes via ion channels, transporters, pumps and exchangers that underlie regulation of V m at rest and during the time course of a stimulus (e.g. ACh). A detailed discussion of how the mathematical modelling was performed now follows, with a list of the parameters used in Table 2.
Figure 7. Schematic diagram of ion channels and pumps in computational model of lymphatic endothelium.

A variety of channels and pumps are present in LECs; see ‘Methods’ for detailed description. K+ currents are in green, Na+ currents are in red, Cl− currents are in black and Ca2+ currents are in blue. ACh stimulation leads to IP3 formation and activation of the IP3R on the endoplasmic reticulum. Calcium ions in the cytosol and endoplasmic reticulum are buffered with calmodulin (CaM) and calreticulin (CRT).
Table 2.
Model parameters for lymphatic endothelium
| Parameter | Description | Initial estimate | Model value* | Reference | |
|---|---|---|---|---|---|
| C m | Plasma membrane capacitance | 14 pF | Silva et al. (2007) | ||
| A m | Plasma membrane surface area | 584 μm2 | Voets et al. (1999) | ||
| k B | Boltzmann constant | 1.38 × 10−23 m2 kg s−2 K−1 | — | ||
| voli | Intracellular volume | 1.173 pl | Silva et al. (2007) | ||
| volCa | Cytosolic volume available to Ca2+ | 0.912 pl | Silva et al. (2007) | ||
| volIS | Endoplasmic reticulum volume | 0.335 pl | Silva et al. (2007) | ||
| R | Gas constant | 8.314 J mol K−1 | — | ||
| T | Temperature | 303 K | Experiments | ||
| F | Faraday constant | 96485.33 C mol−1 | — | ||
| [K+]o | Extracellular K+ concentration | 4.7 mm | Experiments | ||
| [Na+]o | Extracellular Na+ concentration | 151.1 mm | Experiments | ||
| [Ca2+]o | Extracellular Ca2+ concentration | 2 mm | Experiments | ||
| [Cl−]o | Extracellular Cl− concentration | 155.6 mm | Experiments | ||
|
|
Maximum IP3R current | 4.67 × 106 pA mm −1 | 3.16 × 106 pA mm −1 | Silva et al. (2007) | |
|
|
[IP3] for half‐maximal IP3R current | 1.6 μm | Silva et al. (2007) | ||
|
|
[Ca2+]i for half‐maximal Ca2+‐dependent inhibition of IP3R current | 1.0 μm | Silva et al. (2007) | ||
|
|
Maximum SERCA current | 0.88 pA | Silva et al. (2007) | ||
| K SERCA, IS | [Ca2+]i for quarter‐maximal SERCA current | 0.15 μm | Silva et al. (2007) | ||
| k leak, IS | ER leak constant for Ca2+ | 0.0176 pA mm −2 | 3.52 pA mm −2 | Silva et al. (2007) | |
|
|
Maximum NaK current | 22.6 pA | Silva et al. (2007) | ||
|
|
[K+]o for half‐maximal NaK activation | 1.32 mm | Silva et al. (2007) | ||
|
|
[Na+]i for half‐maximal NaK activation | 14.5 mm | Silva et al. (2007) | ||
|
|
Maximum PMCA current | 2.67 pA | 4.30 pA | Silva et al. (2007) | |
|
|
[Ca2+]i for half‐maximal PMCA activation | 260 nm | Silva et al. (2007) | ||
| n PMCA | Hill coefficient for [Ca2+]i dependency of PMCA current | 1.4, 1.7, 4.4 | 3.0 | Silva et al. (2007), Wiesner et al. (1996) | |
| P TRPV4, Na, max | Maximum permeability of TRPV4 to Na+ | 3.95 × 10−7 cm s−1 | 2.08 × 10−7 cm s−1 | Silva et al. (2007) | |
| P ratio, TRPV4 | Permeability ratio of Ca2+ to Na+ for TRPV4 (P Ca/P Na) | 6 | Gees et al. (2010) | ||
| K TRPV4 | ER depletion for half‐maximal TRPV4 activation | 0.25 mm | Experiments | ||
| n TRPV4 | Hill coefficient for [Ca2+]IS dependency of TRPV4 current | 3 | Experiments | ||
| P Orai, max | Maximum permeability of Orai to Ca2+ | 1.15 × 10−7 cm s−1 | 1.25 × 10−6 cm s−1 | Silva et al. (2007) | |
| K Orai | ER depletion for half‐maximal Orai activation | 0.25 mm | Experiments | ||
| n Orai | Hill coefficient for [Ca2+]IS dependency of Orai current | 3 | Experiments | ||
| P TRP | Permeability of TRP to Na+ and Ca2+ | (1.3–5.3) × 10−8 cm s−1 | 2.01 × 10−8 cm s−1 | Experiments | |
| P Kleak | Permeability of leak channels to K+ | 1.3 × 10−7 cm s−1 | 1.26 × 10−7 cm−1 | Experiments | |
| G Kir | Kir conductance constant | 0.142 nS mm −0.5 | 0.051 nS mm −0.5 | Silva et al. (2007) | |
| n Kir | Kir power constant | 0.5 | Silva et al. (2007) | ||
|
|
K+ driving force at half Kir channel conductance | 39.4 mV | Silva et al. (2007) | ||
| νKir | Slope factor of Kir conductance | 7.08 mV | Silva et al. (2007) | ||
| G VRAC | VRAC conductance | 0.381 nS | 0.0381 nS | Silva et al. (2007) | |
| G CaCC | Maximum CaCC conductance | 37.4 nS | 3.74 nS | Silva et al. (2007) | |
| K CaCC | [Ca2+]i at which CaCC Ca2+‐dependent open probability is 50% | 287 nm | Silva et al. (2007) | ||
| n CaCC | Hill coefficient of CaCC Ca2+‐dependent open probability function | 1.89 | Silva et al. (2007) | ||
| V CaCC, h | Membrane potential for half CaCC conductance | 662 mV | Silva et al. (2007) | ||
| νCaCC | Slope factor of CaCC conductance | 132 mV | Silva et al. (2007) | ||
| k B, on | Forward binding rate for Ca2+–calmodulin binding | 100 mm −1•ms−1 | Silva et al. (2007) | ||
| k B, off | Backward binding rate for Ca2+–calmodulin binding | 0.3 ms−1 | Silva et al. (2007) | ||
| B T | Total concentration of Ca2+ binding sites in the cytosol | 0.12 mm | Silva et al. (2007) | ||
|
|
Total concentration of Ca2+ binding sites in the ER stores | 8.8 mm | Prins & Michalak (2011) | ||
| K CSQN | Dissociation constant for CSQN | 2 mm | Prins & Michalak (2011) | ||
|
|
Degradation rate for IP3 | 2 × 10−3 ms−1 | Silva et al. (2007) | ||
|
|
Agonist‐mediated steady state IP3 generation rate | 5.5 × 10−8 mm ms−1 | Silva et al. (2007) | ||
|
|
Time constant for IP3 generation rate change | 33 s | Silva et al. (2007) |
*If the parameter value used in the model is different from the initial estimates from references. CSQN, calsequestrin.
The LECT plasma membrane is assumed to act as a capacitor, and the ion channels and pumps are regarded as parallel paths (Diep et al. 2005). The membrane potential (V m) is calculated using Kirchhoff's law by:
| (1) |
where C m is membrane capacitance and the currents in the parentheses are trans‐plasmalemmal ionic currents. The Nernst Potential (E) of each individual ion was calculated using:
| (2) |
where Y indicates K+, Na+, Ca2+ or Cl− and z Y is +1, +1, +2 and −1 for K+, Na+, Ca2+ and Cl−, respectively. [Y]i indicates intracellular and [Y]o extracellular concentrations. A lumped approach was used to model the reactions and transports inside the LECT. The intracellular space was divided into two compartments: the cytosol and the endoplasmic reticulum (ER). Each ion or protein was assumed to be homogenously distributed in the cytosolic space, ER compartment, or both. K+, Na+ and Cl− can all diffuse through the intracellular space while the ER membrane is assumed to be impermeable to Ca2+, IP3, calmodulin and other proteins such that their flux is defined through membrane channels and transporters while IP3–IP3 receptor (IP3R) interactions were assumed to be the main pathway for Ca2+ release from the ER. Although ryanodine–ryanodine receptor interactions have been shown to release Ca2+ from ER stores, particularly in excitable cells (Fill & Copello, 2002), they were excluded from the present model because of the lack of supporting data from lymphatic endothelium. Thus, the ER stores denote IP3‐sensitive Ca2+ stores, which were modelled using the same equations as Silva and colleagues (2007).
