

Abstract
Background and Purpose
Phytocannabinoids are produced in Cannabis sativa L. in acidic form and are decarboxylated upon heating, processing and storage. While the biological effects of decarboxylated cannabinoids such as Δ9‐tetrahydrocannabinol have been extensively investigated, the bioactivity of Δ9‐tetahydrocannabinol acid (Δ9‐THCA) is largely unknown, despite its occurrence in different Cannabis preparations. Here we have assessed possible neuroprotective actions of Δ9‐THCA through modulation of PPARγ pathways.
Experimental Approach
The effects of six phytocannabinoids on PPARγ binding and transcriptional activity were investigated. The effect of Δ9‐THCA on mitochondrial biogenesis and PPARγ coactivator 1‐α expression was investigated in Neuro‐2a (N2a) cells. The neuroprotective effect was analysed in STHdh Q111/Q111 cells expressing a mutated form of the huntingtin protein and in N2a cells infected with an adenovirus carrying human huntingtin containing 94 polyQ repeats (mHtt‐q94). The in vivo neuroprotective activity of Δ9‐THCA was investigated in mice intoxicated with the mitochondrial toxin 3‐nitropropionic acid (3‐NPA).
Key Results
Cannabinoid acids bind and activate PPARγ with higher potency than their decarboxylated products. Δ9‐THCA increased mitochondrial mass in neuroblastoma N2a cells and prevented cytotoxicity induced by serum deprivation in STHdh Q111/Q111 cells and by mutHtt‐q94 in N2a cells. Δ9‐THCA, through a PPARγ‐dependent pathway, was neuroprotective in mice treated with 3‐NPA, improving motor deficits and preventing striatal degeneration. In addition, Δ9‐THCA attenuated microgliosis, astrogliosis and up‐regulation of proinflammatory markers induced by 3‐NPA.
Conclusions and Implications
Δ9‐THCA shows potent neuroprotective activity, which is worth considering for the treatment of Huntington's disease and possibly other neurodegenerative and neuroinflammatory diseases.
Abbreviations
- 3‐NPA
3‐nitropropionic acid
- CBD
cannabidiol
- CBDA
cannabidiol acid
- CBG
cannabigerol
- CBGA
cannabigerol acid
- GFAP
glial fibrillary acidic protein
- HD
Huntington's disease
- mHtt
mutant huntingtin
- MTT
3‐(4,5‐dimethyl‐2‐thiazolyl)‐2,5‐diphenyl‐2H‐tetrazolium bromide
- PGC1‐α
PPARγ coactivator 1‐α
- SPPARM
selective PPARγ modulator
- Δ9‐THC
Δ9‐tetahydrocannabinol
- Δ9‐THCA
Δ9‐tetahydrocannabinol acid
Introduction
The first phytocannabinoid (cannabinol) was isolated from Cannabis in 1896 (Wodd et al., 1896), but more than 50 years elapsed before it was realized that these compounds are produced and stored in the plant as their acidic precursors (cannabinoid acids or pre‐cannabinoids) (Krejci and Santavy, 1955). Decarboxylation requires heating, but can take place also at room temperature upon prolonged storage of Cannabis (Wang et al., 2016). Interestingly, decarboxylation does not take place to an appreciable extent after absorption, and this observation has found application in forensic science to distinguish between the presence of Δ9‐tetrahydrocannabinol (Δ9‐THC) following recreational consumption of marijuana products and that following the medicinal use of dronabinol (Marinol®), a formulation of Δ9‐THC in sesame oil. Surpisingly, the acidic precursor of Δ9‐THC, Δ9‐tetahydrocannabinol acid (Δ9‐THCA) is not psychotropic, and its binding to cannabinoid receptors is still a matter of debate (Ahmed et al., 2008; Rosenthaler et al., 2014). Our interest in native phytocannabinoids was particularly taken by this separation between the narcotic and the molecular properties of Δ9‐THCA and by the discovery that amorfrutins, a series of phenethyl‐type phytocannabinoids from liverworts and some leguminous plants, potently modulate the activity of PPARγ. Moreover, carboxylated amorfrutins are more potent PPARγ agonists than their decarboxylated neutral analogues (Fuhr et al., 2015).
