

Abstract
Great efforts have been devoted to effective delivery of therapeutics into cells for cancer therapy. The exploration of nanoparticle based drug delivery systems (DDSs) faces daunting challenges in low efficacy of intracellular delivery. Herein, a localized drug delivery device consisting of photoluminescent mesoporous silica nanoparticles (PLMSNs) and photothermal fibrous matrix was investigated. Specifically, PLMSNs modified with a pH-sensitive polydopamine (PDA) ‘gatekeeper’ served as a doxorubicin (DOX) carrier and could release DOX once the PLMSNs were up-taken by the cancer cells. The PLMSNs were electrostatically assembled on the surface of electrospun biodegradable poly(ε-caprolactone)/gelatin fibrous mesh incorporated with photothermal carbon nanoparticles (CNPs), leading to an implantable patch used as localized delivery platform. Comparing to free particulate DDSs, this implantable composite patch device was found to significantly enable superior cell up-taking effect and consequently enhance in-vitro therapeutic efficacy against tumor cells. Namely, under near infrared irradiation, the photothermal effect of CNPs in the implantable patch weakens the electrostatic interaction between the PLMSNs and poly(ε-caprolactone)/gelatin/CNP fibrous mesh, resulting in the controlled release of the PLMSNs and subsequent internalization into the tumor cells for more effective cancer cell killing. This implantable therapeutic device may therefore inspire another way of developing localized cancer therapy.
1. Introduction
Over the past decades, nanoparticles have been extensively explored to deliver anticancer therapeutics to tumors owing to their preferential accumulation at tumor sites via enhanced permeability and retention (EPR) effect.1–3 In general, nanoparticles that remain at the tumor site are internalized by cells, and intracellular drug release occurs subsequently.4, 5 In consequence, enhanced therapeutic efficacy as well as reduced systematic toxicity, comparing to conventional drug administration, are achieved. This has led researchers to engineer a myriad of drug delivery systems (DDSs) exhibiting unique physicochemical properties (i.e. size, shape and surface chemistry) and programmed biomedical functions.6, 7 Further, nanoparticles have been endowed with contrast agents,8, 9 triggering factors (i.e. pH, enzymatic catalysis etc.) for controlled drug release,10, 11 or other therapeutic agents.12–14 Studies have demonstrated the utility of nanoparticles for detecting and killing cancer cells both in vitro and in vivo. However, the clinical translation of particulate DDSs hangs back. The main challenge is the low delivery efficacy and cell up-taking effect of the nanoparticles at the tumor site induced by rapid filtration in the kidney, clearance via the reticuloendothelial system and overreliance on EPR effect during blood circulation.15–17 In fact, the recent controversial meta-analysis shows that, on average, as low as 0.7% of the injected dose (ID) of intravenously administered nanoparticles accumulates in tumors.18 Tackling this vital problem is being endeavored in the community of drug delivery and cancer therapy.
Localized delivery of anticancer drugs directly to the tumor site was proposed as a promising alternative approach.19–21 Current localized drug delivery systems (LDDSs) are in forms of drug-eluting films, hydrogels, wafers, rods, and microspheres. They are mainly biodegradable polymer based platforms in order to avoid repeated removal surgery.22–24 Alt hough the nanoscale two-dimensional (2D) mesh reported recently can be effectively internalized by the cells after localized injection, it may also possibly to migrate away from the tumor site due to its low dimension in an in vivo environment.25 Recently, a series of implantable electrospun nanofibrous LDDSs have shown superior therapeutic performance in cancer treatment due to their unique strength in ensuring therapeutic dosage levels at the tumor site for extended period while maintaining low systematic drug exposure.26–28 However, the drugs loaded at the surface of LDDSs intend to present burst releasing at the initial stage of drug delivery. Direct incorporation of such biomolecules (drugs) within the polymeric matrix during synthesis may induce drug degradation owing to organic solvents or heating procedures involved.29 More importantly, drug molecules released from LDDSs can hardly penetrate into the tissue or cell membranes in an effective manner. Instead, free drug molecules released are cleared by rapid metabolism, resulting in insufficient dosage at the diseased site and undesired side effect.4 To improve the performance of LDDSs, particulate carriers were recently incorporated into electrospun nanofibers for effective dosage delivery. Prolonged drug release and tunable release kinetics were achieved.30–32 The findings also showed that nanoparticles loaded with macromolecules tend to aggregate within the polymeric precursors, so that unstable electrospinning or heterogeneous fibrous matrix is induced.33 Furthermore, the particle and drug release are highly dependent on the degradation rate of LDDSs,34 leading to an uncontrolled kinetics.
In this study, an implantable delivery system combining photoluminescent mesoporous silica nanoparticles (PLMSNs) and electrospun poly(ε-caprolactone)/gelatin/carbon nanoparticles (PGC) fibers was developed for doxorubicin (DOX, an anticancer drug model) delivery. Carbon nanoparticles (CNPs) were incorporated as a photothermal agent. PLMSNs which are Eu incorporated MSNs modified with dopamine (PDA, a pH-triggering ‘gatekeeper’ agent35) were used for encapsulating and delivering DOX molecules. These particulate carriers were assembled on the surface of fibrous matrix by the electrostatic interaction (scheme 1). The drug release profiles and cell up-taking behavior were systematically revealed via an in-vitro study using human Hep G2 liver cancer cells. This composite ‘tumor patch’ with superior cell up-taking effect and anticancer properties may suggest another promising therapeutic option for curing cancer.
Scheme 1.