IP3R current:
| (3) |
| (4) |
Sarco(endo)plasmic reticulum Ca2+‐ATPase (SERCA) current:
| (5) |
ER leak current:
| (6) |
Mathematical model: transmembrane currents of lymphatic endothelium
Ions can pass through the plasma membrane either through specific channels or via pumps driven by consumption of ATP. Among the ion pumps, we included, Na+–K+‐ATPase (I NaK) and Ca2+‐ATPase (I PMCA), and removed the Na+/Ca2+ exchanger (I NCX) and the Na+–K+–Cl− cotransporter (I NKCC) due to lack of evidence for their presence in the LECTs.
Na+–K+‐ATPase current:
| (7) |
Ca2+‐ATPase current:
| (8) |
The most important difference between blood ECs and lymphatic ECs is our evidence for the lack of Ca2+‐activated K+ channel currents (I SK and I IK) in LECTs, which led to removal of I SK and I IK from the model. Additionally, we have replaced the non‐selective cation channel (I NSC) and store‐operated cation channel (I SOC) in the model of Silva and colleagues (2007) with the Ca2+ release‐activated Ca2+ channel (I Orai; Ruhle & Trebak, 2013), the transient receptor potential vanilloid 4 channel (I TRPV4), a composite term for the remaining transient receptor potential channels (I TRP; Hill‐Eubanks et al. 2014) and the K+ leak channel (I Kleak). In part, these modifications were made to implement recent data on the molecular mechanisms underlying store‐operated and non‐selective cation currents. Herein, we explain those currents that are new while adapting equations developed previously (Silva et al. 2007; Hald et al. 2015).
Results from electrophysiological experiments in the current study suggest that in addition to the sustained increase in [Ca2+]i, the LECT membrane most likely depolarizes due to a subset of TRP channels (including TRPV4) being activated through ACh or a downstream signalling event. The secondary sustained increase in Ca2+ can be explained by stromal interaction molecule 1 (STIM1)‐mediated Orai current. Importantly, two recent studies provide strong evidence for the expression of Orai1 in murine lymphatic endothelium (Choi et al. 2017a,b). However, the level of depolarization we have observed requires involvement of TRP channels. The V m of LECs (∼−70 mV) is closer to the equilibrium potential of K+ than that of Cl− channels. P TRPV4,Na,max was chosen to fit the depolarization of V m relative to our experimental data. Based on the evidence on cooperation of TRP channels in store‐operated current, Orai channel permeability (P Orai,max) was selected to have similar currents to TRPV4 during ER depletion (Liao et al. 2008; Ma et al. 2011). TRP channels have a wide range of P Ca/P Na from <1 to >100 (Clapham et al. 2005; Gees et al. 2010). We have assumed P Ca/P Na of 1 for the basal TRP channels and P Ca/PNa of 6 for TRPV4 (P ratio,TRPV4) (Gees et al. 2010). The Hill coefficient and half‐maximal activation concentration of ER depletion for both TRPV4 and Orai was selected to capture the dynamics of the second phase of Ca2+ entry. Adjustment of the K+ leak channel permeability (P Kleak) ensured that our model matches the consistent observation of ∼11 mV depolarization after I Kir blockade by BaCl2 (see Fig. 10 H). Finally, the effect of GSK101 for TRPV4 activation was modelled by gradually opening the TRPV4 channels until they reached maximum permeability.
Figure 10. Computational modelling: dynamics in ionic fluxes and open probability of key channels, exchangers, and pumps.

A, V m under baseline conditions (time < 0 s) and during ACh (starting at time 0 s). B, as in A for the equilibrium potential for Ca2+ (E Ca), equilibrium potential for Na+ (E Na), equilibrium potential for K+ (E K) and equilibrium potential for Cl− (E Cl). C, for [K+], [Na+], [Cl−] and [Ca2+]IS. D, for [Ca2+]i and [IP3]i. E, for I IP3R, I SERCA and I leak. F, I PMCA, I TRP,Ca, I TRPV4,Ca and I Orai. G, for 3 × I NaK, I TRP,Na, I TRPV4,Na. H, for −2 × I NaK (negative sign is to account for inward K+ current by NaK), I Kir and I leak,K. I, for I CaCC and I VRAC. In C, [Na+] is plotted multiplied by a factor of 10, and [Ca2+]IS by a factor of 100 for visualization. Note that negative currents are inward and positive currents are outward. V m is shown in cyan and [IP3] is in magenta while concentration and currents for K+ are in green, Na+ are in red, Cl− are in black and Ca2+ are in blue. For direct comparison, this figure contains similar types of data to Fig. 7 from Silva et al. (2007), which shows the model for mesenteric artery endothelium in response to ACh.
Data analysis
Analyses included: (1) fluorescence emission collected at 510 nm and expressed as the ratio during excitation at 340 and 380 nm (F 340/F 380); (2) change in F 340/F 380 ratio (∆F 340/F 380) = peak response F 340/F 380 − preceding baseline F 340/F 380; (3) resting V m (mV); (4) change in V m (ΔV m) = peak response V m − preceding baseline V m. All summary data reflect values averaged over 10 s during stable recordings. Statistical analyses (GraphPad Prism 5; GraphPad Software, Inc., La Jolla, CA, USA) included Student's paired and unpaired t test. Differences were accepted as statistically significant with P < 0.05. Summary data are presented as means ± SEM unless specified otherwise. One EC tube (either arteriolar or lymphatic) or intact lymphatic vessel was studied from each mouse for a given experimental protocol with ‘n’ reflecting the number of independent EC tubes or lymphatic vessels.
Results
The present experiments were designed to determine whether the requisite machinery of the EDH pathway (e.g. SKCa/IKCa activation) was present for transducing [Ca2+]i increases into hyperpolarization in native intact lymphatic endothelium. Initial experiments addressed whether pharmacological agonists and antagonists of SKCa/IKCa as well as other K+ channels associated with blood vessel endothelium (e.g. inward‐rectifying K+ (KIR) channels, ATP‐sensitive K+ (KATP) channels; Jackson, 2005) influenced the resting V m of lymphatic endothelium. Initial findings prompted ensuing analyses of the dynamics of both [Ca2+]i and V m in response to the well‐defined M3 receptor agonist ACh (intracellular ER Ca2+ release followed by Ca2+ influx; Himmel et al. 1993) and the TRPV4 agonist GSK101 (Ca2+ influx; Thorneloe et al. 2008). These functional data were complemented by computational modelling to resolve the roles of TRP channels and ions (e.g. Na+, K+, Cl−) underlying the dynamics of Ca2+ and electrical signalling in lymphatic endothelium.
