

Abstract
Single‐cell RNA sequencing is a powerful technology for assessing heterogeneity within defined cell populations. Here, we describe the heterogeneity of a B220+CD117intCD19−NK1.1− uncommitted hematopoietic progenitor having combined lymphoid and myeloid potential. Phenotypic and functional assays revealed four subpopulations within the progenitor with distinct lineage developmental potentials. Among them, the Ly6D+SiglecH−CD11c− fraction was lymphoid‐restricted exhibiting strong B‐cell potential, whereas the Ly6D−SiglecH−CD11c− fraction showed mixed lympho‐myeloid potential. Single‐cell RNA sequencing of these subsets revealed that the latter population comprised a mixture of cells with distinct lymphoid and myeloid transcriptional signatures and identified a subgroup as the potential precursor of Ly6D+SiglecH−CD11c−. Subsequent functional assays confirmed that B220+CD117intCD19−NK1.1− single cells are, with rare exceptions, not bipotent for lymphoid and myeloid lineages. A B‐cell priming gradient was observed within the Ly6D+SiglecH−CD11c− subset and we propose a herein newly identified subgroup as the direct precursor of the first B‐cell committed stage. Therefore, the apparent multipotency of B220+CD117intCD19−NK1.1− progenitors results from underlying heterogeneity at the single‐cell level and highlights the validity of single‐cell transcriptomics for resolving cellular heterogeneity and developmental relationships among hematopoietic progenitors.
Keywords: hematopoiesis, heterogeneity, lineage priming, multipotentiality, single‐cell RNA sequencing
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Development & Differentiation; Immunology
Introduction
The well‐established “classical” model of hematopoiesis (Abramson et al, 1977), together with other versions (Katsura & Kawamoto, 2001), proposes a hierarchical decision‐making process whereby early multipotent progenitors make an irrevocable decision to differentiate toward either lymphoid or myeloid lineages (Kawamoto et al, 2010) through so‐called common lymphoid (Kondo et al, 1997) and common myeloid progenitor (Akashi et al, 2000) intermediates, respectively. However, the proposals for alternative differentiation pathways (Guimaraes et al, 1982; Fogg et al, 2006; Ishikawa et al, 2007) and the description of progenitor cells that contradict the lympho‐myeloid dichotomy (Adolfsson et al, 2005; Balciunaite et al, 2005) have prompted multiple revisions of the classical model. For instance, the pairwise model (Ceredig et al, 2009; Brown et al, 2015) suggests that hematopoiesis is a more versatile and less strictly compartmentalized process than previously thought. Recently developed technologies now permit the study of hematopoiesis from different perspectives and provide significant findings that expand our current knowledge. For instance, studies using different single‐cell lineage tracing strategies in vivo (Naik et al, 2013; Sun et al, 2014; Busch et al, 2015) conclude that in the steady state, progenitor cells downstream of HSCs are the major drivers of adult hematopoiesis and that diverse lineage imprinting occurs earlier than previously anticipated. By performing time‐lapse imaging of single cells in vitro, Hoppe et al challenge the current prevailing model of early myeloid lineage choice (Hoppe et al, 2016).
The emergence of high‐throughput sequencing methods enabling the investigation of single‐cell whole‐transcriptome profiles provides an exceptional tool for interrogating with unprecedented resolution the degree of genotypic heterogeneity among phenotypically homogeneous progenitors. Multiple studies have already reported the use of single‐cell RNA sequencing (scRNA‐seq) to successfully dissect cellular heterogeneity and identify novel subpopulations in the hematopoietic system (Mahata et al, 2014; Shalek et al, 2014; Gren et al, 2015; Kowalczyk et al, 2015; Paul et al, 2015; Drissen et al, 2016) as well as in other fields such as oncology (Kim et al, 2015; Min et al, 2015) or neurobiology (Zeisel et al, 2015). In hematopoiesis, Paul et al (2015) report heterogeneity in myeloid progenitors; Kowalczyk et al (2015) find extensive transcriptome variability among HSCs; and Drissen et al (2016) subdivide the pre‐GM population into a Gata1 + pre‐GM fraction generating mast cells, eosinophils, megakaryocytes, and erythroid cells and a Gata1 − pre‐GM subset generating monocytes, neutrophils, and lymphocytes.
We have previously characterized a phenotypically homogeneous B220+CD117intCD19−NK1.1− hematopoietic progenitor with combined lymphoid and myeloid potential that we called early progenitor with lymphoid and myeloid potential (EPLM) (Balciunaite et al, 2005). EPLMs represent about 0.2% of all nucleated bone marrow cells in wild‐type (WT) mice. This progenitor was identified in a search for a WT counterpart of Pax5 −/− pro‐B cells (Nutt et al, 1999; Rolink et al, 2002). Phenotypically, EPLMs are closely related to the common lymphoid progenitor (CLP) with the marked difference that they are B220+ whereas CLP is B220−. EPLMs also partially overlap with the so‐called Fraction A cells described by Hardy and co‐workers (Li et al, 1996). Functionally, EPLMs showed potent B‐cell developmental potential and strong‐to‐moderate differentiation potential for T and myeloid cells (mostly macrophages). This suggested that under physiological conditions, the developmental fate of EPLM was mainly to become B cells.
In line with the description of individual progenitor cells having multiple lineage potentials, there is an increasing debate regarding their heterogeneity. In the present study, we investigated the multipotentiality of EPLM in detail and further assessed their potential heterogeneity. Firstly, based on the expression of the surface markers Ly6D, SiglecH, and CD11c, we were able to fractionate EPLM into four subpopulations with distinct lineage potentials. Subsequently, we further studied the transcriptional heterogeneity of the two subsets having B‐cell potential by performing scRNA‐seq. Using this approach, we found five additional subgroups each with a different gene expression signature that revealed a clear lympho‐myeloid separation. This conclusion was strengthened by functional experiments at the single‐cell level. Based on these results, we propose that the bifurcation of lymphoid and myeloid molecular priming occurs before the EPLM stage. Furthermore, we suggest that a herein identified subgroup of cells within the Ly6D+SiglecH−CD11c− EPLM subpopulation is the direct precursor of the first B‐cell committed stage, the pro‐B cell.
Results
The EPLM progenitor population can be divided into at least four subpopulations with different sets of potentials
We have previously characterized an uncommitted B220+CD117intCD19−NK1.1− progenitor with combined lymphoid and myeloid potential (EPLM) (Balciunaite et al, 2005). With the aim of determining whether EPLMs represent a homogeneous multipotent population or a mixture of individual lineage‐restricted cells, we examined their expression of Ly6D, SiglecH, and CD11c, markers known to be associated with different hematopoietic lineages. Ly6D has been shown to identify B‐cell‐biased CD19− progenitors (Inlay et al, 2009; Mansson et al, 2010). SiglecH is a specific marker for plasmacytoid dendritic cells (pDC), which also express B220 (Blasius et al, 2006; Zhang et al, 2006). Previously, it was shown that SiglecH was also expressed by a fraction of CLP and pre‐pro‐B cells (Medina et al, 2013), two populations partially overlapping with EPLM (von Muenchow et al, 2016). Since pDC also express the general dendritic cell marker CD11c (Singh‐Jasuja et al, 2013), we chose this as a third surface marker. Staining EPLM for Ly6D and SiglecH expression resulted in the identification of three (Ly6D+SiglecH−, Ly6D+SiglecH+, and Ly6D−SiglecH−) EPLM fractions in WT mice (Appendix Fig S1A).
Subsequently, we investigated CD11c expression within these three fractions. Results showed that the Ly6D+SiglecH− fraction only contained 5.1 ± 0.5% CD11c+ cells, whereas the Ly6D+SiglecH+ fraction was mostly (85.2 ± 2.7%) CD11c+. Interestingly, Ly6D−SiglecH− cells were heterogeneous for CD11c expression, with about one‐third (28.1 ± 2.7%) being CD11c+ (Appendix Fig S1A middle cytogram). This result indicates that the latter fraction can be further subdivided into two, CD11c+ and CD11c−, fractions resulting in four major EPLM subpopulations. In Fig 1A and in subsequent experiments, we represent the four EPLM subsets in a simplified manner by staining for SiglecH and CD11c using antibodies conjugated with the same fluorochromes. We can thus distinguish four EPLM subpopulations hereafter called Ly6D+ (Ly6D+SiglecH−CD11c−), SiglecH+ (Ly6D+SiglecH+CD11c+), TN (triple negative) (Ly6D−SiglecH−CD11c−), and CD11c+ (Ly6D−SiglecH−CD11c+) subpopulations (Fig 1A–C).
Figure 1. Phenotypic and functional heterogeneity of the EPLM progenitor population (B220+CD117intCD19−NK1.1−) based on expression of Ly6D, SiglecH, and CD11c.