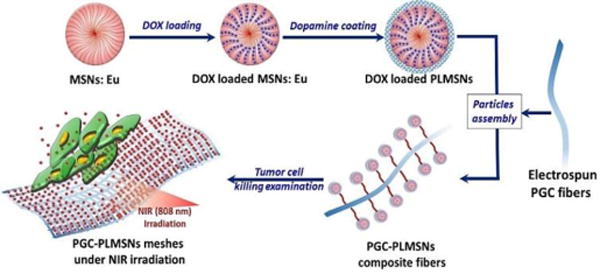
Schematic illustration of assembled DOX loaded PLMSNs on the surface of PGC fibers and using the DOX loaded PGC-PLMSNs fiber meshes to kill cancer cells
2. Results and discussion
2.1 Characteristics of PLMSNs with pH-responsive DOX release
During the immersion in europium nitrate solution, Eu ions entered the nanochannels of MSNs with ease due to the capillarity effect of mesopores at the particle surface. Eu incorporated MSNs (MSNs:Eu) were formed after vacuum drying and annealing process. As shown in Figure 1a & b, MSNs before and after Eu incorporation present a monodispersed spherical morphology with uniform size of ~100 nm. No apparent morphological or dimension changes due to Eu incorporation was observed. The element mapping analysis demonstrated that Eu element uniformly distributed within the matrix of MSNs: Eu (Figure 1c). The XRD patterns of both MSNs and MSNs:Eu samples showed a broad peak centered at ~22°, which is attributed to amorphous silica phase,37 and no impurities were observed. It is noted that the characteristic broad peak at ~22° were weakened after Eu incorporation due to the decrease of bridge-oxygen bonds and glass network integrity.
Figure 1.
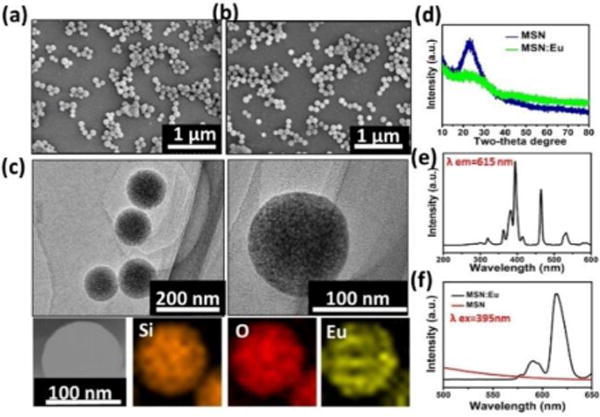
Characterization of MSNs. (a–b) SEM images of (a) MSNs (b) MSNs:Eu. (c) TEM images and elemental mapping pattern of MSNs:Eu. (d) XRD patterns of MSNs before and after Eu incorporation. (e–f) Excitation spectrum (e) and PL spectra under 395 nm excitation (f) of PLMSNs.
Rare earth elements have been widely employed to prepare photoluminescent (PL) nanomaterials.38 Europium (Eu), as an important rare earth element, has been widely used as cell labeling imaging.39, 41 In this study, europium was incorporated into the structure of MSNs to fabricate the drug delivery particles with PL imaging functionalities.40 In the excitation spectra of MSNs: Eu monitored by the Eu3+ 5D0–7F2 transition at 615 nm, sharp peaks at ~395 nm, ~465 nm and ~543 nm were observed owing to f–f transitions of Eu3+ (Figure 1e).41, 42 When the particles were excited using the spectrum at 395 nm, characteristic transition lines from the excited 5D0 level of Eu3+ were detected in the emission spectra. Two main characteristic peaks from 5D0–7F1 (~590 nm) and 5D0–7F2 (~615 nm) are dominant (Figure 1f). This phenomenon has also been reported elsewhere.39, 41 Meanwhile, red emission was also observed under the excitation at 543 nm (Figure S1). No clear photoluminescent phenomenon was observed from MSNs samples without Eu incorporation under excitation at 395 nm.
Previous studies have demonstrated immersing MSNs in a neutral dopamine solution resulted in oxidative self-polymerization dopamine (PDA).35 The formation of PDA coating at the surface of MSNs blocks the pores and thus confines drug molecules stored in the pores. In this study, after DOX loading procedure, a PDA coating was modified at the surface of MSNs: Eu (PLMSNs) following a previous approach reported.35 As shown in Figure 2a & b, the surface of PLMSNs roughens comparing with that of MSNs: Eu, and the TEM image confirms a typical core-shell structure of PLMSNs. The shell thickness is approximately 10 nm. The bands located at ~1612 cm−1, ~1560 cm−1 in the FTIR spectra of PLMSNs attribute to C=C groups and N-H groups, confirming the formation of PDA shell (Figure S2) which serves as the “gatekeepers” for pH-responsive drug releasing. The UV-vis spectrum of DOX solution presents a characteristic peak at ~480 nm (Figure S2b). After drug loading procedure, the UV-vis spectra of PLMSNs presented the same characteristic peak, suggesting the successful loading of DOX. The DOX loading capacity of PLMSNs was analyzed by TG curves. As shown in Figure 2c, the weight variation between nanoparticles before (MSNs:Eu) and after PDA coating (PLMSNs) is ~37.6 %. After loading with DOX, the weight difference of the particles increases by ~17.1%, reflecting its drug loading capacity.
Figure 2.
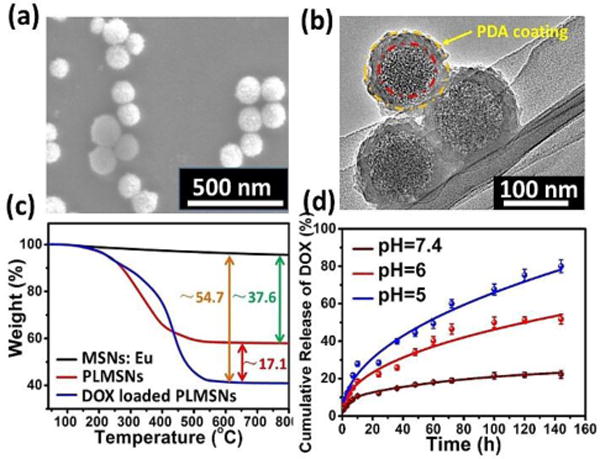
Characterization of PLMSNs. SEM (a) and (b) TEM images of PLMSNs. (c) TG curves of MSNs: Eu, PLMSNs and DOX loaded PLMSNs. (d) Cumulative DOX release profiles of PLMSNs in solutions with different pH values.