KIR channels contribute to resting V m of lymphatic endothelium but KCa and KATP channels do not
Application of agents known to activate KCa channels and thereby evoke robust hyperpolarization in arterial endothelium caused a pronounced depolarization in lymphatic endothelium. As summarized in Fig. 1 A, ACh (1 μm) depolarized lymphatic ECs from a resting V m of −60 ± 7 mV to −33 ± 4 mV (P < 0.05), while the TRPV4 agonist GSK101 (100 nm) depolarized lymphatic ECs from −59 ± 3 to −13 ± 3 mV (P < 0.05). The KIR channel blocker BaCl2 (100 μm) depolarized V m from −60 ± 5 to −49 ± 5 mV (P < 0.05). In contrast, neither the SKCa/IKCa opener NS309 (1 μm) nor inhibition with the combination of apamin (300 nm) + charybdotoxin (100 nm) altered resting V m significantly (ΔV m, 1–3 mV). The large‐conductance KCa channel (BKCa) blocker paxilline (1 μm), the KATP channel opener levcromakalim (10 μm) and the KATP channel inhibitor glyburide (10 μm) were also without significant effect on resting V m (ΔV m, 1–2 mV). Collectively, these findings suggested that KCa channels were not expressed by lymphatic endothelium. We therefore performed endpoint RT‐PCR to determine whether the genes encoding SKCa/IKCa channels (i.e. Kcnn3 and Kcnn4, respectively) were transcribed. As shown in Fig. 1 B, whole popliteal arteries and whole lymphatic vessels expressed mRNA for Kcnn3 and Kcnn4. In contrast, the isolated lymphatic endothelium was devoid of detectable message for Kcnn3 or Kcnn4, while the isolated arterial endothelium showed abundant expression for both of these genes.
Figure 1. Lack of KCa channel activity governing V m in lymphatic endothelium and absence of SKCa and IKCa transcript expression.

A, respective bars indicate changes in resting V m in response to acetylcholine (ACh; 1 μm), TRPV4 activation (GSK101; 100 nm), inward rectifying K+ channel blockade (BaCl2; 100 μm), SKCa/IKCa activation (NS309; 1 μm), SKCa/IKCa blockade (apamin (Ap; 300 nm) + charybdotoxin (ChTx; 100 nm)), large conductance Ca2+‐activated K+ (BKCa) channel block (paxilline (Pax); 1 μm), ATP‐sensitive K+ (KATP) channel activation (levcromakalim (Levcro); 10 μm) and KATP channel blockade (glyburide (Gly; 10 μm)). Depolarization (P < 0.05) occurred in response to ACh, GSK and BaCl2 for ‘(n)’ experiments. Resting V m of LECTs varied from ∼−50 to −80 mV, and therefore respective treatment effects are reported as changes (ΔV m) from resting V m preceding treatment. B, endpoint PCR images indicating mRNA expression of SKCa (Kcnn3), IKCa (Kcnn4), smooth muscle α‐actin (Acta2) and vascular endothelial cadherin (Cdh5) for individual popliteal artery, collecting lymphatic vessel, arterial endothelial cell tube (AECT) and lymphatic endothelial cell tube (LECT). The intact artery and intact lymphatic vessel both expressed mRNA for SKCa and IKCa. In contrast, the LECT did not express mRNA for either SKCa or IKCa, while the AECT expressed abundant mRNA for both of these genes. Brain and thoracic aorta were used as positive controls for KCa channels and cell type‐specific markers (i.e. smooth muscle α‐actin and endothelial cadherin), respectively. For negative controls (Control), no template was loaded.
Lack of EDH in lymphatic endothelium: depolarization occurs with increased [Ca2+]i in response to ACh
We next evaluated [Ca2+]i dynamics simultaneously with V m responses induced by ACh in LECTs while confirming EDH signalling in the endothelium of resistance arteries isolated from the same anatomical site (Fig. 2 A and B). Figure 2 C illustrates a classic arterial endothelial response of [Ca2+]i dynamics to ACh, whereby an initial rapid rise in F 340/F 380 (due to intracellular release from the ER) is followed by a ‘plateau’ phase attributable to Ca2+ influx through the plasma membrane (Cohen & Jackson, 2005; Socha et al. 2012; Behringer & Segal, 2015). Furthermore, arterial endothelium rapidly hyperpolarized from −31 ± 5 to −69 ± 4 mV during peak [Ca2+]i (Fig. 2 E and H, n = 4), with changes in V m mirroring those of [Ca2+]i in both magnitude and time course (Fig. 2 C and E). In contrast, whereas the dynamics of [Ca2+]i responses to ACh in LECTs were similar to that in AECTs (Fig. 2 D and G), V m in LECTs underwent a gradual depolarization from −66 ± 3 to −45 ± 3 mV in response to ACh exposure (Fig. 2 F and H). Thus, during exposure to ACh, hyperpolarization occurs simultaneously with an increase in [Ca2+]i in arterial endothelium, whereas a slow depolarization occurs in lymphatic endothelium.
Figure 2. Lack of EDH in lymphatic endothelium.

A, brightfield image of an AECT isolated from a popliteal artery. B, as in A for a LECT isolated from a popliteal lymphatic vessel; note the valve (arrow) in B that is absent in A. Scale bars (white) in A and B indicate 50 μm. C, recording of Fura‐2 fluorescence response to ACh for an AECT. D, as in C for a LECT. In both traces, note rapid increase in the F 340/F 380 ratio (‘peak’; ≤20 s of ACh onset) that was sustained (i.e. ‘plateaued’) for the duration of ACh exposure. E, simultaneous recording of V m during ACh from an AECT. F, as in E from a LECT. Note that the change V m in E is hyperpolarization that mirrors dynamics in [Ca2+]i in C. In contrast a gradual depolarization occurs in in F that begins approximately during the [Ca2+]i peak phase in D. G, summary of changes in F 340/F 380 values for AECTs and LECTs in response to ACh during the peak (≤20 s of ACh onset) and plateau (90 s following peak) phase of responses. H, as in G for corresponding changes in V m. Data represent n = 4 AECTs and n = 13 LECTs. * P < 0.05, responses different from artery.
To address whether depolarization in response to ACh occurred in LECs of intact vessels, the endothelial V m in isolated and pressurized Ing‐Ax lymphatic vessels was measured. From a resting V m of −64 ± 3 mV (n = 3), LECs depolarized by 51 ± 2 mV to −13 ± 2 mV in response to ACh (1 μm; Fig. 3 A and B). These data illustrate that LEC V m depolarization to ACh occurs irrespective of surrounding smooth muscle cells that exhibit spontaneous action potentials (Von der Weid et al. 2014).
Figure 3. Acetylcholine depolarizes endothelium of intact lymphatic vessels.

A, recording of V m in the endothelial layer of a pressurized (3 cmH2O) Ing‐Ax lymphatic collecting vessel at rest and during ACh (1 μm). Note ∼50 mV depolarization to ACh treatment. B, summary of lymphatic endothelium resting V m in pressurized Ing‐Ax vessels and in response to ACh. * P < 0.05, response different from control. Data represent n = 3 Ing‐Ax lymphatic vessels, each from a separate C57BL/6 mouse.
Depolarization of lymphatic endothelium during ACh stimulation requires Ca2+ influx
As observed for arterial endothelium, removal of 2 mm [Ca2+]o (Cohen & Jackson, 2005; Socha et al. 2011; Behringer & Segal, 2015) or treatment with the non‐selective cation channel blocker LaCl3 (Cohen & Jackson, 2005) eliminated the plateau phase of the [Ca2+]i response in lymphatic endothelium during exposure to ACh, while the initial peak [Ca2+]i response was preserved (Fig. 4 A, B, E (left panel) and F (left panel); compare to Fig. 2 D and G). In reference to Figure 2 D and F, depolarization to ACh still occurred along with the peak phase of the [Ca2+]i response and persisted despite the absence of the plateau phase; however, removal of extracellular Ca2+ reduced depolarization by ∼50% (Fig. 4 C and E (right panel)). When cation influx was blocked with LaCl3, depolarization was eliminated (Fig. 4 D and F (right panel)). These findings suggest that progressive depolarization to ACh in LECs depends upon a sustained increase in [Ca2+]i. The residual depolarization during the absence of [Ca2+]o may be due to Na+ influx that can be abolished by blocking TRP channels permeable to both Ca2+ and Na+ (e.g. TRPV4) by LaCl3. This hypothesis was tested using the TRPV4 opener GSK101 in LECTs from wild‐type (WT) mice and with LECTs isolated from Trpv4 −/− mice.
Figure 4. Role of [Ca2+]o during depolarization of V m to ACh in lymphatic endothelium.
