

Publisher's Note: There is an Inside Blood Commentary on this article in this issue.
Key Points
Alterations of SETD2, a histone 3 lysine 36 trimethyl (H3K36me3) transferase leads to resistance to DNA damaging-chemotherapy in leukemia.
Low H3K36me3 levels impair DNA damage response and increase mutation rate, which may be targeted by H3K36me3 demethylase inhibition.
Abstract
Mutations in SETD2, encoding the histone 3 lysine 36 trimethyltransferase, are enriched in relapsed acute lymphoblastic leukemia and MLL-rearranged acute leukemia. We investigated the impact of SETD2 mutations on chemotherapy sensitivity in isogenic leukemia cell lines and in murine leukemia generated from a conditional knockout of Setd2. SETD2 mutations led to resistance to DNA-damaging agents, cytarabine, 6-thioguanine, doxorubicin, and etoposide, but not to a non–DNA damaging agent, l-asparaginase. H3K36me3 localizes components of the DNA damage response (DDR) pathway and SETD2 mutation impaired DDR, blunting apoptosis induced by cytotoxic chemotherapy. Consistent with local recruitment of DDR, genomic regions with higher H3K36me3 had a lower mutation rate, which was increased with SETD2 mutation. Heterozygous conditional inactivation of Setd2 in a murine model decreased the latency of MLL-AF9–induced leukemia and caused resistance to cytarabine treatment in vivo, whereas homozygous loss delayed leukemia formation. Treatment with JIB-04, an inhibitor of the H3K9/36me3 demethylase KDM4A, restored H3K36me3 levels and sensitivity to cytarabine. These findings establish SETD2 alteration as a mechanism of resistance to DNA-damaging chemotherapy, consistent with a local loss of DDR, and identify a potential therapeutic strategy to target SETD2-mutant leukemias.
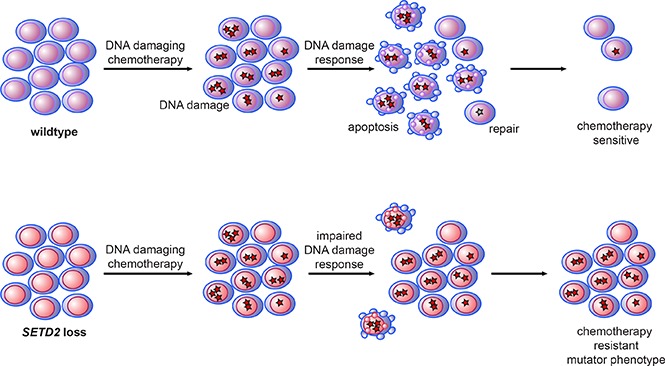
Visual Abstract
Introduction
Relapsed acute leukemia is resistant to chemotherapy and outcomes are poor.1-4 Despite recent efforts to develop novel therapies, the mainstay of treatment of both acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) remains chemotherapy with DNA-damaging agents. We and others have reported an enrichment of mutations in epigenetic regulators in relapsed acute leukemia,5-7 suggesting an association with chemotherapy resistance.
Among genes encoding epigenetic regulators, we identified recurrent mutations in SETD2, the major histone 3 lysine 36 trimethyltransferase (H3K36me3),8 in 10% of pediatric patients with B-cell ALL. The vast majority of mutations were heterozygous, and approximately half of the SETD2 lesions were nonsense, frameshift, or splice site mutations or deletions, consistent with loss-of-function alterations.6 Other studies have reported a high frequency (∼20%) of SETD2 alterations in chemotherapy-resistant, MLL-arranged ALL and AML and hepatosplenic T-cell lymphoma.9 A lower frequency of SETD2 alterations are found in other hematological malignancies, including early T-cell precursor ALL,10 non–MLL-rearranged AML,11 and chronic lymphocytic leukemia (CLL)12 (Figure 1A; supplemental Table 1, available on the Blood Web site) and multiple solid tumors.13-17 SETD2 loss has been associated with adverse clinical outcomes in clear cell renal cell carcinoma,18,19 and CLL12 and has been shown to accelerate disease and increase cell proliferation in some experimental models.9,20
Figure 1.

SETD2 loss leads to resistance to DNA-damaging chemotherapy agents. (A) Diagram of SETD2 nonsense, frameshift, or missense mutations reported in pediatric ALL (Dana-Farber Cancer Institute6 and St. Jude Children's Research Hospital46 cohorts, top) or in cancers of hematological or lymphoid tissue origin (Catalogue of Somatic Mutations in Cancer [COSMIC],47 bottom). (B) Western blot for H3K36me3 of MOLM-13 subclones with SETD2 deletions and isogenic control. The H3K36me3 level normalized to actin is shown. Full western blot with additional subclones are shown in supplemental Figure 1C. (C) H3K36me3 levels for highly expressed and nonexpressed genes in MOLM-13 isogenic clones, normalized over gene body length between the vertical lines. (D) CellTiter-Glo was used to determine the percentage of cell viability after 72 hours of cytarabine, (E) 6-TG, (F) doxorubicin, (G) etoposide, and (H) l-asparaginase. (I) MOLM-13 SETD2 clone 1 was transduced with an RFP+ lentivirus, competed with isogenic control 3 in a 1:20 ratio, and treated with cytarabine, 6-TG, or vehicle for 18 days and assessed by flow cytometry every 3 days, where RFP was used as a read-out for the percentage of SETD2-mutant cells. (J) A similar competition experiment was performed as described in panel I with cells treated with l-asparaginase or vehicle for 18 days and followed by flow cytometry. TES, transcriptional end site; TSS, transcriptional start site.
In this article, we investigate the role of SETD2 alterations on chemotherapy resistance in leukemia models. We provide evidence that cells with SETD2 loss have diminished activation of the DNA damage response (DDR) after exposure to cytotoxic chemotherapy, leading to diminished apoptosis. Consistent with a role in functionally localizing DDR, genomic regions with higher H3K36me3 levels sustained less chemotherapy-induced mutations, and deletions in SETD2 led to a higher mutation rate globally. Lastly, treatment with JIB-04, an inhibitor of the H3K9/36me3 demethylase KDM4A, restored H3K36me3 levels and chemotherapy sensitivity in vitro and in vivo, providing a potential therapeutic strategy.