The in vitro drug release profiles of PLMSNs immersed in PBS solutions with different pH values were examined. At a neutral pH (7.4), ~12% of DOX content was released in the initial 24 h, and as low as ~20 % of total drug contents was released in 6 days. In comparison, in acidic conditions (pH=6.0, 5.0) that simulated those of endosome and lysosome, approximately 22% and 30% of drug contents were released within 24 h, respectively. The main mechanism is that the PDA coating, which prevents the liberation of DOX molecules from the particle pores, tends to detach from the surface of PLMSNs in the acidic condition. Therefore, the PDA coating avoids premature drug release from the nanoparticles in a normal tissue environment with a neutral pH. Once the particles are internalized by tumor cells and reached acidic endosomes by endocytosis, the drug can be effectively released into lysosomes.
2.2 Characteristics of PLMSNs assembled composite (PGC-PLMSNs) fibers
The electrospun PGC fibers present in uniform morphology with a mean diameter ~1μm (Fig. 3a). The water contact angle of the as-prepared PGC fiber mesh is ~63°, indicating its hydrophilic nature (insert of Figure. 3a). After the assembly procedure, large quantity of particles covered the surface of PGC nanofibers (Figure 3b&c). PDA molecules contain –OH groups which interact favorably with -NH2 groups in the Gel-incorporated PGC fibers through electrostatic attractions. In consequence, the water contact angle decreased to 26.8°. This hydrophilic transaction of fiber meshes may facilitate its water affinity after implantation. The FTIR analysis revealed that the characteristic peaks of the composite meshes consist of all peaks from PGC fibers and PLMSNs particles, confirming the formation of PGC-PLMSNs composite fibers. The TG curves (Figure S3) reflect the amount of particles grafted is ~ 9.15%. It is considered that the controlled assembly of therapeutic DDSs on the surface of meshes implanted is an effective strategy in ensuring the high concentration of DDSs at the diseased site while preventing its unexpected aggregation or excessive circulation.
Figure 3.
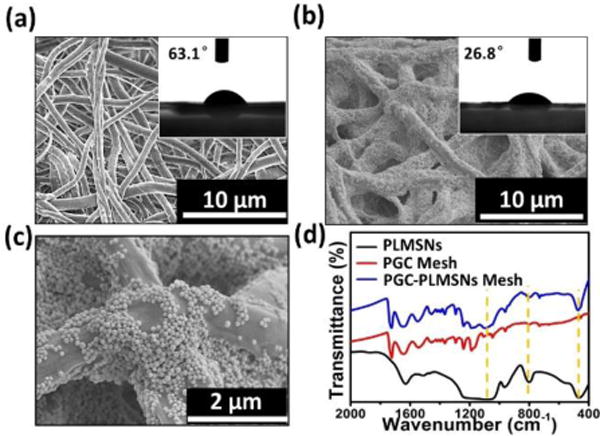
Characterization of meshes. (a) SEM and water contact angle images of electrospun PGC fibers. (b–c) SEM and water contact angle images of PGC-PLMSNs composite fibers. (d) FTIR spectra of PGC fibers, PLMSNs and PGC-PLMSNs composite meshes.
Furthermore, the in vitro study using NIH3T3 fibroblasts confirmed that all samples (PLMSNs, PGC-PLMSNs) without loading DOX contents presented over 90% relative cell viability (to blank control) after culturing for 1, 2 and 3 day, reflecting their expected cytocompatibility. It is noted that the mild NIR irradiation (0.6 W/cm2) for triggering photothermal effect from PGC-PLMSNs meshes did not induce clear negative effect to the cell proliferation (Figure S4).
2.3 In vitro anti-cancer performance
After loading DOX drug, the in vitro anticancer properties of PGC-PLMSNs composite meshes without and with NIR excitation (808 nm, 0.6 W/cm2) were analyzed using Hep G2 cancer cells. As shown in Figure 4, the samples without loading DOX, even after NIR excitation, present high cell viability, as expected. In comparison, the cell viability of all DOX loaded samples is decreased with incubation time. On day 1, free PLMSNs present similar cell viability to the PGC-PLMSNs composite meshes without NIR irradiation. The composite meshes after NIR irradiation induce the lowest cell viability. After incubation for 2 and 3 days, the most significant reduction in cell viability was observed for the composite meshes irradiated with NIR comparing to other two sample groups with similar cell viability.
Figure 4.
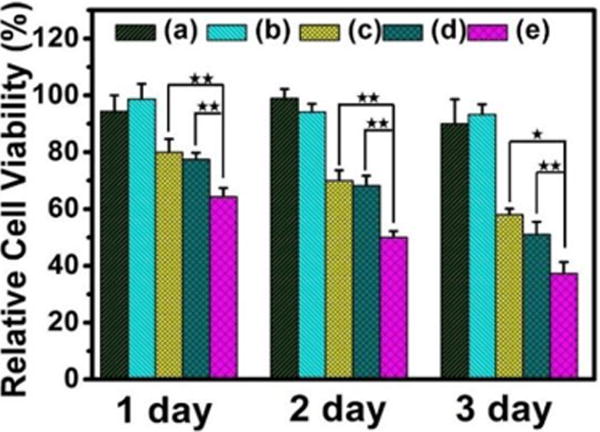
Relative cell viability (to blank control) of different delivery systems at different incubation time points. (a) PGC-PLMSNs meshes (no DOX). (b) PGC-PLMSNs (no DOX) after NIR irradiation (808 nm, 0.6 W/cm2). (c) DOX loaded PLMSNs. (d–e) PGC-PLMSNs composite meshes without (d) and with (e) NIR irradiation (808 nm, 0.6 W/cm2).
