

Abstract
Background
We performed a genomic study in lung adenocarcinoma cases with discordant anaplastic lymphoma kinase (ALK) status by fluorescent in situ hybridization (FISH) and immunohistochemistry (IHC).
Methods
DNA from formalin-fixed paraffin embedded (FFPE) tissues of 16 discordant (4 FISH+/IHC− and 12 FISH−/IHC+) cases by Vysis ALK Break Apart FISH and ALK1 IHC testing were subjected to whole gene capture and next generation sequencing (NGS) of nine genes including ALK, EML4, KIF5B, SND1, BRAF, RET, EZR, ROS1, and TERT. All discordant cases (except one FISH−/IHC+ without sufficient tissue) underwent IHC by D5F3 antibody. In one case with fresh frozen (FF) tissue, whole transcriptome sequencing (RNAseq) was also performed. Twenty-six concordant (16 FISH+/IHC+ and 10 FISH−/IHC−) cases were included as controls.
Results
In four ALK FISH+/IHC− cases, no EML4-ALK fusion gene was observed by NGS but in one case using FF tissue, we identified EML4-BIRC6 and AAK1-ALK fusion genes. RNAseq revealed a highly expressed EML4-BIRC6 fusion transcript and a minimally expressed AAK1-ALK transcript. Among the 12 FISH−/IHC+ cases, no evidence of ALK gene rearrangement was detected by NGS. Eleven of 12 FISH−/IHC+ cases by ALK1 clone were concordant by a repeat ALK IHC with D5F3 antibody (i.e. FISH−/IHC− by D5F3 clone). Among the 16 ALK FISH+/IHC+ positive controls, whole gene-capture identified ALK gene fusion in 15 cases including one case with HIP1-ALK. No ALK fusion gene was observed in all 10 FISH−/IHC− cases. Other fusion genes involving ROS1, EZR, BRAF and SND1 were also found.
Conclusions
ALK FISH results appeared to be false positive in 3 of 4 FISH+/IHC− cases, while no false negative ALK FISH case was identified among 12 ALK FISH−/IHC+ cases by ALK1 clone, in keeping with the concordant FISH−/IHC− status by D5F3 clone. Our targeted whole gene capture approach using FFPE samples was effective for detecting rearrangements involving ALK and other actionable oncogenes.
Introduction
Fluorescent in situ hybridization (FISH) using break-apart probe for the anaplastic lymphoma kinase (ALK) gene has been generally regarded as the gold standard for determining ALK status of lung cancers for using ALK inhibitors. However, the true sensitivity and specificity of the FISH method for detecting ALK gene rearrangement are not well known. Various genomic abnormalities involving the ALK gene could be associated with abnormal signal patterns seen in ALK FISH and with the abnormal protein expression detected by ALK immunohistochemistry (IHC). Neither FISH nor IHC method is perfect and a third method using other molecular techniques may be required to determine the true ALK fusion status in some cases. Clinical experience has indicated that ALK status determined by FISH may not always correlate with responsiveness to ALK inhibitors and that a testing algorithm with combination of FISH, IHC, and reverse transcriptase-polymerase chain reaction (RT-PCR) might be a better approach to select the patients for ALK inhibitor therapy.1
ALK analysis by FISH using the Vysis LSI ALK break apart probe (Abbott Molecular, Abbott Park, IL) has shown that ALK FISH signal patterns can be quite complicated. It is especially difficult to prove the positive ALK FISH result as false positive when a case is positive for FISH and negative for IHC or RT-PCR, due to the limited sensitivity of RT-PCR and IHC; ALK RT-PCR would not detect previously unidentified fusion transcript(s) and ALK protein expression in lung cancers might be low. Thus, it is usually ruled that the negative RT-PCR/IHC result as false negative in those with a positive ALK FISH result. On the other hand, a few cases of false-negative FISH results have been documented in the literature.2–5
There has been an increasing need to develop clinically applicable multiplex assays that could maximize the yield of molecular tests on FFPE tissues from small lung biopsies. Although multiplexed genomic assays have been applied to determine ALK fusions at the DNA and RNA levels, published reports are still sparse in the literature.6,7 The rapid development of technologies for large-scale sequencing (such as NGS) has facilitated high-throughput molecular analysis for large numbers of genes in a single test for a simultaneous detection of deletions, insertions, copy number alterations, rearrangements, and single base substitutions (hot-spot mutations) in all known cancer-related genes.8 Currently, NGS platforms, including whole genome, whole exome, and targeted gene sequencing, represent emerging diagnostic methodologies for the detection of oncogene fusions and mutations in FFPE as well as in FF tumor tissue specimens.9,10
The primary purpose of this study was to examine the molecular basis for cases with discordant ALK status by FISH and IHC. We also wished to establish a scalable method to detect all ALK-rearranged and other targetable fusion events in patients who may benefit from targeted gene specific inhibitor therapies. Herein, we report the use of a NGS-based whole gene capture approach using DNA from FFPE samples and comprehensive bioinformatics analyses to successfully detect rearrangements involving ALK and other oncogenes in lung cancers as a promising diagnostic method applicable to routine clinical practice.