Methods
Cell lines
MOLM-13 (American Type Culture Collection) cells were obtained and verified by DNA short tandem repeat profiling with Promega’s GenePrint System in February 2011 and viably frozen. Cells were thawed and grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin G and 100 μg/mL streptomycin at 37°C in a humidified atmosphere under 5% CO2. We maintained HEK293T cells under similar conditions in Dulbecco’s modified Eagle medium. No short tandem repeat profiling was performed on HEK293T cells. MLL-AF9 Setd2fl/+ Mx1-cre and MLL-AF9 Setd2+/+ Mx1-cre cells were grown in Iscove’s modified Dulbecco medium supplemented with 15% FBS, 100 U/mL penicillin G, 100 μg/mL streptomycin, 10 ng/mL murine interleukin-6, 1.25 ng/mL murine interleukin-3, and 12.5, ng/mL murine stem cell factor.
Generation of gene-edited subclones
The sequences of all small guide RNAs (sgRNAs) and the sequencing primers used in this study are provided in supplemental Table 2. sgRNAs were designed, cloned into the pL-CRISPR.EFS.GFP vector (Addgene #57818), and transduced into cell lines, as previously described.21 MOLM-13 cells were then sorted as GFP+ single cells into RPMI 1640 with 20% FBS and grown into single cell colonies. Next-generation sequencing was used to confirm the presence of insertions or deletions in infected cell lines. The genomic DNA was polymerase chain reaction (PCR)-amplified, with amplicons ranging in size from 200 to 500 bp, which were PCR purified. Barcoded sequencing adapters were ligated to the amplicons and sequenced on an Illumina MiSeq. FASTQ files were aligned to the appropriate genome (human hg19 or mouse mm10) with Burrows-Wheeler Aligner and insertion or deletion size and location were determined from the CIGAR alignment parameter.
Western blot, subcellular fractionation, and antibodies
Western blot analysis was performed using antibodies to H3K36me3 (Cell Signaling #4909), total H3 (Cell Signaling #12648), β-actin (Abcam ab20272), Chk1 phospho-S345 (Cell Signaling #2348), MSH6 (Cell Signaling #12988), HSP90 (Cell Signaling #4874), and HDAC1 (Cell Signaling #2062) according to the manufacturers’ instructions. Subcellular fractions were obtained with the Subcellular Protein Fraction Kit for Cultured Cells (Thermo Fisher Scientific) according to the manufacturer’s instructions.
In vitro chemotherapy resistance by cell viability and competition assay
We determined cell viability using the luminescent cell viability assay CellTiter-Glo (Promega) according to the manufacturer’s instructions. All experiments were performed in triplicate. We analyzed cell viability 72 hours after the initiation of treatment with 6-thioguanine (6-TG), cytarabine, doxorubicin, and l-asparaginase with the indicated concentrations. All experiments were done in triplicate wells and repeated ≥1 to 2 times. A representative experiment is shown.
In vitro competition assays were used to study the presence of chemotherapy resistance. Subcloned MOLM-13 cells with the desired mutations were transduced with a lentiviral vector that results in overexpression of tdTomato. Mutant, tdTomato-positive MOLM-13 cells were mixed in different ratios with subcloned MOLM-13 isogenic control cells or MSH6-mutant cells. Cells were split to a concentration of 250 000 cells/mL, and fresh media and chemotherapy were added every 72 hours. The relative abundance of the cell populations was analyzed using a BD Canto II.
Apoptosis analysis
Annexin V staining was used to assess the presence of apoptosis. Cells were washed in Annexin Binding Buffer and stained for 20 minutes with an antibody against Annexin V PE-Cyanine7 (eBioscience 88-8103-74). 4′,6-Diamidino-2-phenylindole or propidium iodide was used as dead cell marker.
Immunofluorescence staining
Cytospins of cells were prepared at 500 rpm for 5 minutes, permeabilized, and fixed in 4% paraformaldehyde/phosphate-buffered saline. γH2AX phospho-S139 (Abcam) primary antibody was used at 1:400 and incubated overnight at 4°C. After washing, a secondary antibody, anti-rabbit Alexa647 (Cell Signaling Technology), was applied for 30 minutes at room temperature. Samples were incubated with 4′,6-diamidino-2-phenylindole (1 μg/mL) and covered with ProLong Gold antifade reagent. Slides were analyzed by confocal microscopy (Leica TCS SP5), and foci were quantified with Image J software (National Institutes of Health).
Generation of Setd2 knockout mice
The Setd2fl/+ mice were developed by Beijing Biocytogen, Co., Ltd. Setd2 exon 3 was targeted by homologous recombination with a pGK-Neo cassette, resulting in a premature stop codon of Setd2 protein. A C57BL/6 ES cell line was used to generate knockout mice. Mice were bred to Mx1-cre and maintained in a pure C57BL/6 background.
Generation of primary and secondary MLL-AF9 leukemia
Lin-Kit+Sca1+ hematopoietic stem cells were sorted from mice 6 to 8 weeks of age by harvesting of bone marrow cells from the femurs, tibias, hips, and spines of Setd2fl/+ Mx1-cre or Setd2+/+ Mx1-cre mice and transducing with a retrovirus from 293T cells transfected with a MSCV-IRES-MLL-AF9-GFP construct.22 Cells were maintained in 15% Iscove’s modified Dulbecco medium plus penicillin-streptomycin and murine interleukin-6, murine stem cell factor, and murine interleukin-3 (as above) for 2 days before fluorescence-activated cell sorting for GFP+ cells. A total of 200 000 to 300 000 GFP+ preleukemic cells were injected into lethally irradiated C57BL/6 (Taconic) recipients. Mice were monitored daily for clinical symptoms and killed when they appeared moribund or showed any sign of sickness. Whole bone marrow and spleens from 10 mice with primary leukemias were mixed, and red blood cells were lysed and frozen viably for all secondary leukemia experiments. All mouse experiments were approved by the Institutional Animal Care and Use Committee at Memorial Sloan Kettering Cancer Center or Boston Children’s Hospital.
