

Abstract
The extent of genome divergence and the evolutionary events leading to speciation of marine bacteria have mostly been studied for (locally) abundant, free-living groups. The genus Phaeobacter is found on different marine surfaces, seems to occupy geographically disjunct habitats, and is involved in different biotic interactions, and was therefore targeted in the present study. The analysis of the chromosomes of 32 closely related but geographically spread Phaeobacter strains revealed an exceptionally large, highly syntenic core genome. The flexible gene pool is constantly but slightly expanding across all Phaeobacter lineages. The horizontally transferred genes mostly originated from bacteria of the Roseobacter group and horizontal transfer most likely was mediated by gene transfer agents. No evidence for geographic isolation and habitat specificity of the different phylogenomic Phaeobacter clades was detected based on the sources of isolation. In contrast, the functional gene repertoire and physiological traits of different phylogenomic Phaeobacter clades were sufficiently distinct to suggest an adaptation to an associated lifestyle with algae, to additional nutrient sources, or toxic heavy metals. Our study reveals that the evolutionary trajectories of surface-associated marine bacteria can differ significantly from free-living marine bacteria or marine generalists.
Keywords: population genomics, gene flow, gene transfer agent, horizontal gene transfer, ecological niche, bacterial adaptation
Introduction
The marine environment sustains a high diversity of bacteria (Zinger et al. 2011; Sunagawa et al. 2015). So far, the genetic divergence of evolutionary and ecologically distinct but cooccurring marine microbial populations and the underlying drivers have been elucidated for only few, mostly free-living, bacterial groups (Coleman et al. 2006; Johnson et al. 2006; Hunt et al. 2008; Carlson et al. 2009; Swan et al. 2013). Yet, a considerable fraction of marine bacteria occur, grow and evolved attached to surfaces of particles and organisms, where they can constitute up to 20% of the total bacterial biomass in ocean water (Azam et al. 1983) and up to 66% of bacterial biomass during coastal algae blooms (Becquevort et al. 1998). In addition, the contribution of attached bacteria to overall prokaryotic substrate turnover is disproportionally high due to their higher specific activity (Crump et al. 1998; Stocker 2012) and the species diversity of associated bacteria significantly exceeds that of free-living bacteria (Bižic-Ionescu et al. 2015). Compared with free-living bacteria, the high cell density and proximity in surface biofilms may facilitate an increased gene transfer and spread of traits (Balcazar et al. 2015). Despite these particular features of surface-associated marine bacteria, only few studies have actually addressed their genome diversity and evolution. Previous studies focused on the genus Vibrio which, however, is a bacterial generalist not specifically adapted to the attached lifestyle (Hunt et al. 2008; Shapiro et al. 2012; Kirchberger et al. 2016), as opposed to most other species detected on marine surfaces (Bižic-Ionescu et al. 2015).
The Roseobacter group is adapted to the marine environment (Simon et al. 2017) and comprises chemotrophic bacteria often associate with eukaryotes (Buchan et al. 2005). Members of the group have a much higher metabolic and ecological versatility than other dominant marine bacteria (Brinkhoff et al. 2008; Newton et al. 2010). The genus Phaeobacter colonizes artificial and biotic surfaces such as different algae, bryozoan, molluscs, crustaceans, and fish, and is often associated with aquaculture systems and harbors (Rao et al. 2005; Porsby et al. 2008; Prado et al. 2009; Thole et al. 2012; Frank et al. 2015; Gram et al. 2015; Segev et al. 2015). Phaeobacter may exert a probiotic effect due to the production of the antibiotic tropodithietic acid (Brinkhoff et al. 2004; Porsby et al. 2008; Prado et al. 2009; D'Alvise et al. 2012). In association with senescent algae, Phaeobacter can switch from a symbiotic to a pathogenic lifestyle through the induction of algaecide synthesis and subsequent lysis of algal cells (Seyedsayamdost et al. 2011). Phaeobacter spp. are metabolically highly flexible and simultaneously metabolize multiple substrates under nutrient-rich conditions (Zech et al. 2013). They catabolize algal osmolytes like dimethylsulfoniopropionate, and contain the sox genes for sulfur oxidation (Dickschat et al. 2010; Newton et al. 2010; Thole et al. 2012). In their natural habitat, Phaeobacter reach low abundances (Gram et al. 2015; Freese et al. 2017) but nevertheless are ecologically significant through their antilarval and antibacterial activities, preventing biofouling even at low cell densities (Rao et al. 2007). While gene sequences of Phaeobacter are only barely detectable by molecular methods in standard surveys of biofilms or eukaryotes and even entirely absent in the open ocean sequence databases, selective cultivation methods could recover Phaeobacter also from open ocean zooplankton (Gram et al. 2015; Freese et al. 2017). Previous comparative analysis of few isolates suggested that the genus Phaeobacter is distributed worldwide (Thole et al. 2012) but comprises disjunct, exclusively surface-associated populations (Freese et al. 2017).
A considerable number of closely related Phaeobacter strains have recently become available through direct isolation from different geographic regions and habitats (e.g., Hjelm et al. 2004; Porsby et al. 2008; Prado et al. 2009). In the present study, the available isolates were subjected to a detailed population genomic and phenotypic analysis to reveal the mechanisms of incipient diversification and the potential ecological niches of this surface-associated marine model bacterium.
Materials and Methods
Origin and Cultivation of Strains
The 88 Phaeobacter strains originated from aquacultures in Denmark, France and Spain (Hjelm et al. 2004; Porsby et al. 2008; Prado et al. 2009) and coastal marine environments in Australia, France, and Germany (Brinkhoff et al. 2004; Rao et al. 2005) (supplementary fig. S1 and table S1, Supplementary Material online). Further details like isolation conditions are found in the references cited. All strains produce the typical brown pigment that is associated with the formation of tropodithietic acid. Strains were grown with marine broth medium (MB, Difco 2216) and preserved in liquid nitrogen after addition of 10% (v/v) glycerine.
Sequencing of the 16S rRNA Gene and the Internal Transcribed Spacer (ITS) Region
DNA was extracted from 1 ml culture with the DNeasy Blood&Tissue Kit (Qiagen) and the whole 16S rRNA gene plus the ITS region was amplified using the primers 27f (5′-AGA GTT TGA TCM TGG CTC AG-3′; Lane 1991) and 23S-130r (5′-GGG TTB CCC CAT TCR G-3′; Fisher and Triplett 1999). PCR products were purified with the DNA Clean & Concentrator (Zymo) and sequenced by Sanger sequencing. The 16S rRNA sequences were aligned using the aligner tool implemented in ARB 5.1 database SSURef_108_Silva_NR_99_11_10_11 (Ludwig et al. 2004) and the alignment was manually refined based on secondary structure information. ITS sequences were aligned using ClustalW implemented in MEGA 5.05 (Tamura et al. 2011). After testing for the best nucleotide substitution model within MEGA (Jukes-Cantor, 16S rRNA genes; Kimura 2 with gamma distribution, ITS region), pairwise distance matrices were calculated and Maximum Likelihood phylogenetic trees were reconstructed in MEGA. Sequences were deposited in NCBI GenBank (accession numbers: KY357362–KY357447).
Genome Sequencing, Assembly and Annotation
Genomic DNA from 28 cultures in the late exponential phase was extracted with the JETFLEX Genomic DNA Purification Kit (Genomed). For preparation of SMRTbell™ template libraries, 8 µg of genomic DNA were sheared (g-tubes™, Covaris, Woburn, MA, USA), the size range monitored by pulse field gel electrophoresis, and DNA fragments end-repaired and ligated to hairpin adapters using P2 or P4 chemistry (Pacific Biosciences, Menlo Park, CA, USA). SMRT sequencing was carried out on the PacBio RSII (Pacific Biosciences). Illumina libraries were prepared with the TruSeq DNA Sample Prep Kit v2 (lllumina Inc., San Diego, CA, USA) and paired-end sequencing was performed on the HiSeq 2500 for 100 cycles (∼8 million reads per genome).
PacBio reads were assembled de novo in SMRT Portal version 2.0.1 using the RS_HGAP_Assembly.1 and HGAP_Assembly_Advanced.1 protocols. Indel errors were corrected by mapping of Illumina reads using the Burrows–Wheeler Aligner (BWA) (Li and Durbin 2009) and the CLC Genomics Workbench 7.0.1 (CLC bio QIAGEN, Germany) for subsequent variant and consensus calling. The final assembly was trimmed, circularized and adjusted to the replication system as start point (Bunk 2016). Genome sequences were automatically annotated using Prokka 1.8 (Seemann 2014). The genomes of P. inhibens DSM 17395, P. gallaeciensis DSM 26640, and P. inhibens 2.10 (DSM 24558) (Thole et al. 2012; Frank et al. 2014) were retrieved from NCBI GenBank and used as primary database for annotation. P. inhibens T5 (DSM 16374) was available as permanent draft from IMG (Integrated Microbial Genomes & Microbiomes) (Dogs et al. 2013) and closed in this study. In total, this yielded 32 high quality, closed genomes for analysis. Genome sequences were deposited in NCBI GenBank (accession numbers: CP010588–CP010775, CP010784–CP010791, CP010805–CP010810).