The findings above show that, only when PGC-PLMSNs meshes were loaded with DOX contents, NIR irradiation (808 nm, 0.6 W/cm2) induces clear negative effect to cell viability. It is thus considered the evolution of PGC-PLMSNs composite meshes under this NIR irradiation plays a vital role in this phenomenon. Carbon nanomaterials have been widely used as photothermal agents due to the impressive ability to convert near-infrared radiation into heat, as well as their good biocompatibility. As expected, when irradiated by an 808 nm NIR laser (1.0 W/cm2), the carbon nanoparticles solution exhibited a clear concentration-dependent heating effect.(Figure S5). Therefore, under the low power density NIR irradiation, the temperature of a PBS solution containing PGC-PLMSNs meshes presented mild heating progress. The thermal images captured by an infrared camera confirmed a temperature increase by ∼5°C during irradiation with NIR (808 nm, 0.6 W/cm2) for 10 min. In comparison, the temperature variation of pure PBS solution under the same condition was not observed (Figure 5a). We have previous demonstrated the release profile of MSNs: Eu from the assembled composite (PGC-MSNs: Eu) fibers (Figure S6). It is well known that MSNs: Eu also containing -OH groups which interact favorably with -NH2 groups in the Gel-incorporated PGC fibers through electrostatic attractions. Under the irradiation with NIR (808 nm, 0.6 W/cm2), the particles release was enhanced. As a result, during the immersion in PBS solution, a significantly increased amount of nanoparticles assembled was liberated from PGC-PLMSNs meshes (Figure 5b&c). This is attribute to the mild heating effect which weakens the electrostatic interaction between the particles and PCG fiber matrix. Similar principle was reported on NIR-triggered drug delivery platforms.36
Figure 5.
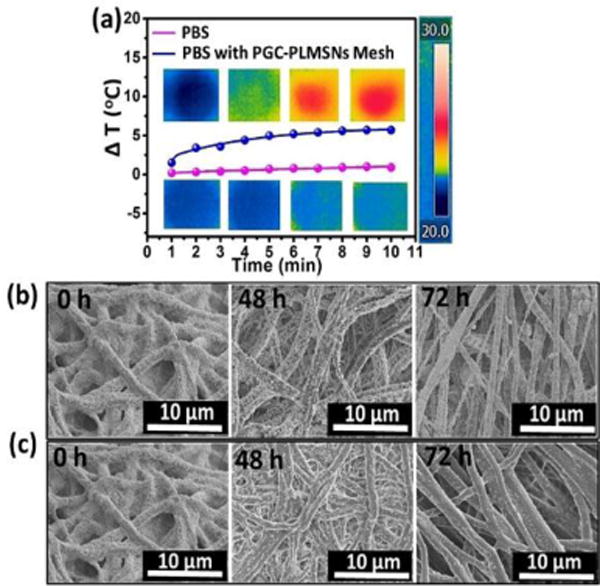
Photothermal behaviors of PGC-PLMSNs. (a) Photothermal heating curves of PBS solution containing PGC-PLMSNs composite meshes and pure PBS solution during irradiation with 808 NIR at 0.6 W/cm2. (b–c) SEM images of the PGC-PLMSNs fibers during immersion in PBS solution (pH=7.4) without (b) and with (c) NIR laser irradiation.
Intra-cellar delivery of DDSs loaded with therapeutics is crucial factor to the efficacy of chemotherapy. The cell up-taking efficiency of PLMSNs by Hep G2 cancer cells was evaluated on three groups of samples, including free PLMSNs, PGC-PLMSNs meshes with and without NIR irradiation. During examination using confocal microscopy, PLMSNs presented red color due to Eu incorporation. After 1-day cell culture, free PLMSNs hardly induced the presence of particles within cancer cells. PGC-PLMSNs meshes without NIR irradiation induced a certain degree of intracellular presence of particles, and the quantity of particles was enhanced on the meshes after NIR irradiation. When cultured for 3 days, the presence of PLMSNs with the strongest red fluorescence was observed for PGC-PLMSNs meshes after NIR irradiation compared to other two sample groups, implying its highest particle internalization by cells (Figure 6). This observation was analyzed quantitatively via flow cytometry. The cell up-taking rates of Free PLMSNs, PGC-PLMSNs without and with NIR irradiation were determined to be ~6.3 %, ~7.6 and ~8.6%, respectively, after 1 day incubation. The up-taking efficiency was increased in synchronization with incubation time. After cell culture for 3 days, the up-taking rate of free PLMSNs group increased to ~24 %, while PGC-PLMSNs meshes exhibited 28.6% up-taking rate. After NIR irradiation, the intracellular delivery rate of PLMSNs from composite meshes reached as high as ~37.5%, which is 1.5 times that of the group of free particles (Figure 7). It is noted that this enhanced cell up-taking phenomenon is induced in an in-vitro cell incubation which is of relatively static microenvironment. Considering the systematic circulation involved in an in vivo circumstance, the enhancement of cell up-taking efficiency can be remarkably amplified due to the immobilization effect of nanoparticles by composite meshes.
Figure 6.