PPARγ, a nuclear receptor, is a master regulator of lipid metabolism and glucose homeostasis (Tontonoz and Spiegelman, 2008). However, PPARγ is expressed in many different tissues and cell types and plays a key role in inflammatory processes and neurodegenerative diseases including Huntington's disease (HD) (Quintanilla et al., 2014). In this sense, it has been shown that glitazones, a class of PPARγ ligands used as anti‐diabetic drugs, are neuroprotective in mutant huntingtin (mHtt)‐expressing cells, reduce mHtt aggregates in the brain, protect from mHtt‐induced striatal neurodegeneration, attenuate neuroinflammation and decrease oxidative damage (Chiang et al., 2012, 2015; Jin et al., 2013), thus supporting the concept that PPARγ may be a valid target for the management of HD (Skerrett et al., 2014). Moreover, the cannabigerol (CBG) derivative VCE‐003.2 exerted a pro‐survival action in progenitor cells during neuronal differentiation through a PPARγ‐dependent pathway. This synthetic cannabinoid also prevented the loss of medium spiny neurones in Huntington's‐like disease models in mice, improving motor deficits, reactive astrogliosis and microglial activation (Diaz‐Alonso et al., 2016). It has also been suggested that an impaired activity of PPARγ coactivator‐1α (PGC‐1α), a transcriptional master coregulator of mitochondrial biogenesis and cellular metabolism, may be a pathological factor causing mitochondrial dysfunction in HD (Johri et al., 2013). Taken together, these studies support the view that PPARγ agonists may have beneficial effects on mitochondrial dysfunction, contributing to the prevention of neurodegeneration in HD (Skerrett et al., 2014; Agarwal et al., 2017).
Here, we have compared the three major phytocannabinoids from Cannabis [Δ9‐THC, cannabidiol (CBD) and CBG] and their corresponding acidic precursors [Δ9‐THCA, cannabidiol acid (CBDA) and cannabigerol acid (CBGA), respectively] as agonists of PPARγ. Our results have highlighted the therapeutic potential of Δ9‐THCA and botanical preparations containing acidic cannabinoids for the treatment of HD disease and possibly other neurodegenerative, metabolic and inflammatory diseases.
Methods
Cannabinoids and botanical preparations
Δ9‐THC and Δ9‐THCA were purified from the Cannabis variety MONIEK (CPVO/20160114), and CBDA was purified from the variety SARA (CPVO/20150098) using a countercurrent chromatography. CBD was also purified from SARA variety and CBG and CBGA from the variety AIDA (CPVO/20160167) following a method described previously (Nadal, 2016). All the cannabinoids have a purity >95%. An extract containing acidic cannabinoids was prepared from the variety MONIEK (100 g dry weight) by n‐hexane extraction (1 × 1 L and 2 × 0.75 L), filtration and evaporation. A portion of the extract was decarboxylated in an oven at 120°C for 1 h to obtain the corresponding extract based on neutral cannabinoids. The content of cannabinoids was evaluated by GC on an Agilent 7890B GC apparatus interfaced with a 5977B mass selective detector. The latter was equipped with a 15 m × 0.25 mm i.d. Rxi‐35Sil_MS capillary column (0.25 μm film thickness). For the simultaneous measure of neutral and acidic cannabinoids, a derivatization process was carried out. Thus, aliquots of the hexane extracts were transferred to a clean tube, evaporated to dryness and then derivatized with bis(trimethylsilyl)trifluoroacetamide containing 2% trimethylchlorosilane (TMCS) at 70°C for 60 min. After cooling to room temperature, the TMCS derivatives were analysed by GC‐MS. The cannabinoid content in both extracts is shown in Table 1. Cannabinoids were dissolved in DMSO to provide stock solutions of 50 mM and were stored at −80°C.