Phylogenomics
Maximum likelihood and maximum parsimony phylogenomic trees were constructed from amino acid supermatrices as described by Simon et al. (2017) employing the JTT model of amino acid evolution (Jones et al. 1992) in conjunction with gamma-distributed substitution rates (Yang 1993) and empirical amino acid frequencies. To assess the stability of the phylogenomic branching, three different supermatrices were compiled from the concatenated orthologs which were 1) present in at least four sequences (4,336 genes, 1,269,031 characters), 2) present after removing uninformative genes using MARE (Meusemann et al. 2010) (3,169 genes, 1,031,284 characters), and 3) present in all 32 genomes (2,919 genes and 941,788 characters). For phylogenetic network analysis, single copy ortholog genes were identified, aligned based on their amino acid sequences and concatenated employing the ODoSE pipeline (Vos et al. 2013). The resulting matrix contained 2,821,782 characters and was fed into a NeighborNet analysis by SplitsTree 4.13.1 (Huson and Bryant 2006).
Evolution of Phaeobacter Chromosomes
Whole chromosome alignments were done with Mauve (Darling et al. 2010) and Easyfig (Sullivan et al. 2011). The gene order conservation was calculated as fraction of orthologous genes that are syntenous based on at least one shared neighbour (allowing for a gene insertion of one) for all pairwise chromosome comparisons (Yelton et al. 2011). Functions of chromosomal proteins were determined according to the COG (Clusters of Orthologous Groups of proteins) database via WebMGA (Wu et al. 2011). All phylogenetic distances and the digital DNA–DNA hybridization were inferred from pairwise comparisons of complete chromosome sequences via Genome BLAST Distance Phylogeny implemented in the GGDC 2.1 web service (Henz et al. 2005; Meier-Kolthoff et al. 2013). The resulting tree topology corresponds to that of the tree shown in figure 1 and the three main clusters are equally well supported. To test for biogeographic clustering of strains, a Pearson’s product-moment correlation between pairwise genomic distances and pairwise geographic distances of sampling sites of the strains was calculated in R (R Core Team 2015). To determine the extent of the core and pan-chromosome, a gene content matrix listing the presence or absence of a gene within a certain chromosome was extracted from the OrthoMCL group files. Subsets with increasing numbers n of strains were randomly selected 100 times, and the number of orthologs present in n strains (core chromosome) as well as the numbers of orthologs present in at least one and at maximum n-1 strains (pan-chromosome) were determined using R (R Core Team 2015). Results were visualized using ggplot2 (Wickham 2009).
Fig. 1.

—Phylogenomics of Phaeobacter. (a) Maximum likelihood (ML) phylogenetic tree inferred from a supermatrix of 1,269,031 aligned amino acid characters. Branches scaled according to the expected number of substitutions per site. Bold edges indicated branches with 100% bootstrapping support from all types of analysis (ML supermatrix; maximum parsimony (MP) supermatrix; ML MARE‐filtered supermatrix; MP MARE‐filtered supermatrix; ML core‐genes matrix and MP core‐genes). The numbers of inferred gene gains (black) and losses (red) are given next to the corresponding branch. Gene number of the inferred ancestral node is given at the midpoint root. (b) Phylogenetic network inferred by NeighborNet from the 2,821,782 aligned nucleotide characters of the concatenated single copy orthologs of 32 Phaeobacter strains. Scale bar, 0.01 changes per nucleotide site.
Gene gains and losses were quantified employing BadiRate (Librado et al. 2012). Gene families were determined using the TribeMCL algorithm (Enright et al. 2002) and a gene content matrix generated which was applied to an ultrametric tree calculated with r8s version 1.80 (Sanderson 2003). The goodness of different gene flow models was evaluated using the Akaike Information Criterion (AIC). For the Phaeobacter strains, the Lambda-Innovation model provided the best fit based on its lowest AIC value and yielded the size of ancestral phylogenetic nodes and the total number of gains and losses per lineage. The Lambda model assumes equal gene birth and death rates whereas innovation explicitly accounts for gene gain by HGT.
Horizontally transferred chromosomal elements were identified for each strain based on deviations from the normalized tetranucleotide frequency. Frequencies of each tetranucleotide were calculated for the whole chromosome and across 5 kb sliding windows with 2.5 kb overlap (Riedel et al. 2013) using the Biostrings package (Pagès et al. 2016) and the similarity between local and global frequencies was calculated as Pearson correlation coefficient. The interquartile-range of the correlation coefficients yielded the threshold below which a region was regarded as horizontally transferred (Riedel et al. 2013). Genomic islands were predicted with the IslandViewer web server (Dhillon et al. 2013) using the three different incorporated methods, SIGI-HMM (measuring codon usage), IslandPath-DIMOB (identifying abnormal sequence composition or the presence of genes related to mobile elements) and IslandPick (a comparative genomics-based method). The function of the proteins was classified according to the COG database via the WebMGA (Wu et al. 2011). A COG was defined as novel for the genus Phaeobacter if it did not occur outside of transferred elements in any Phaeobacter strain.
Prophages were predicted with PHAST (Zhou et al. 2011), but were only considered if they comprised more than just a tail protein and an integrase. If two prophages were located consecutively on the genome and one contained structural genes and terminase and the other replication genes, they were combined. Furthermore, sequences of prophages were compared with identified inducible Phaeobacter prophages characterized by Thole et al. (2012) and if they were identical the start and end of the prophage sequence was corrected accordingly. Similarities of sequences (i.e., identity multiplied by coverage) were calculated using BLAST (Altschul et al. 1990) to define operational taxonomic prophage units (OTU). The OTU threshold was set to 60% since the lowest sequence similarity of the GTA present in all strains was 61.6%. Bacteriophage classification was done by VIRFAM via the head-neck-tail module genes (Lopes et al. 2014).
Phenotyping
Substrate utilization of 190 different carbon sources was determined via the Phenotype MicroArray (OmniLog PM) system using PM01 and PM02-A MicroPlates (AES Chemunex BLG 12111, BLG 12112). Phaeobacter strains were grown on MB agar at 25 °C for 28 h, inoculated into modified medium (Buddruhs et al. 2013) and the respiration kinetics were recorded in the OmniLog Reader at 28 °C for 96 h. GENIII MicroPlates, which require a different inoculation medium did not result in reliable technical replicates for Phaeobacter in contrast to PM01 and PM02-A MicroPlates (data not shown). Based on prior reproducibility tests, two biological parallels were measured for each strain. After aggregation of the curve parameters for each substrate and strain using the R package opm (Vaas et al. 2013), the maximum height (A) as indicator for the metabolic activity was subject to a PCA using the R package vegan (Oksanen et al. 2015).
Statistics
All calculations were performed in R (R Core Team 2015). Significant differences in multiple comparisons of groups were calculated with Tukey procedures (function glht() in the R package multcomp; Hothorn et al. 2008) after an ANOVA (function aov()). Correlations were calculated using Pearson’s product-moment correlation.
Results
Phylogeny, Phylogenomics, and Chromosome Structure
The overall phylogenetic diversity within the recently reclassified (Breider et al. 2014) genus Phaeobacter is very low. Maximum sequence divergence extracted from the pairwise distance matrix of 16S rRNA genes of the 88 available strains reached only 0.44% (supplementary fig. S2, Supplementary Material online). The strains clustered in four clades with an intergroup sequence divergence of ≤5 bp (0.37%). Two of these groups were identified as P. gallaeciensis and P. inhibens based on their phylogenetic affiliation with the respective type strains, DSM 26640 T and DSM 16374 T. The largest group of strains (47%) belonged to P. inhibens and was isolated from all five countries covered by our study (supplementary table S1, Supplementary Material online). Clustering of the ITS sequences yielded three distinct groups and revealed a 20-fold higher nucleotide substitution rate compared with the 16S rRNA genes within the P. inhibens cluster (supplementary fig. S2, Supplementary Material online). Strain P88 and DSM 16374 contain different ITS therefore they occur more than once in the tree but the P. inhibens strains themselves fall into 11 different lineages with high bootstrap support. Although the substitution rate within another cluster, here designated “Phaeobacter piscinae” was lower for the ITS than the 16S rRNA genes, four separate ITS lineages could still be differentiated.
Strains representing unique ITS lineages or isolates from different habitats were selected for genomic comparisons (marked in bold face in supplementary fig. S2, Supplementary Material online). The size of the 32 chromosomes ranged from 3.588 to 3.896 Mb and the gene counts from 3,347 to 3,713. The GC-content of all chromosomes lies between 59.9 and 60.4 mol% G + C (supplementary table S2, Supplementary Material online). All strains contained four nearly identical rRNA operons with exception of P. inhibens DSM 16374 and P88 which possess phylogenetically differing ITS (supplementary fig. S2, Supplementary Material online). The analysis of the core chromosome yielded an asymptotic saturation curve indicating that it is robustly predicted based on this set of 32 strains. In contrast, the pan-chromosome, that is, the whole chromosomal gene repertoire of Phaeobacter did not reach saturation (supplementary fig. S3, Supplementary Material online). As much as 78–87% of the gene content of the Phaeobacter strains fell into the large core chromosome that comprised 2,920 core genes. Even considering the extrachromosomal elements the average core genome still amounted to 78.1% (3,160 core genes) and the extrachromosomal elements of Phaeobacter contain only 11% (449) of all functional genes.
