

Abstract
Since she was 4 years old, the patient had exhibited frequent convulsive seizures, and she experienced severe headaches and depression in adulthood. At the age of 37 years, cerebral calcifications were detected, but she exhibited no cognitive or motor problems. She suffered a cerebral haemorrhage at 49 years old and experienced cognitive dysfunction, dysarthria, dysphagia, and left-hemiparesis as sequelae. After undergoing gastrostomy, she exhibited very slow cognitive deterioration associated with speech disturbance over more than 10 years. She also gradually developed limb spasticity with Babinski signs. Repeated computerised tomography scans revealed unexpected changes including 2 cysts that appeared separately after small haemorrhages, an intracerebral haemorrhage, and intra-cyst bleeding. These longitudinal scans also showed progressive ventricular dilatation and expansion of the leukoencephalopathy, but there were no apparent changes in the intracranial calcifications. Magnetic resonance imaging revealed numerous microbleeds, and magnetic resonance angiography revealed irregularity of the cerebral artery walls with stoppage. Her SNORD118 gene exhibited compound heteromutation of c.38C > G and c.116G > C on different alleles. She was finally diagnosed with leukoencephalopathy with brain calcifications and cysts (Labrune syndrome) at the age of 61 years. Past reports have suggested that diffuse cerebral microangiopathy underlies Labrune syndrome's pathogenesis, but we speculate that cerebral macroangiopathy may also underlie it.
Keywords: SNORD118, Leukoencephalopathy, Calcification, Cyst, Labrune syndrome
Highlights
-
•
Our patient presented with a long history of neurological complaints.
-
•
Computerised tomography scans over many years revealed haemorrhages and cysts.
-
•
She was diagnosed with leukoencephalopathy with brain calcifications and cysts.
-
•
We detected a compound heteromutation in her SNORD118 gene.
1. Introduction
Leukoencephalopathy with brain calcifications and cysts (LCC), also known as Labrune syndrome, is a neuro-radiologically characterised neurodegenerative genetic disorder [1], [2], [3]. Patients with LCC may present with mental retardation, convulsions, corticospinal tract and extrapyramidal signs, movement disorders, and cerebellar ataxia without extracranial abnormalities such as retinal vascular abnormalities [2], [3], [4]. However, these symptoms and signs are not LCC-specific, their presence and timing varies [2], [3], [4], and their temporal development remains poorly understood [2], [3], [4]. Jenkinson et al. [2] recently identified LCC-related mutations in the gene encoding the box C/D small nucleolar RNA U8 (SNORD118), thus indicating that LCC may represent a novel ribosomopathy. For more than 10 years, our hospital has treated a woman with LCC exhibiting a compound SNORD118 heteromutation. We herein report her clinical course and longitudinal neuro-radiological findings.
2. Patient characteristics
The patient is currently 61 years old and had non-consanguineous Japanese parents. A similar disease was suspected in a sister 2 years her senior who exhibited epilepsy from a young age and was later diagnosed with a brain tumour. Further details are unavailable because the sister is deceased and no autopsy or gene analyses were performed. Our patient exhibited no abnormalities at birth but experienced generalised convulsive seizures from the age of 4 years. Her paediatricians diagnosed her with epilepsy and prescribed anti-epileptic drugs, but she did not take them as directed, which is partly why she experienced seizures more than monthly during adolescence. She still managed to achieve average grades in school, including in physical education. The convulsions gradually became less frequent from the age of 15 years and disappeared before adulthood, but she began frequently complaining of severe headaches. Although she regularly used pain-killers in her 20s, she experienced no problems in daily living or in her work in a service trade. She was diagnosed with depression in her early 30s and prescribed anti-depressants.
At 37 years old, she visited a neurologist at a nearby hospital to exclude organic disease, and computerised tomography (CT) scans revealed cerebral calcifications. However, she exhibited no cognitive or motor dysfunctions, so no causative disease was diagnosed. At 49 years old, she developed a right putaminal haemorrhage and received conservative treatment. She nevertheless became bed-ridden with sequelae including cognitive dysfunction, dysarthria, left-hemiparesis, and dysphagia, for which she underwent a gastrostomy. She was transferred to a disability care centre and consulted physicians at Oyamada Memorial Spa Hospital at 50 years old. Head CT revealed intracranial calcifications and ventricular dilatation with leukoencephalopathy but no cysts (Fig. 1A). Over the next 10 years, she exhibited slow cognitive deterioration with speech disturbances including aphasia and gradually developed limb spasticity with Babinski signs. Although she did not exhibit any sudden neurological changes indicative of stroke, repeated CT scans identified significant changes, including 2 cysts that appeared separately after small haemorrhages and exhibited intra-cyst bleeding (Fig. 1B-G). At the age of 60 years, magnetic resonance (MR) imaging revealed numerous microbleeds, and MR angiography of the cerebral arteries revealed wall irregularities and stoppage (Fig. 2). One year later, a right occipital lobe haemorrhage was detected (Fig. 3). The longitudinal CT scans also revealed progressive ventricular dilatation and expanding leukoencephalopathy but no apparent changes in the intracranial calcifications. SNORD118 analysis revealed compound heteromutation of c.38C > G and c.116G > C on different alleles, and the patient was finally diagnosed with LCC at 61 years old. Although she has since been maintained in stable general condition, neurological symptoms still progress very slowly. She can nod in response to questions but cannot provide meaningful speech responses. She exhibits left-side dominant spastic quadriplegia but no retinal vascular disease.