In vivo chemotherapy treatment
C57/BL6 male and female mice (Taconic), 6 to 8 weeks of age, were injected IV with 300 000 GFP+ primary MLL-AF9 Setd2+/+ Mx1-cre or MLL-AF9 Setd2fl/+ Mx1-cre bone marrow cells. The number of mice used in each arm is noted in the legends of Figures 3 and 4. Mice were bled under anesthesia on a weekly basis, and peripheral blood (PB) was assessed for GFP+ cells by flow cytometry. For 1-day chemotherapy response experiments, mice were allocated to therapy once PB GFP reached 20% to 50% to maintain a similar mean GFP percentage value in each arm. Mice with >50% PB GFP were excluded from the experiment. Mice received a dose of cytarabine (100 mg/kg intraperitoneally [IP]), JIB-04 (Tocris; 110 mg/kg in 10% dimethyl sulfoxide [DMSO] and 90% sesame oil IP), AZD 1775 (MK 1775, Selleckchem; 60 mg/kg in 10% DMSO and 90% methylcellulose per os [PO] gavage) or vehicle (10% DMSO and 90% sesame oil), and then were bled and euthanized 12 to 16 hours later. PB GFP percentage and Annexin V staining were assessed by flow cytometry. For survival curves, mice were transplanted as above, and when PB GFP reached 3% to 10%, mice were allocated to therapy to maintain a similar mean GFP percentage value in each arm. Mice with >10% PB GFP were excluded from the 1-day experiment. Five days of therapy with each treatment was given similarly to as above, and survival was measured until death or euthanization due to lethargy or hind limb paralysis. Each experiment was replicated in an independent transplantation cohort.
Figure 3.

Setd2 heterozygous loss leads to decreased leukemia latency and cytarabine resistance in vivo. (A) Schema for conditional knockout of the third exon of Setd2. (B) PCR of genomic DNA of bone marrow from mice with wild-type, homozygous, or heterozygous exon 3 of Setd2 with the flanking loxP site crossed with Mx1-cre, with or without the induction of excision by polyinosinic:polycytidylic acid, as indicated. (C) Lin-Kit+Sca1+ bone marrow cells from Setd2fl/fl, Setd2fl/+, or Setd2+/+ Mx1-cre mice were transduced with a MSCV-IRES-MLL-AF9-GFP construct and 200 000 GFP+ preleukemic cells were injected into lethally irradiated C57BL/6 recipients. Mice were monitored daily and killed when they appeared moribund or showed signs of sickness. (D) Western blot for H3K36me3 in secondary MLL-AF9 Setd2+/+, Setd2fl/+, or Setd2fl/fl Mx1-cre leukemia. H3K36me3 levels were quantified and normalized to the level of total H3 in each lane. (E) A total of 200 000 bone marrow cells from mice with primary MLL-AF9 Setd2fl/+ Mx1-cre or Setd2+/+ Mx1-cre leukemia were injected into secondary C57BL/6 recipients. Mice with secondary MLL-AF9 Setd2+/+ Mx1-cre (n = 9) or Setd2fl/+ Mx1-cre (n = 24) leukemia received a single dose of cytarabine when their PB GFP was ∼30% and were rebled 12 to 16 hours later to determine the reduction in cell number, expressed as a percentage of pretreatment GFP or (F) stained with AnnexinV to determine the amount of apoptosis after treatment. (G) Mice with secondary MLL-AF9 Setd2+/+ Mx1-cre leukemia initiated treatment with 5 days of cytarabine (n = 5) or vehicle (n = 6) when PB GFP was ∼5%. (H) Mice with secondary MLL-AF9 Setd2fl/+ Mx1-cre leukemia initiated treatment with 5 days of cytarabine (n = 5) or vehicle (n = 6) when PB GFP was ∼5%.
Figure 4.

KDM4 inhibition increases sensitivity to cytarabine in Setd2-mutant leukemia cells. (A) MOLM-13 isogenic control 3 or SETD2 clone 1 were treated with JIB-04 or DMSO and western blotted for H3K36me3. (B) MLL-AF9 Setd2+/+ or Setd2fl/+ Mx1-cre cells were treated with JIB-04 or DMSO and western blotted for H3K36me3. (C) Cell titer Glo was used to determine the percentage of cell viability after 72 hours of JIB-04 for MOLM-13 isogenic control 3 or SETD2 clone 1. (D) Cell titer Glo was used to determine the percentage of cell viability after 72 hours of cytarabine for MOLM-13 isogenic control 3 or SETD2 clone 1, with or without JIB-04 (10 nM). (E) Mice with secondary MLL-AF9 Setd2fl/+ Mx1-cre leukemia received a single dose of vehicle (n = 4), cytarabine (n = 5), JIB-04 (n = 5), and cytarabine with JIB-04 (n = 6) when their PB GFP was ∼40% and were rebled 12 to 16 hours later to determine the reduction in cell number, expressed as a percentage of pretreatment GFP, or (F) stained with AnnexinV to determine the amount of apoptosis after treatment.
Exome sequencing
Genomic DNA was extracted with the DNeasy (Qiagen) kit as per the manufacturer’s instructions. DNA was fragmented (Covaris sonication) to 250 bp and additionally purified using AMPure XP beads (Agentcourt). Size-selected DNA was then ligated to specific adaptors during library preparation as per the manufacturer’s instructions (Kapa Library Prep). Each library was made with sample-specific barcodes, quantified using the Illumina MiSeq, and libraries were pooled at equal mass to a total of 750 ng for Exome v5 enrichment using the Agilent SureSelect hybrid capture kit. All capture pools were then pooled together and sequenced on a HiSeq 3000 in Rapid Run Mode at a final equivalent of 4 exomes per lane. Pooled sample reads were deconvoluted and sorted using the Picard tools. Reads were aligned to the reference sequence b37 edition from the Human Genome Reference Consortium using BWA, and duplicate reads were identified and removed using the Picard tools. Pileup files were generated using Samtools, and single nucleotide variant and indel calls were made using Varscan23 version 2.3.3 in paired mode with the matched pretreatment sample as the germ line control. High-quality variants with a >30% variant allele fraction were gathered across all samples and then force called in every sample. Only high-quality variants contained within the coordinates of the capture baitset, with coverage >20 reads in every sample were used. Variants present in the pretreatment sample were removed from consideration. Synonymous mutations were considered in the mutation rate analysis.