Results of the phylogenomic analyses of the chromosomal genes were very robust and congruent with the 16S rRNA gene and ITS phylogenies. The major clades could be distinguished in all types of analysis and were supported by high bootstrap values (fig. 1). A third clade was clearly separated from P. gallaeciensis and P. inhibens which was confirmed by low values for digital DNA–DNA hybridization (39.2–45.5%, median 41.0% for chromosomes; 38.9–46.9%, median 41.0% for whole genomes). Consequently, the novel species name “P. piscinae” was assigned to the third clade which encompasses two divergent subclusters. The phylogenetic network inferred from nucleotide sequences of the core genes showed almost no conflicting phylogenies (fig. 1b), indicating a low recombination between the different strains. Strains of P. gallaeciensis had nearly identical genomes with only 317 polymorphic sites in the Phaeobacter core chromosome although they were isolated from different geographic regions and associated with different eukaryotes (algae, Pecten maximus, Ostrea edulis, Venerupis philippinarum; supplementary table S1, Supplementary Material online). The intraspecific diversification in the other clades was much more pronounced (P. inhibens: 191,198 sites) (fig. 1b). Overall, genomic distances of the strains did not reflect geographic distances (Pearson Correlation test r = −0.07, 95%CI(−0.16, 0.01), P > 0.5) indicating that clustering of strains is independent of their geographic distribution (supplementary fig. S4, Supplementary Material online). Furthermore, the genomic distance of Phaeobacter strains originating from the same habitat is not lower than of strains originating from different habitats (supplementary fig. S5a, Supplementary Material online). Nearly all habitats contained strains from different clades and genomic distances did not differ significantly for any habitat (supplementary fig. S5b, Supplementary Material online). Thus, genomic clades did not show specificity for the different habitats as they are presently distinguished based on standard environmental data.
All chromosomes were largely syntenic (supplementary fig. S6a, Supplementary Material online). The high synteny was further confirmed by a strong pairwise gene order conservation (0.99–1.0, median 0.997). Distinct chromosomal rearrangements were due to inversions of long (>100 kb) chromosomal segments and were also detected among the chromosomes of very closely related strains such as P. gallaeciensis strain P73 versus strains DSM 26640, P11, P63, P75. Inversions were located at seven different breakpoints and mostly close to one of the rRNA operons or a tRNA (supplementary fig. S6a, Supplementary Material online). They never disrupted a gene; rather they occurred adjacent to, or replaced, a horizontally transferred element (supplementary fig. S6b, Supplementary Material online).
A highly homogenous tetranucleotide frequency distribution was also observed among all chromosomes. Their median of the local to global tetranucleotide frequency similarity ranged between 0.83 and 0.84 which was similar to the closely related Leisingera methylohalidivorans (NC_023135; 0.83) and Ruegeria pomeroyi (NC_003911; 0.84) but significantly higher than for another more distant member of the Roseobacter group, Dinoroseobacter shibae (CP000830; 0.81), and for species with high ratios of recombination to point mutation like “Candidatus Pelagibacter ubique” (CP000084; 0.76) and Flavobacterium psychrophilum (AM398681; 0.71) (Vos and Didelot 2009) (Tukey, P < 0.001). This corroborates the low rate of recombination in Phaeobacter.
Pan-Chromosome Structure and Evolution
Phaeobacter chromosomes have expanded continuously during the divergence of the genus and gene gains occurred over all branches. Since the last common ancestor, an average of 47 genes were gained (1.4% of the gene content) and three genes were lost (0.08% of the gene content) (fig. 1a). Both values are significantly different from zero (t-test, P < 0.001). The highest gene gain (458, 12.7% of the gene content) was detected for the branch of P. gallaeciensis, indicating a considerable evolutionary divergence of its pan-genome. Gene gains and losses on terminal branches were in the same range (median gains, 1.8%; median losses, 0.04%). The numbers of gained genes were positively correlated with the pairwise phylogenomic distance and the number of amino acid substitutions per site (Pearson correlation r = 0.71, 95%CI(0.67, 0.75) and r = 0.62, 95%CI(0.43, 0.75), both P < 0.001) suggesting that the pan-chromosome expanded constantly and in parallel with the diversification of the nucleotide and amino acid sequences. The best fit model (lowest AIC values) explaining the gene dynamics was the Lambda-Innovation model where innovation rates (1.6 × 10−3 innovations per branch [relative age]) significantly exceeded Lambda rates (2.9 × 10−4 births/deaths per gene and branch [relative age]) by one order of magnitude (Mann–Whitney Rank Sum Test, P < 0.001). This indicates that expansion of the pan-chromosome was predominantly caused by horizontal gene acquisition and/or de novo gene origin rather than by gene duplication events.
Between 15 and 25 elements per genome were predicted to be of foreign origin based on tetranucleotide frequency (9–20 elements), the analysis of prophages (1–5 elements) and genomic islands (7–17 elements) (supplementary fig. S6b, Supplementary Material online). On average these elements together constituted 7.5% of the chromosomal gene content and contained an average of ten genes. The size of the nonprophage elements were on average 7.5 kb. The elements were distributed over the whole chromosome without clade specific patterns (supplementary fig. S6b, Supplementary Material online). The majority (71%) of the gained genes estimated via the Lambda-Innovation model were detected in the transferred elements. In addition, the numbers of gained genes were tightly correlated to their numbers in transferred elements (fig. 2), emphasizing HGT as the major source for the genome expansion in all lineages of Phaeobacter alike. Most transferred proteins were closely related to those of the related genera Ruegeria, Leisingera, and Roseobacter, which represent typical generalists within the Rhodobacteraceae (supplementary table S3, Supplementary Material online).
Fig. 2.
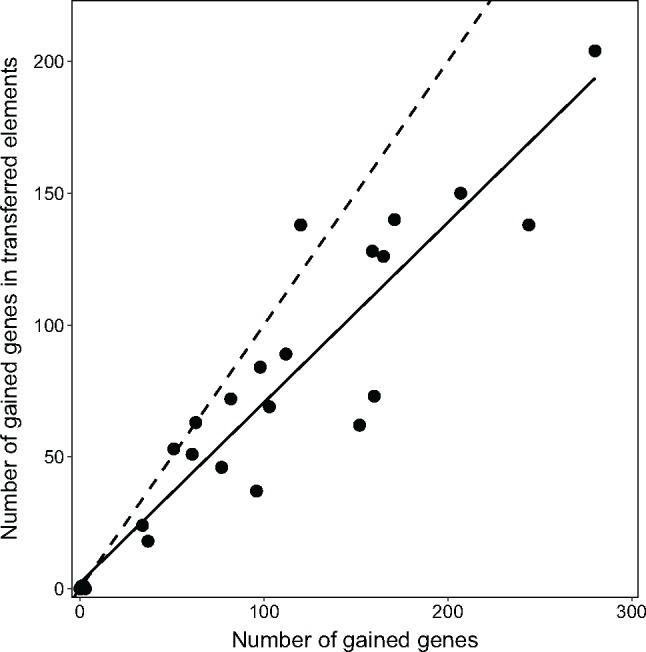
—Comparison between the number of gained genes for each Phaeobacter strain (numbers at terminal branches in fig. 1a) and the number of gained genes occurring in predicted horizontally transferred elements. Solid line indicate the calculated linear regression (slope 0.68, pearson correlation r = 0.95, 95%CI(0.89, 0.97), P < 0.001). Dashed line depicts theoretical correlation of r = 1, that is, if all genes were gained by detectable HGT.
A total of 84 putative prophages were detected. One type of prophage was present in all 32 chromosomes and identified as the gene transfer agent (GTA) (cf. Thole et al. 2012). The phylogeny of GTA largely corresponded to the branching pattern of the core genome (supplementary fig. S7, Supplementary Material online), indicating an early acquisition of GTA before the divergence of the three Phaeobacter species and a subsequent coevolution with their chromosomes. In addition, all strains carried the essential competence genes required for GTA-mediated gene uptake (Brimacombe et al. 2015) (supplementary table S4, Supplementary Material online). The 52 remaining prophages were classified as Myoviridae, Siphoviridae, and Podoviridae. Based on the established OTU threshold, they clustered into 20 OTUs of which seven (Pro1–Pro7) occurred in multiple strains and 13 were unique (supplementary table S5, Supplementary Material online). In contrast to GTA, the prophage pattern of the 32 Phaeobacter strains did not follow their genome phylogeny (supplementary fig. S8, Supplementary Material online). Pro1, the second most frequent prophage type in Phaeobacter, occurred in 12 genomes and could therefore be analyzed in detail. The nucleotide phylogeny of Pro1 was entirely congruent with the chromosome tree for members of P. inhibens, suggesting an integration of this phage before the radiation of this species and subsequent repeated losses (supplementary fig. S9, Supplementary Material online). However, the occurrence of two different Pro1 prophages in the genomes of “P. piscinae” suggests at least two independent transfers of Pro1 into the chromosome of “P. piscinae” (colored branches in supplementary fig. S9, Supplementary Material online). In the host chromosome, Pro1 always occurred at the same position with the exception of strains DSM16374 and P48. No Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) were detected in any of the Phaeobacter strains indicating low selection pressure from lytic phages.