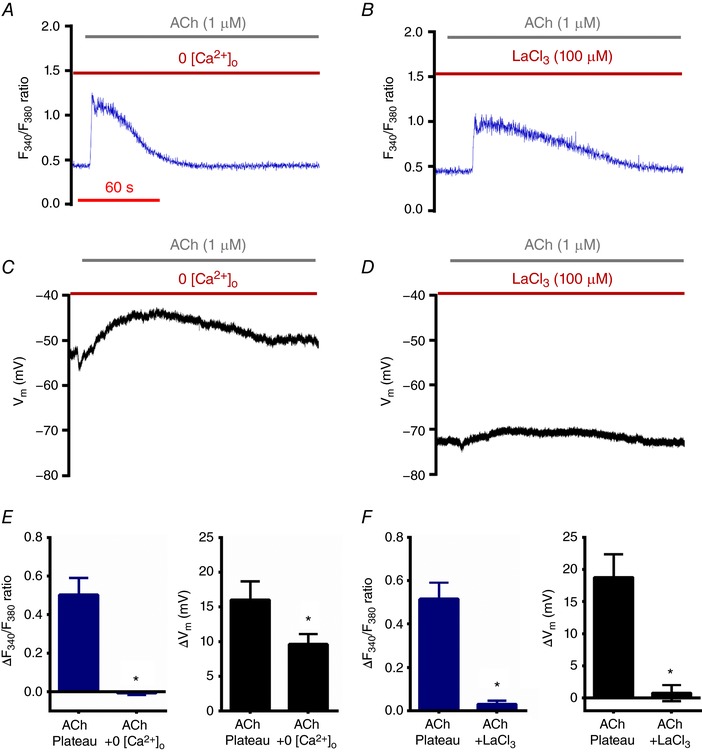
A, recording of Fura‐2 fluorescence during ACh in the absence of [Ca2+]o in a LECT. B, as in A, in the presence of 2 mm [Ca2+]o and during treatment with LaCl3, a broad‐spectrum blocker of Ca2+‐permeable channels in the plasma membrane. Note that the plateau phase is abolished in both A and B (compare to Fig. 2 D). C, as in A, with recording of V m. D, as in B, with recording of V m. Note that depolarization is impaired in C and D (compare to Fig. 2 F). E, summary of changes in F 340/F 380 ratio (left) and V m (right) during the plateau phase (90 s) of the response to ACh in the presence and absence of 2 mm [Ca2+]o. F, as in E, indicating treatment with LaCl3 (+2 mm [Ca2+]o). Values were obtained at the same time points in paired experiments (ACh versus ACh + LaCl3) corresponding to the abolished F 340/F 380 plateau (∆F 340/F 380 ≈ 0 recorded, ∼ 2.5 min after ACh onset) via treatment with LaCl3. Data represent n = 5 paired experiments with ACh ± 2 mm [Ca2+]o and n = 4 paired experiments with ACh ± LaCl3. * P < 0.05, responses different from ACh (with 2 mm [Ca2+]o) alone.
Depolarization of lymphatic endothelium occurs via TRPV4 channels
TRPV4 channels are integral to agonist‐mediated Ca2+ influx across plasma membranes of arterial endothelium (Zhang et al. 2009; Sonkusare et al. 2012; Qian et al. 2014). However, these channels are also permeable to Na+ and are only moderately more selective for Ca2+ over Na+ (P Ca/P Na ≈ 6) compared to other Ca2+‐permeable cation channels such as TRPV5 and TRPV6 (P Ca/P Na > 100) (Clapham et al. 2005). Membrane permeability to Na+ is functionally important given its integral role during excitation and depolarization of mammalian cells (Hille, 2001). In response to opening TRPV4 channels with GSK101, wild‐type LECTs responded with a robust increase in [Ca2+]i (Fig. 5 A and G) coincident with depolarization (Fig. 5 D and H). A key finding from these experiments is that depolarization to GSK101 was sustained (Fig. 5 E) in the absence of extracellular Ca2+ or of a rise in [Ca2+]i (Fig. 5 B) but was eliminated in the presence of LaCl3 (Fig. 5 F).
Figure 5. Opening of TRPV4 channels leads to depolarization of lymphatic endothelium.

A, recording of Fura‐2 fluorescence during GSK101 (with 2 mm [Ca2+]o) in lymphatic endothelium. B, as in A, with 0 [Ca2+]o. C, as in A, with LaCl3. Note that increases in F 340/F 380 are abolished in B and C. D, as in A, with recording of V m. E, as in B, with recording of V m. F, as in C, with recording of V m. Note that the depolarization in E (GSK101 + 0 [Ca2+]o) is absent in F (GSK101 + LaCl3). G, summary of changes in F 340/F 380 ratio at 3 min following onset of GSK101 with 2 mm [Ca2+]o present, GSK101 + 0 [Ca2+]o and with GSK101 + LaCl3. H, as in G, for changes in V m. * P < 0.05, responses different from GSK101 with 2 mm [Ca2+]o. Data represent n = 5 experiments for GSK101 with 2 mm [Ca2+]o), n = 4 for GSK101 with 0 [Ca2+]o and n = 4 for GSK101 with LaCl3.
In LECTs isolated from Trpv4−/− mice, both the rise in [Ca2+]i and the depolarization to ACh (∆F 340/F 380: 0.35 ± 0.12, ∆V m: 6 ± 2 mV, n = 4) were attenuated significantly (P < 0.05) compared to wild‐type C57BL/6 mice (∆F 340/F 380: 0.72 ± 0.07, ∆V m: 17 ± 2 mV, n = 13) (Fig. 6 A, C and E vs. Fig. 2 D, F, G and H). Further, [Ca2+]i and depolarization responses to GSK were absent in LECTs from Trpv4 −/− mice (Fig. 6 B, D and F; ∆F 340/F 380: −0.01 ± 0.02, ∆V m: −2 ± 3 mV; n = 5 LECTs isolated from 4 mice).
Figure 6. Lack of TRPV4 channels reduces [Ca2+]i and eliminates depolarization to ACh and GSK101 in lymphatic endothelium.

A, recording of Fura‐2 fluorescence during ACh with 2 mm [Ca2+]o in a LECT isolated from a Trpv4 −/− mouse. B, as in A, with GSK101. C, as in a A, with recording of V m. D, as in B, with recording of V m. Note that the plateau in F 340/F 380 and depolarization are diminished during ACh treatment (A and C) and are abolished during GSK101 treatment (B and D) when compared to LECTs from C57BL/6 (WT) mice. D, as in B, with recording of V m. E, summary of changes in F 340/F 380 values (left) and V m (right) during the ACh plateau phase (90 s) in LECTs isolated from WT (n = 13) vs. Trpv4 −/− mice (n = 4). F, as in E, indicating data at 3 min following onset of GSK101 treatment (WT and Trpv4 −/−; n = 5 each). For E, WT data during ACh treatment were taken from Fig. 2 G (∆[Ca2+]i) and Fig. 2 H (∆V m). For F, WT data during GSK101 treatment were taken from Fig. 5 G (∆[Ca2+]i) and Fig. 5 H (∆V m). * P < 0.05, responses different between LECTs isolated from WT versus Trpv4 −/− mice.
These findings collectively illustrate that TRPV4 channels play an integral role in lymphatic endothelium signalling, with both Ca2+ and Na+ influx contributing to membrane depolarization in response to ACh. We used computational modelling to resolve the contributions of Ca2+ and Na+ with respect to other ions (e.g. K+, Cl−) and membrane channels (e.g. Orai, volume‐regulated anion Cl− channels) in lymphatic endothelium.
Mathematical modelling: ionic fluxes and electrical dynamics in lymphatic endothelium
Computational modelling for ion channels in LECs was adapted from models published for ion channels in blood vessel endothelial cells (Fig. 7) (Wiesner et al. 1996; Silva et al. 2007; Gees et al. 2010; Prins & Michalak, 2011).