Materials and Methods
Cases and ALK assessment
This study was approved by Institutional Review Board of Mayo Foundation. All discordant cases were identified from our historical cohort of resected lung adenocarcinoma cases. Selected concordant cases were included as the positive and negative controls for NGS. A total of 45 cases were initially were analyzed further for fusions involving ALK and other targeted oncogenes. Three of these 45 cases were excluded due to failed gene capture and the remaining 42 cases were included in the present report. ALK IHC was performed with ALK1 antibody (Dako, 1:100 dilution) and scored as 0, 1, 2 or 3 as previously described.11 ALK IHC score 0 and 1 were regarded as negative (IHC−) and ALK IHC score 2 and 3 as positive (IHC+) as in our previous study.11 Interphase molecular cytogenetic studies using Vysis Break Apart ALK Probe Kit (Abbott Molecular) were performed on FFPE sections and scored as either positive (≥15%) or negative (<15%) as previously described.11 Hundred tumor cells were counted in each case.
Discordant cases underwent repeat IHC with a different ALK antibody (clone D5F3, Cell Signaling Technology, Danvers, MA). Four micron sections cut from formalin-fixed, paraffin-embedded blocks were placed on charged slides; slides were then dried and melted in a 62°C oven for twenty minutes. Immunohistochemical staining was performed as follows: slides were placed on a Ventana BenchMark XT (Ventana Medical Systems Inc., Tucson, AZ) for staining. The staining protocol included on-line deparaffinization, HIER (Heat Induced Epitope Retrieval) with Ventana Cell Conditioning 1 for 32 minutes, primary ALK antibody incubation for 32 minutes at 37°C (clone D5F3, a rabbit monoclonal antibody, 1:100 dilution, Cell Signaling Technology, Danvers, MA). Antigen-antibody reactions were visualized using Ventana Optiview Universal DAB Detection Kit. Counterstaining was performed on the Ventana BenchMark XT using Ventana Hematoxylin II for eight minutes, followed by bluing reagent for four minutes. The positive control was a known ALK-rearranged lung adenocarcinoma and the negative control was a mouse IgG1 serum substitution for the primary ALK antibody.
Whole gene capture for NGS
A custom capture kit (Agilent) was designed to span the entire genomic sequence of 9 genes (based upon hg 19) including ALK, EML4, KIF5B, SND1, BRAF, RET, EZR, ROS1, and TERT for target-specific gene capture and fusion identification by NGS. The total captured region was approximately 1.6 Mb. DNA was extracted from lung adenocarcinoma using FFPE tissues where two to three 10μ-thick sections per case with tumor purity of 40–95% were used to extract the genomic DNA using the Qiagen AllPrep DNA/RNA FFPE kit (Quiagen). Paired-end indexed libraries were prepared using the NEBNext Ultra Library prep protocol (NEB). Briefly, up to 500 ng of FFPE genomic DNA in 50 μl TE buffer was fragmented using the Covaris LE210 sonicator to generate double-stranded DNA fragments with blunt or sticky ends at a fragment size mode of between 150–200bp. The ends were repaired using the NEB End-prep enzyme mix after which looped Multiplex NEBNext Adaptors for Illumina were added using the NEB Blunt/TA Ligase Master Mix and linearized with USER enzyme. Adapter-ligated DNA fragments were size-selected to enrich for 200 bp inserts (~320 bp total library size) using a double SPRI bead purification method followed by 6 cycles of PCR using NEB Universal PCR Primer and NEB Index Primer. The concentration and size distribution of the libraries were determined using Agilent Bioanalyzer DNA 1,000 chips.
Custom capture was carried out using the Agilent Bravo liquid handler according to a modified protocol for Agilent’s SureSelect XT. Four samples were pooled equally for a total of 750 ng of the prepped libraries. The pools were incubated with the custom biotinylated capture baits supplied in the kit for 24 hours at 65°C. The captured hybrids were recovered using Dynabeads MyOne Streptavidin T1 from Dynal. The DNA was eluted from the beads and purified using Ampure XP beads from Agencourt. The purified capture products were then amplified using the SureSelect Post-Capture Indexing forward and Index PCR reverse primers (Agilent) for 12 cycles. Libraries were quantified and sequenced as 101 × 2 paired end reads on an Illumina HiSeq 2000 using TruSeq SBS sequencing kit version 3 and HiSeq data collection version 2.0.12.0 software. On average, 73 million reads were generated for each sample, the minimal unique reads per sample ranged between 10 to 80 million with most samples having 20–40 million reads each.
Whole transcriptome sequencing using fresh frozen tissue
We used 100ng of RNAs from both tumor and matched adjacent normal tissue to generate libraries using TruSeq™ RNA sample preparation V2 protocol (Illumina, San Diego). Sequencing was performed on a HiSeq 2000 instrument with a 51 cycles pair-end read. FASTQ formatted raw files were mapped and aligned to hg19. Fusion transcripts were identified using SnowShoes-FTD version 2.0.12 A fusion was called when there was at least two reads that span the fusion junction and three reads that encompassing the two fusion transcripts.
Data Analysis and Fusion Detection in Targeted Sequencing
A. CREST – identification of fusions utilizing split reads
Alignment and pre-processing
We aligned the reads to the hg19 reference genome using two different alignment programs, Burrows Wheeler Aligner (BWA,v 0.5.9)13 and NovoAlign (VN:V2.08.01).14 The default options were used for BWA. NovoAlign penalties were set for structural variation (120), gap opening (40), and gap extend (5). NovoAlign fragment length mean (425) and standard deviation (80) were set. Duplicate reads were marked by Picard.15
Detection and filtering of Structural Variants
Structural variants were detected using a localized search option implemented in CREST.16 The CREST algorithm detects structural variation by identifying split reads (reads that can be split into two segments that align to disjoint genomic locations) from soft-clipped reads. For each alignment and each gene, we ran the CREST program with the localized search interval set according to gene definition. Duplicate reads, events with coverage greater than 1,000,000 reads, and redundant structural variation predictions were removed. To identify the top candidates from among the structural variants detected by CREST, we considered the quantity and robustness of support, requiring that either there was at least 1 supporting soft-clipped read for each breakpoint in one alignment or that the event was detected in both the BWA and NovoAlign alignments. Further, we reviewed each of the events satisfying these criteria by applying the BLAT (BLAST-like alignment tool) to the consensus sequences determined by CREST.