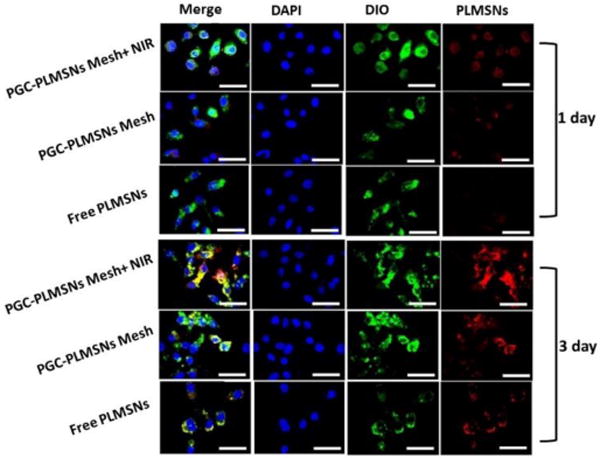
Laser scanning confocal microscopy (LSCM) images of Hep G2 cells cultured with free PLMSNs, PGC-PLMSNs meshes and PGC-PLMSNs meshes exposed to laser irradiation (NIR 808 nm, 0.6 W/cm2). For each panel, images from left to right show the Hep G2 cell nuclei stained by DAPI (blue), cell membrane stained by DIO (green), and particles fluorescence in cells (red). The scale bar is 50 μm.
Figure 7.
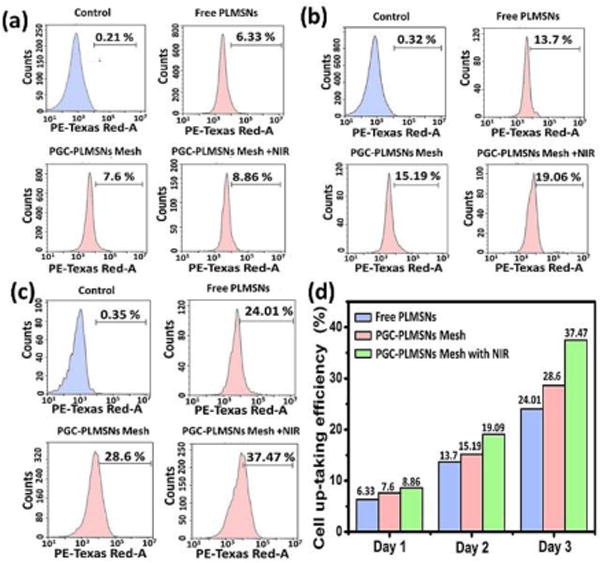
Cell up-taking efficiency of PGC-PLMSNs. (a–c) Evaluation of cell up-taking efficiency of free PLMSNs, PGC-PLMSNs composite meshes without and with laser irradiation (NIR 808 nm, 0.6 W cm−2) for (a) 1 day, (b) 2 days, (c) 3 days by flow cytometry. (d) The summary of cell up-taking efficacy between three samples.
The enhanced in-vitro up-taking efficiency induced by PGC-PLMSNs meshes after NIR irradiation is attributed to several mechanisms. The first mechanism is related to the unique microstructure characteristics of composite meshes. PLMSNs are assembled on the surface of fibrous meshes. Cells seeded tend to adhere, spread, and penetrate into the fibrous structure through micropores. The nanoparticles, assembled with high concentration comparing with free-particle sample group, are internalized by the cells during proliferation process. In contrast, free nanoparticles (suspended in solution), which lack the proximity with tumor cells, lead to certain remnant during cell culture process. The second mechanism arises from the mild photothermal effect. It is clear that a relatively fast particle liberation occurs during 808 nm NIR irradiation. Meanwhile, it has been reported the local mild heating effect enhances the permeability of cell membrane.43 The intracellular endocytosis is enhanced, and thus the up-taking activity of cancer cells is increased. Therefore, the PGN-PLMSNs composite meshes after NIR excitation induce the highest cell up-taking efficiency comparing to other typical drug delivery platforms, and consequently presents the strongest performance of intracellular DOX delivery as well as in vitro anticancer effect.
3 Conclusions
To endow effective intracellular delivery of therapeutics for chemotherapy, a localized drug delivery platform (PGC-PLMSNs composite patch), consisting of PLMSNs and photothermal electrospun PGC fibrous matrix, was designed and synthesized. PLMSNs presented effective pH-triggered drug release kinetics owing to the coverage of PDA shell. After the irradiation of 808 nm NIR, PLMSNs can rapidly be liberated from the composite patches. The preliminary in vitro study using cancer cells demonstrated that this PGC-PLMSNs composite patch, after NIR irradiation, induced a high cell up-taking rate of ~37.5% after culturing for 3 days and enhanced anti-cancer efficacy. The main mechanisms include its intrinsic localized delivery characteristics, which maintains the sufficient on-site dosage, and mild photothermal properties, which induces enhanced cell up-taking phenomenon. Compared to the current intravenous injection of DDSs, this implantable composite patch enables the effective localized delivery and enhanced cell up-taking efficiency of therapeutic nanoparticles while avoiding excessive systemic drug circulation. This research has therefore been anticipated to trigger a series of following exploration of hybrid drug delivery platforms, which combine advantages of DDSs and LDDSs for more effective and safer cancer chemotherapy.
4 Experimental Section
4.1 Synthesis of europium incorporated mesoporous silica nanoparticles (MSNs: Eu)
MSNs were synthesized using the surfactant templating method according to previous study.35 In a typical procedure, 0.2 g cetyltrimethylammonium bromide (CTAB, ≥99%, Sigma–Aldrich Inc.), 25 mL deionized water, 7 mL ethanol, 50 μL of triethanolamine (TEA, Sinopharm Chemical Reagent) were mixed and stirred at 60 °C for 30 min. Subsequently, 2 mL tetraethylorthosilicate (TEOS, Sigma Aldrich Inc., USA) was added into the solution under stirring for 2 h. When white precipitate appeared, the solution was cooled down to the ambient temperature, and the precipitate was collected by centrifugation. Following air-drying at 80 °C, the particles were calcined at 550 °C for 5 h to remove organic additives. Subsequently, a precursor solution was prepared by dissolving europium nitrate in deionized water. The concentration of Eu3+ was controlled at 0.5 mol L−1. 400 μL of the precursor solution was mixed with 200 mg silica nanoparticles, and the mixture was dried in a vacuum oven at 50 °C for 12 h. Finally, the samples were sintered at 600 °C for 3 h to obtain the europium incorporated MSNs: Eu.