ACh treatment
Figure 8 A illustrates a two‐phase response (i.e. ‘peak’ and ‘plateau’) similar to what we resolved experimentally for lymphatic endothelium (Fig. 2 D), whereby depolarization occurs in parallel (compare Figs 2 F and 8 D). When extracellular Ca2+ is removed, the plateau is abolished (compare Figs 4 A and 8 B) but there is a discrepancy in how depolarization is affected (compare Figs 4 C and 8 E), attributable to the manner by which TRPV4 and Orai activation is modelled. When [Ca2+]o falls, ER calcium is released leading to opening of TRPV4 and the storage‐operated Ca2+ channel, Orai (Ruhle & Trebak, 2013). TRPV4 channels then allow sodium entry, resulting in the depolarization in the model (Fig. 8 E). Computational blocking of TRPV4 and Orai mimics treatment with the non‐selective cation blocker LaCl3, whereby depolarization is effectively abolished (Fig. 8 F) in the presence of a significantly reduced [Ca2+]i plateau during stimulation with ACh (compare Figs 4 B and 8 C). Similar to data obtained from Trpv4 −/− mice (see Fig. 6 A, C and E), the model also shows that the absence of TRPV4 alone is sufficient to abolish the depolarization (Fig. 8 F) while both TRPV4 and Orai currents need to be eliminated to remove the Ca2+ plateau in response to ACh (Fig. 8 C).
Figure 8. Computational modelling: intracellular Ca2+ changes with V m during stimulation with ACh.

A, [Ca2+]i changes during ACh stimulation in lymphatic endothelium. The pattern of rapid increase in [Ca2+]i followed by a sustained plateau is similar to the one observed in Fura‐2 experiments (Fig. 2 D). B, as in A, in the absence of [Ca2+]o in lymphatic endothelium, or C, in the presence of 2 mm [Ca2+]o and during virtual blockade of TRPV4 and Orai channels. Note that removal of the Ca2+ from outside or blockade of membrane Ca2+ channels in the model also results in an abolished plateau phase (compare to Fig. 4 A and B). D, V m during ACh for lymphatic endothelium showing the depolarization similar to experimental recordings (Fig. 2 F). E, as in D, with V m calculated during ACh in the absence of [Ca2+]o, or F, in the presence of 2 mm [Ca2+]o and during virtual blockade of TRPV4 and Orai channels (compare to Fig. 4 C and D). Note that depolarization is abolished only with LaCl3 and not in the absence of [Ca2+]o, suggesting a role for Na+ currents in depolarization.
GSK101 treatment
Figure 9 illustrates the model's prediction of a parallel increase in [Ca2+]i (Fig. 9 A) and depolarization (Fig. 9 D) in response to TRPV4 activation with GSK101, which emulate Fig. 5 A and D respectively. Despite eliminating the rise in [Ca2+]i during GSK101 treatment when extracellular Ca2+ is absent (Figs 5 B and 9 B), depolarization is not significantly decreased (Figs 5 E and 9 E). However, elimination of Orai and TRPV4 currents effectively abolish any increase in [Ca2+]i (Figs 5 C and 9 C) or depolarization of V m (Figs 5 F and 9 F) during treatment with GSK101 (note similarity to data obtained from Trpv4 −/− mice during GSK101 treatment; Fig. 6 B, D and F).
Figure 9. Computational modelling: intracellular Ca2+ changes with V m during continuous stimulation with GSK101.

A, [Ca2+]i changes during GSK101 (with 2 mm [Ca2+]o) in lymphatic endothelium. Opening TRPV4 channels leads to an increase in [Ca2+]i similar to that observed in Fura‐2 experiments (Fig. 5 A). B, as in A, in the absence of [Ca2+]o in lymphatic endothelium, or C, in the presence of 2 mm [Ca2+]o and during virtual blockade of TRPV4 and Orai channels. Note that removal of external Ca2+ or blockade of membrane Ca2+ channels in the model abolished any change in [Ca2+]i (compare to Fig. 5 B and C). D, V m during GSK (with 2 mm [Ca2+]o) for lymphatic endothelium showing depolarization similar to experimental recording (Fig. 5 D). E, as in D, calculated V m during GSK101 in the absence of [Ca2+]o in lymphatic endothelium, or F, in the presence of 2 mm [Ca2+]o and during virtual blockade of TRPV4 and Orai channels. Note that depolarization is present in E (GSK + 0 [Ca2+]o) but not in F (GSK + LaCl3) similar to the experimental recording (compare to Fig. 5 E and F). Further, note similarities in C and F compared to Fig. 5 C and F, respectively, consistent with recordings obtained from LECTs of Trpv4 −/− mice.
Dynamics in ionic fluxes and open probability of key channels, exchangers and pumps
The strength of the mathematical model is the simultaneous calculation of currents contributed by ion channels, pumps, transporters and exchangers that underlie the depolarization response in lymphatic endothelium, a task that is not feasible using experimental approaches alone within a single study. Figure 10 A illustrates the integrated depolarization response at rest (t < 0 s) and during ACh stimulation (t > 0 s) (compare to Fig. 2 F). In response to ACh, the model predicts that IP3R is activated, releasing Ca2+ from the ER (Fig. 10 E), which is then removed from the cytosol by the plasma membrane Ca2+‐ATPase (PMCA; Fig. 10 F). As the ER Ca2+ drops in response to ACh (Fig. 10 C), TRPV4 and Orai channels open (Fig. 10 F and G) leading to a plateau increase in [Ca2+]i (Fig. 10 D); however, the permeability of TRPV4 to Na+ leads to depolarization (Fig. 10 A). In this model, TRP channels other than TRPV4 appear to play a negligible role in depolarization (Fig. 10 F and G). The Na+–K+‐ATPase (NaK) pump maintains Na+ and K+ concentration gradients and currents (Fig. 10 G for Na+ and Fig. 10 H for K+) across the plasma membrane at rest and during ACh (Fig. 10 C). Basal Cl− currents (Fig. 10 I) through Ca2+‐activated chloride channels (CaCCs) and volume‐regulated anion channels (VRACs) are negligible in the absence of Na+–K+–Cl− cotransporters (NKCCs); however, membrane depolarization in response to ACh results in transient Cl− currents through CaCCs and VRACs.
Altogether, ACh in LECTs triggers internal Ca2+ release from the ER which in turn activates non‐selective cation channels (or TRPs) in the plasma membrane and, in particular, TRPV4. While Ca2+ influx through TRPV4 channels is observed, it is the concomitant influx of Na+ that primarily underlies the integrated V m response to ACh in the form of depolarization.
Discussion
The present study has simultaneously monitored membrane potential and intracellular Ca2+ in freshly isolated lymphatic endothelial cell tubes for the first time. Complementary experiments and computational modelling have revealed a two‐phase response to ACh (i.e. ‘peak’ and ‘plateau’) in [Ca2+]i dynamics that is mediated through activation of TRPV4 channels and mimics the behaviour reported for endothelium of arterioles and resistance arteries (Cohen & Jackson, 2005; Socha et al. 2011; Behringer & Segal, 2015). However, in striking contrast to the hyperpolarization of blood vessel endothelium attributable to the efflux of K+, we find that the rise in [Ca2+]i in LECs is associated with depolarization attributable to the influx of Ca2+ and Na+. The depolarization observed in LEC tubes in response to ACh was confirmed in intact collecting lymphatic vessels that were pressurized and contracting. Moreover, the absence of SKCa/IKCa activity and gene expression in LECs is in contrast to the robust expression of these channels in arterial endothelium (Jackson, 2005; Ledoux et al. 2006; Behringer & Segal, 2012). Whereas sustained elevation of [Ca2+]i was abolished by removing extracellular Ca2 + or blocking the entry of Ca2+ through TRPV4, depolarization persisted until Na+ was prevented from entering cells through non‐selective cation channels with LaCl3. These findings build on previous electrophysiological studies of LECs in guinea pig mesenteric lymphatic vessels (von der Weid & Van Helden, 1997).