RNA sequencing and alignment
Total RNA was extracted from cultured cells using Trizol followed by RNeasy purification (Qiagen) and then sent to the Center for Cancer Computational Biology at Dana-Farber Cancer Institute (Boston, MA) for all library construction and sequencing. RNA was first put through quality control using Qubit (Life Technologies) and the Bioanalyzer (Agilent). RNA quantity was determined on the Qubit using the Qubit RNA Assay Kit (Life Technologies), and RNA quality was determined on the Bioanalyzer using the RNA Pico Kit (Agilent). Using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB), 50 to 100 ng of total RNA was converted into a DNA library following the manufacturer’s protocol, with no modifications. Following library construction, DNA libraries were then put through quality control. Library quantity was determined using the Qubit High Sensitivity DNA Kit (Life Technologies), and library size was determined using the Bioanalyzer High Sensitivity Chip Kit (Agilent). Finally, libraries were put through quantitative PCR using the Universal Library Quantification Kit for Illumina (Kapa Biosystems) and run on the 7900HT Fast quantitative PCR machine (ABI). Libraries passing quality control were diluted to 2 nM and sequenced on the NextSequation 500 (Illumina) at a final concentration of 2 pM on a single-read flow cell with 75 sequencing cycles, following all manufacturer protocols. Raw reads were aligned to the transcriptome derived from University of California Santa Cruz genome assembly hg19 using STAR24 with default parameters. Read counts for each transcript was measured using featureCounts.25
Chromatin immunoprecipitation-sequencing and analysis
MOLM-13 cells were washed twice in phosphate-buffered saline and incubated with 1% methanol-free formaldehyde for 5 minutes at ambient temperature followed by cytoplasm lysis and Covaris sonication to yield fragmented chromatin in the 150- to 400-bp range. The concentration of cross-linked chromatin was measured by double-stranded DNA content using PicoGreen (Invitrogen). Reference chromatin from S2 cells (Drosophila melanogaster) was mixed with chromatin from MOLM-13 cells at a 1:100 mass (∼1:5 molar) ratio, similar to that previously described.26 Immune complexes precipitated with anti-H3K36me3 antibody (Cell Signaling) were washed, eluted from magnetic beads, and de–cross-linked as previously described.27 The resultant DNA fragments were quantified and used for TruSeq adapter ligation (NEBNext chromatin immunoprecipitation sequencing [ChIP-seq]) following amplification for HiSeq2500 (Illumina) sequencing to ∼3 × 107 reads per sample.
Reads were trimmed for quality and Illumina adapter sequences using trim_galore and discarded if the length was <20 nucleotides. Filtered reads were then aligned to a jointly indexed human–Drosophila melanogaster genome (hg19-dm3) using bowtie2 with default parameters. Ambiguously mapped reads were discarded, and the resultant bam file was de-multiplexed into alignments for hg19 only or dm3 only. Aligned reads with the same start site and orientation were collapsed using the Picard tool MarkDuplicates. The total aligned read count for dm3 (Nd) was then used to calculate the normalization constant of the hg19 ChIP signal, using α = 1/Nd, as described previously.26 The H3K79me2 signal over gene bodies was then normalized to this factor as well as gene body length for all downstream analyses.
Determination of 6-TG–induced local mutation rate, H3K36me36 and expression
Pretreatment populations of MOLM-13 isogenic sgRNA control 3 (isogenic control 3) and MOLM-13 SETD2 clone 1 were subjected to exome sequencing, poly A–primed RNA sequencing (RNAseq), and ChIP for H3K36me3 with Drosophila melanogaster chromatin spike-in control (described above). MOLM-13 isogenic control 3 and SETD2 clone 1 cells were then separately subjected to 14 days of 6-TG treatment, similar to that described above, in competition assays with 200 nM of 6-TG or vehicle. After 14 days, surviving cells were diluted to single cells and subcloned as similar to what was described in the “Generation of gene-edited subclones” section to obtain pure populations of cells for subsequent exome sequencing.
Using custom R scripts, exons were sorted by the average amount of normalized H3K36me3. To determine the average mutation rate, a moving window average function was used to determine the total number of mutations present in all sequenced exomes in the window, divided by the number of base pairs in the window, divided by the number of exomes sequenced. The window was 14 Mb in size. The average normalized H3K36me6 was determined for each window in a similar manner. Because the RNAseq was 3′ biased, each exon was assigned the average expression of the entire gene, and the moving window analysis was repeated as above.
Statistical analysis
For Figures 2D,F, 3E-F, and 4E-F and supplemental Figure 3D, the Student t test was used to determine the statistical significance between each group. For Figure 3G-H, the log rank (Mantel-Cox) test was used to determine if the differences in survival between each group were statistically different. Calculations for these figures were performed in GraphPad Prism (GraphPad Software). For Figure 2G, Pearson’s correlations were used to determine the statistical significance in mutational frequency and average normalized H3K36me3 level for each moving window. These calculations were performed in R (R Foundation).
Figure 2.

SETD2 alteration impairs the DNA damage response and increases mutation rate at sites of diminished H3K36me3. (A) MOLM-13 SETD2 clone 1 and MOLM-13 isogenic control 3 were treated with cytarabine (100 nM), 6-TG (200 nM), or l-asparaginase (0.001 IU/μL) and assessed for apoptosis by flow cytometry after staining for AnnexinV and propidium iodide (PI) 24 to 48 hours later. (B) Western blots for Chk1 phospho-S345 of MOLM-13 SETD2 clone 1 and MOLM-13 isogenic control 3 treated with vehicle, 6-TG, (200 nM) or cytarabine (100 nM) for 4 hours. (C) MOLM-13 isogenic lines were treated with cytarabine or vehicle and examined for γ-H2A.X foci. Representative images shown. Primary stain: rabbit anti-gamma H2AX phospho S139; secondary stain: anti-rabbit Alexa647; counterstain: DAPI. Original magnification ×40. (D) The number of foci per cell were quantified in ImageJ for each condition. The red threshold is twice the standard deviation above the mean number of foci in the vehicle-treated isogenic control. A Student t test was performed between each pair of treatments. (E) Schema of experimental approach. MOLM-13 SETD2 clone 1 or isogenic control 3 cells were treated with 6-TG (200 nM) or vehicle for 14 days and then single-cell sorted to obtain subclones with clonal mutations. Whole exome sequencing (WES) was performed on 10 6-TG–treated subclones (4 SETD2-mutant and 6 isogenic control) and 1 each of vehicle-treated SETD2-mutant and isogenic control subclones. The pretreatment SETD2-mutant or isogenic control clones were subjected to WES, H3K36me3 ChIP-Seq, and total RNAseq. (F) The number of novel mutations for each 6-TG–treated MOLM-13 SETD2-mutant or isogenic control subclone compared with its matched pretreatment clone was determined based on WES. The total mutations and expected GA/CT transitions are shown. (G) Exons were ordered by the amount of normalized H3K36me3 for both the SETD2-mutated and isogenic control clones. Mutation rates were calculated using a moving window average, dividing the total number of mutations by the number of base pairs for each window. The average H3K36me3 level for each window was also determined, in each clone. A Pearson’s correlation was used to calculate the R2 value for each clone. (H) Moving window average of H3K36me3 levels arranged by gene expression level on RNAseq. (I) Mutation rates were calculated using a moving window average of genes by expression profile. The average expression level for each window was also determined and a Pearson’s correlation was used to calculate the R2 value for the MOLM-13 isogenic control and the MOLM-13 SETD2-mutant clone.