Diversification of Functional Genes and Phenotypes
In all Phaeobacter chromosomes, functional genes of amino acid transport and metabolism (COG category E) were dominant, followed by genes encoding transcription proteins (category K; 81% of which represent regulatory proteins) and by genes encoding energy production and conversion (category C) (fig. 3a). All chemotaxis genes belonging to COG category T and numerous genes involved in biofilm formation (e.g., motility, polysaccharide metabolism, and export) were found in the strains as expected based on their phenotypic properties. The quantitative distribution of COGs across different categories was similar (Mann–Whitney Rank Sum Test: no significant differences, P > 0.33; Pearson’s test: significantly positive correlation, P > 0.88, P < 0.001) to that of other Alphaproteobacteria available in IMG like the facultatively anaerobic soil bacterium Paracoccus denitrificans or the oligotrophic free-living marine Planktomarina temperata (fig. 3a). The distribution of COG categories within the foreign, transferred elements in Phaeobacter was significantly dependent of the chromosomal COG category distribution in Alphaproteobacteria (Chi-square tests, P 0.02–<0.001). Aside from poorly characterized COGs, most COGs fell also into characterized categories C, E, and K (fig. 3a). In contrast to the chromosomal COG distribution in Phaeobacter, a significantly higher proportion of category L (replication, recombination and repair) was present in transferred elements (paired t-test, P < 0.001) but 29% of the COGs were identified as transposases which can be expected in transferred elements. In order to quantify HGT events leading to evolutionary innovations, the proportion of COGs in transferred elements, which were novel for all Phaeobacter chromosomes was determined (fig. 3b, triangles). Only few of the COGs represent innovations (supplementary table S6, Supplementary Material online). The percentages correspond to an average of 5.1 and 3.4 novel COGs of categories L and K, respectively, that were transferred per Phaeobacter genome, and one novel COG of categories D (cell cycle control, cell division, chromosome partitioning), E and M (cell wall/membrane/envelope biogenesis) transferred per genome.
Fig. 3.

—Abundance of different COGs present in the 32 Phaeobacter chromosomes in comparison to Paracoccus denitrificans PD1222 and Planktomarina temperata RCA23, DSM 22400, both from IMG, sorted by categories (a) and number of COGs gained through HGT (b). Open triangles indicate percentage of COGs gained by HGT that are novel for the genus Phaeobacter. Box plots show median, 25% and 75% percentiles, whiskers the 1.5*interquartile range and all values outside the range are shown as outliers. Letters indicate COG categories: A (RNA processing and modification), B (chromatin structure and dynamics), C (energy production and conversion), D (cell cycle control, cell division, chromosome partitioning), E (amino acid transport and metabolism), F (nucleotide transport and metabolism), G (carbohydrate transport and metabolism), H (coenzyme transport and metabolism), I (lipid transport and metabolism), J (translation, ribosomal structure and biogenesis), K (transcription), L (replication, recombination and repair), M (cell wall/membrane/envelope biogenesis), N (cell motility), O (posttranslational modification, protein turnover, chaperones), P (inorganic ion transport and metabolism), Q (secondary metabolites biosynthesis, transport and catabolism), R (general function prediction only), S (function unknown), T (signal transduction mechanisms), U (intracellular trafficking, secretion, and vesicular transport), and V (defense mechanisms).
Principal component analysis (PCA) of functional gene content identified sets of specific functional genes commensurate with the clustering of Phaeobacter species (fig. 4). In particular, P. gallaeciensis contains an ABC type cobalt transport system, additional catalases, a complete Type IV secretion system previously described for plasmid pDSHI10 of D. shibae (Petersen et al. 2013), and a 1-aminocyclopropane-1-carboxylate (ACC) deaminase (gr8, gr10; fig. 4, supplementary table S7, Supplementary Material online). In most cases these genes were found on transferred genomic islands. Outside of P. gallaeciensis, the functional genes for the Type IV secretion system and ACC deaminase were only detected in the most deeply branching strains of P. inhibens (DSM 17395, P10) where it occurred in an element of foreign origin. A functional gene unique for P. inhibens is a divalent cation transporter (gr4). Heme utilization proteins (gr2), which are characteristic for other members of the Roseobacter group (Roe et al. 2013) are missing in this species. “P. piscinae” lacks a beta-lactamase class A and a Na+/glutamate symporter (gr3). Notably, members of this group that fell into two separate phylogenetic subclades also differed with respect to the presence of copper resistance proteins (gr6, gr12) in one subgroup (strains P14, P23, P36, P42; compare fig. 1) and the absence of polyketides and nonribosomal peptides synthases (COG2091, gr16) in the second subgroup (strains P71, P13, P18). Again, the copper resistance genes were located on genomic islands.
Fig. 4.

—Principal component analysis of the presence of COGs in the 32 Phaeobacter chromosomes. Strains were colored according to their phylogenetic lineage (P_gal: P. gallaeciensis, P_inh: P. inhibens, P_pis: “P. piscinae”). Only COGs differing significantly between the clades (Tukey test with P ≤ 0.02) are indicated, COGs at the same position were grouped (gr10: COG0310, 0338, 0376, 0412, 0543, 0619, 1122, 1231, 1269, 1508, 1757, 2072, 3284, 3437, 3531, 3886, 4206, 5266; gr12: COG1276, 2132; gr13: COG2957, 2994, 5183; gr14: COG1144, 1271, 3203; gr16: COG3319, 3321; gr17: COG0423, 3448; gr2: COG3720, 4558, 4559, 4771; gr20: COG2932, 3409; gr3: COG0786, 2367, 3290; gr4: COG0598, 3457, 4067; gr6: COG2610, 3667; gr8: COG0417, 0501, 2515, 2948, 3451, 3504, 3702, 3704, 3736, 3838; gr9: COG1816, 2198, 2746).
Phaeobacter strains were also phenotypically compared based on their metabolic activity utilizing 190 different carbon substrates. The analysis of these data by PCA revealed distinct substrate utilization patterns of Phaeobacter clades (fig. 5). P. gallaeciensis was separated from the other strains based on its significantly higher metabolic activity utilizing the different Tween compounds, α-hydroxybutyric acid, melibionic acid, and α-methyl-D-galactoside. In addition, group wise (Tukey) tests revealed a preferential utilization of α-ketobutyric acid, sorbitol, methyl lactate, and unfavored utilization of tyramine and glucosamine of this species (P < 0.05). “P. piscinae” utilized N-acetylglutamate significantly better than the other strains, but α-ketobutyric acid and L-homoserine enabled a lower metabolic activity. P. inhibens strain P88 was a strong outlier because it exclusively was capable of utilizing xylitol and L-arabitol (fig. 5, insert) probably due to the exclusive presence of a D-xylulose reductase (PhaeoP88_01862, K05351) and xylokinase (PhaeoP88_01866, K00854).
Fig. 5.

—Principal component analysis of the utilization patterns of 190 substrates determined for the 32 Phaeobacter strains on OmniLog PM01 and PM02A plates. Strains were colored according to their phylogenetic lineage (P_gal: P. gallaeciensis, P_inh: P. inhibens, and P_pis: “P. piscinae”). PCA based on two biological replicates for each strain which mean position is shown. Substrates depicted contribute more than average to the ordination of data (Borcard et al. 2011). Results for principal component (PC) 1 and PC 3 are shown. Insert shows analysis for PC1 and PC2; its full scale plot, and an analysis along PC1 and PC4 are shown in supplementary fig. S10, Supplementary Material online).
Discussion
Evolution of the Phaeobacter Genome
The high fraction of the core genome of all three Phaeobacter species and its high synteny was rather unexpected based on the genome size and high metabolic versatility of Phaeobacter (Brinkhoff et al. 2008; Seyedsayamdost et al. 2011; Gram et al. 2015). So far, high values for synteny (>0.9) have been reported for the SAR11 subclade Ia which is characterized by a small streamlined genome (Grote et al. 2012). A high relative fraction of the core genome (∼80%) was only determined for bacterial species with specialised lifestyles or ecological niches, such as the subclade Ia of the free-living SAR11 (Grote et al. 2012), the obligate intracellular Chlamydia psittaci group (Voigt et al. 2012) and symbiotic Vibrio fischerii (Bongrand et al. 2016). In bacterial generalists with a comparable high 16S rRNA gene similarity (>99%) and genomic similarity the core genome constitutes a much smaller fraction, e.g., 52% in Pseudomonas syringae or 38–44% in Vibrio cholerae (Thompson et al. 2009; Nowell et al. 2014).