Physiological relevance
The EDH response in arterial endothelium is associated with hyperpolarization through activation of SKCa/IKCa channels, which is then conducted along the endothelial layer via homocellular gap junctions and into surrounding SMCs through myoendothelial gap junctions to coordinate dilatation along the vessel (Bagher & Segal, 2011, Garland et al. 2011). In the present study, our data suggest that LECs lack SKCa/IKCa channels responsible for EDH. Taken together with our previous findings that popliteal collecting lymphatic vessel dilatation to ACh is blocked completely by inhibiting NO production (Scallan & Davis, 2013), it appears that muscular lymphatic vessels do not possess an EDH pathway. Further, it appears likely that mouse lymphatic endothelium does not exhibit conducted hyperpolarization, given that its resting V m of ∼−70 mV is already quite negative as shown here in both LECTs and in intact lymphatic endothelium, which is in agreement with a previous report (von der Weid & Van Helden, 1997). Thus, LECs are poised to depolarize rather than hyperpolarize and depolarization would attenuate the driving force for Ca2+ entry and subsequent NO production through eNOS. This interpretation is consistent with LEC depolarization serving to permit LMC contraction.
ECs and SMCs of blood vessels are coupled electrically (Emerson & Segal, 2000), but there is so far a lack of evidence that LECs and LMCs are coupled to each other in this manner. In fact, because these two cell layers differ in their respective V m at rest (LMCs: −45 mV and LECs: −70 mV), there must be a barrier to the passage of substantial electrical charge between them (von der Weid & Van Helden, 1997), despite the fact that LECs are known to express several connexin isoforms, including Cx43 and Cx37 (Kanady et al. 2011; Sabine et al. 2012; Munger et al. 2016). The lack of electrical current passage between the LECs and LMCs was confirmed in the present study by recording a stable lymphatic endothelial membrane potential in intact lymphatic vessels that were contracting, and thus were coated with smooth muscle cells that were firing repetitive action potentials. One outcome from this lack of direct coupling is that the lymphatic endothelium is unable to regulate LMC contraction through electrical signals. The lack of coupling between respective cell layers would prevent loss of charge from LMCs into LECs, thereby facilitating conduction of a contraction wave (i.e. depolarization) along the LMC layer. Therefore, we propose that collecting lymphatic vessels are optimized for the initiation and conduction of depolarization, in contrast to blood vascular endothelium, which is optimized for the initiation and conduction of hyperpolarization signals that drive vasodilatation.
TRPV4 currents predominate in lymphatic endothelium
Previous studies have established a role for TRPV4 channels in promoting Ca2+ influx into the endothelium of resistance arteries, whereby activation of SKCa/IKCa and hyperpolarization produces vasodilatation (Zhang et al. 2009; Sonkusare et al. 2012; Qian et al. 2014). In the present study, mice genetically lacking Trpv4 show a ∼70% reduction in depolarization to ACh during the plateau phase as compared to wild‐type C57BL/6 mice (see Fig. 6 E, right panel). To our knowledge, this is the first study to indicate a functional role for TRPV4 in governing cation influx in lymphatic endothelium. The robust entry of Ca2+ and Na+ combined with a lack of SKCa/IKCa channel activity results in an accumulation of positive charge along the intracellular side of the plasma membrane, thereby leading to depolarization of V m (Hille, 2001) (Fig. 2). As the absence of extracellular Ca2+ reduced depolarization by nearly half during ACh stimulation (Fig. 4 E, right panel), our findings suggest an equal role for Na+ entry in depolarizing LECs. Furthermore, as the direct activation of TRPV4 channels with GSK101 elicits a full depolarization response in the absence of [Ca2+]o (Figs 5 and 9), the influx of Na+ alone can fulfil this physiological role of TRPV4 in membrane depolarization during the regulation of lymphatic function. Consistent with this interpretation is that blocking non‐selective cation channels with LaCl3 (Fig. 5 F and H) or genetic deletion of Trpv4 (Fig. 6 D and F, right panel) eliminated depolarization to GSK101.
Mathematical modelling analysis
The mathematical model presented here is based on a well‐developed model of mesenteric artery endothelium (Silva et al. 2007) that could reproduce our experimental measurement of [Ca2+]i and V m in mouse popliteal arteries (Fig. 2 C and E). Removal of the SKCa/IKCa currents was based on the present experiments in which blocking agents were applied without significant effects (Fig. 1) and the lack of LECT hyperpolarization to either ACh or GSK. With approximation of the model parameters to reflect the more negative V m in LECs compared to vascular ECs, we matched [Ca2+]i and V m dynamics to our experimental data for ACh and GSK (compare Figs 4 and 5 to Figs 8 and 9). To further substantiate our application of this model to LECs, we compared the results from blocking ion channels and found a similar degree of depolarization after Kir blockade (∼11.3 mV computationally versus 11 ± 1 mV experimentally). Additionally, NaK blockade in guinea pig lymphatic endothelium with ouabain (von der Weid & Van Helden, 1997) produced a 6 mV depolarization, similar to the predictions of our model (6.7 mV). In a previous in vitro study of cultured LECs with Fura‐2 measurement of [Ca2+]i, the SERCA inhibitor thapsigargin elicited rapid ER depletion followed by a sustained [Ca2+]i increase from TRPV4 and Orai activation (Jafarnejad et al. 2015). We observed a similar dynamic in our model when SERCA current was removed. We conclude that our model of LECs demonstrates the details of ionic current balance in the resting condition as well as during ACh treatment and can therefore be utilized to generate novel hypotheses by investigating scenarios that include blockade of ion channels for which selective pharmacological tools are lacking. First, the model predicts that PMCA channels are required for Ca2+ extrusion after stimulation with ACh and therefore their blockade would lead to a considerable increase in [Ca2+]i, a point that should be investigated experimentally (Fig. 10 F). Second, the model predicts that the Na+ permeability of TRPV4 channels is essential for ACh‐mediated depolarization of LECTs, which is consistent with the central finding of the present experiments. Third, the model identifies the Ca2+:Na+ permeability ratio of TRPV4 as a primary parameter in determining the balance between sustained influx of Ca2+ versus V m depolarization. Fourth, it predicts that after ACh stimulation, 56% of the total Na+ TRP current is through TRPV4, which is in agreement with arteriolar endothelium (Sonkusare et al. 2012). Overall, the model provides a comprehensive picture of all requisite transmembrane currents, ionic concentrations, and potentials, which are otherwise difficult or impractical to measure experimentally. For instance, the model offers a tantalizing prediction as to why the resting V m is significantly more negative in lymphatic endothelium versus blood endothelium: the potential absence of significant NKCC channel expression would prevent the inward Cl− current that underlies the steady‐state depolarization in blood endothelium (Russell, 2000).
Summary and conclusions
The function of endothelial Ca2+‐activated K+ channels as an interface between intracellular Ca2+ dynamics and production of hyperpolarization has long been recognized (Busse et al. 1988). An integral component of resistance blood vessels is to coordinate smooth muscle cell relaxation and vasodilatation in accord with metabolic demand (Segal, 2015). Conversely, collecting lymphatics also undergo active Ca2+ and electrical dynamics (von der Weid & Van Helden, 1997; Ferrusi et al. 2004) but do so to generate rhythmic contractions and unidirectional pumping of fluid (von der Weid et al. 1996; Mizuno et al. 1997). We sought to resolve the underlying ionic mechanisms of lymphatic endothelial function working from our established understanding of endothelium‐derived hyperpolarization demonstrated in blood vessel endothelium. Overall, we found that while [Ca2+]i dynamics were similar to arterial endothelium in response to agonists such as ACh, depolarization (rather than hyperpolarization) ensued in lymphatic endothelium. This striking difference can be explained by the lack of Ca2+‐activated K+ channels in LECs while Ca2+ and Na+ ions enter through non‐selective TRPV4 cation channels. Thus, the lack of an EDH pathway in lymphatic endothelium is likely a crucial adaptation to ensure the efficient conduction of contraction waves in the adjacent lymphatic muscle cells that are required for propulsive lymph flow.