Table 1.
Analysis of cannabinoid content in MONIEK extracts before and after decarboxylation
| MONIEK | Decarboxylated MONIEK | |
|---|---|---|
| CBDA | 5.35 ± 0.00 | N.D. |
| CBD | N.D. | N.D. |
| CBGA | 8.08 ± 0.02 | N.D. |
| CBG | 1.34 ± 0.00 | 2.12 ± 0.11 |
| Δ9‐THCA | 64.0 ± 0.65 | 7.99 ± 0.01 |
| Δ9‐THC | 4.00 ± 0.01 | 62.3 ± 0.7 |
Data represent the percentage of the content of cannabinoids in the dry extract (w/w). N.D., not detected.
Cell lines
HEK‐293T, Neuro‐2a (N2a) (ATCC, Manassas, VA, USA), STHdh Q7/Q7 and STHdh Q111/Q111 (Prof. Javier Fernandez‐Ruiz, Universidad Complutense de Madrid, Spain) cells were cultured in DMEM supplemented with 10% FBS, 2 mM l‐glutamine and 1% (v/v) penicillin/streptomycin. HEK‐293T and N2a cells were maintained at 37°C in a humidified atmosphere containing 5% CO2 and STHdh Q7/Q7 and STHdh Q111/Q111 cells, which express either a wild‐type or a mutated form of the huntingtin protein, were cultured at 33°C (Trettel et al., 2000).
PPARγ binding and transcriptional assays
PPARγ binding activity was studied using the PolarScreen™ PPARγ Competitor Assay kit (Life Technologies, Carlsbad, CA, USA). Experiments were performed in triplicate and IC50 values were calculated using GraphPad Prism. K i values were calculated using a previously described web‐server tool to calculate K i values from IC50 values given the total receptor and ligand concentrations and the K D of the target‐ligand reaction (Cer et al., 2009). To investigate PPARγ transcriptional activity, HEK‐293T cells were seeded in 24‐well plates and transiently co‐transfected with the expression vector GAL4‐PPARγ and the luciferase reporter vectors GAL4‐luc (firefly luciferase) and pRL‐CMV (Renilla luciferase) using Roti©‐Fect (Carl Roth, Karlsruhe, Germany) following the manufacturer's instructions. After stimulation, the luciferase activities were quantified using the Dual‐Luciferase Assay (Promega, Madison, WI, USA).
Western blots
Cells were seeded at 2 × 105 cells per well in 60 mm plates and, after 24 h, treated with the indicated concentrations of the compounds for 6 h. Then, cells were washed with PBS, and proteins extracted in 50 μL of lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 10% glycerol and 1% NP‐40) supplemented with 10 mM NaF, 1 mM Na3VO4, 10 μg·mL−1 leupeptin, 1 μg·mL−1 pepstatin and aprotinin and 1 μL·mL−1 saturated PMSF. Protein concentration was determined by the Bradford assay (Bio‐Rad, Hercules, CA, USA), and samples (30 μg protein) were boiled at 95°C in Laemmli buffer and analysed by electrophoresis in 10% SDS/PAGE gels. Separated proteins were transferred to PVDF membranes (20 V for 30 min), and after blocking with non‐fat milk or BSA in TBST buffer, primary antibodies were added. The washed membranes were incubated with appropriate secondary antibodies coupled to HRP that were detected by an enhanced chemiluminescence system (USB Corporation, Cleveland, OH, USA). Rabbit antibody against PPARγ (C26H12) and mouse anti‐β‐actin antibody (AC‐74) were obtained from Cell Signalling Technology (Beverly, MA, USA) and Sigma‐Aldrich respectively.