Results
SETD2 loss leads to resistance to DNA-damaging agents and outgrowth of mutant cells
To test whether SETD2 alteration leads to chemotherapy resistance, we used CRISPR/Cas9 gene editing to generate isogenic leukemia cell lines with or without frameshift deletions in SETD2, close to previously reported mutations in leukemia. Because SETD2 alterations are most frequently associated with MLL rearrangement in ALL and AML,20 we selected the MLL-rearranged MOLM-13 AML cell lines, which are wild-type for TP53 and the H3K36 methyltransferases SETD2, NSD2, and ASH1L. MOLM-13 clones with heterozygous and compound heterozygous SETD2 frameshift mutations had a decrease in H3K36me3 by western blot (Figure 1B; supplemental Figure 1A,C) as well as a local decrease as assessed by ChIP-seq (Figure 1C; supplemental Figure 2). Interestingly, heterozygous clones had >75% H3K36me3 loss, and compound heterozygotes had residual H3K36me3, leading both types of clones to have a similar amount of residual H3K36me3. SETD2 transcript by RNA sequencing showed no evidence of nonsense-mediated decay.
We subjected SETD2-mutant clones and isogenic nontargeting sgRNA control clones to 72 hours of treatment with chemotherapy agents used in leukemia therapy, the DNA-damaging agents cytarabine, 6-TG, doxorubicin, etoposide, and the non–DNA-damaging agent l-asparaginase. The half-maximal inhibitory concentration for the DNA-damaging agents was increased threefold to fivefold in SETD2-mutant cells compared with isogenic controls (Figure 1D-G). In contrast, SETD2-mutant cells were equally as sensitive as isogenic control cells to l-asparaginase, which does not induce DNA damage (Figure 1H).
To test whether SETD2-mutant cells have a competitive advantage when exposed to longer-term chemotherapy, we mixed a fluorescently labeled SETD2-mutant clone with isogenic control cells at a 1:20 ratio, and treated the cells with chemotherapy or vehicle control. Treatment with either cytarabine or 6-TG led to a striking selection of the SETD2-mutant cells, from 5% of cells to 70% to 100% of the population (Figure 1I). Treatment with l-asparaginase resulted in little to no selective advantage for the SETD2-mutant clone over a similar long-term competitive assay (Figure 1J).
To confirm these findings in another cell line, we performed the same in vitro competition experiment using murine leukemia cells driven by MLL-AF922 with CRISPR-Cas9 editing of the Setd2 gene. As observed with human cells, the MLL-AF9 Setd2-mutant murine leukemia cell line had a competitive advantage in the presence of cytarabine or 6-TG, but not l-asparaginase (supplemental Figure 3A-B). These studies demonstrate that SETD2 loss leads to resistance to the commonly used DNA-damaging agents cytarabine, 6-TG, doxorubicin, and etoposide in leukemia cells, but not l-asparaginase.
SETD2 loss impairs the DNA damage response and increases the mutation rate at sites of diminished H3K36me3
We next examined apoptosis, as measured by Annexin V binding, and found that apoptosis to cytarabine and 6-TG was impaired, but the apoptotic response to l-asparaginase was intact in SETD2-mutant cells (Figure 2A). We found that phosphorylation of Chk1, a critical member of DDR, was impaired in SETD2-mutant cells treated with cytarabine or 6-TG (Figure 2B). Consistent with an early defect DDR, the generation of γ-H2A.X foci is impaired in SETD2-mutant cells after cytarabine treatment (Figure 2C-D). These results indicate that SETD2-mutant cells have impaired DDR.
Several groups have examined the relationship between SETD2 and DNA damage recognition and repair, finding that the H3K36me3 mark localizes proteins involved with homologous recombination28-30 (LEDGF) and mismatch repair31 (MSH6). SETD2 loss has been suggested to lead to a hypermutator phenotype.31 We confirmed that MSH6, a member of the mismatch repair complex, did indeed bind chromatin and was delocalized with our model of SETD2 loss. Also, consistent with this mechanism, MSH6 homozygous loss in the MOLM-13 isogenic model (supplemental Figure 1B,D) recapitulates resistance to cytarabine, doxorubicin, and etoposide similar to SETD2 mutations (supplemental Figure 4).
We sought to explore the functional consequences of localizing MSH6 to specific regions of the genome, and we hypothesized that localization of the H3K36me3 mark might prioritize DDR in some parts of the genome over others, leading to lower mutation rates at regions with high H3K36me3 levels. To study this experimentally, we aimed to induce a large number of tolerable, detectable mutations randomly throughout the genome and compare the local mutation rate to patterns of H3K36me3 found by ChIP-Seq in isogenic cell lines with or without SETD2 mutation. We treated SETD2-mutant MOLM-13 cells and isogenic control cells with 6-TG, which leads to G to A and C to T transitions, for 14 days. After treatment, we generated single-cell subclones and performed whole exome sequencing on treated and pretreated clonal populations to determine the sites of novel 6-TG–induced mutations (Figure 2E). 6-TG induced twice as many mutations in SETD2-mutant cells as in isogenic control cells (39.25 vs 21, P = .08), particularly the expected G to A and C to T transitions (31.5 vs 13, P = .05) (Figure 2F).
We performed spike-in normalized ChIP-Seq to determine the level of H3K36me3 for each exonic region (supplemental Figure 2) and correlate them to the mutated positions found on exome sequencing. We combined variants from sequenced subclones and compared the number of mutations per megabase to the level of H3K36me3 using a moving window analysis. In isogenic control cells, we found a strong inverse correlation between normalized H3K36me3 level and mutational frequency (R2 = −0.95, Pearson’s correlation). In SETD2-mutant cells, H3K36 levels were markedly lower, but still present, and a similar inverse relationship with mutation rate was present at these areas of low but detectable H3K36me3 (Figure 2G). Regions of similar H3K36me3 had a similar mutation rate in both models.