Annotation, Summarization, and Prioritization of Structural Variants
The CREST program indicates the type of structural variant detected (e.g. translocation, inversion, insertion, duplication, deletion) as well as orientation, breakpoints, and consensus sequence across the breakpoint. We used the UCSC refFlat gene annotation table to further annotate the breakpoint regions according to the transcriptional boundaries of the RefSeq genes and the RefSeq gene region (e.g. intron, exon, intergenic, etc).17 For structural variant predictions involving two breakpoints that are within the transcript regions of separate genes, a fusion transcript could potentially result from the structural variant. From the consensus sequence provided by CREST, we determined the orientation of the fusion transcript. Essentially, when two segments of genomic DNA are joined, with each segment from a gene region, there are four possibilities with respect to the two gene segments. For genes A and B, the first case is that the 3′ end of gene A segment is joined to the 5′ end of the gene B segment. We use the notation A->B in this case. The second case is that the 5′ end of gene A segment is joined to the 3′ end of the gene B segment. This is represented by B->A. The third case is that the 5′ end of gene A segment is joined to the antisense strand of the 5′ end of the gene B segment. This is represented in our notation as <-A:B->. This is essentially equivalent to <-B:A-> in that if the DNA SV supports <-B:A-> it also supports <-A:B->. The fourth case is that the 3′ end of gene A segment is joined to the antisense strand of the 3′ end of the gene B segment. This is represented in our notation as A-><-B. This is equivalent to B-><-A, ie if the DNA SV supports B-><-A it also supports A-><-B. The orientations of the detected DNA SV’s in this work are provided in Supplemental Table 1. For all detected DNA SV’s involving two genes, we report an orientation class as defined by these 4 cases described above. In the case of a detected DNA SV’s for which one breakpoint is in a gene region and the other is in an intergenic region, there is no orientation class to assign, but we do report these events as well.
B. Structural Variant Analysis tools (SVAtools) identification of fusions utilizing both discordant read pairs and split reads
Alignment and pre-processing
Using a binary indexing mapping algorithm18 (BIMA), we mapped all read-pairs to the reference genome GRCh38 using a concordant insert size limit of 5000 bps. BIMA transforms the reference genome to a binary index allowing for rapid mapping. High accuracy is achieved by simultaneous mapping of both reads in a read-pair.18 BIMA is tuned to detect and report read-pairs that map to two discontinuous genomic areas, including when a single read crosses a breakpoint (split read) or a biotin-junction (common in NGS mate-pair library preparation).
Detection and filtering of Structural Variants
Structural variants were detected using a suite of algorithms, SVAtools, designed specifically for detecting structural variants in read-pair data mapped by BIMA.19,20 SVAtools is an in-house R package, developed by the Biomarker Discovery Lab at Mayo Clinic. Briefly, all read-pairs are sorted by mapped chromosome and position for rapid indexing. Replicate read-pairs are removed. A customized rapid clustering algorithm detects read-pairs and split reads supporting a common breakpoint. A minimum of three read-pairs is required to form a cluster. These clusters must pass a system of masks and filters. The mask eliminates normal structural variants not present in the reference genome, eliminates mapping artifacts due to repeat or un-sequenced genomic regions and eliminates artifacts due to NGS library preparation. The filters use cluster size and BIMA mapping scores to identify poorly qualified breakpoints and false positives. Default settings for the filters were applied except as follows: –protocol pairedEnd –radius 3000 –mismatch_lim 10 – –homCountmin 20 –homf 1.75. Only clusters with at least 8 read-pairs were considered for structural variant detection, and only breakpoints involving genes, with at least one target gene were considered for this study. Genomic regions of suspected structural variation can be inspected and visualized via junction plots (illustrations of all read-pairs mapping within and between two genomic regions). Junction plots served as a useful tool to double-check for false-negatives, to reveal clusters of read-pairs that were incorrectly removed during masking or filtering. All structural variants detected via these methods are listed in supplement table 2. The interpretation of each rearrangement follows the notation described above in the CREST annotation.