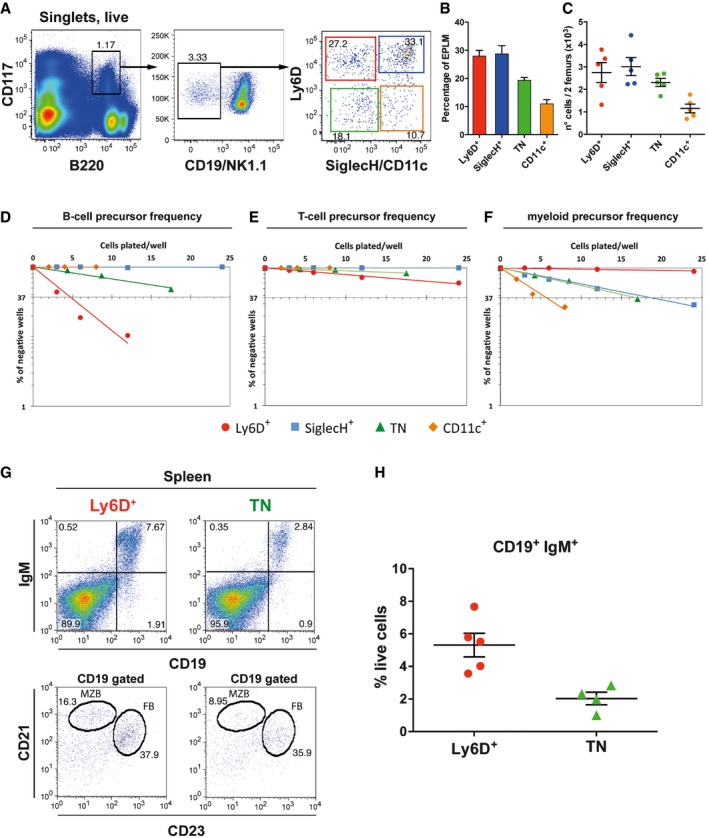
-
ARepresentative FACS plots of EPLM from the bone marrow of WT mice with the addition of Ly6D, SiglecH, and CD11c identifying four subpopulations.
-
B, CPercentages (B) and absolute numbers (C) of WT EPLM subpopulations (n = 5).
-
D–FIn vitro limiting dilution analysis of Ly6D+, TN, SiglecH+, and CD11c+ for B‐cell (D), T‐cell (E), or myeloid (F) potentials.
-
G, HB‐cell reconstitution of sub‐lethally irradiated Rag2‐deficient mice with 4 × 103 Ly6D+ (n = 5) or TN (n = 4) cells from WT. (G) Representative FACS plots from spleens of reconstituted mice. (H) Quantification of CD19+IgM+ splenocytes.
We next assessed whether the heterogeneous expression of Ly6D, SiglecH, and CD11c cell surface markers by EPLM was correlated with distinct developmental potentials. For that purpose, graded numbers of sorted EPLM subsets were plated on stromal cells and the appropriate cytokines that support B‐cell (OP9 + IL‐7), T‐cell (OP9‐DL1 + IL‐7), or myeloid (ST2) differentiation. Cell growth was scored at day 10 for OP9 and at day 15 for OP9‐DL1 and ST2 cell cultures by use of an inverted microscope. FACS analysis was performed to confirm the identity of the growing cells (Appendix Fig S1B). Under B‐cell differentiation conditions, Ly6D+ and TN subpopulations generated colonies, thus revealing B‐cell potential. The B‐cell precursor frequency was higher among Ly6D+ (on average 1 in 5) than in TN (on average 1 in 20) cells (Fig 1D). CD11c+ and SiglecH+ subpopulations did not generate colonies under either B‐ or T‐cell conditions (Fig 1D and E), whereas Ly6D+ and TN cells generated T cells at low frequencies (Fig 1E). When EPLM subpopulations were plated on ST2 stromal cells, the Ly6D+ subset did not generate myeloid clones, whereas all other EPLM subsets possessed myeloid potential (Appendix Fig S1B), although at different frequencies (Fig 1F). Therefore, the lymphoid potential of EPLM seems to reside only within the Ly6D+ and TN subpopulations. As the physiological role of EPLM appears to be the generation of B cells in the bone marrow, further analyses were focused on Ly6D+ and TN cells.
We therefore tested the in vivo capacity of Ly6D+ and TN progenitors to reconstitute the B‐cell compartment of lymphocyte‐deficient mice. The two EPLM subsets were sorted from WT mice and 4 × 103 cells transferred into sub‐lethally irradiated Rag2‐deficient recipient mice. Flow cytometry of the spleens at 3 weeks following transfer revealed that B‐cell compartments were significantly reconstituted in all mice. Both Ly6D+ and TN progenitors generated CD19+IgM+ B cells (Fig 1G upper panels and H). Further analysis of the spleen CD19+ cells revealed the presence of both CD21highCD23− marginal zone B cells and CD21intCD23+ follicular B cells (Fig 1G lower panels). Therefore, although TN cells present less B‐cell precursor frequency in vitro, both populations have in vivo B‐cell developmental potential. Additional analysis of the thymus showed that in line with the in vitro observations, EPLM subpopulations had limited T‐cell in vivo developmental potential. Only the Ly6D+ subset had any T‐cell reconstitution potential (1/5 mice) whereas TN cells were unable to reconstitute the thymus (0/5) (Appendix Fig S1C and D).
In conclusion, the EPLM progenitor population is phenotypically and functionally heterogeneous and based on the differential expression of Ly6D, SiglecH, and CD11c, can be further divided into at least four subpopulations with distinct developmental potential biases.
A TN fraction is the direct precursor of the Ly6D+ EPLM subpopulation
As a population, the TN subset of EPLM would appear to have multilineage developmental potential prompting the question whether it is still composed of a mixture of lineage‐restricted cells. Therefore, we further explored the heterogeneity of EPLM subpopulations by performing single‐cell RNA sequencing (scRNA‐seq). In order to enable the transcriptomic analysis, and since EPLM subpopulations are present in limited numbers in WT mice (Fig 1C), we turned to a mouse model where EPLM cells are more abundant. We have previously shown that the total EPLM compartment of Flt3Ltg mice (Tsapogas et al, 2014) or WT mice injected with Flt3L (Ceredig et al, 2006) is dramatically expanded without major alterations in their developmental potentials. However, because these analyses were performed on total EPLM, we first investigated whether Flt3Ltg EPLM subpopulation percentages and developmental potentials differed significantly from their WT counterparts.
Analysis of Ly6D, SiglecH, and CD11c expression by Flt3Ltg EPLM showed only minor differences in their relative frequencies compared to WT (Fig 2A and B, and Appendix Fig S2A), whereas their absolute numbers were increased by almost two orders of magnitude (Fig 2C). Moreover, in vitro differentiation assays revealed the same set of developmental potentials as in WT mice: the Ly6D+ being lymphoid‐restricted (1 in 11 ± 1.3 B cell, 1 in 5.2 ± 0.6 T cell, and < 1 in 500 myeloid progenitors), the TN having trilineage developmental potential (1 in 70 ± 13.2 B cell, 1 in 8.6 ± 2.2 T cell, and 1 in 15 ± 6.72 myeloid progenitors), and the SiglecH and CD11c subpopulations being devoid of lymphoid potential (Fig 2D–F and Appendix Fig S2B). In terms of frequency, the B‐cell potential of Ly6D+ and TN from Flt3Ltg mice was almost twofold lower than that of their WT counterparts (Appendix Fig S2B), whereas their T‐cell potential was significantly increased (Appendix Fig S2B), probably as a result of reduced expression of Pax5 (Appendix Fig S2C) (Holmes et al, 2006; von Muenchow et al, 2016). However, the presence of all EPLM subpopulations in Flt3Ltg mice at similar ratios to WT and with a comparable set of developmental potentials indicates that the Flt3Ltg mouse may be a valid model to further evaluate the heterogeneity of EPLM subpopulations.
Figure 2. Identification and analysis of Flt3Ltg EPLM subpopulations.
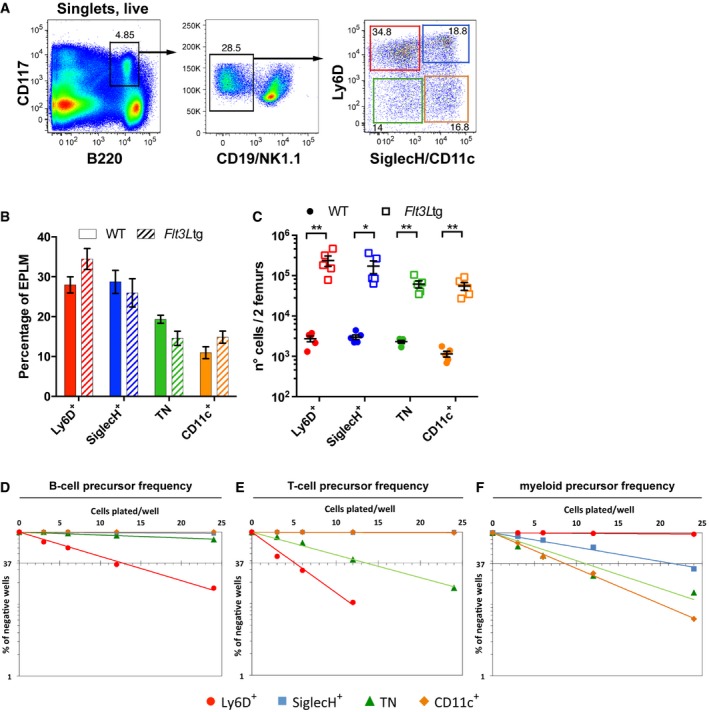
-
ARepresentative FACS plots of EPLM from the bone marrow of Flt3Ltg mice with the addition of Ly6D, SiglecH, and CD11c identifying four subpopulations.
-
B, CComparison of EPLM subpopulations from WT (n = 5, circles) and Flt3Ltg (n = 5, squares) mice in percentages (B) or absolute numbers (C).
-
D–FB‐cell, T‐cell, and myeloid precursor frequencies of EPLM subpopulations from Flt3Ltg mice obtained by limiting dilution performed as in Fig 1.