H3K36me3 is deposited in the bodies of expressed genes32 due to the interaction of SETD2 with hyperphosphorylated RNA polymerase 2.7,18 Consistent with this, we found a strong correlation of H3K36me3 level and expression by RNAseq (Figure 2H); however, H3K36me3 levels plateaued in highly expressed genes. As expected, SETD2-mutant cells had decreased H3K36me3 at all expression levels. We found a strong inverse correlation between gene expression and mutation rate in control and SETD2-mutant cells, consistent with high H3K36me3 in highly expressed genes (Figure 2I).
Setd2 conditional murine knockout and in vivo validation of chemotherapy resistance
To model heterozygous and homozygous SETD2 loss more precisely and to study the consequent effects of SETD2 loss in vivo, we generated a conditional Setd2 knockout mouse with a floxed third exon (Figure 3A). These mice were crossed to the Mx1-cre strain and induced to excise the loxP-flanked cassette in the hematopoietic compartment by treatment with polyinosinic:polycytidylic acid (poly I:C) (Figure 3B). Bone marrow from polyinosinic:polycytidylic acid–treated Setd2fl/+ Mx1-cre, Setd2fl/fl Mx1-cre, and Setd2+/+ Mx1-cre mice were then transformed with an MLL-AF9–expressing retrovirus marked with GFP. Transduced bone marrow cells were sorted for GFP and then transplanted into sublethally irradiated recipient C57BL/6 mice, which subsequently developed primary leukemia. Setd2fl/+ accelerated leukemia onset, whereas Setd2fl/fl delayed leukemia onset significantly compared with Setd2+/+ cells (Figure 3C). MLL-AF9 Setd2fl/+ primary leukemias had approximately half of the amount of H3K36me3 compared with MLL-AF9 Setd2+/+ leukemia, and MLL-AF9 Setd2fl/fl leukemia had undetectable amounts of H3K36me3 as determined by western blot analysis (Figure 3D), consistent with a loss-of-function allele. The MLL-AF9 Setd2fl/fl leukemia had fewer cells in S-phase than the MLL-AF9 Setd2+/+ or MLL-AF9 Setd2fl/+ leukemias, and the MLL-AF9 Setd2fl/fl leukemia was not transplantable into a secondary recipient mice in contrast with the leukemias with wild-type or heterozygous loss of Setd2 (supplemental Figure 5A-B). Because the vast majority of SETD2 mutations in human leukemia are heterozygous and homozygous mutations are rare, we focused on assessing the MLL-AF9 Setd2fl/+ leukemia for chemotherapy resistance.
Compared with MLL-AF9 Setd2+/+ leukemia, MLL-AF9 Setd2fl/+ leukemia was resistant to cytarabine in vitro, and had impaired apoptosis, consistent with our data from human cell lines (supplemental Figure 3C-D). To assess these effects in vivo, we performed secondary transplantation of MLL-AF9 Setd2fl/+ or MLL-AF9 Setd2+/+ leukemia and monitored the mice for the development of leukemia. We treated cohorts of mice with a mean of 30% GFP in the PB with 1 dose of cytarabine (100 mg/kg IP) and harvested cells 12 to 16 hours later. Mice with MLL-AF9 Setd2fl/+ leukemia had significantly less leukemia clearance as measured by PB GFP percentage (Figure 3E) and less apoptosis (Figure 3F) compared with MLL-AF9 Setd2+/+ leukemia. To assess for an effect on survival, we began treatment at a mean of 5% GFP in the PB and injected mice IP with cytarabine (100 mg/kg) or vehicle control once per day for 5 days. Treatment with cytarabine significantly increased survival in mice with MLL-AF9 Setd2+/+ leukemia (Figure 3G), but not in mice with MLL-AF9 Setd2fl/+ leukemia (Figure 3H). These studies demonstrate that heterozygous Setd2 loss, which is often seen in human leukemia, causes an aggressive disease with decreased leukemia latency and chemotherapy resistance.
Chemotherapy resistance due to SETD2 loss can be targeted by inhibition of KDM4A
Because loss of the H3K36me3 mark led to abrogation of DNA damage recognition and chemotherapy resistance, we hypothesized that increasing the H3K36me3 mark might restore chemotherapy sensitivity in SETD2-mutant cells. Although it is difficult to restore SETD2 methyltransferase function in cells with inactivating mutations, inhibition of KDM4A, an H3K9 and H3K36 demethylase, is a potential therapeutic strategy. Although no selective inhibitor of KDM4A exists, the small molecule JIB-04 inhibits multiple histone demethylases, including KDM4A.33 In both human SETD2-mutant cell lines and in murine MLL-AF9 Setd2fl/+ Mx1-cre leukemia, we confirmed that JIB-04 increases H3K36me3 levels within 24 hours of treatment (Figure 4A-B). The secondary loss of SETD2 in the MOLM-13 isogenic clones did not alter the sensitivity of the cells to JIB-04 (Figure 4C); however, the addition of JIB-04 shifts the cytarabine half-maximal inhibitory concentration curve of the resistant MOLM-13 SETD2-mutant clone back to the sensitive MOLM-13 isogenic control clone curve (Figure 4D). Lastly, the combination of cytarabine (100 mg/kg IP) and JIB-04 (110 mg/kg IP) led to a significant increase in apoptosis and leukemia clearance in secondary MLL-AF9 Setd2fl/+ Mx1-cre leukemias, whereas cytarabine or JIB-04 treatment alone did not (Figure 4E-F).
Another potential route of targeting SETD2 loss is synthetic lethal interactions with impaired downstream effects of SETD2 loss. Pfister and colleagues34 found that solid tumor cells with SETD2 loss were sensitive to Wee1 inhibition with the clinical compound AZD1775 through dual transcriptional and proteolytic downregulation of RRM2. In isogenic MOLM-13 cells and the Setd2 conditional knockout model, SETD2 loss did not reduce RRM2 expression levels as assessed by RNAseq, and AZD1775 alone did not decrease the number of leukemia cells or increase Annexin V significantly after a single dose. However, the combination of AZD1775 with cytarabine was significantly more effective than cytarabine alone (supplemental Figure 5C-D). This combination has previously been shown to be synergistic in leukemia, in a variety of cell lines and patient xenografts, without regard to SETD2 mutation status.35 Although combination treatment with cytarabine and JIB-04 or AZD1775 was more effective than single agents in the short term, 1 week of combination therapy did not extend survival with the doses and schedules we attempted, highlighting the aggressive nature of Setd2-mutant leukemia (supplemental Figure 5E).