Results
Fusion Identification by Whole Gene Capture
Discordant cases were comprised of 4 cases that were positive by FISH but negative by IHC using ALK1 clone (all IHC score 1) and 12 cases that were negative for FISH but positive for IHC using ALK1 clone (all IHC score 2) (Table 1). ALK IHC using D5F3 was also negative in all 4 FISH+/IHC− cases as using ALK1, while the ALK IHC+ cases by ALK1 clone in 11 of 12 FISH−/IHC+ cases tested by D5F3 clone were IHC− (i.e. the 11 tested cases were concordant by D5F3 clone as FISH−/IHC−) (Table 2). The percentages of cells with abnormal signals in FISH were 15–27% in the 4 FISH+/IHC− cases and 0–7% in the 12 FISH−/IHC+ cases (Table 2). Concordant cases (16 FISH+/IHC+ and 10 FISH−/IHC−) were used as controls (Table 1). No fusion gene involving either ALK or EML4 was identified in the 3 FISH+/IHC− cases using FFPE DNA for NGS while 1 case using FF tissue showed AAK1-ALK and EML4-BIRC6 fusion genes by both fusion detection algorithms (Figure 1). Whole gene capture and sequencing analysis with alignment algorithms revealed no ALK fusion in any of the 12 FISH−/IHC+ cases. Of note, one of 12 FISH−/IHC+ cases showed CD74−>ROS1 (Table 2). Of the 16 FISH+/IHC+ cases, 13 cases showed DNA structural variant support for EML4->ALK fusions by whole gene capture, while 1 case showed support for the reciprocal fusion ALK->EML4 fusion. One other case had a fusion involving Huntington-interacting protein-1 (HIP1) and ALK genes. The remaining one FISH+/IHC+ case did not show any evidence for a fusion by gene capture; this case had a borderline FISH positivity (15%) and IHC was also weakly positive (score 2). Repeated IHC with D5F3 clone was negative. Thus, both the original FISH and IHC results appeared to be a false positive based on the absence of identifiable ALK fusion by targeted gene capture and NGS. There was one FISH+/IHC+ case that revealed a novel SND1-BRAF fusion gene. No DNA support was found for a fusion involving the ALK gene in any of the 10 ALK FISH−/IHC− cases. However, we detected an SND1->BRAF fusion gene involving exons in two and EZR->ROS1 in one of these 10 cases. All identified fusions are listed in Table 2.
Table 1.
Summary of ALK IHC and FISH
| Score 3 IHC+ | Score 2 IHC+ | Score 1 IHC− | Score 0 IHC− | Total | |
|---|---|---|---|---|---|
| FISH + | 11 | 5 | 4 | 0 | 20 |
| FISH − | 0 | 12 | 6 | 4 | 22 |
| Total | 11 | 17 | 10 | 4 | 42 |
Values in bold indicate samples with discordant FISH/IHC scores.
Table 2.
Fusion identification by CREST and SVAtools.
| Sample ID | Tumor Percent | ALK FISH (%) | ALK IHC (ALK1) | ALK IHC (D5F3) | CREST* | SVAtools** |
|---|---|---|---|---|---|---|
| Y31 | N/A | − (1) | 1 | N.T | – | – |
| Y32 | N/A | − (6) | 1 | N.T | – | EZR-><-RAB3C |
| Y33 | N/A | − (2) | 0 | N.T | – | – |
| Y34 | 70% | − (N/A) | 0 | N.T | – | – |
| Y35 | N/A | − (9) | 1 | N.T | EZR->ROS1 (10) | EZR->ROS1 (4) |
| Y38 | 80% | − (2) | 0 | N.T | – | – |
| Y39 | 90% | − (N/A) | 1 | N.T | – | – |
| Y65 | 40% | − (N/A) | 1 | N.T | – | – |
| Y68 | 95% | − (N/A) | 1 | N.T | SND1->BRAF not detected by BWA-Novoalign/CREST; detected by BWAMEM/CREST (17) | SND1-> BRAF (11) |
| Y69 | 90% | − (N/A) | 0 | N.T | SND1->BRAF (9) | SND1-> BRAF (21) |
|
| ||||||
| Y56 | 60% | − (N/A) | 2 | N.T | – | – |
| Y63 | 50% | − (0) | 2 | 0 | – | – |
| Y72 | 60% | − (3) | 2 | 0 | – | – |
| Y73 | 80% | − (1) | 2 | 0 | CD74->ROS1 (40) | CD74-> ROS1 (79); ROS1->CD74 (11) |
| Y74 | 90% | − (7) | 2 | 0 | – | – |
| Y75 | 50% | − (5) | 2 | 0 | – | – |
| Y76 | 70% | − (0) | 2 | 0 | – | – |
| Y77 | 90% | − (5) | 2 | 0 | – | – |
| Y78 | 90% | − (1) | 2 | 0 | – | – |
| Y79 | 70% | − (N/A) | 2 | 0 | – | – |
| Y80 | 80% | − (1) | 2 | 0 | – | – |
| Y81 | 70% | − (5) | 2 | 0 | – | – |
|
| ||||||
| 891A | N/A | + (27) | 1 | 0 | AAK1->ALK (94); EML4->BIRC6 (160) | AAK1->ALK (152); EML4->BIRC6 (187) |
| Y20 | N/A | + (15) | 1 | 0 | – | – |
| Y53 | 70% | + (15) | 1 | 0 | – | – |
| Y60 | 60% | + (18) | 1 | 0 | – | – |
|
| ||||||
| Y02 | 90% | + (N/A) | 2 | N.T | HIP1->ALK (20) | HIP1->ALK (33) |
| Y04 | 95% | + (29) | 2 | N.T | EML4->ALK (42) | EML4->ALK (94) |
| Y05 | 90% | + (60) | 3 | N.T | ALK->EML4 (29);<-ALK:EML4-> (24) | <-ALK EML4->(25); ALK->EML4 (21) |
| Y06 | 80% | + (25) | 2 | N.T | EML4->ALK (13);ALK->EML4 (60) | EML4->ALK (43); ALK->EML4 (48) |
| Y07 | 60% | + (35) | 2 | N.T | EML4->ALK (56) | EML4->ALK (99) |
| Y08 | 90% | + (N/A) | 3 | N.T | EML4->ALK (25);ALK->EML4 (25) | EML4->ALK (46); ALK->EML4 (31) |
| Y12 | N/A | + (52) | 3 | N.T | EML4->ALK (13) | EML4->ALK (13) |
| Y15 | 60% | + (N/A) | 3 | N.T | EML4->ALK (13) | EML4->ALK (9) |
| Y18 | 60% | + (N/A) | 3 | N.T | EML4->ALK (87); ALK->EML4 (58) | EML4->ALK (98); ALK->EML4 (52) |
| Y22 | 50% | + (70) | 3 | N.T | EML4->ALK (19) | EML4->ALK (42) |
| Y26 | N/A | + (72) | 3 | N.T | EML4->ALK (43) | EML4->ALK (49) |
| Y27 | 70% | + (68) | 3 | N.T | EML4->ALK (23) | EML4->ALK (36) |
| Y52 | N/A | + (N/A) | 3 | N.T | EML4->ALK (28);ALK->EML4 (41) | EML4->ALK (76); ALK->EML4 (49) |
| Y54 | 70% | + (15) | 2 | 0 | – | – |
| Y59 | 80% | + (N/A) | 3 | N.T | EML4->ALK (30); SND1->BRAF not detected by BWA-Novoalign/CREST; detected by BWAMEM/CREST (17) | EML4->ALK (13); SND1-> BRAF(13) |
| Y61 | 80% | + (N/A) | 3 | N.T | EML4->ALK (79) | EML4->ALK (56) |
Number of supporting split reads is shown in parentheses.