Bulk RNA sequencing was performed to identify the genes characteristic of the Ly6D+ and TN subsets from Flt3Ltg mice. This analysis revealed 1,008 differentially expressed genes (DEG, Fig 3A and Dataset EV1), with 493 genes being more highly expressed by Ly6D+ compared to TN cells (Fig 3A red), and 515 genes expressed at lower levels (Fig 3A blue). Notably, Flt3 was not among the DEG (Dataset EV1). For scRNA‐seq, single Ly6D+ and TN cells from the same Flt3Ltg mice (see Materials and Methods) were captured with the C1 Fluidigm system (Appendix Fig S3A and B) and only cells with more than 60% of mapped reads, at least 2 × 105 counts, and more than 800 detected genes were selected for further analysis (Appendix Fig S3C–F); thus, 152 Ly6D+ and 213 TN single cells were analyzed. In principal component analysis (PCA), cells did not cluster according to the C1 chip they were captured, thus revealing that there was no major batch effect whereas the main source of variation (PC1) was the number of genes detected per cell (Appendix Fig S3G and H, and Dataset EV4). In order to maximize the biological signal, we made use of the DEG between Ly6D+ and TN cells obtained from the bulk RNA‐seq experiment (Fig 3A). PCA with that subset of genes partially segregated the TN from the Ly6D+ cells (PC1 axis, Fig 3B). Remarkably, some cells still overlapped (Fig 3B and Dataset EV5), thus revealing that the two EPLM subpopulations are to some extent transcriptionally similar and therefore suggesting that they might be developmentally related.
Figure 3. Ly6D+ and TN EPLM subpopulations are transcriptionally and developmentally related.
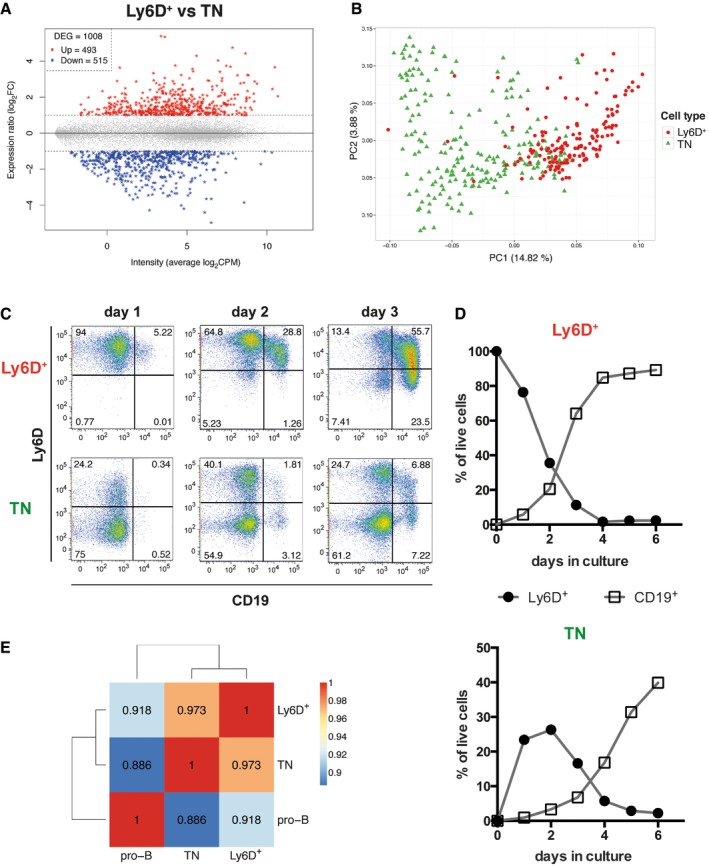
-
AGene expression ratio of Ly6D+ versus TN cells from Flt3Ltg (vertical axis) plotted against the average expression intensity (horizontal axis), showing 1,008 differentially expressed genes (DEG, stars): 493 highly expressed in Ly6D+ (Up, red) and 515 highly expressed in TN (Down, blue) identified by bulk RNA‐seq (n = 4).
-
BPCA of 152 Ly6D+ and 213 TN single cells from Flt3Ltg using as gene set the DEG identified in (A) and colored according to the cell type. Average gene expression was centered to zero.
-
C, DKinetics of CD19+ and Ly6D+ EPLM generation in vitro. (C) Ly6D/CD19 FACS plots of the in vitro progeny of Ly6D+ (upper row) and TN EPLM (lower row) at days 1–3 after initiation of culture. Cells shown are SiglecH−CD11c−NK1.1−. (D) Kinetics of Ly6D+ EPLM and CD19+ cell generation in vitro from Ly6D+ (top graph) and TN EPLM (bottom graph).
-
EHeatmap with pairwise Pearson's transcriptome correlation of Ly6D+, TN, and pro‐B averaged populations (bulk RNA‐seq, n = 4).
To address a potential precursor–product relationship between the two subsets, we plated highly purified (> 99%) Ly6D+ and TN EPLMs and assessed whether the TN could initially give rise to the Ly6D+ fraction and subsequently to CD19+ committed B cells in vitro. After 24 h in medium containing Flt3L and IL‐7, we already observed a significant number of Ly6D single positive cells in cultures initiated with sorted Ly6D‐negative cells (Fig 3C lower left panel), indicating that the TN EPLM subpopulation can differentiate into the Ly6D+ subset. B cells expressing CD19 were initially detected at day 2 reaching 40% of live cells in culture at day 6, whereas by this time, Ly6D expression had decreased (Fig 3C and D lower panels). As expected, Ly6D+ cells differentiated faster into B cells, reaching 85% CD19+ cells already at day 4 (Fig 3C and D upper panels), thus confirming that they have higher B‐cell precursor frequency compared with TN, as shown in Fig 1D. The slower kinetics observed in the TN cells might be explained by their transition through a Ly6D+CD19− stage before differentiating to CD19+ B cells. Moreover, the different ability of TN and Ly6D+ cells to generate B cells is reflected in their transcriptome. Thus, when comparing the gene expression profile (RNA‐seq) of bulk Ly6D+ and TN with CD19+CD117int pro‐B cells from Flt3Ltg mice, we observed a higher transcriptome correlation of the Ly6D+ subset to pro‐B cells (r = 0.918), than that of the TN subset (r = 0.886) (Fig 3E).
Single‐cell RNA sequencing identifies two Ly6D+ and three TN subgroups with distinct transcriptional signatures
Apart from the identification of a developmental relationship between Ly6D+ and TN cells, the latter PCA showed that Ly6D+ cells were relatively homogeneous, whereas the TN cells were more heterogeneous (Fig 3B and Dataset EV5). We quantified the degree of cell‐to‐cell heterogeneity by calculating the cell pairwise Pearson's correlation coefficients. Ly6D+ single cells showed an overall higher and seemingly homogeneous transcriptome correlation (predominant yellow/orange color and mean correlation of 0.42, Appendix Fig S4A left) compared with the TN single cells (predominant blue color and mean correlation of 0.32, Appendix Fig S4A right), indeed indicating that the TN EPLM subpopulation has a more heterogeneous transcriptome and might be composed of a mixture of more transcriptionally diverse cells. Subsequent cell clustering using the Partitioning Around Medoids (PAM) algorithm revealed two distinct groups (G1 and G2) of Ly6D+ and three (G3, G4, and G5) of TN cells as illustrated in the PCA plots colored accordingly (Fig 4A, Appendix Fig S4D and Datasets EV6 and EV7). Thus, the Ly6D+ population is further subdivided into G1 Ly6D+, composed of 56 cells and G2 Ly6D+, composed of 82 cells, whereas the TN population is subdivided into G3 TN with 85 cells, G4 TN composed of 52 cells, and G5 TN with 56 cells. In order to explore the degree of similarity among the clustered groups of cells, we calculated their pairwise transcriptome correlation, which revealed that the two Ly6D+ subgroups had higher transcriptome association (r = 0.696) than those observed between any of the TN subgroups (r = 0.582 G3/G4, r = 0.662 G3/G5, r = 0.666 G4/G5) (Fig 4B). Interestingly, the two groups of cells with the highest transcriptome correlation (r = 0.721) are part of different EPLM subpopulations, namely the G2 Ly6D+ and G3 TN (Fig 4B), thereby suggesting that the G3 TN group could be the fraction of the TN population that, as we have observed in culture, is the precursor of the Ly6D+ cells. Finally, the subgroup having the most distinct transcriptome profile is the G4 TN (Fig 4B left column).
Figure 4. Cell clustering identifies three TN and two Ly6D+ subgroups with distinct genetic signatures.
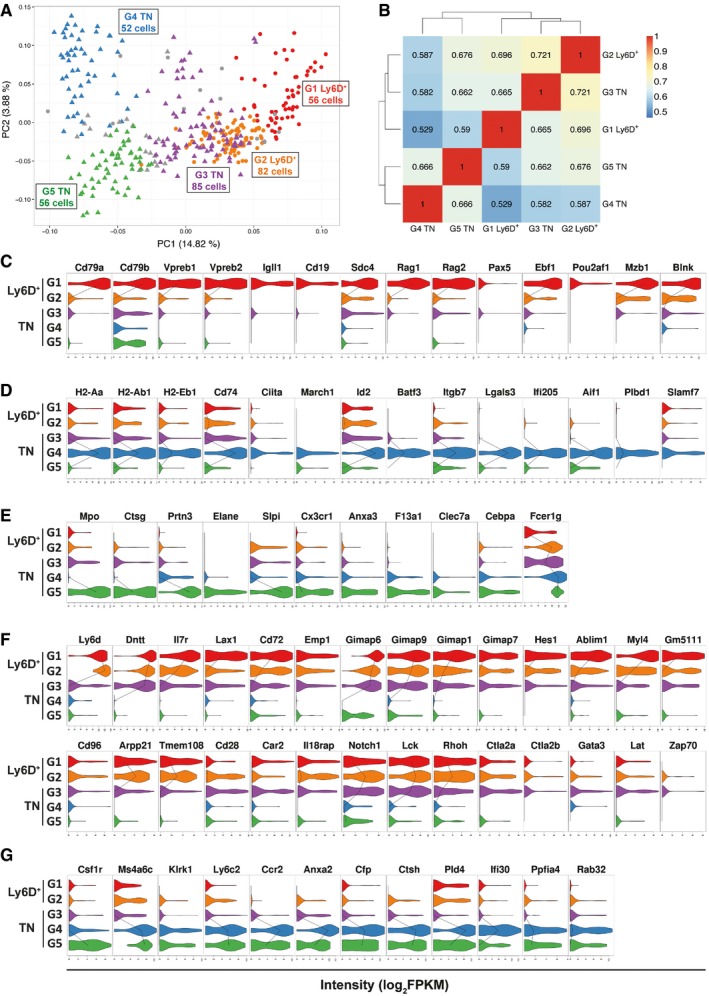
-
APCA generated as in Fig 3B showing the subgroups revealed by PAM clustering method (see Materials and Methods). Circles: Ly6D+, triangles: TN. G1 Ly6D+ (n = 56), G2 Ly6D+ (n = 82), G3 TN (n = 85), G4 TN (n = 52), G5 TN (n = 56).