Discussion
Using both in vitro and in vivo models of leukemia, we found that alterations of the epigenetic regulator SETD2, which is recurrently mutated in acute leukemia, leads to resistance to the DNA-damaging agents that are commonly used to treat leukemia. Our data indicate that SETD2 heterozygous loss accelerates leukemogenesis and leads to chemotherapy resistance primarily through delocalization of DDR. Whereas complete SETD2 loss is detrimental to leukemia formation, clones with compound heterozygous mutations can be isolated, which retain residual H3K36me3 activity and retain chemotherapy resistance.
Defects in the DNA mismatch recognition and repair pathway lead to resistance to DNA-damaging agents in vitro and poor outcomes in patients.36,37 Silencing of the mismatch repair protein MSH2 has been associated with chemotherapy resistance in leukemia.38 Histone marks have been known to regulate the DNA damage response and mismatch repair, including H3K36me3 localizing MSH6. We show that SETD2 loss impacts resistance to clinically relevant DNA-damaging chemotherapy agents, in vitro and in vivo, and, importantly, that such resistance can be mitigated in the short term with the use of KDM4A inhibition in combination with cytarabine, providing a potential therapeutic avenue to investigate. Mismatch repair defects predict sensitivity to immune checkpoint blockade,39 and homologous recombination defects sensitize cells to poly(adenosine 5'-diphosphate-ribose) polymerase (PARP) inhibition,40 providing additional potential therapeutic strategies. Total loss of Setd2 delayed leukemia formation in vivo, suggesting that targeting SETD2 itself could be a vulnerability in SETD2-mutant and perhaps even SETD2–wild-type leukemia. However, it is unknown if this dependence is limited to MLL-rearranged leukemia, and there would be concerns about partial inhibition leading to chemotherapy resistance. SETD2 mutation status, decreased SETD2 expression, and/or decreased H3K36me3 levels have been linked to poor clinical outcomes in CLL12 and clear-cell renal cell carcinoma; however, the effect of SETD2 mutation status on prognosis in acute leukemia remains under study. If shown to be adverse in leukemia, SETD2 mutation could be used in risk stratification and help guide therapy toward agents without DNA-damaging mechanisms.
Deficits in homologous recombination, which may occur in complete knockdown of SETD2,30 have generally led to sensitivity to DNA-damaging agents, such as cisplatin,41 however, defects in DNA double-strand break repair can be successfully overcome in resistant cancer cells by bypassing, tolerating, and using alternate mechanisms to repair damage,42,43 analogous to the model we propose. That being said, the impact of SETD2 heterozygous loss on homologous recombination is unclear.
The local mutation rate is not even throughout the genome,44 and a lower somatic mutation rate has been observed in regions of high gene expression, open chromatin, and high H3K36me3 as well as other chromatin marks.45 In this article, we demonstrate, in an experimental setting, that the local mutation rate is inversely correlated to H3K36me3 levels. Accurate DNA repair requires significant cellular energy and delays DNA synthesis and the cell cycle. Through its interaction with RNA polymerase,32 SETD2 would provide a mechanism to mark critical expressed genes and regions of the genome with H3K36me3 that should be prioritized for more accurate DNA fidelity by recruiting the DDR machinery involved with mismatch repair and homologous recombination, but, lastly, also by inducing apoptosis (Figure 5A). Conversely, this model predicts that cells would tolerate more mutations and use lower fidelity repair, such as nonhomologous end joining, in genomic regions without H3K36me3.
Figure 5.

Proposed model of H3K36me3 and DNA damage repair, SETD2 loss, and chemotherapy resistance. (A) The interaction of SETD2 with RNA polymerase leads to high levels of H3K36me3 on expressed genes, which localize several known DNA damage–recognition proteins to the region, even in the absence of DNA damage. DNA damage in those regions is rapidly recognized by local DDR machinery, leading to downstream signaling, including Chk1 phosphorylation and DNA repair and/or apoptosis, and consequently the acquisition of few mutations. In regions of nonexpressed genes, H3K36me3 is low and there is relatively less localization of DDR machinery. This leads to relatively less DDR activation, and DNA damage goes unrecognized and unrepaired, leading to cell survival with concurrent mutations. (B) In wild-type cells, chemotherapy-induced DNA damage triggers DDR, resulting in some repair, but significant apoptosis. In SETD2-mutant cells, the relative lack of DDR results in less repair, but also less apoptosis, leading to the survival of mutated cells.
In summary, we have shown that SETD2 alteration leads to an aggressive leukemia with resistance to DNA-damaging chemotherapy agents through regional impairment of DDR and apoptosis (Figure 5B) and that targeting H3K36me3 demethylases may provide a therapeutic strategy to reverse chemotherapy resistance.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the Dana-Farber Cancer Institute Center for Cancer Computational Biology, the Dana-Farber Cancer Institute Center for Cancer Genome Discovery, and the Massachusetts General Hospital Center for Computational and Integrative Biology DNA Core for next-generation sequencing.
This work was supported by the William Lawrence and Blanche Hughes Foundation, the National Institutes of Health, National Cancer Institute (grant P01 CA066996), the Leukemia and Lymphoma Society, and the Howard Hughes Medical Institute. B.G.M. was supported by a St. Baldrick’s Foundation Scholar grant, an Alex's Lemonade Stand Foundation Young Investigator grant, and K12 and K08 (K08 CA184419) grants from the National Institutes of Health, National Cancer Institute. S.H.C. is a Damon Runyon-Sohn Pediatric Fellow supported by the Damon Runyon Cancer Research Foundation (DRSG-5-13). J.D.K. is supported by the NWO Diamond Program and the Prins Bernard Cultural Fund.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: B.G.M., J.D.K., B.L.E., J.J.H., S.H.C., and S.A.A. designed the studies; B.G.M., J.D.K., S.H.C., A.V.K., R.K., J.L.K., R.L.Z., C.A.C., M.E.M., J.C., R.C., and P.V.G. performed the experiments; and B.G.M., J.D.K, S.H.C. S.A.A., and B.L.E. analyzed the data and wrote the manuscript with input and editing from all authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Benjamin L. Ebert, Division of Hematology, Brigham and Women’s Hospital, 77 Ave Louis Pasteur, HIM 743, Boston, MA 02115; e-mail: bebert@partners.org.