Number of supporting read-pairs and split reads is shown in parentheses.
“+” FISH Positive ≥ 15%, “ − ” FISH or Fusion negative, N.T; not tested, N/A; not available
Figure 1.
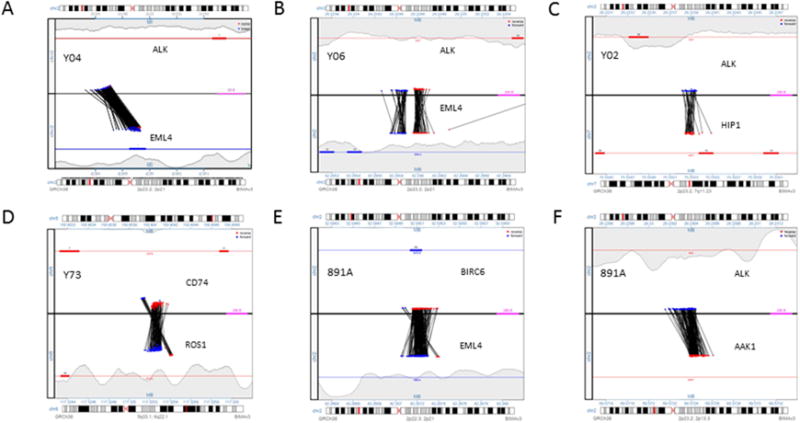
Junction plots of identified fusion genes by SVAtools. EML4-ALK (A), balanced EML4-ALK (B), HIP1-ALK (C), CD74-ROS1 (D), EML4-BIRC6 (E), and AAK1-ALK (F). Junctions plots are a 2-panel representation of the junction of both breakpoints in a chromosomal rearrangement. Reads from paired-ends mapping to breakpoint A are plotted on the top panel, connected by a line to the corresponding paired-end read mapping to breakpoint B in the bottom panel. Genes and reads mapping to the (−) DNA strand are shown in red, while genes and reads mapping to the (+) DNA strand are shown in blue. The color and pattern of discordant read-pairs can be used to identify different types of rearrangements, including balanced or unbalanced translocations, deletions, and inversions.
Molecular Rearrangements Identified by RNAseq Using Fresh Frozen Tissue in a FISH+/IHC− Case
Whole transcriptome profiling (RNAseq) of the mRNA revealed a strong expression of the EML4-BIRC6 fusion gene (Figure 2A), in support of the results of the DNA-based whole gene capture. In contrast, RNAseq showed a low level AAK1-ALK fusion gene expression (Figure 2B), which would explain positive ALK FISH test by a break-apart probe as well as little or no ALK protein expression by ALK IHC (score 1 by ALK1; score 0 by D5F3). Both RNAseq and the gene capture did not identify the EML4-ALK fusion in this case. As was observed in our previous study on this case,21 evidence of EGFR mutation was observed in the expressed transcripts by RNAseq (Figure 2C).
Figure 2.
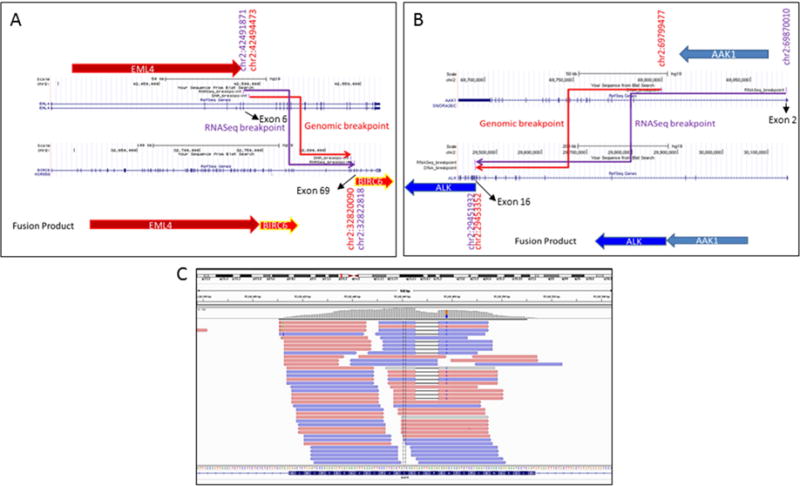
Fusion structures of EML4-BIRC6 (A) and AAK1-ALK (B) genes by RNASeq and custom capture DNA sequencing. EGFR gene mutation is shown in (C).