-
BHeatmap with pairwise Pearson's transcriptome correlation of Ly6D+ and TN subgroups. The number of cells (n) per subgroup is specified in (A).
-
C–GViolin plots with genes highly expressed in G1 Ly6D+ (C), G4 TN (D), G5 TN (E), (G1, G2) Ly6D+ and G3 TN (F), or G4 and G5 TN (G) subgroups compared to the other subgroups. The median expression level is shown with a line when more than 50% of the cells express the indicated gene.
Next, we performed differential gene expression analysis among the clustered groups of cells (Appendix Fig S4B and Dataset EV2). This revealed that only 95 genes were differentially expressed when comparing the two Ly6D+ subgroups (Appendix Fig S4B first row) and with overall low significance levels (Appendix Fig S4C, left). In contrast, comparisons among the TN subgroups yielded more DEG (170–823, Appendix Fig S4B second to fourth rows), and with overall higher significance levels and fold changes (Appendix Fig S4C). Of note, there were only 25 DEG when comparing the two subgroups with the highest transcriptome correlation (Appendix Fig S4B lower row), therefore confirming that the G2 Ly6D+ and G3 TN subgroups of cells sorted as two phenotypically distinct EPLM subpopulations (Ly6D+ and Ly6D−) are highly related cells.
We identified several distinct gene expression patterns for the subgroups and after a detailed screening based on the DEG lists, we show in Fig 4C–G a manually curated selection of genes representative of each expression pattern. Compared to the other subgroups, the G1 Ly6D+ have higher expression levels of genes related to B‐cell biological processes (BP) (Appendix Fig S5A), such as Cd79a (Igα), Cd79b (Igβ), VpreB1/2, Igll1 (λ5), Ebf1, Pax5, or Blnk (Fig 4C). Therefore, although the entire Ly6D+ population is lymphoid‐restricted and has a strong B‐cell developmental potential (Fig 1), single‐cell transcriptomic analysis reveals that the B‐cell signature is mostly contained within the G1 Ly6D+ subgroup. The G4 TN cluster of cells expresses genes characteristic of the conventional dendritic cell (cDC) lineage such as H2‐Aa, H2‐Ab1, H2‐Eb1, Cd74 (Ii), Ciita, March1, Id2, or Batf3 (Fig 4D). Apart from antigen processing and presentation, they are also involved in actin cytoskeleton organization, leukocyte adhesion, actin polymerization and depolymerization, protein complex assembly, and regulation of cellular component size (Appendix Fig S5B). This indicates that this group, which is the most transcriptionally different to the rest, might already express the intracellular machinery necessary to acquire DC morphology and the antigen presenting function characteristic of mature cDC. The G5 TN subgroup is characterized by expression of myeloid‐related genes such as Cebpα, Mpo, Elane, Ctsg, Prtn3, Fcer1g, Clec7a, or Cx3cr1 (Fig 4E) involved in innate biological processes (Appendix Fig S5C). The myeloid signature of G5 TN suggests that this might be the fraction of TN largely containing the observed myeloid potential.
Interestingly, the G2 Ly6D+ and G3 TN cells show a similar gene expression pattern (Fig 4F and G), which is linked to that of the G1 Ly6D+ subset. Quantitatively, some genes are highly expressed in the G1 Ly6D+ cells (Fig 4F upper panels) whereas others in the G2 Ly6D+ and G3 TN cells (Fig 4F lower panels). Among the latter, there are T‐cell‐related genes such as Notch1, Lck, Rhoh, Ctla2a, Ctla2b, Gata3, Lat, or Zap70 (Fig 4F lower right panels), thereby indicating that the G2 Ly6D+ and G3 TN cells might retain T‐cell developmental potential. As a conclusion, these two groups have a lymphoid genetic profile that is not resolved into any lineage, being enriched for B and T biological processes (Appendix Fig S5D) and co‐expressing B, T, and lymphoid genes (Il7r, Dntt, or Lax1, Fig 4F upper left panels). Finally, another identified expression pattern corresponds to mostly myeloid‐related genes (Csfr1, Ccr2, Ifi30, or Ctsh) expressed in both G4 and G5 TN cells (Fig 4G).
In summary, the single‐cell transcriptomic analysis of Ly6D+ and TN EPLM subpopulations reveals that (i) the clustered groups of cells have distinct transcriptional signatures (represented in Fig 5A) and (ii) the degree of heterogeneity in the entire Ly6D+ and TN populations is reflected by their subgroups’ expression profiles. Thus, both Ly6D+ subsets present a lymphoid transcriptional profile, with the G1 Ly6D+ cells showing evidence of B‐cell priming, whereas those of the TN present signatures of both lymphoid (G3 TN) and myeloid (G4 and G5 TN) lineages, including some with a cDC lineage profile (G4 TN).
Figure 5. Mixed‐ and opposed‐lineage states at the single‐cell level.
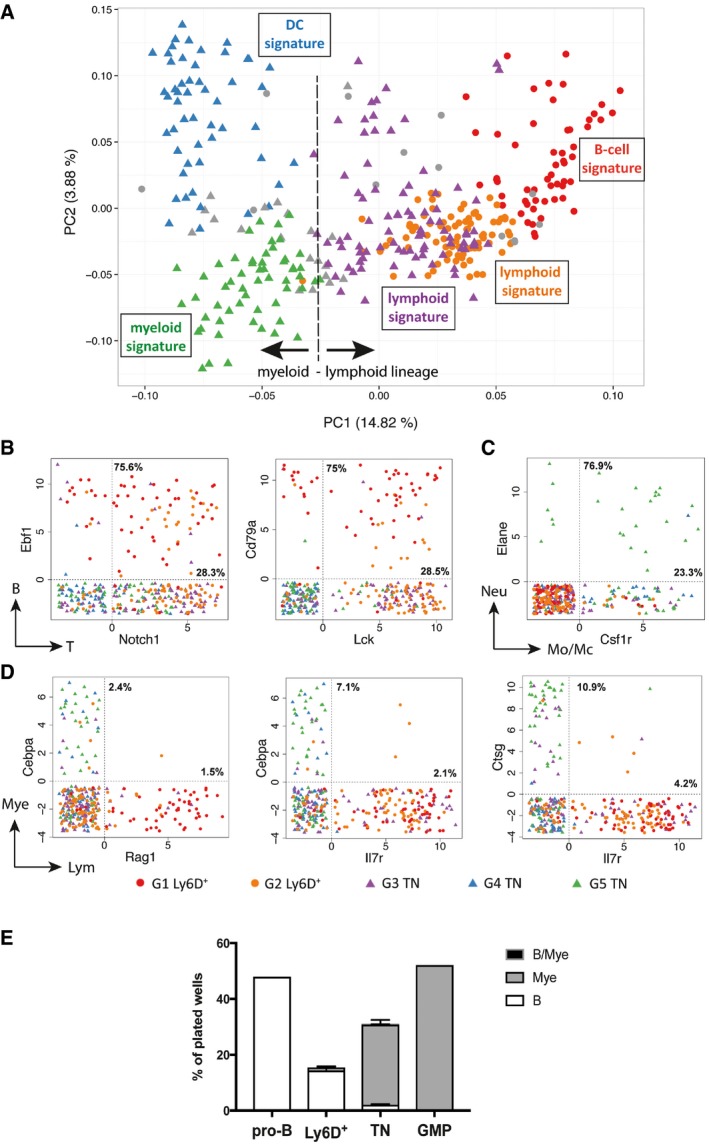
-
ASame PCA plot as in Fig 4A summarizing the genetic signatures of the Ly6D+ and TN subgroups revealed by our in silico analysis.
-
B–DScatter plots showing the expression levels in log2FPKM of selected B and T (B), neutrophil (Neu) and monocyte/macrophages (Mo/Mc) (C) or myeloid (Mye) and lymphoid (Lym) (D) lineage marker pairs in the Ly6D+ and TN subpopulations. Dotted vertical and horizontal lines delimit when the transcript of the indicated gene is detected (> 0). Percentages within the double‐positive area of the plot indicate the fraction of cells co‐expressing both genes to the number of cells expressing only one gene (top: gene on vertical axis; bottom: gene on horizontal axis).
-
EB‐cell, myeloid, and bipotent (B/Mye) developmental potential of the indicated single‐cell sorted populations from Flt3Ltg. Three independent experiments were performed for Ly6D+ and TN cells and one for the control pro‐B and GMP cells. Shown is mean ± SEM.