Fig. 1.

Longitudinal head CT findings.
(A) At the age of 50 years, the patient exhibited several calcifications in her basal ganglia and lateral ventricular dilatation with moderate leukoencephalopathy but no apparent cyst formation. (B) At the age of 56 years, she exhibited a small high-signal region in the leukoencephalopathy area of the left frontal lobe (arrow) that we suspected was a small haemorrhage. (C) At the age of 57 years, she exhibited cystic formation within the small high-signal area in B (arrow) with some intra-cyst bleeding. A distinct low-density area in the right corona radiata was also observed (arrowhead). (D) At the age of 58 years, she exhibited a small high-signal region in the leukoencephalopathy area of the right temporo-parietal lobe (arrow) that we suspected was a small haemorrhage. (E) At the age of 59 years, she exhibited a small low-density area within the small-high signal area in D (arrow). (F) At the age of 60 years, she developed a large cystic formation in the right temporo-parietal lobe. (G) When she was 61 years old, we observed bleeding in her cyst in F. These CT scans obtained over 10 years revealed progressive ventricular dilatation and expansion of the leukoencephalopathy but no apparent changes in the intracranial calcifications. Abbreviations: CT, computerised tomography; R, right side.
Fig. 2.

Head MR imaging findings obtained at the age of 60 years.
(A) T2-weighted imaging revealed 2 cysts with clear boundaries and extensive leukoencephalopathy. (B) Susceptibility-weighted imaging revealed numerous bleeds of various size. (C) Diffusion-weighted imaging revealed several high-signal regions. (D) MR angiography of the intracranial arteries revealed wall irregularities and stoppage, particularly on the right side.
Abbreviations: MR, magnetic resonance; R, right side.
Fig. 3.
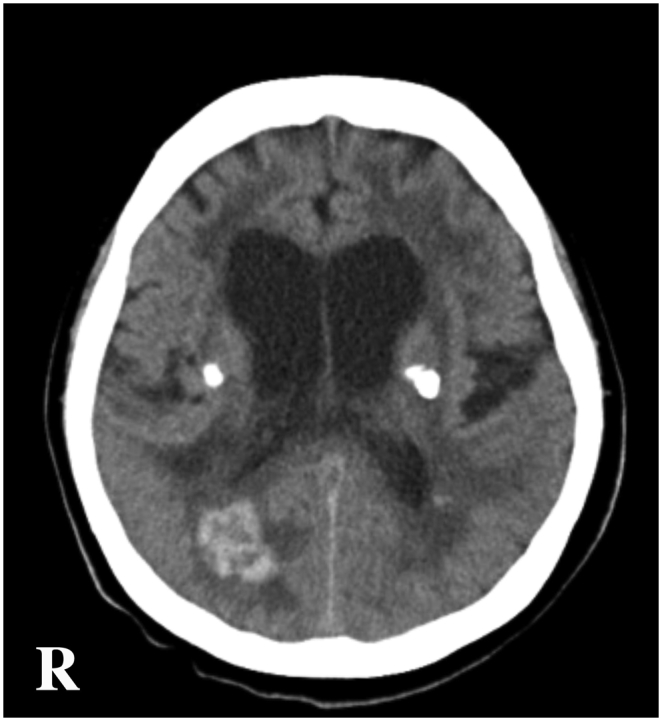
Head CT findings obtained at the age of 61 years.
Cerebral haemorrhage was incidentally found in the right occipital lobe during a regular follow-up scan.
Abbreviations: CT, computerised tomography; R, right side.