Determination of mitochondrial biogenesis
N2a cells were seeded in 96‐well plates (3.5 × 103 cells per well), and after 24 h, stimulated in quadruplicate wells with Δ9‐THC or Δ9‐THCA at the indicated concentrations for 72 h. Rosiglitazone (10 μM) was used as positive control. Then, Mitotracker Green (100 nM; Thermo Fisher Scientific, Waltham, MA, USA) was added to culture medium for 30 min. Cells were washed with PBS, and fresh culture medium was added. Images were taken, and fluorescence was measured using the cell imaging system IncuCyte HD (Essen BioScience, Inc., Hertfordshire, UK).
Striatal neuroprotection in vitro
STHdh Q7/Q7 and STHdh Q111/Q111 cells (104 cells per well) were seeded in DMEM supplemented with 10% FBS in 96‐well plates, and after 24 h, the culture medium was changed to DMEM containing 0.5% FBS for 4 h (serum deprived). Then, Δ9‐THC or Δ9‐THCA was added to culture medium in the absence or the presence of 5 μM GW9662, a PPARγ antagonist for 48 h. Cell viability was measured by 3‐(4,5‐dimethyl‐2‐thiazolyl)‐2,5‐diphenyl‐2H‐tetrazolium bromide (MTT) assay. Briefly, 50 μL of 3‐MTT (5 mg·mL−1) from a mixture solution of MTT : DMEM (1:2) per well was added for 4 h at 33°C in darkness. Supernatants were removed, and 100 μL DMSO was added to each well. Absorbance was measured at 550 nm using a TriStar LB 941.
Animals
All animal care and experimental procedures were performed in accordance with European Union guideline and approved by the Animal Research Ethic Committee of Córdoba University and the Andalusian Committee for Animal Experimentation (2014PI/017). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). A total of 70 adult (16 weeks old), C57BL/6 male mice weighing between 23 and 25 g (Envigo, Valencia, Spain) were used in the studies. Mice were housed in the Animal Facilities of Córdoba University in groups of 5–6 in polycarbonate cages (300 × 180 × 150 mm) with access to food and water ad libitum. A 12 h light/dark cycle was maintained, with controlled temperature (20 ± 2°C) and relative humidity (40–50%).
Mouse model of striatal neurodegeneration
Systemic administration of 3‐nitropropionic acid (3‐NPA), an inhibitor of the mitochondrial complex II, results in a progressive locomotor deterioration and striatal degeneration resembling HD in several different mice strains (Borlongan et al., 1997). In our work, we had five experimental groups‐ control (PBS+vehicle); 3NPA (3NPA+vehicle); 3NPA+THCA; 3NPA+THCA+T0070907; THCA (PBS+THCA). Each group had 9 animals and there were no experimental losses. All treatments were given by i.p.injection of 100μL each. Striatal neurodegeneration was induced in C57BL/6 mice, by seven i.p. injections of 3‐NPA (50 mg·kg−1) every 12 h, over 4 days. Control mice received seven PBS injections. Vehicle (1:1:18 ethanol : Cremophor : saline) or Δ9‐THCA (20 mg·kg−1) was injected 30min before the PBS or 3NPA every 24h, over 4 days. The PPARγ antagonist T0070907 (5 mg·kg−1).was injected 15min before THCA, every 24h, over 4 days. Twelve hours after the last administration of 3‐NPA, behavioural analyses were carried out by measuring hindlimb clasping, hindlimb dystonia, truncal dystonia and general locomotor activity, as previously described (Fernagut et al., 2002). Each mice was given a score 0, 1 or 2 for each test, where 0 corresponds to normal behaviour and 2 with the maximum motor disorder. The analysis of symptomatology was carried out in a blinded manner by two independent observers. Animals were killed by cervical dislocation, and brains were removed. The right hemispheres were used to dissect the striatum to study mRNA expression for Tnf‐α, Inos, Il‐6 and Cox‐2. The other hemisphere was fixed in 4% formaldehyde for histological analysis.