Expression of lymphoid and myeloid genes is mutually exclusive in single EPLM cells
We have observed that the same group of cells can co‐express genes of different lymphoid or myeloid lineages. For instance, the G2 Ly6D+ cells express both the early B‐cell transcription factor Ebf1 and the T‐cell master regulator Notch1 (Fig 4C and F), whereas the G5 TN cells express both the neutrophil marker Elane and the macrophage colony‐stimulating factor receptor Csf1r (Fig 4E and G). In order to elucidate whether these expression patterns also occur at the single‐cell level, we plotted the expression levels of representative pairs of transcripts, each characteristic of different lineages. We observed that a large proportion of the Ebf1 + cells also expressed Notch1 (75.6%, Fig 5B left). The co‐expression level was also high when comparing the immunoglobulin α chain (Igα or Cd79a) with the T‐cell tyrosine kinase Lck (75% of the Cd79a + and 28.5% of the Lck + cells, Fig 5B right). Moreover, when examining neutrophil–monocyte/macrophage lineages, a high proportion of single cells co‐expressed Elane and Csfr1 (76.9% of the Elane + and 23.3% of the Csfr1 + cells, Fig 5C). Therefore, the EPLM progenitor population contains single cells with mixed‐lineage states within the lymphoid (B and T) and myeloid (granulocyte and monocyte/macrophage) lineages.
In contrast, we detected distinct lymphoid and myeloid specification for the EPLM subgroups, with the cells on the left part of the PCA plot being myeloid primed (G4 and G5 TN), whereas those in the center (G2 Ly6D+ and G3 TN) and on the right (G1 Ly6D+) being lymphoid primed (Fig 5A). This marked lympho‐myeloid separation was confirmed at the single‐cell level since we did not encounter significant co‐expression of early myeloid (Cebpa and Ctsg) and lymphoid (Rag1 and Il7r) specification genes (Fig 5D). The mutually exclusive expression of lymphoid and myeloid genes indicates that EPLM might be composed of a mixture of cells with either lymphoid or myeloid molecular priming and that the multilineage developmental potential observed for the TN EPLM subpopulation is possibly not contained in the same single cell. In order to address that, we sorted single Ly6D+ and TN cells and cultured them under conditions promoting both lymphoid and myeloid lineages (OP9 stromal cells supplemented with IL‐7 and MCSF). Ly6D+ mostly generated B‐cell colonies (14.3%, 166/1,161), albeit at a lower frequency compared with pro‐B cells (48%, 92/192), whereas TN mainly differentiated into myeloid cells (28%, 584/2,088), even though at lower frequency than granulocyte–macrophage progenitors (GMP) (52.1%, 100/192), and only 2.1% of TN generated B cells (Fig 5E). These frequencies resemble the ones obtained in limiting dilution assays (Fig 2D and F). Interestingly, only three out of 2,088 (0.15%) TN‐containing wells resulted into mixed lympho‐myeloid colonies (Fig 5E). Therefore, based on both the molecular and functional data presented herein, we conclude that single EPLMs are not bipotent for lymphoid and myeloid lineages.
Discussion
The emergence of high‐throughput sequencing methods enabling the investigation of single‐cell whole‐transcriptome profiles generates data that contribute to the active debate regarding the molecular heterogeneity of phenotypically homogenous progenitors having multiple lineage potentials. In this study, we present a comprehensive characterization of the heterogeneity of EPLM, a previously described uncommitted and lympho‐myeloid multipotent hematopoietic B220+CD117intCD19−NK1.1− progenitor (Balciunaite et al, 2005).
First, we found that phenotypically, EPLM expressed heterogeneous levels of the cell surface markers Ly6D, SiglecH, and CD11c, resulting in the subdivision of the progenitor into at least four subpopulations (Fig 1A). These markers have been already reported to be associated with different hematopoietic lineages. Therefore, it is not surprising that when we assessed the developmental potential of the above‐identified EPLM subsets at the population level by in vitro differentiation assays, we observed that the phenotypic heterogeneity of EPLM was reflected in functional heterogeneity with different subpopulations having different sets of potentials (Fig 1D–F). We found that SiglecH+ and CD11c+ subsets could not generate lymphoid cells, suggesting that in agreement with their cell surface marker profile, they could be (at least a fraction of them) already committed to the pDC and cDC lineages. Ly6D+ cells were lymphoid‐restricted, and notably, the TN subset, which lacks expression of the three specification markers, showed trilineage (B, T, and myeloid) developmental potential although with lower B‐ and T‐cell precursor frequencies compared with their Ly6D+ counterparts.
Next, we performed scRNA‐seq to further explore the heterogeneity of EPLM subpopulations having B‐cell developmental potential (Ly6D+ and TN) and sought to identify a B‐cell specified subset prior to commitment. For that, we made use of a mouse model generated in our laboratory, the Flt3Ltg mouse line, which shows a significant but proportional increase of all EPLM subsets (Fig 2). Given the technical limitations of the system used (C1 Fluidigm platform) for the scRNA‐seq experiment, the Flt3Ltg mouse is currently the most suitable model to isolate EPLM subpopulations in large numbers for scRNA‐seq and for functional and molecular experiments. The C1 Fluidigm system requires a loading of at least 3,000 cells and captures a maximum of 96 per run. Therefore, to investigate rare populations, new commercial platforms with higher capture efficiency and throughput such as the recently reported GemCode Technology [10× Genomics (Zheng et al, 2017)] are needed. The study reported herein captured 365 single‐cell gene expression snapshots of the Ly6D+ (152) and TN (213) transcriptional landscapes.
Our initial analysis indicated that the TN EPLM subset is more heterogeneous than their Ly6D+ partners, as suggested by their branching structure in the PCA and their low cell‐to‐cell transcriptome correlation (Fig 3B and Appendix Fig S4A). Moreover, the PAM clustering based on the selected subset of genes partitioned Ly6D+ EPLM into two major clusters, whereas the TN subset was divided into three robust groups. Importantly, when comparing the two EPLM subpopulations, we found that they are transcriptionally (Fig 3B) and developmentally related, with a fraction of the TN subset being the precursor of Ly6D+ EPLM (Fig 3C and D). This result explains the slower kinetics and decreased efficiency of the latter progenitor in differentiating to CD19+ B cells. Single‐cell clustering was able to identify a fraction of the TN cells, namely G3 TN that is almost transcriptionally identical to G2 Ly6D+ cells (Fig 4A). These two subgroups have the highest transcriptome correlation and the lowest number of DEG. Therefore, although they are sorted as two phenotypically distinct populations, the G2 Ly6D+ and G3 TN subgroups could be considered as one subset. This finding highlights the limitations of accurately defining complete cellular identities by relying on expression of a few cell surface markers, the so‐called top‐down approach (Satija & Shalek, 2014). This notion has been manifested in other studies such as when Paul et al (2015) suggested that the standard gating for sorting megakaryocyte–erythrocyte progenitors (MEP) might be better‐termed EP gating. In terms of validation, a limitation of the scRNA‐seq approach is that it implies a subsequent prospective strategy, which, although useful for some research studies (Mahata et al, 2014; Drissen et al, 2016), depends on the identification of cell surface markers or the existence of reporter mice. Unfortunately, from the transcriptome profile, we could not find a robust cell surface marker defining the G3 TN cells that would enable us to prospectively isolate the newly identified subgroup.
We examined whether our cell clustering reflects gene expression signatures of progenitors from distinct hematopoietic lineages. For that, we performed our analysis on all (14,814) detected genes across the 365 single cells and examined the DEG and enriched biological processes defining each subgroup. With this approach, we were able to unravel marked transcriptional biases between individual EPLM subsets indicative of molecular priming toward distinct fates. The G1 Ly6D+ subgroup showed a strong B‐cell transcriptional signature (red in Fig 5A) with robust expression of B‐cell‐related genes characteristic of the pro‐B‐cell stage (Cd79a, Vpreb genes, Igll1, Cd19, Ebf1, or Blnk Fig 4C) and B‐cell enriched biological processes (Appendix Fig S5A). Therefore, we propose the herein newly identified G1 Ly6D+ subset, phenotypically closely related to PDCA‐1− BLP and PDCA‐1− pre‐pro‐B cells (Medina et al, 2013), as the direct precursor of the first B‐cell committed stage, namely the CD117intCD19+ pro‐B cell. The G2 Ly6D+/G3 TN subset (orange and purple, respectively, in Fig 5A) showed a lymphoid transcriptional signature (Fig 4C and F) with a B‐cell specification less marked than that of the G1 Ly6D+, revealing a B‐cell priming gradient from G2 Ly6D+/G3 TN to G1 Ly6D+. Similarly, Paul et al (2015) reported a gradient of erythrocyte transcription. In addition, the latter subset might be, as reflected by their central location in the PCA, in an intermediate state while exhibiting promiscuous expression of B‐ and T‐cell genes. The G4 TN subgroup, which has the most distant transcriptome, revealed a cDC transcriptional signature (blue in Fig 5A) with consistent expression of genes (H2 genes, Cd74, Ciita, Id2, or Batf3 Fig 4D) and enriched biological processes (Appendix Fig S5B) either related to antigen processing and presentation or necessary for cDC development. Finally, the G5 TN subgroup exhibited a myeloid transcriptional signature (green in Fig 5A), expressing myeloid genes (Mpo, Ctsg, Prtn3, Elane, Cx3cr1, Cebpa, or Csfr1 Fig 4E) related to innate processes (Appendix Fig S5C). Taken together, these data suggest functional heterogeneity among the EPLM subpopulations and validate single‐cell RNA sequencing as a powerful technology to identify biologically meaningful subgroups and unravel transitional states.