Genomic Rearrangements Accompanying Oncogene Fusions
By whole gene capture of 9 genes, we were able to observe associated DNA rearrangements that accompanied the functional oncogenic fusions (Supplemental tables 1–3). Several junction plots were also illustrated (Figure 1). At least 4 cases had detectable reciprocal DNA rearrangements from the same genetic event that likely led to the EML4->ALK fusion while others involved ALK->EML4 or a fusion involving 5′ ALK and another gene on either chromosome 2 or another chromosome. Details of genetic events resulting in the HIP1->ALK fusion gene found in a FISH+/IHC+ positive control (case Y2) are listed in the supplemental Tables 2 and 3.
Several other tumors had DNA rearrangements involving one or more targeted oncogene but appeared to not generate an active oncogene fusion of the target genes. As described earlier, one of the 16 FISH+/IHC+ positive cases did not demonstrate any evidence of EML4 ->ALK but there was strong evidence of ALK->EML4 fusion indicating the presence of genomic rearrangements in the region leading to the ALK rearrangement and protein expression (supplemental Tables 1–3).
Discussion
A French multicentric study reported that discordances in ALK status between FISH and IHC were mainly found in the cases with FISH results ranging from 10% to 20% of rearranged cells.22 Three of our 4 FISH+/IHC− cases also fell in this range (15–18%), in keeping with the results in that study. A recent study showed that approximately 6% of ALK FISH cases belong to a borderline group for which ALK FISH evaluation has of limited reliability possibly due to sampling effects.23 Thus, those cases should be considered equivocal and therapy decisions should include additional tests and clinical considerations.23
A recent multicenter ALK IHC testing of non-small cell cancer showed that a high concordance after standardization of techniques and interpretation criteria.24 ALK IHC using ALK1 clone might not be reliable, especially without using an appropriate detection system, but ALK IHC using D5F3 or other clones can provide reliable and reproducible results as shown in the previous studies.11,25–30 There are many different clones of ALK antibodies for IHC.
Discordant cases in the present study were based on the cohort of cases stained with ALK1 clone in our database predating D5F3 clone. We repeated immunostaining on all discordant cases (except one case without available tissue) with D5F3 antibody, which has been shown to correlate better with ALK FISH.27,31 In all four FISH+/IHC− cases, the negative IHC result by ALK1 antibody was confirmed by D5F3 antibody. On the other hand, the positive IHC result (all score 2) by ALK1 antibody reversed as negative by D5F3 antibody in 11 of 11 FISH−/IHC+ cases, where sufficient tissue was available for repeat staining. Thus, this FISH−/IHC+ group most likely have been concordantly ALK negative (i.e. FISH−/IHC−), which would explain the absence of detectable ALK gene rearrangements by NGS in this group.
Three of four FISH+/IHC− cases were negative for any ALK gene rearrangement by the genomic approach, which challenged the positive ALK FISH result in these cases. As mentioned earlier, the positive ALK FISH results in these 3 cases were borderline (15–18%), which might explain the discordances as observed in the recent French study.22,23 ALK FISH in the remaining 1 FISH+/IHC− case showed break apart probes in 27% of tumor cell nuclei; this case showed BIRC6->EML4 and AAK->ALK fusions by both NGS of DNA and RNAseq. In our previous study21, we found EML4–ALK fusion by RT-PCR followed by sequencing in this case using FFPE. However, RNAseq of the mRNA on the FF tissue in the current study did not detect any EML4-ALK gene rearrangement and only demonstrated a minimal AAK-ALK mRNA expression. Rather, we identified a strong mRNA expression of the EML4-BIRC6 fusion gene (Figure 2A), which was not available at that time. As was shown in our previous study21, RNAseq in this study analyses supported EGFR del L747-S752 in exon 19 (Figure 2C). Given the questionable state of EML4-ALK gene rearrangement as well as the lack of ALK protein expression (i.e. IHC-by both ALK1 and D5F3 clones), EGFR mutation was most likely the driver mutation for the adenocarcinoma in this case.
As controls, we examined 26 concordant (16 FISH+/IHC+ and 10 FISH−/IHC−) cases by targeted gene capture-based NGS approach. The results of concordant ALK positive cases (FISH+/IHC+) were confirmed by NGS except one case that did not show any fusion by gene capture. This case had a borderline FISH positivity (15%). The original ALK IHC staining with ALK1 clone was also weakly positive (score 2) and a repeat IHC with D5F3 clone was negative. Thus, the original results of both FISH and IHC were likely false positive in this case, which would explain the absence of identifiable ALK fusion gene on NGS. Moreover, this case has been tested for KRAS gene and showed mutation in G12D (data not shown), which further supports the negative NGS result as ALK gene rearrangement and KRAS mutation are known to be mutually exclusive. NGS confirmed the results of all concordant ALK negative control cases (FISH−/IHC−).
Our study demonstrated the potential of NGS using archival FFPE samples as a diagnostic tool in detecting rearrangement involving multiple genes. We used two bioinformatics approaches by CREST and SVAtools to detect rearrangement(s) involving multiple genes with more detailed and comprehensive information as to the identity and the orientation of gene rearrangement. While independently analyzed, both methods were highly concordant and provided us with high confidence on discordant as well as concordant cases for ALK status by FISH and IHC.