The accessory genome typically contains the genes relevant for the adaptation to particular environments (Kettler et al. 2007). Commensurate with the large core genome, the net gain of Phaeobacter genomes in accessory genes was small (average gains and losses, 1.8% and 0.04% of the chromosome, respectively) and lower than in other bacteria (up to 20% and 9.2% net gain in Prochlorococcus and Pseudomonas syringae, respectively) (Kettler et al. 2007; Nowell et al. 2014). The majority of the accessory genes of Phaeobacter were gained through transferred elements and likely were drawn from other members of the Roseobacter group. Like other members of the Roseobacter group, all Phaeobacter strains investigated contained GTAs, which can mediate high rates of interspecific gene transfer (Lang and Beatty 2007; McDaniel et al. 2010; Luo and Moran 2014). The GTA was acquired early before divergence of Phaeobacter and in contrast to prophages was always maintained in and continuously evolved with the chromosome. GTAs transfer small DNA fragments randomly generated from the host genome to recipient bacteria containing the necessary competence genes (Hynes et al. 2012; Brimacombe et al. 2015). The small size of the horizontally acquired elements in Phaeobacter, their origin in other Roseobacter genera containing GTA, as well as the presence of competence genes in Phaeobacter all are commensurate with HGT events mediated by GTAs. These findings indicate that GTAs constitute an important driver of HGT, diversification and niche adaptation in the genus Phaeobacter. One third of the accessory genome was identified as prophages including a Myoviridae phage. Obviously, the diversity of phages within the Roseobacter group extends well beyond the few documented Podoviridae and Siphoviridae (Huang et al. 2011; Ji et al. 2015; Liang et al. 2016). In contrast to GTA, vertical transmission of prophages seems to be rare in Phaeobacter and loss of prophages may have occurred repeatedly, in contrast to other bacterial groups such as the Enterobacteriacae (Bobay et al. 2014). The rapid turnover of prophages may also be caused by deletion during the transformation events mediated by GTA (Brimacombe et al. 2015; Rocha 2016).
Despite their comparatively limited acquisition of functional genes and high genome conservation, Phaeobacter encompasses three clearly separated phylogenomic clusters that are only marginally affected by homologous recombination. This finding is not due to sampling artifacts or cultivation bias as demonstrated by our phylogenetic analysis of all the 12 environmental 16S rRNA gene sequences that are available in public databases. All these sequences were found to group with one of the three Phaeobacter clusters but did not branch off between them (not shown). Even P. gallaeciensis strains isolated from the open ocean zooplankton fell into the existing sequence cluster (Freese et al. 2017). Since we did not find evidence for geographic isolation as the driver of diversification, the three Phaeobacter clades likely constitute distinct ecotypes (Cohan and Perry 2007). Different ecotypes with different physiologies and cluster-specific functional genes have been identified in the marine Prochlorococcus and the SAR11 clade (Venter et al. 2004; Kettler et al. 2007; Rusch et al. 2007), but in the latter cases differed by as much as > 4% (Carlson et al. 2009) and only rarely by <1% (Hunt et al. 2008) in their 16S rRNA gene sequences. Since the differentiation in Phaeobacter occurred on a lower level of phylogenetic divergence (<0.5%) we sought to identify potential mechanisms of this micro-diversification.
Potential Drivers of Speciation
While no habitat preference was obvious for the Phaeobacter clades, specific gene functions and physiological traits distinguished the clades from each other, suggesting that they are linked to the evolutionary diversification within the genus. Due to the low population census sizes, and the failure to detect Phaeobacter at its low abundances by cultivation-independent approaches in most marine samples (see Introduction) prompted us to apply a reverse ecology approach in combination with a laboratory analysis of phenotypic properties to predict differences in the ecology of the clades. The large phylogenetic distance of P. gallaeciensis to the other clusters enabled the identification of a particularly large number of gained genes for this species. Algae, unlike copepods or fish, rapidly take up cobalt (Nolan et al. 1992). Therefore, the presence of the ABC-type cobalt transporter to acquire sufficient amounts of the essential trace element (Rodionov et al. 2006) may give P. gallaeciensis a competitive edge when growing in association with algae. In line with this hypothesis, the high number of catalases detected in P. gallaeciensis would protect the cells against damage and inhibition by reactive oxygen species produced by algae (Palenik et al. 1987; Oda et al. 1997). The complete, chromosomal type IV secretion system in P. gallaeciensis strains may allow the transfer of effector macromolecules to the host (Christie et al. 2005). Plant-associated terrestrial bacteria degrade the plant hormone precursor 1-aminocyclopropane-1-carboxylate (ACC) which led to promoted plant growth (Nascimento et al. 2014). In marine algae, ethylene is also produced via ACC (Maillard et al. 1993; Plettner et al. 2005) and the acquisition of an ACC deaminase by P. gallaeciensis as well as its superior utilization of the deamination product α-ketobutyric acid suggest that this species may exert a growth promoting effect on algae. P. gallaeciensis is also more competitive to utilize the algal osmoprotectant sorbitol as well as methyl lactate which may act as an antimicrobial component produced by microalga (Santoyo et al. 2009). Although these traits also appear in bacteria from other environments, their combined occurrence suggest that P. gallaeciensis has an advantage in algal associations over the other clades. However, our data indicate that the traits determined do not confer exclusive habitat specificity as for instance P. gallaeciensis can also occur with clam larvae (Ruiz-Ponte et al. 1998; supplementary table S1, Supplementary Material online). However, a metagenomics study of Roseobacter group members associated with an Emiliania huxleyi bloom revealed highest relative abundances of P. gallaeciensis which were 3.5 times higher than of P. inhibens (Segev et al. 2016), supporting our conclusions on the selective advantages of P. gallaeciensis in algal associations.
The other Phaeobacter ecotypes were characterized by a faster utilization of glucosamine and of the biogenic amine tyramine. Tyramine occurs in many organisms but elevated concentrations occur by oxygen-limited decomposition of protein-rich organic matter like fish or other seafood (Prester 2011), whereas glucosamine is most abundant in chitinous organisms like crustaceae (Benner and Kaiser 2003). Marine animals or environments rich with animal resource patches may thus constitute preferred environments for P. inhibens and “P. piscinae.” Selective advantages of an association of “P. piscinae” with animals is further suggested by the lack of a specific transporter for glutamate, since the latter is mainly produced by plant and algae (Matsunaga et al. 1988; Tapiero et al. 2002), and also by the significantly more rapid utilization of N-acetylglutamate by “P. piscinae.” N-acetylglutamate is a metabolic intermediate in the arginine synthesis and hence occurs in many organisms, but particularly high levels are present in fish where it acts as an important cofactor in the urea cycle (Caldovic and Tuchman 2003). Notably, one of the two “P. piscinae” subclusters has exclusively acquired genes homologous to CopABCD. These genes facilitate copper homeostasis and resistance in copper rich environments (Bondarczuk and Piotrowska-Seget 2013) and may represent a specific adaptation of members of the subcluster to copper compounds introduced in aquaculture environments through antifouling coatings or feeding supplements (Lorentzen et al. 1998; Stickney and McVey 2002). The distinct patterns of functional genes in the two “P. piscinae” subclusters indicate an incipient diversification of two different ecotypes in this particular cluster.
The loss of the heme acquisition/degradation system that distinguishes P. inhibens from the majority of the Roseobacter group (Roe et al. 2013) and also from the other Phaeobacter clusters suggests an adaptation to the utilization of iron ions through siderophores (Thole et al. 2012; investigated strains were reclassified from P. gallaeciensis to P. inhibens). Within P. inhibens the 20 strains did not reveal a prevalence of any particular functional gene in different strains, suggesting that the ongoing radiation within this particular cluster was selectively neutral.
It has to be emphasized that most of the phenotypic differences detected in carbon substrate utilization concerned the efficiency of substrate utilization. These quantitative rather than qualitative differences seem to be of particular relevance for the evolution of Phaeobacter, but would have remained undetected by conventional testing. Our approach allowed the prediction of potential niches of different Phaeobacter clades that can now be tested in targeted phenotypic or environmental approaches.
Hallmarks and Implications of the Phaeobacter Population Genomics
Whereas the evolution of the entire Roseobacter group has been characterized by a steady net genome reduction (Luo et al. 2013), our data revealed that the more recent evolution of the three Phaeobacter species occurred through a slow, but continuous expansion of their chromosomes. They evolve in a manner different from free-living marine bacteria like the SAR11 clade and Prochlorococcus which were shaped by genome streamlining (Luo et al. 2011; Grote et al. 2012). The large core genome, high level of genome synteny, and the low proportion of chromosomal gene flow indicates that the three clusters of Phaeobacter have to be considered as “young” bacterial species (Nowell et al. 2014) which nevertheless already may constitute potential ecotypes. The traits for their versatile life-style are still conserved among all strains investigated. Within the surface-associated Phaeobacter, the evolutionary processes thus differ from that of marine generalists which are also capable of growing in the attached mode, such as Vibrio which maintains a large flexible genome leading to strong variations in gene content between closely related strains of the same population (Polz et al. 2006). Additional research is needed to better understand if these are general principles of evolution and niche adaptation of surface-associated bacteria which can constitute up to 66% of bacterial biomass in coastal marine environments (Becquevort et al. 1998).