We also investigated lineage priming at the single‐cell level and found a significant proportion of single cells co‐expressing early B‐ and T‐cell (Fig 5B) or granulocyte and monocyte/macrophage (Fig 5C) specification genes. This is consistent with other studies where mixed lymphoid (Miyamoto et al, 2002; Mansson et al, 2010) or myeloid (Hu et al, 1997; Naik et al, 2013; Olsson et al, 2016) lineage patterns of gene expression are reported in single cells. However, whereas heterogeneity is well studied in myeloid progenitors, we are not aware of other reports addressing mixed lymphoid priming at the single‐cell level and whole‐transcriptome scale. Strikingly, and in agreement with other reports (Sakhinia et al, 2006; Ng et al, 2009; Schlenner & Rodewald, 2010; Schlenner et al, 2010; Guo et al, 2013), we did not observe single cells with mixed lymphoid and myeloid gene expression profiles (Fig 5D). Further supporting these findings, Schlenner et al (2010) made use of an Il7r fate mapping mouse line to determine the non‐lymphoid origin of thymic myeloid cells. Nevertheless, we cannot exclude that due to the “snapshot” nature of the transcriptomic analysis, as well as the medium throughput of cells analyzed, we are missing a transient and presumably rare intermediate state with promiscuous lympho‐myeloid gene expression. Our functional results, with only 0.15% (below the impurity sorting threshold) of lympho‐myeloid mixed clones derived from single EPLM, argue that the bifurcation of the lymphoid and myeloid molecular priming and developmental potential occurs before the EPLM stage. Therefore, the common or separate origin of the Ly6D+/G3 TN (lymphoid primed) versus the G4/G5 TN (myeloid primed) EPLM fractions is of interest and requires further investigation. In line with our findings, there is an increasing body of evidence supporting the notion that priming occurs much earlier in development than previously thought. Indeed, expression of lineage‐affiliated genes has been reported as early as in the HSC stage, with various analyses indicating biases at the apex of hematopoiesis (Benz et al, 2012; Guo et al, 2013; Moignard et al, 2013, 2015; Naik et al, 2013; Ema et al, 2014; Tsang et al, 2015; Nestorowa et al, 2016; Notta et al, 2016). The lack of lympho‐myeloid bipotency on single EPLMs also suggests that the lymphoid‐primed TN subset identified by scRNA‐seq analysis, namely the G3 TN, might be the fraction of TN cells that retains B‐cell potential and gives rise to Ly6D+ cells in culture.
In summary, our study first identifies four phenotypically and functionally distinct subpopulations of the previously reported EPLM hematopoietic progenitor and subsequently provides a comprehensive study of the molecular and functional heterogeneity of two EPLM subsets, Ly6D+ and TN cells, which retain B‐cell developmental potential. Whereas the Ly6D+ subset is composed of two lymphoid specified subgroups with a B‐cell priming gradient, the TN subset is composed of three groups of cells with lymphoid or myeloid transcriptional signatures and developmental potentials, including some cells with a cDC lineage profile. Finally, we favor the concept that the lympho‐myeloid potential of the EPLM progenitor is not maintained at the single‐cell level, thus providing another good example to support the finding that previously characterized multipotent progenitor populations are in fact composed of mixtures of cells with differently restricted differentiation capacities. Ultimately, this study makes a significant contribution in the characterization of phenotypic and transcriptomic heterogeneity and lineage priming of progenitors during early stages of lymphoid development.
Materials and Methods
Mice
C57BL/6 (WT), B6 Rag2‐deficient (Shinkai et al, 1992), B6 Flt3L transgenic [Flt3Ltg (Tsapogas et al, 2014)], and Pax5‐reporter (Fuxa & Busslinger, 2007) (in WT or Flt3Ltg background) mice used herein were 6–11 weeks old and matched by age and sex for each experiment. All mice were bred and maintained in our animal facility under specific pathogen‐free conditions. All animal experiments were carried out according to institutional guidelines (authorization numbers 1886 and 1888 from Kantonales Veterinäramt, Basel).
Flow cytometry and cell sorting
Bone marrow cell suspensions were obtained from both femurs of individual mice as indicated in Appendix Supplementary Methods. For flow cytometry or cell sorting, the following antibodies were used (from BD Pharmingen, eBioscience, BioLegend, or produced in‐house): anti‐B220 (RA3‐6B2), anti‐CD117 (2B8), anti‐CD19 (1D3), anti‐NK1.1 (PK136), anti‐SiglecH (551), anti‐CD11c (HL3), anti‐Ly6D (49‐H4), anti‐Thy1.2 (53‐2.1), anti‐F4/80 (F4/80) conjugated with FITC, PE, PE/Cy7, APC, BV421, or biotin. Biotin‐labeled antibodies were revealed using streptavidin‐BV650. Analytical flow cytometry was performed using a BD LSR Fortessa (BD Biosciences) and data were analyzed using FlowJo v9.8 Software (Treestar). For cell sorting, a FACS Aria IIu (BD Biosciences) was used and in all instances, sorted bulk cells were > 98% pure.
In vitro cultures
Limiting dilution assay
ST2, OP9, and OP9 stromal cells expressing the Notch ligand Delta‐like 1 (OP9‐DL1) were co‐cultured with sorted progenitor cells as previously described [(Ceredig et al, 2006) and Appendix Supplementary Methods].
Bulk cultures with cytokines
Thirty‐five thousand sorted hematopoietic progenitors from Flt3Ltg mice were cultured with 50 ng/ml Flt3L prepared in‐house and 100 U/ml IL‐7 in a 24‐well flat‐bottom plate. Cells were maintained as previously described (Ceredig et al, 2006) and from day 1 to day 6, one well containing cells from each population was analyzed by flow cytometry for Ly6D and CD19 expression.
Single‐cell cultures
Single Ly6D+ and TN cells from two pooled Flt3Ltg mice were sorted on 96‐well plates and co‐cultured with OP9 stromal cells supplemented with 100 U/ml IL‐7 and 10 ng/μl MCSF (PreproTech). Wells were scored as B‐cell clones (after 10 days), myeloid clones (at day 15), or mixed clones (at day 15) using an inverted microscope and flow cytometry staining.
In vivo reconstitution assay
Recipient Rag2‐deficient mice were γ‐irradiated using a Cobalt source (Gammacell 40, Atomic Energy of Canada, Ltd) at a dose of 400 rad 4 h prior to reconstitution. Indicated numbers of sorted hematopoietic progenitors from donor mice (WT or Flt3Ltg) were injected intravenously. After 3 weeks, spleen and thymus of recipient mice were separately analyzed by flow cytometry.
Statistical analysis
Statistical analysis was performed with GraphPad Prism v6.0f software. Two‐tailed unpaired Student's t‐tests were used for statistical comparisons. Data are presented as mean values ± SEM (n.s. not significant or P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001). The exact P‐value is indicated in the figure legend.
Bulk RNA sequencing
Ly6D+ and TN EPLM subpopulations as well as CD19+CD117int pro‐B cells were sorted from femurs of two male Flt3Ltg mice (6–8 weeks of age) in quadruplicates. After each sort, 100 μl containing ~3 × 104 cells from the Ly6D+ and TN samples was used for the capture of single cells. The remaining cells were centrifuged, resuspended in 0.5 ml of TRIzol reagent, and stored at −80°C for later total RNA extraction and bulk RNA sequencing (Appendix Supplementary Methods).
Single‐cell RNA sequencing
Capture of single cells
Single cells were captured from ex vivo sorted hematopoietic progenitors on a small‐sized (5–10 μm) “C1 Single‐Cell Auto Prep IFC for mRNA sequencing” (Fluidigm) using the Fluidigm C1 system as explained in Appendix Supplementary Methods. A total of three chips per population were used yielding to 178 Ly6D+ and 232 TN single cells captured (Appendix Fig S3A and B). Subsequently, cells were lysed, the polyA containing mRNA molecules were hybridized to oligo‐dT, and whole‐transcriptome full‐length amplified cDNA was prepared by template switching on the C1 chip using the SMARTer Ultra Low RNA kit for the Fluidigm C1 System (Clontech). Quantification of cDNA was done with Quant‐iT PicoGreen dsDNA Assay Kit; TEcan instrument.
Library preparation and sequencing
Illumina single‐cell libraries were constructed in 96‐well plates using the Nextera XT DNA Library Preparation Kit (Illumina) following the protocol supplied by Fluidigm (“Using C1 to Generate Single‐Cell cDNA Libraries for mRNA Sequencing” and Appendix Supplementary Methods). Indexed DNA libraries originated from single cells captured in three different chips (288 libraries) were pooled in equal volumes and loaded on one NextSeq 500 High Output flow cell (Illumina). Single‐end sequencing was performed on the Illumina NextSeq™ 500 Sequencing System (D‐BSSE, Basel) for 76 cycles. Only FastQ files corresponding to C1 chambers with a single cell were selected (Appendix Supplementary Methods). We obtained a total of 360 and 371 millions of reads for the Ly6D+ and TN cells, respectively. The average number of reads per cell was 2 × 106 for the Ly6D+ and 1.6 × 106 for the TN (Appendix Fig S3C).
Pre‐processing of sequencing data
All downstream analysis was performed using the open‐source R software accessed via RStudio server (R version 3.2.0). Sequencing reads were aligned and count table was generated as explained in Appendix Supplementary Methods. Approximately 80% of total reads were successfully mapped for each sample (Appendix Fig S3D). Total counts per cell were approximately 8.1 × 105 for the Ly6D+ and 7.2 × 105 for the TN (Appendix Fig S3E). Genes with no counts across all samples were excluded from the analysis. At least one read was detected for a total of 14,814 genes across all 410 captured cells, corresponding to approximately 3,500 expressed genes per cell in both Ly6D+ and TN (Appendix Fig S3F). During the quality control, cells having < 60% of mapped reads, < 2 × 105 counts, or < 800 detected genes were filtered out from further analysis (dotted red lines in Appendix Fig S3). In total, 89% of the cells (152 Ly6D+ and 213 TN) passed these criteria. Raw counts were normalized between cells and genes, expressed as fragments per kilobase of transcript per million mapped reads (FPKM) and transformed to the log2‐scale (log2FPKM).