However, the gene capture methods may still have false calls or missed rearrangements that are present at low frequencies and could not be distinguished from potential fusion artifacts, low level contamination, or background noise associated with NGS library prep and passing the filters set forth by the bioinformatics calling algorithms as might have been the case in one of our FISH+/IHC− case (case 891A); on examination of IGV of RNAseq, there might be some expressions in the exon 20 of ALK fused with EML4 (in addition to the exon 16 of ALK fused with AAK), which both bioinformatics in the present study might have failed to detect. The heterogeneity of mutation status within the same tumor sample may also present challenges to genomic based analysis32.
Finally, it is worth noting that our gene-capture approach identified fusions associated with genomic rearrangements for the genes of interest, regardless of their ability to give rise to an overexpressed oncogene. While this approach requires further evaluation in a much larger cohort of samples, the accompanying genomic rearrangements we identified in this study provided additional clues to enable more sensitive and specific assessment of the fusion status in a clinical sample. Among the cases with genomic evidence supporting the EML4->ALK gene fusions, we observed the reciprocal fusion ALK->EML4 fusion in four cases (Table 2). In one ALK FISH+/IHC+ case, however, gene capture only observed fusions involving ALK-> EML4 and/or <-ALKEML4-> but not the EML4->ALK fusion; the genomic evidence from the other four cases with both EML4->ALK and the reciprocal fusion ALK->EML4 fusions suggests that this case is probably also ALK positive. Unfortunately, the patients in our historic cohort have not been treated with ALK inhibitors, which did not allow us to evaluate its biologic implication as an oncogenic driver or relation to responsiveness to the therapy.
By NGS approach, we found several other oncogene fusions that have not yet been reported or only recently identified. We found 1 of 16 ALK FISH+/IHC+ tumors harboring the Huntington-interacting protein-1 (HIP1)-ALK fusion gene, which has been recently identified as a new fusion partner of ALK in non-small cell lung cancer33–35, adding to the four currently known fusion partners (EML-4, KIF5B, TFG, KLC1). Both CREST and SVAtools identified a CD74-ROS1 fusion among the 12 ALK FISH−/IHC+ group and an EZR-ROS1 fusion among the 10 FISH−/IHC− negative group. Finally, we observed an SND1-BRAF fusion in three cases, one of which also showed an EML4-ALK fusion gene. A previous study also has reported an SND1-BRAF fusion along with other driver gene mutations including EGFR and ERBB2.36
Taken together, our genomic study demonstrated that our 3 ALK FISH+ (all at borderline 15–18%)/ IHC− cases were likely false positive by FISH. Both gene capture with NGS and RNAseq methods demonstrated that the remaining FISH+/IHC− case harbored AAK1-ALK and EML4-BIRC6 fusions, both of which have not been documented in the literature. While requiring further evaluation using a much larger cohort of samples, our DNA based genomic approach using hybrid gene capture using FFPE tissues could be a powerful tool to detect ALK gene rearrangement as well as other molecular alterations of multiple genes as a single test.
Supplementary Material
Acknowledgments
Supported in part by Investigator-Initiated Research grant by Pfizer, Inc. New York, NY, Center for Individualized Medicine, Mayo Clinic, and the Hillsberg Award for Cancer Research, National Foundation for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: No conflict of interest
References
- 1.Chihara D, Suzuki R. More on crizotinib. The New England journal of medicine. 2011;364:776–7. doi: 10.1056/NEJMc1013325. author reply 778. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida A, et al. Bright-field dual-color chromogenic in situ hybridization for diagnosing echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase-positive lung adenocarcinomas. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2011;6:1677–86. doi: 10.1097/JTO.0b013e3182286d25. [DOI] [PubMed] [Google Scholar]
- 3.Murakami Y, Mitsudomi T, Yatabe Y. A Screening Method for the ALK Fusion Gene in NSCLC. Frontiers in oncology. 2012;2:24. doi: 10.3389/fonc.2012.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peled N, et al. Next-generation sequencing identifies and immunohistochemistry confirms a novel crizotinib-sensitive ALK rearrangement in a patient with metastatic non-small-cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2012;7:e14–6. doi: 10.1097/JTO.0b013e3182614ab5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pekar-Zlotin M, et al. Fluorescence in situ hybridization, immunohistochemistry, and next-generation sequencing for detection of EML4-ALK rearrangement in lung cancer. The oncologist. 2015;20:316–22. doi: 10.1634/theoncologist.2014-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li T, Kung HJ, Mack PC, Gandara DR. Genotyping and genomic profiling of non-small-cell lung cancer: implications for current and future therapies. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31:1039–49. doi: 10.1200/JCO.2012.45.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng Z, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nature medicine. 2014;20:1479–84. doi: 10.1038/nm.3729. [DOI] [PubMed] [Google Scholar]
- 8.Ross JS, Cronin M. Whole cancer genome sequencing by next-generation methods. American journal of clinical pathology. 2011;136:527–39. doi: 10.1309/AJCPR1SVT1VHUGXW. [DOI] [PubMed] [Google Scholar]
- 9.Lipson D, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nature medicine. 2012;18:382–4. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeuchi K, et al. RET, ROS1 and ALK fusions in lung cancer. Nature medicine. 2012;18:378–81. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 11.Yi ES, et al. Correlation of IHC and FISH for ALK gene rearrangement in non-small cell lung carcinoma: IHC score algorithm for FISH. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2011;6:459–65. doi: 10.1097/JTO.0b013e318209edb9. [DOI] [PubMed] [Google Scholar]