References
- 1.Bhojwani D, Pui C-H. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013;14(6):e205-e217. [DOI] [PubMed] [Google Scholar]
- 2.Thol F, Schlenk RF, Heuser M, Ganser A. How I treat refractory and early relapsed acute myeloid leukemia. Blood. 2015;126(3):319-327. [DOI] [PubMed] [Google Scholar]
- 3.Ofran Y, Rowe JM. Treatment for relapsed acute myeloid leukemia: what is new? Curr Opin Hematol. 2012;19(2):89-94. [DOI] [PubMed] [Google Scholar]
- 4.Locatelli F, Moretta F, Rutella S. Management of relapsed acute lymphoblastic leukemia in childhood with conventional and innovative approaches. Curr Opin Oncol. 2013;25(6):707-715. [DOI] [PubMed] [Google Scholar]
- 5.Mullighan CG, Zhang J, Kasper LH, et al. . CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471(7337):235-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mar BG, Bullinger LB, McLean KM, et al. . Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat Commun. 2014;5:3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao H, Wang L-M, Luo Y, et al. . Mutations in epigenetic regulators are involved in acute lymphoblastic leukemia relapse following allogeneic hematopoietic stem cell transplantation. Oncotarget. 2016;7(3):2696-2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun X-J, Wei J, Wu X-Y, et al. . Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J Biol Chem. 2005;280(42):35261-35271. [DOI] [PubMed] [Google Scholar]
- 9.McKinney M, Moffitt AB, Gaulard P, et al. . The Genetic Basis of Hepatosplenic T-cell Lymphoma. Cancer Discov. 2017;7(4):369-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Ding L, Holmfeldt L, et al. . The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masetti R, Castelli I, Astolfi A, et al. . Genomic complexity and dynamics of clonal evolution in childhood acute myeloid leukemia studied with whole-exome sequencing. Oncotarget. 2016;7(35):56746-56757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parker H, Rose-Zerilli MJ, Larrayoz M, et al. . Genomic disruption of the histone methyltransferase SETD2 in chronic lymphocytic leukaemia. Leukemia. 2016;30(11):2179-2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerlinger M, Rowan AJ, Horswell S, et al. . Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883-892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varela I, Tarpey P, Raine K, et al. . Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469(7331):539-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duns G, van den Berg E, van Duivenbode I, et al. . Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res. 2010;70(11):4287-4291. [DOI] [PubMed] [Google Scholar]
- 16.Fontebasso AM, Schwartzentruber J, Khuong-Quang D-A, et al. . Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013;125(5):659-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huether R, Dong L, Chen X, et al. . The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun. 2014;5:3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Liu L, Qu Y, et al. . Prognostic value of SETD2 expression in patients with metastatic renal cell carcinoma treated with tyrosine kinase inhibitors. J Urol. 2016;196(5):1363-1370. [DOI] [PubMed] [Google Scholar]
- 19.Liu W, Fu Q, An H, et al. . Decreased expression of SETD2 predicts unfavorable prognosis in patients with nonmetastatic clear-cell renal cell carcinoma. Medicine (Baltimore). 2015;94(45):e2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu X, He F, Zeng H, et al. . Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet. 2014;46(3):287-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heckl D, Kowalczyk MS, Yudovich D, et al. . Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat Biotechnol. 2014;32(9):941-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krivtsov AV, Twomey D, Feng Z, et al. . Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442(7104):818-822. [DOI] [PubMed] [Google Scholar]
- 23.Koboldt DC, Zhang Q, Larson DE, et al. . VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dobin A, Davis CA, Schlesinger F, et al. . STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923-930. [DOI] [PubMed] [Google Scholar]
- 26.Orlando DA, Chen MW, Brown VE, et al. . Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Reports. 2014;9(3):1163-1170. [DOI] [PubMed] [Google Scholar]
- 27.LaFave LM, Béguelin W, Koche R, et al. . Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med. 2015;21(11):1344-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aymard F, Bugler B, Schmidt CK, et al. . Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol. 2014;21(4):366-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfister SX, Ahrabi S, Zalmas L-P, et al. . SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Reports. 2014;7(6):2006-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carvalho S, Vítor AC, Sridhara SC, et al. . SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. eLife. 2014;3:e02482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li F, Mao G, Tong D, et al. . The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell. 2013;153(3):590-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Almeida SF, Grosso AR, Koch F, et al. . Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat Struct Mol Biol. 2011;18(9):977-983. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Chang J, Varghese D, et al. . A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. 2013;4:2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfister SX, Markkanen E, Jiang Y, et al. . Inhibiting WEE1 selectively kills histone H3K36me3-deficient cancers by dNTP starvation. Cancer Cell. 2015;28(5):557-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porter CC, Kim J, Fosmire S, et al. . Integrated genomic analyses identify WEE1 as a critical mediator of cell fate and a novel therapeutic target in acute myeloid leukemia. Leukemia. 2012;26(6):1266-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clin Cancer Res. 1998;4(1):1-6. [PubMed] [Google Scholar]
- 37.Cohn DE, Frankel WL, Resnick KE, et al. . Improved survival with an intact DNA mismatch repair system in endometrial cancer. Obstet Gynecol. 2006;108(5):1208-1215. [DOI] [PubMed] [Google Scholar]
- 38.Diouf B, Cheng Q, Krynetskaia NF, et al. . Somatic deletions of genes regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nat Med. 2011;17(10):1298-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le DT, Uram JN, Wang H, et al. . PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509-2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mateo J, Carreira S, Sandhu S, et al. . DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697-1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tebbs RS, Zhao Y, Tucker JD, et al. . Correction of chromosomal instability and sensitivity to diverse mutagens by a cloned cDNA of the XRCC3 DNA repair gene. Proc Natl Acad Sci USA. 1995;92(14):6354-6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guillemette S, Serra RW, Peng M, et al. . Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev. 2015;29(5):489-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ray Chaudhuri A, Callen E, Ding X, et al. . Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535(7612):382-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Supek F, Lehner B. Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature. 2015;521(7550):81-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Polak P, Karlić R, Koren A, et al. . Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015;518(7539):360-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou X, Edmonson MN, Wilkinson MR, et al. . Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet. 2016;48(1):4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Forbes SA, Beare D, Boutselakis H, et al. . COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45(D1):D777-D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.