Data analysis
If not otherwise specified, the downstream analysis was performed using the 1,008 DEG [false discovery rate (FDR) < 0.05 and abs|log2(FoldChange)| > 1] from the bulk RNA‐seq experiment when comparing Ly6D+ with TN populations.
Dimensionality reduction was performed with PCA. Average gene expression was centered to zero, and PCA plots were generated with the ggplot2 v2.1.0 R package. To visualize the degree of cell‐to‐cell heterogeneity, an annotated heatmap of sample pairwise Pearson's correlation coefficients was produced using the NMF v0.20.6 R package. Eight Ly6D+ cells were not considered for subsequent clustering because of their very low transcriptome correlation to any other cell, on average < 0.3 (Appendix Fig S4A left). Cell clustering was performed using the PAM method implemented in the cluster v2.0.4 R package (Reynolds et al, 2006) as explained in Appendix Supplementary Methods. The optimal number of clusters was K = 2 for Ly6D+ (with average silhouette width of 0.10) and K = 3 for the TN (with average silhouette width of 0.13). Cells with negative silhouette width values were excluded while the other 331 cells were assigned to one of the five groups. Average expression across all detected genes was calculated for each of the five clusters of single cells, and a heatmap with Pearson's correlation coefficients was generated with the top 50% of genes with highest variance across analyzed datasets (calculated as inter‐quartile range) and visualized with the NMF v0.20.6 R package.
Differential gene expression analysis to compare the clustered groups of cells was performed using the 14,528 detected expressed genes across the 331 single cells with edgeR v3.12.1 (Robinson et al, 2010). Genes with a FDR < 0.05 and abs|log2(FoldChange)| > 1 were considered as differentially expressed. Volcano, violin, and scatter plots were produced using custom R scripts.
Gene Ontology enrichment analysis was performed with the DAVID 6.8 bioinformatics database, based on Fisher's exact method (Huang da et al, 2009a,b).
Data availability
The bulk RNA‐seq as well as the single‐cell RNA‐seq data from this publication has been deposited in NCBI's Gene Expression Omnibus database (Edgar et al, 2002; Barrett et al, 2013) (https://www.ncbi.nlm.nih.gov/geo/) and assigned the GEO Series accession number GSE102456 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE102456).
Author contributions
LA‐S performed experiments, analyzed the bioinformatics data, and wrote the manuscript; LvM and PT performed experiments, analyzed data, and revised the manuscript; GC performed experiments; KE performed the single‐cell capture, prepared the libraries, and performed the sequencing; CB provided the Fluidigm C1 system and technical advice; RC designed experiments and revised the manuscript; RI analyzed the bioinformatics data and revised the manuscript. AR designed and performed experiments, revised the manuscript, and supervised the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Review Process File
Acknowledgements
We dedicate this work to the memory of our dear mentor, colleague and friend Prof. Antonius Rolink. We thank members of the Rolink laboratory and the DECIDE network for critical discussions, and Julien Roux for reviewing the manuscript. We thank Meinrad Busslinger for providing the Pax5‐reporter mice. A.R. was holder of the chair in immunology endowed by L. Hoffmann–La Roche Ltd, Basel. This study was supported by the Swiss National Science Foundation and by the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007–2013 under Research Executive Agency grant agreement number 315902. R.C. was supported by Science Foundation Ireland under grant numbers SFI09/SRC/B1794 and SFI07/SK/B1233b. Calculations were performed at sciCORE (https://scicore.unibas.ch/) scientific computing core facility at University of Basel.
See also: G Karlsson et al (December 2017)
References
- Abramson S, Miller RG, Phillips RA (1977) The identification in adult bone marrow of pluripotent and restricted stem cells of the myeloid and lymphoid systems. J Exp Med 145: 1567–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adolfsson J, Mansson R, Buza‐Vidas N, Hultquist A, Liuba K, Jensen CT, Bryder D, Yang L, Borge OJ, Thoren LA, Anderson K, Sitnicka E, Sasaki Y, Sigvardsson M, Jacobsen SE (2005) Identification of Flt3+ lympho‐myeloid stem cells lacking erythro‐megakaryocytic potential a revised road map for adult blood lineage commitment. Cell 121: 295–306 [DOI] [PubMed] [Google Scholar]
- Akashi K, Traver D, Miyamoto T, Weissman IL (2000) A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404: 193–197 [DOI] [PubMed] [Google Scholar]
- Balciunaite G, Ceredig R, Massa S, Rolink AG (2005) A B220+ CD117+ CD19− hematopoietic progenitor with potent lymphoid and myeloid developmental potential. Eur J Immunol 35: 2019–2030 [DOI] [PubMed] [Google Scholar]
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A (2013) NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res 41: D991–D995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz C, Copley MR, Kent DG, Wohrer S, Cortes A, Aghaeepour N, Ma E, Mader H, Rowe K, Day C, Treloar D, Brinkman RR, Eaves CJ (2012) Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell 10: 273–283 [DOI] [PubMed] [Google Scholar]
- Blasius AL, Cella M, Maldonado J, Takai T, Colonna M (2006) Siglec‐H is an IPC‐specific receptor that modulates type I IFN secretion through DAP12. Blood 107: 2474–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G, Mooney CJ, Alberti‐Servera L, Muenchow L, Toellner KM, Ceredig R, Rolink A (2015) Versatility of stem and progenitor cells and the instructive actions of cytokines on hematopoiesis. Crit Rev Clin Lab Sci 52: 168–179 [DOI] [PubMed] [Google Scholar]
- Busch K, Klapproth K, Barile M, Flossdorf M, Holland‐Letz T, Schlenner SM, Reth M, Hofer T, Rodewald HR (2015) Fundamental properties of unperturbed haematopoiesis from stem cells in vivo . Nature 518: 542–546 [DOI] [PubMed] [Google Scholar]
- Ceredig R, Rauch M, Balciunaite G, Rolink AG (2006) Increasing Flt3L availability alters composition of a novel bone marrow lymphoid progenitor compartment. Blood 108: 1216–1222 [DOI] [PubMed] [Google Scholar]
- Ceredig R, Rolink AG, Brown G (2009) Models of haematopoiesis: seeing the wood for the trees. Nat Rev Immunol 9: 293–300 [DOI] [PubMed] [Google Scholar]
- Drissen R, Buza‐Vidas N, Woll P, Thongjuea S, Gambardella A, Giustacchini A, Mancini E, Zriwil A, Lutteropp M, Grover A, Mead A, Sitnicka E, Jacobsen SE, Nerlov C (2016) Distinct myeloid progenitor‐differentiation pathways identified through single‐cell RNA sequencing. Nat Immunol 17: 666–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ema H, Morita Y, Suda T (2014) Heterogeneity and hierarchy of hematopoietic stem cells. Exp Hematol 42: 74–82 e72 [DOI] [PubMed] [Google Scholar]
- Fogg DK, Sibon C, Miled C, Jung S, Aucouturier P, Littman DR, Cumano A, Geissmann F (2006) A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 311: 83–87 [DOI] [PubMed] [Google Scholar]
- Fuxa M, Busslinger M (2007) Reporter gene insertions reveal a strictly B lymphoid‐specific expression pattern of Pax5 in support of its B cell identity function. J Immunol 178: 8222–8228 [DOI] [PubMed] [Google Scholar]
- Gren ST, Rasmussen TB, Janciauskiene S, Hakansson K, Gerwien JG, Grip O (2015) A single‐cell gene‐expression profile reveals inter‐cellular heterogeneity within human monocyte subsets. PLoS One 10: e0144351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes JE, Francis GE, Bol SJ, Berney JJ, Hoffbrand AV (1982) Differentiation restriction in the neutrophil‐granulocyte, macrophage, eosinophil‐granulocyte pathway: analysis by equilibrium density centrifugation. Leuk Res 6: 791–800 [DOI] [PubMed] [Google Scholar]
- Guo G, Luc S, Marco E, Lin TW, Peng C, Kerenyi MA, Beyaz S, Kim W, Xu J, Das PP, Neff T, Zou K, Yuan GC, Orkin SH (2013) Mapping cellular hierarchy by single‐cell analysis of the cell surface repertoire. Cell Stem Cell 13: 492–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes ML, Carotta S, Corcoran LM, Nutt SL (2006) Repression of Flt3 by Pax5 is crucial for B‐cell lineage commitment. Genes Dev 20: 933–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe PS, Schwarzfischer M, Loeffler D, Kokkaliaris KD, Hilsenbeck O, Moritz N, Endele M, Filipczyk A, Gambardella A, Ahmed N, Etzrodt M, Coutu DL, Rieger MA, Marr C, Strasser MK, Schauberger B, Burtscher I, Ermakova O, Burger A, Lickert H, et al (2016) Early myeloid lineage choice is not initiated by random PU.1 to GATA1 protein ratios. Nature 535: 299–302 [DOI] [PubMed] [Google Scholar]
- Hu M, Krause D, Greaves M, Sharkis S, Dexter M, Heyworth C, Enver T (1997) Multilineage gene expression precedes commitment in the hemopoietic system. Genes Dev 11: 774–785 [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA (2009a) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA (2009b) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Inlay MA, Bhattacharya D, Sahoo D, Serwold T, Seita J, Karsunky H, Plevritis SK, Dill DL, Weissman IL (2009) Ly6d marks the earliest stage of B‐cell specification and identifies the branchpoint between B‐cell and T‐cell development. Genes Dev 23: 2376–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa F, Niiro H, Iino T, Yoshida S, Saito N, Onohara S, Miyamoto T, Minagawa H, Fujii S, Shultz LD, Harada M, Akashi K (2007) The developmental program of human dendritic cells is operated independently of conventional myeloid and lymphoid pathways. Blood 110: 3591–3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura Y, Kawamoto H (2001) Stepwise lineage restriction of progenitors in lympho‐myelopoiesis. Int Rev Immunol 20: 1–20 [DOI] [PubMed] [Google Scholar]