- 12.Asmann YW, et al. A novel bioinformatics pipeline for identification and characterization of fusion transcripts in breast cancer and normal cell lines. Nucleic acids research. 2011;39:e100. doi: 10.1093/nar/gkr362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Novocraft. Novoalign: Powerful tool designed for mapping of short reads onto a reference genome from Illumina, Ion Torrent, and 454 NGS platforms. 2014 2.08.01 edn. [Google Scholar]
- 15.Institute, B., editor. BroadInstitute. Picard: A set of tools (in Java) for working with next generation sequencing data in the BAM format. 2014. 1.96 edn. [Google Scholar]
- 16.Wang J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods. 2011;8:652–4. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karolchik D, Hinrichs AS, Kent WJ. The UCSC Genome Browser. Curr Protoc Bioinformatics. 2012;4 doi: 10.1002/0471250953.bi0104s40. Chapter 1, Unit1. [DOI] [PubMed] [Google Scholar]
- 18.Drucker TM, et al. BIMA V3: an aligner customized for mate pair library sequencing. Bioinformatics. 2014;30:1627–9. doi: 10.1093/bioinformatics/btu078. [DOI] [PubMed] [Google Scholar]
- 19.Murphy SJ, et al. Identification of independent primary tumors and intrapulmonary metastases using DNA rearrangements in non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32:4050–8. doi: 10.1200/JCO.2014.56.7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vasmatzis G, et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood. 2012;120:2280–9. doi: 10.1182/blood-2012-03-419937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boland JM, et al. MET and EGFR mutations identified in ALK-rearranged pulmonary adenocarcinoma: molecular analysis of 25 ALK-positive cases. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2013;8:574–81. doi: 10.1097/JTO.0b013e318287c395. [DOI] [PubMed] [Google Scholar]
- 22.Lantuejoul S, et al. French multicentric validation of ALK rearrangement diagnostic in 547 lung adenocarcinomas. The European respiratory journal. 2015;46:207–18. doi: 10.1183/09031936.00119914. [DOI] [PubMed] [Google Scholar]
- 23.von Laffert M, et al. ALK-FISH borderline cases in non-small cell lung cancer: Implications for diagnostics and clinical decision making. Lung cancer. 2015;90:465–71. doi: 10.1016/j.lungcan.2015.09.022. [DOI] [PubMed] [Google Scholar]
- 24.von Laffert M, et al. Multicenter immunohistochemical ALK-testing of non-small-cell lung cancer shows high concordance after harmonization of techniques and interpretation criteria. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2014;9:1685–92. doi: 10.1097/JTO.0000000000000332. [DOI] [PubMed] [Google Scholar]
- 25.Kim H, et al. Detection of ALK gene rearrangement in non-small cell lung cancer: a comparison of fluorescence in situ hybridization and chromogenic in situ hybridization with correlation of ALK protein expression. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2011;6:1359–66. doi: 10.1097/JTO.0b013e31821cfc73. [DOI] [PubMed] [Google Scholar]
- 26.McLeer-Florin A, et al. Dual IHC and FISH testing for ALK gene rearrangement in lung adenocarcinomas in a routine practice: a French study. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2012;7:348–54. doi: 10.1097/JTO.0b013e3182381535. [DOI] [PubMed] [Google Scholar]
- 27.Mino-Kenudson M, et al. A novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistry. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:1561–71. doi: 10.1158/1078-0432.CCR-09-2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinez P, et al. Fluorescence in situ hybridization and immunohistochemistry as diagnostic methods for ALK positive non-small cell lung cancer patients. PloS one. 2013;8:e52261. doi: 10.1371/journal.pone.0052261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paik JH, et al. Clinicopathologic implication of ALK rearrangement in surgically resected lung cancer: a proposal of diagnostic algorithm for ALK-rearranged adenocarcinoma. Lung cancer. 2012;76:403–9. doi: 10.1016/j.lungcan.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 30.Sholl LM, et al. Combined use of ALK immunohistochemistry and FISH for optimal detection of ALK-rearranged lung adenocarcinomas. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2013;8:322–8. doi: 10.1097/JTO.0b013e31827db604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minca EC, et al. ALK status testing in non-small cell lung carcinoma: correlation between ultrasensitive IHC and FISH. The Journal of molecular diagnostics : JMD. 2013;15:341–6. doi: 10.1016/j.jmoldx.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 32.Shaw AT, et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. The New England journal of medicine. 2016;374:54–61. doi: 10.1056/NEJMoa1508887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang DD, et al. HIP1-ALK, a novel ALK fusion variant that responds to crizotinib. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2014;9:285–94. doi: 10.1097/JTO.0000000000000087. [DOI] [PubMed] [Google Scholar]
- 34.Hong M, et al. HIP1-ALK, a novel fusion protein identified in lung adenocarcinoma. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2014;9:419–22. doi: 10.1097/JTO.0000000000000061. [DOI] [PubMed] [Google Scholar]
- 35.Ou SH, et al. Identification of a novel HIP1-ALK fusion variant in Non-Small-Cell Lung Cancer (NSCLC) and discovery of ALK I1171 (I1171N/S) mutations in two ALK-rearranged NSCLC patients with resistance to Alectinib. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2014;9:1821–5. doi: 10.1097/JTO.0000000000000368. [DOI] [PubMed] [Google Scholar]
- 36.Jang JS, et al. Common Oncogene Mutations and Novel SND1-BRAF Transcript Fusion in Lung Adenocarcinoma from Never Smokers. Scientific reports. 2015;5:9755. doi: 10.1038/srep09755. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.