Similar to the general evolutionary trend of the entire Roseobacter group (Luo et al. 2013), innovation in the genus Phaeobacter occurred mostly through acquisition of gene families involved in amino acid transport and metabolism (COG category E), gene regulation (K), and replication/recombination/repair (L). Based on our genome analysis, the particular adaptation of members of the Roseobacter group to the utilization of ephemeral nutrient patches (Luo et al. 2013) was further strengthened during the recent and ongoing differentiation of Phaeobacter species. We actually do not know which environmental factors triggered the first steps of incipient diversification. However, our results suggest that, at the present stage of evolutionary diversification, the limited number of clade-specific laterally transferred genes provides adaptive advantage to different niches existing on surfaces of marine organisms and particles and drive ongoing diversification.
Given the distinct, but phylogenetically closely related clusters (>99.5% 16S rRNA gene sequence similarity) that were maintained during the evolution of Phaeobacter, our results not only confirmed that distinct geno- and ecotypic diversity is often hidden in bacterial groups with similar 16S rRNA gene sequences (Jaspers and Overmann 2004) but actually indicate mechanisms how populations with highly similar rRNA gene sequences diversify. Furthermore, the presently employed general cutoff for species delineation (97% or 98.6%) appears too coarse and should only be applied after careful consideration of the overall genomic divergence.
Taken together, the cluster-specific adaptations and genomic diversification revealed by the present work indicates that acquisition of functional genes reinforce speciation that may even be ongoing in “P. piscinae” whereas the recent genome divergence within P. inhibens so far has remained largely neutral. Our research further indicates that GTA likely mediates a large part of HGT and is an important driver of genome expansion in Phaeobacter. However, it remains to be investigated if this is a general characteristic of the surface-dwelling roseobacters.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
The work was supported by the Transregional Collaborative Research Centre “Roseobacter” (TRR 51/2 TA07), funded by the Deutsche Forschungsgemeinschaft. Cisse H. Porsby, Susanna Prado Plana and Jean-Louis Nicolas provided strains and information regarding their habitat. Rüdiger Pukall organized the strains, checked their quality and preserved them. We thank Markus Göker for fruitful comments and discussions regarding the design of phylogenetic analysis. We thank Isabel Vogt for correcting the genome annotations according to the NCBI requirements and Anika Methner, Alicia Geppert, Nicole Heyer, and Simone Severitt for excellent technical assistance.
Literature Cited
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 215(3):403–410. [DOI] [PubMed] [Google Scholar]
- Azam F, et al. 1983. The ecological role of water-column microbes in the sea. Mar Ecol Prog Ser. 10:257–263.http://dx.doi.org/10.3354/meps010257 [Google Scholar]
- Balcazar JL, Subirats J, Borrego CM.. 2015. The role of biofilms as environmental reservoirs of antibiotic resistance. Front Microbiol. 6:1216.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becquevort S, Rousseau V, Lancelot C.. 1998. Major and comparable roles for free-living and attached bacteria in the degradation of Phaeocystis-derived organic matter in Belgian coastal waters of the North Sea. Aquat Microb Ecol. 14:39–48.http://dx.doi.org/10.3354/ame014039 [Google Scholar]
- Benner R, Kaiser K.. 2003. Abundance of amino sugars and peptidoglycan in marine particulate and dissolved organic matter. Limnol Oceanogr. 48(1):118–128.http://dx.doi.org/10.4319/lo.2003.48.1.0118 [Google Scholar]
- Bižic-Ionescu M, et al. 2015. Comparison of bacterial communities on limnic versus coastal marine particles reveals profound differences in colonization. Environ Microbiol. 17(10):3500–3514. [DOI] [PubMed] [Google Scholar]
- Bobay LM, Touchon M, Rocha EPC.. 2014. Pervasive domestication of defective prophages by bacteria. Proc Natl Acad Sci U S A. 111(33):12127–12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarczuk K, Piotrowska-Seget Z.. 2013. Molecular basis of active copper resistance mechanisms in Gram-negative bacteria. Cell Biol Toxicol. 29(6):397–405.http://dx.doi.org/10.1007/s10565-013-9262-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongrand C, et al. 2016. A genomic comparison of 13 symbiotic Vibrio fischeri isolates from the perspective of their host source and colonization behavior. ISME J. 10(12):2907–2917.http://dx.doi.org/10.1038/ismej.2016.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borcard D, Gillet F, Legendre P, editor. 2011. Numerical ecology with R. New York: Springer. [Google Scholar]
- Breider S, et al. 2014. Genome-scale data suggest reclassifications in the Leisingera–Phaeobacter cluster including proposals for Sedimentitalea gen. nov and Pseudophaeobacter gen. nov. Front Microbiol. 5:416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimacombe CA, Ding H, Johnson JA, Beatty JT.. 2015. Homologues of genetic transformation DNA import genes are required for Rhodobacter capsulatus gene transfer agent recipient capability regulated by the response regulator CtrA. J Bacteriol. 197(16):2653–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkhoff T, Giebel HA, Simon M.. 2008. Diversity, ecology, and genomics of the Roseobacter clade: a short overview. Arch Microbiol. 189(6):531–539. [DOI] [PubMed] [Google Scholar]
- Brinkhoff T, et al. 2004. Antibiotic production by a Roseobacter clade-affiliated species from the German Wadden Sea and its antagonistic effects on indigenous isolates. Appl Environ Microbiol. 70(4):2560–2565.http://dx.doi.org/10.1128/AEM.70.4.2560-2565.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan A, González JM, Moran MA.. 2005. Overview of the marine Roseobacter lineage. Appl Environ Microbiol. 71(10):5665–5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buddruhs N, et al. 2013. Molecular and phenotypic analyses reveal the non-identity of the Phaeobacter gallaeciensis type strain deposits CIP 105210T and DSM 17395. Int J Syst Evol Microbiol. 63(Pt 11):4340–4349. [DOI] [PubMed] [Google Scholar]
- Bunk B. Genomefinishing tools. 2016. Available from: https://github.com/boykebunk/genomefinish, last accessed July 2016.