Additional information
Competing interests
The authors have no competing interests to declare or conflicts of interest to disclose. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
E.J.B., J.P.S., J.A.C. and S.D.Z. designed and performed experiments in the laboratories of S.S.S. and M.J.D. M.J. performed computational modelling in the laboratory of J.E.M. E.J.B., J.P.S., M.J.D., J.A.C., S.D.Z. and S.S.S. analysed and interpreted data. E.J.B., J.P.S., M.J., S.D.Z. and J.A.C. drafted the manuscript and/or prepared the figures. S.S.S. and M.J.D. edited the manuscript. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This research was supported by National Institutes of Health grants R01 HL‐122608 and HL‐122578 to M.J.D. and R37‐HL041026 to S.S.S. E.J.B. was supported by NIH grants F32‐HL110701 and K99/R00‐AG047198. J.P.S. was supported by K99/R00‐HL124142. J.E.M. was supported by R01‐HL094269, U01‐HL123420, The Royal Society, The Royal Academy of Engineering and The Sir Leon Bagrit Trust.
Translational perspective
In the blood circulation, endothelial cell function in resistance vessels employs activation of Ca2+‐sensitive K+ channels to hyperpolarize and promote relaxation of vascular smooth muscle cells. Thus, vasodilatation can be triggered through TRPV4 channels that promote Ca2+ influx. However, in the lymphatic circulation it is unknown whether the endothelium uses Ca2+‐sensitive K+ channel activity to govern the activity of lymphatic muscle cells for pumping fluid away from peripheral tissues to prevent oedema. To answer this question, we studied freshly isolated endothelial tubes of collecting lymphatic vessels that are free of such confounding factors as smooth muscle cells and the flow of lymph. We evaluated intracellular Ca2+ levels in conjunction with membrane potential during exposure to pharmacological agents known to activate Ca2+‐sensitive K+ channels and TRPV4 channels to elevate intracellular Ca2+. Our findings indicate that, despite robust increases in intracellular Ca2+, the endothelium of collecting lymphatics lacks Ca2+‐sensitive K+ channels yet expresses TRPV4 channels and depolarizes with Ca2+ influx. Genetic deletion of Trpv4 attenuated Ca2+ influx, confirming the integral role of TRPV4 channels in lymphatic endothelium. Our findings indicate that (1) endothelial Ca2+‐sensitive K+ channel activity is not requisite for lymphatic function and (2) vascular endothelia throughout the body need not share the same genetic and protein composition to perform their physiological roles. These data provide fundamentally new insight for precision medicine, whereby selective treatments are developed for key vascular biomarkers to promote health and cure disease.
Acknowledgements
Min Li provided excellent technical assistance with endpoint PCR.
Linked articles This article is highlighted by a Perspective by Dora & van Helden. To read this Perspective, visit https://doi.org/10.1113/JP275323.
E. J. Behringer and J. P. Scallan contributed equally to this work.
Edited by: Kim Barrett & Kathleen Morgan
Contributor Information
Michael J. Davis, Email: davismj@health.missouri.edu.
Steven S. Segal, Email: segalss@health.missouri.edu.
References
- Aspelund A, Robciuc MR, Karaman S, Makinen T & Alitalo K (2016). Lymphatic system in cardiovascular medicine. Circ Res 118, 515–530. [DOI] [PubMed] [Google Scholar]
- Bagher P & Segal SS (2011). Regulation of blood flow in the microcirculation: role of conducted vasodilation. Acta Physiol (Oxf) 202, 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ & Segal SS (2012). Tuning electrical conduction along endothelial tubes of resistance arteries through Ca2+‐activated K+ channels. Circ Res 110, 1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ & Segal SS (2015). Membrane potential governs calcium influx into microvascular endothelium: integral role for muscarinic receptor activation. J Physiol 593, 4531–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ, Socha MJ, Polo‐Parada L & Segal SS (2012). Electrical conduction along endothelial cell tubes from mouse feed arteries: confounding actions of glycyrrhetinic acid derivatives. Br J Pharmacol 166, 774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R, Edwards G, Félétou M, Fleming I, Vanhoutte PM & Weston AH (2002). EDHF: bringing the concepts together. Trends Pharmacol Sci 23, 374–380. [DOI] [PubMed] [Google Scholar]
- Busse R, Fichtner H, Luckhoff A & Kohlhardt M (1988). Hyperpolarization and increased free calcium in acetylcholine‐stimulated endothelial cells. Am J Physiol Heart Circ Physiol 255, H965–H969. [DOI] [PubMed] [Google Scholar]
- Chakraborty S, Davis MJ & Muthuchamy M (2015). Emerging trends in the pathophysiology of lymphatic contractile function. Semin Cell Dev Biol 38, 55–66.25617600 [Google Scholar]
- Choi D, Park E, Jung E, Seong YJ, Hong M, Lee S, Burford J, Gyarmati G, Peti‐Peterdi J, Srikanth S, Gwack Y, Koh CJ, Boriushkin E, Hamik A, Wong AK & Hong YK (2017a). ORAI1 activates proliferation of lymphatic endothelial cells in response to laminar flow through Kruppel‐like factors 2 and 4. Circ Res 120, 1426–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D, Park E, Jung E, Seong YJ, Yoo J, Lee E, Hong M, Lee S, Ishida H, Burford J, Peti‐Peterdi J, Adams RH, Srikanth S, Gwack Y, Chen CS, Vogel HJ, Koh CJ, Wong AK & Hong YK (2017b). Laminar flow downregulates Notch activity to promote lymphatic sprouting. J Clin Invest 127, 1225–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE, Julius D, Montell C & Schultz G (2005). International Union of Pharmacology. XLIX. Nomenclature and structure‐function relationships of transient receptor potential channels. Pharmacol Rev 57, 427–450. [DOI] [PubMed] [Google Scholar]
- Cohen KD & Jackson WF (2005). Membrane hyperpolarization is not required for sustained muscarinic agonist‐induced increases in intracellular Ca2+ in arteriolar endothelial cells. Microcirculation 12, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Scallan JP, Wolpers JH, Muthuchamy M, Gashev AA & Zawieja DC (2012). Intrinsic increase in lymphangion muscle contractility in response to elevated afterload. Am J Physiol Heart Circ Physiol 303, H795–H808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diep HK, Vigmond EJ, Segal SS & Welsh DG (2005). Defining electrical communication in skeletal muscle resistance arteries: a computational approach. J Physiol 568, 267–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson GG, Neild TO & Segal SS (2002). Conduction of hyperpolarization along hamster feed arteries: augmentation by acetylcholine. Am J Physiol Heart Circ Physiol 283, H102–H109. [DOI] [PubMed] [Google Scholar]
- Emerson GG & Segal SS (2000). Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ Res 87, 474–479. [DOI] [PubMed] [Google Scholar]
- Félétou M (2016). Endothelium‐dependent hyperpolarization and endothelial dysfunction. J Cardiovasc Pharmacol 67, 373–387. [DOI] [PubMed] [Google Scholar]
- Ferrusi I, Zhao J, van Helden D & von der Weid PY (2004). Cyclopiazonic acid decreases spontaneous transient depolarizations in guinea pig mesenteric lymphatic vessels in endothelium‐dependent and ‐independent manners. Am J Physiol Heart Circ Physiol 286, H2287–H2295. [DOI] [PubMed] [Google Scholar]
- Fill M & Copello JA (2002). Ryanodine receptor calcium release channels. Physiol Rev 82, 893–922. [DOI] [PubMed] [Google Scholar]
- Garland CJ & Dora KA (2017). EDH: endothelium‐dependent hyperpolarization and microvascular signaling. Acta Physiol (Oxf) 219, 152–161. [DOI] [PubMed] [Google Scholar]
- Garland CJ, Hiley CR & Dora KA (2011). EDHF: spreading the influence of the endothelium. Br J Pharmacol 164, 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gees M, Colsoul B & Nilius B (2010). The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb Perspect Biol 2, a003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hald BO, Welsh DG, Holstein‐Rathlou NH & Jacobsen JC (2015). Origins of variation in conducted vasomotor responses. Pflugers Arch 467, 2055–2067. [DOI] [PubMed] [Google Scholar]