4.2 Drug loading and releasing
DOX was used as an anticancer model drug. 50 mg MSNs:Eu sample was added into DOX aqueous solution (1mg mL−1, 20 mL) at ambient temperature. The mixed solution was subsequently stirred in dark for 24 h. The DOX-loaded samples were collected and washed with deionized water to remove the DOX content adsorbed, and dried at 37 °C in vacuum. Surface modification with PDA was subsequently carried out. Briefly, 100 mg MSNs: Eu after DOX loading was dispersed in 100 mL Tris–HCl buffer (pH 8.5, 10 mmol L−1), and hydrochloride dopamine (25 mg) was added under stirring at room temperature for 8 h in the dark. The PDA modified MSNs:Eu particles (PLMSNs) were centrifuged, washed with deionized water and stored in 4 °C for further use. PLMSNs without loading DOX drug were also prepared following the same procedure for comparison. The loading capacity of PLMSNs was analyzed by TG method and was calculated by the following equation: DOX loading capacity = (mass of DOX in the PLMSNs)/(mass of DOX loaded PLMSNs)×100%.
The in vitro DOX release test was performed by immersing 50 mg PLMSNs in 20 mL phosphate-buffered saline (PBS, HyClone Laboratories Inc., Logan, Utah) with three different pH values (7.4, 6.0, and 5.0) under stirring at 37 ± 0.1 °C. At each time interval, 10 mL buffer solution was collected and replaced with an equal volume of PBS with same pH. The content of DOX released under different pH was measured using an ultraviolet-visible (UV–vis) spectrophotometer (TU-1810, China) at a wavelength (λmax) of 480 nm. The cumulative release percentage was calculated according to the previous study:44 cumulative release percentage = (cumulative amount of DOX released from PLMSNs at each time interval)/(amount of total DOX loaded on the PLMSNs) ×100%. Three measurements were performed for each sample.
4.3 Preparation of PLMSNs assembled composite fiber meshes
The fibrous meshes were prepared via electrospinning. 10 wt % gelatin (GEL, type B from bovine skin) and 10 wt % poly(ε-caprolactone) (PCL, MW = 70000–90000) solutions in 2,2,2-trifluoroethanol (TFE, CF3CH2OH, ≥99%) were prepared respectively and mixed under magnetic stirring at 40 °C. 5 wt % (with respect to biopolymer weight) carbon nanoparticles (CNPs, purity >97%, Alfa Aesar) were dispersed in the PCL and gelatin mixture solution. The precursor was subjected to an electrospinning setup to prepare PGC fibers. The distance and the applied voltage between the needle tips and the collector were set at 15 cm and 6–8 kV, respectively. The precursor flow rate was set at 1.5 mL h−1. Finally, the electrospun fibrous meshes prepared were aged to evaporate residual solvent and stored in vacuum. Subsequently, the PLMSNs nanoparticles were assembled onto PGC fibers. Briefly, 40 mg PDA coated MSNs:Eu (PLMSNs) was dispersed in 100 ml ethanol solution. 100 mg fiber mesh was immersed in the particle solution at the room temperature. After stirring for 6 h, the particles assembled fiber meshes (PGC-PLMSNs) were collect and stored in vacuum.
4.4 Release of nanoparticles from PGC-PLMSNs meshes
To study the particle release behavior from the fibers composite, the meshes were immersed in PBS (pH=7.4) at 37 °C in an incubator with rotating rate of 100 rpm. At each time interval, a buffer solution was replaced with an equal volume of fresh PBS solution. Meanwhile, PGC-PLMSNs fibers were also exposed to 808 nm laser irradiation at power density of 0.6 W/cm2 for 10 min to reveal the NIR-triggered particles release behavior. At selected time intervals, the fiber meshes were collected and examined using scanning electron microscope (FESEM, Hitachi SU-70, Japan).
4.5 In vitro study
The in vitro biocompatibility of PLMSNs and PGC-PLMSNs composites without loading DOX was assessed using NIH3T3 cells by a typical 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) cell assay. Briefly, fiber meshes (with/without NIR irradiation) and nanoparticles were seeded with 100 μl of NIH3T3 cell solution (5 × 104/ml cells) in a 96-well plate and cultured in DMEM medium in a 5% CO2 incubator at 37 °C. The proliferation of cultured NIH3T3 cells was examined after 24, 48 and 72 hours. To study the influence of photothermal effect on the cell viability, PGC-PLMSNs meshes were irradiated by a NIR laser (808 nm, 0.6 W/cm2) for 10 min prior to cell culturing. After each incubation time interval, 10 μL of MTT solution was added into each well and incubated for 4 hours. Then the original culture medium was replaced with 100 μL of dimethyl sulfoxide (DMSO) per well. The final survival of NIH3T3 cell was measured at 490 nm using a microplate reader (Tecan 50, The Switzerland).