- Kawamoto H, Ikawa T, Masuda K, Wada H, Katsura Y (2010) A map for lineage restriction of progenitors during hematopoiesis: the essence of the myeloid‐based model. Immunol Rev 238: 23–36 [DOI] [PubMed] [Google Scholar]
- Kim KT, Lee HW, Lee HO, Kim SC, Seo YJ, Chung W, Eum HH, Nam DH, Kim J, Joo KM, Park WY (2015) Single‐cell mRNA sequencing identifies subclonal heterogeneity in anti‐cancer drug responses of lung adenocarcinoma cells. Genome Biol 16: 127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo M, Weissman IL, Akashi K (1997) Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 91: 661–672 [DOI] [PubMed] [Google Scholar]
- Kowalczyk MS, Tirosh I, Heckl D, Rao TN, Dixit A, Haas BJ, Schneider RK, Wagers AJ, Ebert BL, Regev A (2015) Single‐cell RNA‐seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res 25: 1860–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YS, Wasserman R, Hayakawa K, Hardy RR (1996) Identification of the earliest B lineage stage in mouse bone marrow. Immunity 5: 527–535 [DOI] [PubMed] [Google Scholar]
- Mahata B, Zhang X, Kolodziejczyk AA, Proserpio V, Haim‐Vilmovsky L, Taylor AE, Hebenstreit D, Dingler FA, Moignard V, Gottgens B, Arlt W, McKenzie AN, Teichmann SA (2014) Single‐cell RNA sequencing reveals T helper cells synthesizing steroids de novo to contribute to immune homeostasis. Cell Rep 7: 1130–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansson R, Zandi S, Welinder E, Tsapogas P, Sakaguchi N, Bryder D, Sigvardsson M (2010) Single‐cell analysis of the common lymphoid progenitor compartment reveals functional and molecular heterogeneity. Blood 115: 2601–2609 [DOI] [PubMed] [Google Scholar]
- Medina KL, Tangen SN, Seaburg LM, Thapa P, Gwin KA, Shapiro VS (2013) Separation of plasmacytoid dendritic cells from B‐cell‐biased lymphoid progenitor (BLP) and Pre‐pro B cells using PDCA‐1. PLoS One 8: e78408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min JW, Kim WJ, Han JA, Jung YJ, Kim KT, Park WY, Lee HO, Choi SS (2015) Identification of distinct tumor subpopulations in lung adenocarcinoma via single‐cell RNA‐seq. PLoS One 10: e0135817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Iwasaki H, Reizis B, Ye M, Graf T, Weissman IL, Akashi K (2002) Myeloid or lymphoid promiscuity as a critical step in hematopoietic lineage commitment. Dev Cell 3: 137–147 [DOI] [PubMed] [Google Scholar]
- Moignard V, Macaulay IC, Swiers G, Buettner F, Schutte J, Calero‐Nieto FJ, Kinston S, Joshi A, Hannah R, Theis FJ, Jacobsen SE, de Bruijn MF, Gottgens B (2013) Characterization of transcriptional networks in blood stem and progenitor cells using high‐throughput single‐cell gene expression analysis. Nat Cell Biol 15: 363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moignard V, Woodhouse S, Haghverdi L, Lilly AJ, Tanaka Y, Wilkinson AC, Buettner F, Macaulay IC, Jawaid W, Diamanti E, Nishikawa S, Piterman N, Kouskoff V, Theis FJ, Fisher J, Gottgens B (2015) Decoding the regulatory network of early blood development from single‐cell gene expression measurements. Nat Biotechnol 33: 269–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Muenchow L, Alberti‐Servera L, Klein F, Capoferri G, Finke D, Ceredig R, Rolink A, Tsapogas P (2016) Permissive roles of cytokines interleukin‐7 and Flt3 ligand in mouse B‐cell lineage commitment. Proc Natl Acad Sci USA 113: E8122–E8130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik SH, Perie L, Swart E, Gerlach C, van Rooij N, de Boer RJ, Schumacher TN (2013) Diverse and heritable lineage imprinting of early haematopoietic progenitors. Nature 496: 229–232 [DOI] [PubMed] [Google Scholar]
- Nestorowa S, Hamey FK, Pijuan Sala B, Diamanti E, Shepherd M, Laurenti E, Wilson NK, Kent DG, Gottgens B (2016) A single‐cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128: e20–e31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SY, Yoshida T, Zhang J, Georgopoulos K (2009) Genome‐wide lineage‐specific transcriptional networks underscore Ikaros‐dependent lymphoid priming in hematopoietic stem cells. Immunity 30: 493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, Kaufmann KB, McLeod J, Laurenti E, Dunant CF, McPherson JD, Stein LD, Dror Y, Dick JE (2016) Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 351: aab2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt SL, Heavey B, Rolink AG, Busslinger M (1999) Commitment to the B‐lymphoid lineage depends on the transcription factor Pax5. Nature 401: 556–562 [DOI] [PubMed] [Google Scholar]
- Olsson A, Venkatasubramanian M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H, Grimes HL (2016) Single‐cell analysis of mixed‐lineage states leading to a binary cell fate choice. Nature 537: 698–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren‐Shaul H, Winter D, Lara‐Astiaso D, Gury M, Weiner A, David E, Cohen N, Lauridsen FK, Haas S, Schlitzer A, Mildner A, Ginhoux F, Jung S, Trumpp A, Porse BT et al (2015) Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell 163: 1663–1677 [DOI] [PubMed] [Google Scholar]
- Reynolds AP, Richards G, de la Iglesia B, Rayward‐Smith VJ (2006) Clustering rules: a comparison of partitioning and hierarchical clustering algorithms. J Math Modelling Algorithms 5: 475–504 [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolink AG, Schaniel C, Bruno L, Melchers F (2002) In vitro and in vivo plasticity of Pax5‐deficient pre‐B I cells. Immunol Lett 82: 35–40 [DOI] [PubMed] [Google Scholar]
- Sakhinia E, Byers R, Bashein A, Hoyland J, Buckle AM, Brady G (2006) Gene expression analysis of myeloid and lymphoid lineage markers during mouse haematopoiesis. Br J Haematol 135: 105–116 [DOI] [PubMed] [Google Scholar]
- Satija R, Shalek AK (2014) Heterogeneity in immune responses: from populations to single cells. Trends Immunol 35: 219–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenner SM, Madan V, Busch K, Tietz A, Laufle C, Costa C, Blum C, Fehling HJ, Rodewald HR (2010) Fate mapping reveals separate origins of T cells and myeloid lineages in the thymus. Immunity 32: 426–436 [DOI] [PubMed] [Google Scholar]
- Schlenner SM, Rodewald HR (2010) Early T cell development and the pitfalls of potential. Trends Immunol 31: 303–310 [DOI] [PubMed] [Google Scholar]
- Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, Lu D, Chen P, Gertner RS, Gaublomme JT, Yosef N, Schwartz S, Fowler B, Weaver S, Wang J, Wang X, Ding R, Raychowdhury R, Friedman N, Hacohen N, Park H et al (2014) Single‐cell RNA‐seq reveals dynamic paracrine control of cellular variation. Nature 510: 363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall AM, Alt FW (1992) RAG‐2‐deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 68: 855–867 [DOI] [PubMed] [Google Scholar]
- Singh‐Jasuja H, Thiolat A, Ribon M, Boissier MC, Bessis N, Rammensee HG, Decker P (2013) The mouse dendritic cell marker CD11c is down‐regulated upon cell activation through Toll‐like receptor triggering. Immunobiology 218: 28–39 [DOI] [PubMed] [Google Scholar]
- Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, Klein A, Hofmann O, Camargo FD (2014) Clonal dynamics of native haematopoiesis. Nature 514: 322–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang JC, Yu Y, Burke S, Buettner F, Wang C, Kolodziejczyk AA, Teichmann SA, Lu L, Liu P (2015) Single‐cell transcriptomic reconstruction reveals cell cycle and multi‐lineage differentiation defects in Bcl11a‐deficient hematopoietic stem cells. Genome Biol 16: 178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsapogas P, Swee LK, Nusser A, Nuber N, Kreuzaler M, Capoferri G, Rolink H, Ceredig R, Rolink A (2014) In vivo evidence for an instructive role of fms‐like tyrosine kinase‐3 (FLT3) ligand in hematopoietic development. Haematologica 99: 638–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Munoz‐Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, Marques S, Munguba H, He L, Betsholtz C, Rolny C, Castelo‐Branco G, Hjerling‐Leffler J, Linnarsson S (2015) Brain structure. Cell types in the mouse cortex and hippocampus revealed by single‐cell RNA‐seq. Science 347: 1138–1142 [DOI] [PubMed] [Google Scholar]
- Zhang J, Raper A, Sugita N, Hingorani R, Salio M, Palmowski MJ, Cerundolo V, Crocker PR (2006) Characterization of Siglec‐H as a novel endocytic receptor expressed on murine plasmacytoid dendritic cell precursors. Blood 107: 3600–3608 [DOI] [PubMed] [Google Scholar]
- Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall‐Levin M, Wyatt PW, Hindson CM, Bharadwaj R et al (2017) Massively parallel digital transcriptional profiling of single cells. Nat Commun 8: 14049 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Review Process File
Data Availability Statement
The bulk RNA‐seq as well as the single‐cell RNA‐seq data from this publication has been deposited in NCBI's Gene Expression Omnibus database (Edgar et al, 2002; Barrett et al, 2013) (https://www.ncbi.nlm.nih.gov/geo/) and assigned the GEO Series accession number GSE102456 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE102456).