3. Discussion
In 1996, Labrune et al. [1] described 3 unrelated children with LCC who had exhibited mental retardation and convulsions from early infancy to adolescence. They proposed that the symptoms of LCC arise from diffuse cerebral microangiopathy because brain biopsy specimens from 2 patients revealed blood vessel calcification, gliosis, Rosenthal fibres, and angiomatous-like microvessel rearrangements. Jenkinson et al. [2] identified biallelic SNORD118 sequence variants throughout their cohort of 40 radiologically stereotyped European patients belonging to 33 families. Iwama et al. [3] identified biallelic SNORD118 variants in 7 out of 8 of Japanese patients. Our patient exhibited the biallelic SNORD118 variants c.38C > G and c.116G > C. c.116G > C is a novel variant, but c.38C > G was documented in 1 Italian patient in Jenkinson et al.’s study [2] and 6 Japanese patients in Iwama et al.’s study [3], which suggests that it may be relatively common in Japanese patients but rare in European patients. The relationships between individual mutations and clinical presentations remain unknown [2], [3]. Jenkinson et al. [2] performed a functional analysis of select SNORD118 variants and obtained evidence that they differentially affect the expression, processing, and protein binding of the U8 non-coding RNA molecule that SNORD118 encodes. This suggests that U8 is essential for cerebral vascular homeostasis.
Epileptic seizure is probably the most common LCC-related sign [1], [2], [3], [4] and was observed in our patient. Another common sign is delayed development [1], [2], [3], [4], but she did not exhibit this. Interestingly, she experienced severe headaches into adulthood, and 2 of Iwama et al.'s [3] patients exhibited headaches as initial symptoms. Tamura et al. [4] found that headache is a relatively frequent LCC symptom. Although our patient required anti-depressant therapy, previous reports did not mention psychiatric symptoms such as depression [1], [2], [3], [4]. She also exhibited various neurological disorders including cognitive dysfunction, aphasia, and spastic tetraparesis, and we suspect that these resulted from the extensive organic cerebral disorders of LCC.
After a cerebral haemorrhage at the age of 49 years, our patient very slowly developed cognitive dysfunction, speech disturbance, and limb spasticity without apparent stroke-like episodes, though repeated CT scans revealed significant changes. Brain calcifications and leukoencephalopathy are usually recognised in LCC, but intracranial cysts are sometimes not [2], [3]. Her CT scans at the age of 50 years revealed calcifications and leukoencephalopathy but no cysts. Two cysts subsequently appeared, each following a small cerebral haemorrhage and exhibiting intra-cyst bleeding. This implies that cerebral small bleeding in the white matter influences cyst formation but that calcification does not.
Our patient suffered a relatively large cerebral haemorrhage in the right putamen at 49 years old and another in the right occipital lobe at 59 years old. Although 1 of Iwama et al.'s [3] patients died of subarachnoid haemorrhage and 1 of Tamura et al.'s [4] died of intracerebral haemorrhage, studies of LCC do not always consider LCC-related haemorrhages. However, Jenkinson et al. [2] found that their patients' brain biopsy specimens exhibited numerous macrophages with haemosiderin deposits occurring both perivascularly and throughout the tissue, which indicated old haemorrhages. Furthermore, susceptibility-weighted MR imaging of our patient revealed numerous microbleeds, and MR angiography of her intracranial arteries revealed wall irregularities and stoppage. The complications from cerebral bleeding in LCC should influence our thinking about LCC pathology. Although Labrune et al. [1] and Jenkinson et al. [2] speculated that diffuse cerebral microangiopathy underlies LCC, our patient's case suggests that cerebral macroangiopathy may also underlie LCC.
Footnotes
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
- 1.Labrune P., Lacroix C., Goutieres F. Extensive brain calcifications, leukodystrophy, and formation of parenchymal cysts: a new progressive disorder due to diffuse cerebral microangiopathy. Neurology. 1996;46:1297–1301. doi: 10.1212/wnl.46.5.1297. [DOI] [PubMed] [Google Scholar]
- 2.Jenkinson E.M., Rodero M.P., Kasher P.R. Mutations in SNORD118 cause the cerebral microangiopathy leukoencephalopathy with calcifications and cysts. Nat. Genet. 2016;48:1185–1192. doi: 10.1038/ng.3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iwama K., Mizuguchi T., Takanashi J.I. Identification of novel SNORD118 mutations in seven patients with leukoencephalopathy with brain calcifications and cysts. Clin. Genet. 2017;92:180–187. doi: 10.1111/cge.12991. [DOI] [PubMed] [Google Scholar]
- 4.Tamura R., Ohira T., Emoto K. Leukoencephalopathy, cerebral calcifications, and cysts: a clinical case involving a long-term follow-up and literature review. J. Neurol. Sci. 2017;373:60–65. doi: 10.1016/j.jns.2016.12.014. [DOI] [PubMed] [Google Scholar]