The in vitro anti-tumor efficacy of free PLMSNs nanoparticles loaded with DOX and its composite fiber meshes (with and without NIR irradiation) was examined against human Hep G2 liver cancer cells via MTT assay following the same procedure above. The DOX loaded samples were irradiated with NIR light (808 nm, 0.6 W/cm2) for 10 min. The relative concentration of DOX drug was varied from 0.17 μg/mL. All tests were performed in six replicates. The data are given as mean ± SD. Statistical analysis was performed with a student’s t test and the difference was considered as significant when *p ˂ 0.05 and **p ˂ 0.01
The cell up-taking properties of nanoparticles was evaluated with a confocal laser scanning microscope (CLSM, Fluoview Olympas Japan) and flow cytometry (FCM, NovoCyte 3130). For CLSM assessment, HepG2 cancer cells were seeded onto coverslips in 6-well plates and cultured with different samples with an equivalent amount of nanoparticles. Then, the cells were fixed with 2.5% glutaraldehyde for 50 min and stained with DAPI for 7 min and DIO for 10 min. For FCM analysis, at each preset time point, cells were treated with trypsin and centrifuged at 1000 rpm for 5 min to collect the cells. Then, the flow cytometer was used to analyze the fluorescence intensity of the cells collected. Cell-up taking efficiency was calculated according to the previous study as follows:45 cellular uptake efficiency= (the number of PLMSNs-labeled tumor cells)/(the total number of tumor cells counted) ×100%.
4.6 Characterization
The morphology and microstructure of particles were characterized by field-emission scanning electron microscopy (FESEM, Hitachi SU-70, Japan) operated at 3 kV and high-resolution transmission electron microscopy (HRTEM, Tecnai F20, FEI, USA) operated at 200 kV. The phase and crystal structure were examined using an X-ray diffraction instrument (XRD, X’Pert PRO, The Netherlands) operated at 40 mA and 40 kV using Cu Kα radiation. The scanning range was set at 10° < 2θ < 80° with a step size of 0.167°. The FTIR spectra of the samples were recorded using a PerkinElmer 580B infrared spectrophotometer on KBr pellets (Tensor 27, Bruker, Germany) in the frequency range of 4000–400 cm−1 with a resolution of 4 cm−1. The photoluminescence (PL) spectra were recorded via a fluorescence spectrophotometer (PL, FLSP920, Edinburgh) under excitation by Xenon lamp at ambient temperature. To minimize experimental uncertainties, the spectra collection and sample positions were maintained in identical conditions.
Supplementary Material
Acknowledgments
This work was financially supported by the National Nature Science Foundation of China (51232006, 51672247, and 51673168), the 111 program of China (No. B16042), the Major State Research Program of China (2016YFC1101900), the National Key Research and Development Program of China (2016YFA0100900), and the Natural Science Foundation of Zhejiang Province (LY15E020005 and LZ16E030001). CBM would also like to thank the financial support from National Institutes of Health (CA195607).
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Peer D, Karp J, Hong S, farokhzad O, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 2.Davis M, Chen Z, Shin D. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 3.Torchilin V. Nat Rev Drug Discov. 2014;13:813–827. doi: 10.1038/nrd4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park K. ACS nano. 2013;7:7442–7447. doi: 10.1021/nn404501g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li X, He Q, Shi J. ACS nano. 2014;8:1309–1320. doi: 10.1021/nn4046985. [DOI] [PubMed] [Google Scholar]
- 6.Nel A, Mädler L, Velegol D, Xia T, Hoek E, Somasundaran P, Klaessig F, Thompson V. Nat Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 7.Albanese A, Tang P, Chan W. Annu Rev Biomed Eng. 2012;14:1–16. doi: 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- 8.Wolfbeis O. Chem Soc Rev. 2015;44:4743–4768. doi: 10.1039/c4cs00392f. [DOI] [PubMed] [Google Scholar]
- 9.Kircher M, Zerda A, Jokerst J, Zavaleta C, Kempen P, Mittra E, Pitter K, Huang R, Campos C, Habte F, Sinclair R, Brennan C, Mellinghoff I, Holland E, Gambhir S. Nat Med. 2012;18:829–834. doi: 10.1038/nm.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng R, Meng F, Deng C, Klok H, Zhong Z. Biomaterials. 2013;34:3647–3657. doi: 10.1016/j.biomaterials.2013.01.084. [DOI] [PubMed] [Google Scholar]
- 11.Rica R, Aili D, Stevens M. Adv Drug Deliver Rev. 2012;64:967–978. doi: 10.1016/j.addr.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 12.Yang W, Guo W, Le W, Lv G, Zhang F, Shi L, Wang X, Wang J, Wang S, Chang J, Zhang B. ACS nano. 2016;10:10245–10257. doi: 10.1021/acsnano.6b05760. [DOI] [PubMed] [Google Scholar]