- Hill‐Eubanks DC, Gonzales AL, Sonkusare SK & Nelson MT (2014). Vascular TRP channels: performing under pressure and going with the flow. Physiology (Bethesda) 29, 343–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B ( 2001). Ion Channels of Excitable Membranes, 3rd edn, Ch. 22, Evolution and origins, pp. 693–722. Sinauer Associates, Sunderland. [Google Scholar]
- Himmel HM, Whorton AR & Strauss HC (1993). Intracellular calcium, currents, and stimulus‐response coupling in endothelial cells. Hypertension 21, 112–127. [DOI] [PubMed] [Google Scholar]
- Jackson WF (2005). Potassium channels in the peripheral microcirculation. Microcirculation 12, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafarnejad M, Cromer WE, Kaunas RR, Zhang SL, Zawieja DC & Moore JE Jr (2015). Measurement of shear stress‐mediated intracellular calcium dynamics in human dermal lymphatic endothelial cells. Am J Physiol Heart Circ Physiol 308, H697–H706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanady JD, Dellinger MT, Munger SJ, Witte MH & Simon AM (2011). Connexin37 and Connexin43 deficiencies in mice disrupt lymphatic valve development and result in lymphatic disorders including lymphedema and chylothorax. Dev Biol 354, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsberger M, Sauser R, Beny JL & Meister JJ (2005). Role of the endothelium on arterial vasomotion. Biophys J 88, 3845–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux J, Werner ME, Brayden JE & Nelson MT (2006). Calcium‐activated potassium channels and the regulation of vascular tone. Physiology (Bethesda) 21, 69–78. [DOI] [PubMed] [Google Scholar]
- Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL & Birnbaumer L (2008). Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM‐regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci USA 105, 2895–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad DS, Murphey CR, Clark JW & Giles WR (1996). A model of the action potential and underlying membrane currents in a rabbit atrial cell. Am J Physiol Heart Circ Physiol 271, H1666–H1696. [DOI] [PubMed] [Google Scholar]
- Ma X, Cheng K‐T, Wong C‐O, O'Neil RG, Birnbaumer L, Ambudkar IS & Yao X (2011). Heteromeric TRPV4‐C1 channels contribute to store‐operated Ca2+ entry in vascular endothelial cells. Cell Calcium 50, 502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno R, Dornyei G, Koller A & Kaley G (1997). Myogenic responses of isolated lymphatics: modulation by endothelium. Microcirculation 4, 413–420. [DOI] [PubMed] [Google Scholar]
- Munger SJ, Geng X, Srinivasan RS, Witte MH, Paul DL & Simon AM (2016). Segregated Foxc2, NFATc1 and connexin expression at normal developing venous valves, and connexin‐specific differences in the valve phenotypes of Cx37, Cx43, and Cx47 knockout mice. Dev Biol 412, 173–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins D & Michalak M (2011). Organellar calcium buffers. Cold Spring Harb Perspect Biol 3, a004069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Francis M, Kohler R, Solodushko V, Lin M & Taylor MS (2014). Positive feedback regulation of agonist‐stimulated endothelial Ca2+ dynamics by KCa3.1 channels in mouse mesenteric arteries. Arterioscler Thromb Vasc Biol 34, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhle B & Trebak M (2013). Emerging roles for native Orai Ca2+ channels in cardiovascular disease. Curr Top Membr 71, 209–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JM (2000). Sodium‐potassium‐chloride cotransport. Physiol Rev 80, 211–276. [DOI] [PubMed] [Google Scholar]
- Sabine A, Agalarov Y, Maby‐El Hajjami H, Jaquet M, Hagerling R, Pollmann C, Bebber D, Pfenniger A, Miura N, Dormond O, Calmes JM, Adams RH, Makinen T, Kiefer F, Kwak BR & Petrova TV (2012). Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic‐valve formation. Dev Cell 22, 430–445. [DOI] [PubMed] [Google Scholar]
- Scallan JP & Davis MJ (2013). Genetic removal of basal nitric oxide enhances contractile activity in isolated murine collecting lymphatic vessels. J Physiol 591, 2139–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scallan JP, Wolpers JH & Davis MJ (2013). Constriction of isolated collecting lymphatic vessels in response to acute increases in downstream pressure. J Physiol 591, 443–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal SS (2015). Integration and modulation of intercellular signaling underlying blood flow control. J Vasc Res 52, 136–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva HS, Kapela A & Tsoukias NM (2007). A mathematical model of plasma membrane electrophysiology and calcium dynamics in vascular endothelial cells. Am J Physiol Cell Physiol 293, C277–C293. [DOI] [PubMed] [Google Scholar]
- Socha MJ, Domeier TL, Behringer EJ & Segal SS (2012). Coordination of intercellular Ca2+ signaling in endothelial cell tubes of mouse resistance arteries. Microcirculation 19, 757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socha MJ, Hakim CH, Jackson WF & Segal SS (2011). Temperature effects on morphological integrity and Ca2+ signaling in freshly isolated murine feed artery endothelial cell tubes. Am J Physiol Heart Circ Physiol 301, H773–H783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socha MJ & Segal SS (2013). Isolation of microvascular endothelial tubes from mouse resistance arteries. J Vis Exp 81, e50759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill‐Eubanks DC & Nelson MT (2012). Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336, 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Dalsgaard T, Bonev AD, Hill‐Eubanks DC, Kotlikoff MI, Scott JD, Santana LF & Nelson MT (2014). AKAP150‐dependent cooperative TRPV4 channel gating is central to endothelium‐dependent vasodilation and is disrupted in hypertension. Sci Signal 7, ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorneloe KS, Sulpizio AC, Lin Z, Figueroa DJ, Clouse AK, McCafferty GP, Chendrimada TP, Lashinger ES, Gordon E, Evans L, Misajet BA, Demarini DJ, Nation JH, Casillas LN, Marquis RW, Votta BJ, Sheardown SA, Xu X, Brooks DP, Laping NJ & Westfall TD (2008). N‐((1S)‐1‐{[4‐((2S)‐2‐{[(2,4‐dichlorophenyl)sulfonyl]amino}‐3‐hydroxypropanoyl)‐1‐piperazinyl]carbonyl}‐3‐methylbutyl)‐1‐benzothiophene‐2‐carboxamide (GSK1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: Part I. J Pharmacol Exp Ther 326, 432–442. [DOI] [PubMed] [Google Scholar]
- Voets T, Droogmans G, Raskin G, Eggermont J & Nilius B (1999). Reduced intracellular ionic strength as the initial trigger for activation of endothelial volume‐regulated anion channels. Proc Natl Acad Sci USA 96, 5298–5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Weid PY, Crowe MJ & Van Helden DF (1996). Endothelium‐dependent modulation of pacemaking in lymphatic vessels of the guinea‐pig mesentery. J Physiol 493, 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Weid PY, Lee S, Imtiaz MS, Zawieja DC & Davis MJ (2014). Electrophysiological properties of rat mesenteric lymphatic vessels and their regulation by stretch. Lymphat Res Biol 12, 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Weid PY & Van Helden DF (1997). Functional electrical properties of the endothelium in lymphatic vessels of the guinea‐pig mesentery. J Physiol 504, 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner TF, Berk BC & Nerem RM (1996). A mathematical model of cytosolic calcium dynamics in human umbilical vein endothelial cells. Am J Physiol Cell Physiol 270, C1556–C1569. [DOI] [PubMed] [Google Scholar]
- Yang J, Clark JW Jr, Bryan RM & Robertson C (2003). The myogenic response in isolated rat cerebrovascular arteries: smooth muscle cell model. Med Eng Physics 25, 691–709. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R, Warltier DC, Suzuki M & Gutterman DD (2009). Transient receptor potential vanilloid type 4‐deficient mice exhibit impaired endothelium‐dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension 53, 532–538. [DOI] [PMC free article] [PubMed] [Google Scholar]