- Caldovic L, Tuchman M.. 2003. N-Acetylglutamate and its changing role through evolution. Biochem J 372(Pt 2):279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CA, et al. 2009. Seasonal dynamics of SAR11 populations in the euphotic and mesopelagic zones of the northwestern Sargasso Sea. ISME J. 3(3):283–295.http://dx.doi.org/10.1038/ismej.2008.117 [DOI] [PubMed] [Google Scholar]
- Christie PJ, Atmakuri K, Krishnamoorthy V, Jakubowski S, Cascales E.. 2005. Biogenesis, architecture, and function of bacterial type IV secretion systems. Annu Rev Microbiol. 59:451–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohan FM, Perry EB.. 2007. A systematics for discovering the fundamental units of bacterial diversity. Curr Biol. 17(10):R373–R386. [DOI] [PubMed] [Google Scholar]
- Coleman ML, et al. 2006. Genomic islands and the ecology and evolution of Prochlorococcus. Science 311(5768):1768–1770.http://dx.doi.org/10.1126/science.1122050 [DOI] [PubMed] [Google Scholar]
- Crump BC, Baross JA, Simenstad CA.. 1998. Dominance of particle-attached bacteria in the Columbia River estuary, USA. Aquat Microb Ecol. 14:7–18.http://dx.doi.org/10.3354/ame014007 [Google Scholar]
- D'Alvise PW, et al. 2012. Phaeobacter gallaeciensis reduces Vibrio anguillarum in cultures of microalgae and rotifers, and prevents vibriosis in cod larvae. PLoS One 7:e43996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT.. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5(6):e11147.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon BK, Chiu TA, Laird MR, Langille MG,I, Brinkman FS.. 2013. IslandViewer update: improved genomic island discovery and visualization. Nucleic Acids Res. 41(W1):W129–W132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickschat JS, Zell C, Brock NL.. 2010. Pathways and substrate specificity of DMSP catabolism in marine bacteria of the Roseobacter clade. Chembiochem 11(3):417–425.http://dx.doi.org/10.1002/cbic.200900668 [DOI] [PubMed] [Google Scholar]
- Dogs M, et al. 2013. Genome sequence of Phaeobacter inhibens type strain (T5(T)), a secondary metabolite producing representative of the marine Roseobacter clade, and emendation of the species description of Phaeobacter inhibens. Stand Genomic Sci. 9(2):334–350.http://dx.doi.org/10.4056/sigs.4448212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA.. 2002. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30(7):1575–1584.http://dx.doi.org/10.1093/nar/30.7.1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher MM, Triplett EW.. 1999. Automated approach for ribosomal intergenic spacer analysis of microbial diversity and its application to freshwater bacterial communities. Appl Environ Microbiol. 65(10):4630–4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank O, et al. 2014. Complete genome sequence of the Phaeobacter gallaeciensis type strain CIP 105210(T) (= DSM 26640(T)=BS107(T). Stand Genomic Sci. 9(3):914–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank O, et al. 2015. Plasmid curing and the loss of grip: the 65-kb replicon of Phaeobacter inhibens DSM 17395 is required for biofilm formation, motility and the colonization of marine algae. Syst Appl Microbiol. 38(2):120–127.http://dx.doi.org/10.1016/j.syapm.2014.12.001 [DOI] [PubMed] [Google Scholar]
- Freese HM, Methner A, Overmann J.. 2017. Adaptation of surface-associated bacteria to the open ocean: a genomically distinct subpopulation of Phaeobacter gallaeciensis colonizes Pacific mesozooplankton. Front Microbiol. 8:1659.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gram L, et al. 2015. Phaeobacter inhibens from the Roseobacter clade has an environmental niche as a surface colonizer in harbors. Syst Appl Microbiol. 38(7):483–493.http://dx.doi.org/10.1016/j.syapm.2015.07.006 [DOI] [PubMed] [Google Scholar]
- Grote J, et al. 2012. Streamlining and core genome conservation among highly divergent members of the SAR11 clade. mBio 3(5):e00252-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henz SR, Huson DH, Auch AF, Nieselt-Struwe K, Schuster SC.. 2005. Whole-genome prokaryotic phylogeny. Bioinformatics 21(10):2329–2335. [DOI] [PubMed] [Google Scholar]
- Hjelm M, Riaza A, Formoso F, Melchiorsen J, Gram L.. 2004. Seasonal incidence of autochthonous antagonistic Roseobacter spp. and Vibrionaceae strains in a turbot larva (Scophthalmus maximus) rearing system. Appl Environ Microbiol. 70(12):7288–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hothorn T, Bretz F, Westfall P.. 2008. Simultaneous inference in general parametric models. Biom J. 50(3):346–363.http://dx.doi.org/10.1002/bimj.200810425 [DOI] [PubMed] [Google Scholar]
- Huang SK, Zhang YU, Chen F, Jiao NZ.. 2011. Complete genome sequence of a marine roseophage provides evidence into the evolution of gene transfer agents in alphaproteobacteria. Virol J. 8(1):124..http://dx.doi.org/10.1186/1743-422X-8-124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt DE, et al. 2008. Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science 320(5879):1081–1085.http://dx.doi.org/10.1126/science.1157890 [DOI] [PubMed] [Google Scholar]
- Huson DH, Bryant D.. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 23(2):254–267.http://dx.doi.org/10.1093/molbev/msj030 [DOI] [PubMed] [Google Scholar]
- Hynes AP, Mercer RG, Watton DE, Buckley CB, Lang AS.. 2012. DNA packaging bias and differential expression of gene transfer agent genes within a population during production and release of the Rhodobacter capsulatus gene transfer agent, RcGTA. Mol Microbiol. 85(2):314–325. [DOI] [PubMed] [Google Scholar]
- Jaspers E, Overmann J.. 2004. Ecological significance of microdiversity: identical 16S rRNA gene sequences can be found in bacteria with highly divergent genomes and ecophysiologies. Appl Environ Microbiol. 70(8):4831–4839.http://dx.doi.org/10.1128/AEM.70.8.4831-4839.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji JD, Zhang R, Jiao NZ.. 2015. Complete genome sequence of Roseophage vB_DshP-R1, which infects Dinoroseobacter shibae DFL12. Stand Genomic Sci. 10(1):6..http://dx.doi.org/10.1186/1944-3277-10-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ZI, et al. 2006. Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science 311(5768):1737–1740.http://dx.doi.org/10.1126/science.1118052 [DOI] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM.. 1992. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 8(3):275–282. [DOI] [PubMed] [Google Scholar]
- Kettler GC, et al. 2007. Patterns and implications of gene gain and loss in the evolution of Prochlorococcus. PLoS Genet. 3(12):2515–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchberger PC, et al. 2016. A small number of phylogenetically distinct clonal complexes dominate a coastal Vibrio cholerae population. Appl Environ Microbiol. 82(18):5576–5586.http://dx.doi.org/10.1128/AEM.01177-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DJ. 1991. 16S/23S rRNA sequencing In: Stackebrandt E, Goodfellow M, editors. Nucleic acid techniques in bacterial systematics. Chichester: John Wiley & Sons; p. 115–175. [Google Scholar]
- Lang AS, Beatty JT.. 2007. Importance of widespread gene transfer agent genes in alpha-proteobacteria. Trends Microbiol. 15(2):54–62.http://dx.doi.org/10.1016/j.tim.2006.12.001 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760.http://dx.doi.org/10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang YT, et al. 2016. Complete genome sequence of the siphovirus Roseophage RDJL Phi 2 infecting Roseobacter denitrificans OCh114. Mar Genomics 25:17–19.http://dx.doi.org/10.1016/j.margen.2015.10.009 [DOI] [PubMed] [Google Scholar]
- Librado P, Vieira F, Rozas J.. 2012. BadiRate: estimating family turnover rates by likelihood-based methods. Bioinformatics 28(2):279–281.http://dx.doi.org/10.1093/bioinformatics/btr623 [DOI] [PubMed] [Google Scholar]
- Lopes A, Tavares P, Petit M-A, Guérois R, Zinn-Justin S.. 2014. Automated classification of tailed bacteriophages according to their neck organization. BMC Genomics 15:1027.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorentzen M, Maage A, Julshamn K.. 1998. Supplementing copper to a fish meal based diet fed to Atlantic salmon parr affects liver copper and selenium concentrations. Aquac Nutr. 4:67–72.http://dx.doi.org/10.1046/j.1365-2095.1998.00046.x [Google Scholar]
- Ludwig W. et al. 2004. ARB: a software environment for sequence data. Nucleic Acids Res. 32:1363–1371.http://dx.doi.org/10.1093/nar/gkh293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Moran MA.. 2014. Evolutionary ecology of the marine Roseobacter clade. Microbiol Mol Biol Rev. 78(4):573–587.http://dx.doi.org/10.1128/MMBR.00020-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Csûros M, Hughes AL, Moran MA.. 2013. Evolution of divergent life history strategies in marine alphaproteobacteria. mBio 4(4):e00373-13.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Friedman R, Tang J, Hughes AL.. 2011. Genome reduction by deletion of paralogs in the marine cyanobacterium Prochlorococcus. Mol Biol Evol. 28(10):2751–2760.http://dx.doi.org/10.1093/molbev/msr081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillard P, Thepenier C, Gudin C.. 1993. Determination of an ethylene biosynthesis pathway in the unicellular green-alga, Haematococcus pluvialis: relationship between growth and ethylene production. J Appl Phycol. 5(1):93–98.http://dx.doi.org/10.1007/BF02182426 [Google Scholar]
- Matsunaga T, Nakamura N, Tsuzaki N, Takeda H.. 1988. Selective production of glutamate by an immobilized marine blue-green alga, Synechococcus sp. Appl Microbiol Biotechnol. 28(4-5):373–376. [Google Scholar]
- McDaniel LD, et al. 2010. High frequency of horizontal gene transfer in the oceans. Science 330(6000):50.http://dx.doi.org/10.1126/science.1192243 [DOI] [PubMed] [Google Scholar]
- Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M.. 2013. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meusemann K, et al. 2010. A phylogenomic approach to resolve the arthropod tree of life. Mol Biol Evol. 27(11):2451–2464.http://dx.doi.org/10.1093/molbev/msq130 [DOI] [PubMed] [Google Scholar]
- Nascimento FX, et al. 2014. New Insights into 1-aminocyclopropane-1-carboxylate (ACC) deaminase phylogeny, evolution and ecological significance. PLoS One 9(6):e99168.,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton RJ, et al. 2010. Genome characteristics of a generalist marine bacterial lineage. ISME J. 4(6):784–798.http://dx.doi.org/10.1038/ismej.2009.150 [DOI] [PubMed] [Google Scholar]
- Nolan CV, Fowler SW, Teyssie J-L.. 1992. Cobalt speciation and bioavailability in marine organisms. Mar Ecol Prog Ser. 88:105–116.http://dx.doi.org/10.3354/meps088105 [Google Scholar]
- Nowell RW, Green S, Laue BE, Sharp PM.. 2014. The extent of genome flux and its role in the differentiation of bacterial lineages. Genome Biol Evol. 6(6):1514–1529.http://dx.doi.org/10.1093/gbe/evu123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T, et al. 1997. Generation of reactive oxygen species by raphidophycean phytoplankton. Biosci Biotechnol Biochem. 61(10):1658–1662.http://dx.doi.org/10.1271/bbb.61.1658 [DOI] [PubMed] [Google Scholar]
- Oksanen J, et al. 2015. vegan: community ecology package. Available from: https://CRAN.R-project.org/package=vegan, last accessed October 2016.