- 13.Gandra N, Abbineni G, Qu X, Huai Y, Wang L, Mao C. Small. 2013;9:215–221. doi: 10.1002/smll.201202090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Q, Wang X, Wang C, Feng L, Li Y, Liu Z. ACS nano. 2015;9:5223–5233. doi: 10.1021/acsnano.5b00640. [DOI] [PubMed] [Google Scholar]
- 15.Sanhai W, Sakamoto J, Canady R, Ferrari M. Nat Nanotechnol. 2008;3:242–244. doi: 10.1038/nnano.2008.114. [DOI] [PubMed] [Google Scholar]
- 16.Petros R, DeSimone J. Nat Rev Drug Discov. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 17.Chauhan V, Jain R. Nat Mater. 2013;12:958–962. doi: 10.1038/nmat3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilhelm S, Tavares A, Dai Q, Ohta S, Audet J, Dvorak H, Chan Warren C. Nat Rev Mater. 2016;1:1–12. [Google Scholar]
- 19.Chen Y, Hou Z, Liu B, Huang S, Li C, Lin J. Dalton Trans. 2015;44:3118–3127. doi: 10.1039/c4dt03113j. [DOI] [PubMed] [Google Scholar]
- 20.Qiu K, He C, Feng W, Wang W, Zhou X, Yin Z, Chen L, Wang H, Mo X. J Mater Chem B. 2013;1:4601–4611. doi: 10.1039/c3tb20636j. [DOI] [PubMed] [Google Scholar]
- 21.Chen Z, Chen Z, Zhang A, Hu J, Wang X, Yang Z. Biomater Sci. 2016;4:922–932. doi: 10.1039/c6bm00070c. [DOI] [PubMed] [Google Scholar]
- 22.Liu R, Wolinsky J, Catalano P, Chirieac L, Wagner A, Grinstaff M, Colson Y, Raut C. Ann Surg Oncol. 2012;19:199–206. doi: 10.1245/s10434-011-1871-4. [DOI] [PubMed] [Google Scholar]
- 23.Lei N, Gong C, Qian Z, Luo F, Wang C, Wang H, Wei Y. Nanoscale. 2012;4:5686–5693. doi: 10.1039/c2nr30731f. [DOI] [PubMed] [Google Scholar]
- 24.Wolinsky J, Colson Y, Grinstaff M. J Control Release. 2012;159:14–26. doi: 10.1016/j.jconrel.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li B, Setyawati MI, Chen L, Xie J, Ariga K, Lim CT, Garaj S, Leong DT. ACS Appl Mater Interfaces. 2017;9:15286–15296. doi: 10.1021/acsami.7b02529. [DOI] [PubMed] [Google Scholar]
- 26.Yuan Z, Pan Y, Cheng R, Sheng L, Wu W, Pan G, Feng Q, Cui W. Nanotechnology. 2016;27:245101. doi: 10.1088/0957-4484/27/24/245101. [DOI] [PubMed] [Google Scholar]
- 27.Sasikala ARK, Unnithan AR, Yun YH, Park CH, Kim CS. Acta Biomater. 2016;31:122–133. doi: 10.1016/j.actbio.2015.12.015. [DOI] [PubMed] [Google Scholar]
- 28.Yang G, Wang J, Li L, Ding S, Zhou S. Macromol Biosci. 2014;14:965–976. doi: 10.1002/mabi.201300575. [DOI] [PubMed] [Google Scholar]
- 29.Hu J, Ma P. Pharm Res. 2011;28:1273–1281. doi: 10.1007/s11095-011-0367-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song B, Wu C, Chang J. Acta Biomater. 2012;8:1901–1907. doi: 10.1016/j.actbio.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 31.Zheng F, Wang S, Shen M, Zhu M, Shi X. Polym Chem. 2013;4:933–941. [Google Scholar]
- 32.Mickova A, Buzgo M, Benada O, Rampichova M, Fisar Z, Filova E, Tesarova M, Lukas D, Amler E. Biomacromolecules. 2012;13:952–962. doi: 10.1021/bm2018118. [DOI] [PubMed] [Google Scholar]
- 33.GhavamiNejad A, Sasikala A, Unnithan A, Thomas R, Jeong Y, Vatankhah-Varnoosfaderani M, Stadler F, Park C, Kim C. Adv Funct Mater. 2015;25:2867–2875. [Google Scholar]
- 34.Yang G, Wang J, Wang Y, Li L, Guo X, Zhou S. ACS nano. 2015;9:1161–1174. doi: 10.1021/nn504573u. [DOI] [PubMed] [Google Scholar]
- 35.Zheng Q, Lin T, Wu H, Guo L, Ye P, Hao Y, Guo Q, Jiang J, Fu F, Chen G. Int J Pharm. 2014;463:22–26. doi: 10.1016/j.ijpharm.2013.12.045. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Zhou Y, Gu T, Wang G, Ren Z, Weng W, Li X, Han G, Mao C. Part Part Syst Charact. 2016;33:896–905. doi: 10.1002/ppsc.201600166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Li B, Xu G, Ahmad Z, Ren Z, Dong Y, Li X, Weng W, Han G. Colloid Surfaces B. 2014;122:785–791. doi: 10.1016/j.colsurfb.2014.08.022. [DOI] [PubMed] [Google Scholar]
- 38.Li X, Li Y, Chen X, Li B, Gao B, Ren Z, Han G, Mao C. Langmuir. 2016;32:3226–3233. doi: 10.1021/acs.langmuir.6b00290. [DOI] [PubMed] [Google Scholar]
- 39.Xue Y, Du Y, Yan J, Liu Z, Ma PX, Chen X, Lei B. J Mater Chem B. 2015;3:3831–3839. doi: 10.1039/c5tb00204d. [DOI] [PubMed] [Google Scholar]
- 40.Miao G, Chen X, Mao C, Li X, Li Y, Lin C. J Sol-Gel Sci Technol. 2014;69:250–259. [Google Scholar]
- 41.Wu C, Xia L, Han P, Mao L, Wang J, Zhai D, Fang B, Chang J, Xiao Y. ACS Appl Mater Interfaces. 2016;8:11342–11354. doi: 10.1021/acsami.6b03100. [DOI] [PubMed] [Google Scholar]
- 42.Kang X, Huang S, Yang P, Ma P, Yang D, Lin J. Dalton Trans. 2011;40:1873–1879. doi: 10.1039/c0dt01390k. [DOI] [PubMed] [Google Scholar]
- 43.Tian B, Wang C, Zhang S, Feng L, Liu Z. ACS nano. 2011;5:7000–7009. doi: 10.1021/nn201560b. [DOI] [PubMed] [Google Scholar]
- 44.Luan J, Wu K, Li C, Liu J, Ni X, Xiao M, Xu Y, Kuang Y, Jiang F. Carbohydrate Polymers. 2017;171:9–17. doi: 10.1016/j.carbpol.2017.04.094. [DOI] [PubMed] [Google Scholar]
- 45.Zhou L, Zheng X, Gu Z, Yin W, Zhang X, Ruan L, Yang Y, Hu Z, Zhao Y. Biomaterials. 2014;35:7666–7678. doi: 10.1016/j.biomaterials.2014.05.051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.