- Pagès H, Aboyoun P, Gentleman R, DebRoy S.. 2016. Biostrings: string objects representing biological sequences, and matching algorithms. Available from: https://bioconductor.org/packages/release/bioc/html/Biostrings.html, last accessed October 2016.
- Palenik B, Zafiriou OC, Morel FMM.. 1987. Hydrogen peroxide production by a marine phytoplankter. Limnol Oceanogr. 32(6):1365–1369.http://dx.doi.org/10.4319/lo.1987.32.6.1365 [Google Scholar]
- Paradis E, Claude J, Strimmer K.. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20(2):289–290.http://dx.doi.org/10.1093/bioinformatics/btg412 [DOI] [PubMed] [Google Scholar]
- Petersen J, Frank O, Göker M, Pradella S.. 2013. Extrachromosomal, extraordinary and essential: the plasmids of the Roseobacter clade. Appl Microbiol Biotechnol. 97(7):2805–2815. [DOI] [PubMed] [Google Scholar]
- Plettner I, Steinke M, Malin G.. 2005. Ethene (ethylene) production in the marine macroalga Ulva (Enteromorpha) intestinalis L. (Chlorophyta, Ulvophyceae): effect of light-stress and co-production with dimethyl sulphide. Plant Cell Environ. 28(9):1136–1145.http://dx.doi.org/10.1111/j.1365-3040.2005.01351.x [Google Scholar]
- Polz MF, Hunt DE, Preheim SP, Weinreich DM.. 2006. Patterns and mechanisms of genetic and phenotypic differentiation in marine microbes. Philos Trans R Soc B Biol Sci. 361(1475):2009–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsby CH, Nielsen KF, Gram L.. 2008. Phaeobacter and Ruegeria species of the Roseobacter clade colonize separate niches in a danish turbot (Scophthalmus maximus)-rearing farm and antagonize Vibrio anguillarum under different growth conditions. Appl Environ Microbiol. 74(23):7356–7364.http://dx.doi.org/10.1128/AEM.01738-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado S, Montes J, Romalde JL, Barja JL.. 2009. Inhibitory activity of Phaeobacter strains against aquaculture pathogenic bacteria. Int Microbiol. 12(2):107–114. [PubMed] [Google Scholar]
- Prester L. 2011. Biogenic amines in fish, fish products and shellfish: a review. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 28(11):1547–1560.http://dx.doi.org/10.1080/19440049.2011.600728 [DOI] [PubMed] [Google Scholar]
- R Core Team. 2015. R: a language and environment for statistical computing. Available from: https://www.R-project.org/, last accessed October 2016. [Google Scholar]
- Rao D, et al. 2007. Low densities of epiphytic bacteria from the marine alga Ulva australis inhibit settlement of fouling organisms. Appl Environ Microbiol. 73(24):7844–7852.http://dx.doi.org/10.1128/AEM.01543-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao D, Webb JS, Kjelleberg S.. 2005. Competitive interactions in mixed-species biofilms containing the marine bacterium Pseudoalteromonas tunicata. Appl Environ Microbiol. 71(4):1729–1736.http://dx.doi.org/10.1128/AEM.71.4.1729-1736.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel T, et al. 2013. Genomics and physiology of a marine Flavobacterium encoding a proteorhodopsin and a xanthorhodopsin-like protein. PLoS One 8(3):e57487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EPC. 2016. Using sex to cure the genome. PLoS Biol. 14(3):e1002417.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov DA, Hebbeln PF, Gelfand MSFAU, Eitinger T.. 2006. Comparative and functional genomic analysis of prokaryotic nickel and cobalt uptake transporters: evidence for a novel group of ATP-binding cassette transporters. J Bacteriol. 188(1):317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe KL, Hogle SL, Barbeau KA.. 2013. Utilization of heme as an iron source by marine alphaproteobacteria in the Roseobacter clade. Appl Environ Microbiol. 79(18):5753–5762.http://dx.doi.org/10.1128/AEM.01562-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Ponte C, Cilia V, Lambert C, Nicolas JL.. 1998. Roseobacter gallaeciensis sp. nov., a new marine bacterium isolated from rearings and collectors of the scallop Pecten maximus. Int J Syst Evol Microbiol. 48(2):537–542. [DOI] [PubMed] [Google Scholar]
- Rusch DB, et al. 2007. The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol. 5:398–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson MJ. 2003. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19(2):301–302.http://dx.doi.org/10.1093/bioinformatics/19.2.301 [DOI] [PubMed] [Google Scholar]
- Santoyo S, et al. 2009. Green processes based on the extraction with pressurized fluids to obtain potent antimicrobials from Haematococcus pluvialis microalgae. LWT: Food Sci Technol. 42(7):1213–1218.http://dx.doi.org/10.1016/j.lwt.2009.01.012 [Google Scholar]
- Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069.http://dx.doi.org/10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- Segev E, et al. 2016. Dynamic metabolic exchange governs a marine algal-bacterial interaction. Elife 5:e17473.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segev E, Tellez A, Vlamakis H, Kolter R.. 2015. Morphological heterogeneity and attachment of Phaeobacter inhibens. PLoS One 10(11):e0141300.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedsayamdost MR, Case RJ, Kolter R, Clardy J.. 2011. The Jekyll-and-Hyde chemistry of Phaeobacter gallaeciensis. Nat Chem. 3(4):331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro BJ, et al. 2012. Population genomics of early events in the ecological differentiation of bacteria. Science 336(6077):48–51.http://dx.doi.org/10.1126/science.1218198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M, et al. 2017. Phylogenomics of Rhodobacteraceae reveals evolutionary adaptation to marine and non-marine habitats. ISME J. 11(6):1483–1499.http://dx.doi.org/10.1038/ismej.2016.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickney RR, McVey JP, editors. 2002. Responsible marine aquaculture. New York: CABI Publishing. [Google Scholar]
- Stocker R. 2012. Marine microbes see a sea of gradients. Science 338(6107):628..http://dx.doi.org/10.1126/science.1208929 [DOI] [PubMed] [Google Scholar]
- Sullivan MJ, Petty NK, Beatson SA.. 2011. Easyfig: a genome comparison visualizer. Bioinformatics 27(7):1009–1010.http://dx.doi.org/10.1093/bioinformatics/btr039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunagawa S, et al. 2015. Structure and function of the global ocean microbiome. Science 348:6237. [DOI] [PubMed] [Google Scholar]
- Swan BK, et al. 2013. Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc Natl Acad Sci U S A. 110(28):11463–11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28(10):2731–2739.http://dx.doi.org/10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapiero H, Mathé G, Couvreur P, Tew KD.. 2002. II. Glutamine and glutamate. Biomed Pharmacother. 56(9):446–457. [DOI] [PubMed] [Google Scholar]
- Thole S, et al. 2012. Phaeobacter gallaeciensis genomes from globally opposite locations reveal high similarity of adaptation to surface life. ISME J. 6(12):2229–2244.http://dx.doi.org/10.1038/ismej.2012.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CC, et al. 2009. Genomic taxonomy of vibrios. BMC Evol Biol. 9(1):258.http://dx.doi.org/10.1186/1471-2148-9-258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaas LAI, et al. 2013. opm: an R package for analysing OmniLog((R)) phenotype microarray data. Bioinformatics 29(14):1823–1824.http://dx.doi.org/10.1093/bioinformatics/btt291 [DOI] [PubMed] [Google Scholar]
- Venter JC, et al. 2004. Environmental genome shotgun sequencing of the Sargasso Sea. Science 304(5667):66–74.http://dx.doi.org/10.1126/science.1093857 [DOI] [PubMed] [Google Scholar]
- Voigt A, Schöfl G, Saluz HP.. 2012. The Chlamydia psittaci genome: a comparative analysis of intracellular pathogens. PLoS One 7(4):e35097.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M, Didelot X.. 2009. A comparison of homologous recombination rates in bacteria and archaea. ISME J. 3(2):199–208.http://dx.doi.org/10.1038/ismej.2008.93 [DOI] [PubMed] [Google Scholar]
- Vos M, et al. 2013. ODoSE: a webserver for genome-wide calculation of adaptive divergence in prokaryotes. PLoS One 8(5):e62447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H, editor. 2009. ggplot2: elegant graphics for data analysis. New York: Springer. [Google Scholar]
- Wu S, Zhu Z, Fu L, Niu B, Li W.. 2011. WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics 12:444.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZH. 1993. Maximum-likelihood-estimation of phylogeny from DNA-sequences when substitution rates differ over sites. Mol Biol Evol. 10:1396–1401. [DOI] [PubMed] [Google Scholar]
- Yelton AP, et al. 2011. A semi-quantitative, synteny-based method to improve functional predictions for hypothetical and poorly annotated bacterial and archaeal genes. PLoS Comput Biol. 7(10):e1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zech H, et al. 2013. Adaptation of Phaeobacter inhibens DSM 17395 to growth with complex nutrients. Proteomics 13(18–19):2851–2868. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS.. 2011. PHAST: a fast phage search tool. Nucleic Acids Res. 39(Suppl):W347–W352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinger L, et al. 2011. Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS One 6(9):224570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.