Gene expression
N2a cells (105 cells per mL) were stimulated with Δ9‐THC, Δ9‐THCA or rosiglitazone for 72 h, and total RNA was extracted using the High Pure RNA Isolation Kit (Roche Diagnostic, Indianapolis, IN, USA). RNA was extracted from the striatum using the Qiagen RNeasy Lipid Kit (Qiagen, Hilden, Germany). Total RNA (1 μg) was retrotranscribed using the iScript cDNA Synthesis Kit, and the cDNA analysed by real‐time PCR using the iQTM SYBR Green Supermix and a CFX96 Real‐time PCR Detection System (Bio‐Rad). The HPRT gene was used to standardize mRNA expression in each sample. Gene expression was quantified using the 2−ΔΔCt method, and the percentage of relative expression against controls (untreated cells or mice) was calculated. The primers used in this study are described in Supporting Information Table S1.
Histological analysis
Brains were embedded in paraffin, and sections (5μm) cut and used for Nissl staining. Immunohistochemical analysis was performed to study activated microglia (Iba‐1+) or astrocytes [glial fibrillary acidic protein (GFAP)+], as described previously (Valdeolivas et al., 2015). A Leica DM2500 microscope and a LeicaDFC420c camera were used for slide observation and photography, and all image processing was done using ImageJ (National Institutes of Health, Bethesda, MD, USA). Multiple sections, selected from levels located approximately 200 μm from the middle of the lesion, were obtained from each brain and used to generate a mean value per mouse. All histological data were obtained in a blinded manner by two independent observers.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The in vitro data are shown as mean ± SD and in vivo results as mean ± SEM. Statistical analysis was performed in all the experiments shown using the SPSS v.19 software for Windows (IBM Corporation, NY, USA). Statistical analysis for multiple groups was performed by one‐way ANOVA followed by Tukey's post hoc test when F achieved P < 0.05, and there was no significant variance in homogeneity. Some results were normalized to control to avoid unwanted sources of variation. Such data were subjected to Kruskal–Wallis non‐parametric test followed by Dunn's post hoc test using the GraphPad Prism v.5 for Windows (GraphPad Software Inc, La Jolla, CA, USA). Statistical significance was set at P < 0.05.
Materials
Rosiglitazone was obtained from Cayman Chemical (Ann Arbor, MI, USA), and all other reagents were purchased from Sigma‐Aldrich (St. Louis, MO, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b)
Results
Cannabinoid acids bind and activate PPARγ
The main phytocannabinoid acids present in fresh Cannabis sativa L. plant material include Δ9‐THCA, CBDA and CBGA, which after decarboxylation generate their active neutral forms, Δ9‐THC, CBD and CBG, that are mainly found in processed plant material. As PPARγ is a potential target for some natural and synthetic cannabinoids, we first wanted to investigate whether neutral and acid cannabinoids were able to bind to PPARγ and compare their binding capacity with that of rosiglitazone. Using a PPARγ competitor‐binding assay, the cannabinoid acids outperformed the neutral cannabinoids in binding to the nuclear receptor. Δ9‐THCA was the most potent cannabinoid with an IC50 of 0.47 μM, in the same range as that of rosiglitazone (0.29 μM) (Figure 1). To further study the ability of these cannabinoids to activate PPARγ transcriptional activity, 293T cells were transfected with a pair of GAL4‐PPARγ/GAL4‐luc plasmids and stimulated with increasing concentrations of the compounds for 6 h. In this assay, Δ9‐THCA was more potent than Δ9‐THC (Figure 2A), and CBDA was more effective than CBD, but only at the higher concentrations (Figure 2B). In contrast, CBGA and CBG showed equal potency in activating PPARγ, indicating that there is not always a direct correlation between binding affinity and transcriptional activity (Figure 2C). Interestingly, a phytoextract of MONIEK, a Cannabis variety containing high concentrations of Δ9‐THCA, activated PPARγ in a concentration‐dependent manner, and this activity was greatly reduced after decarboxylation of the extract (Figure 2D).
Figure 1.
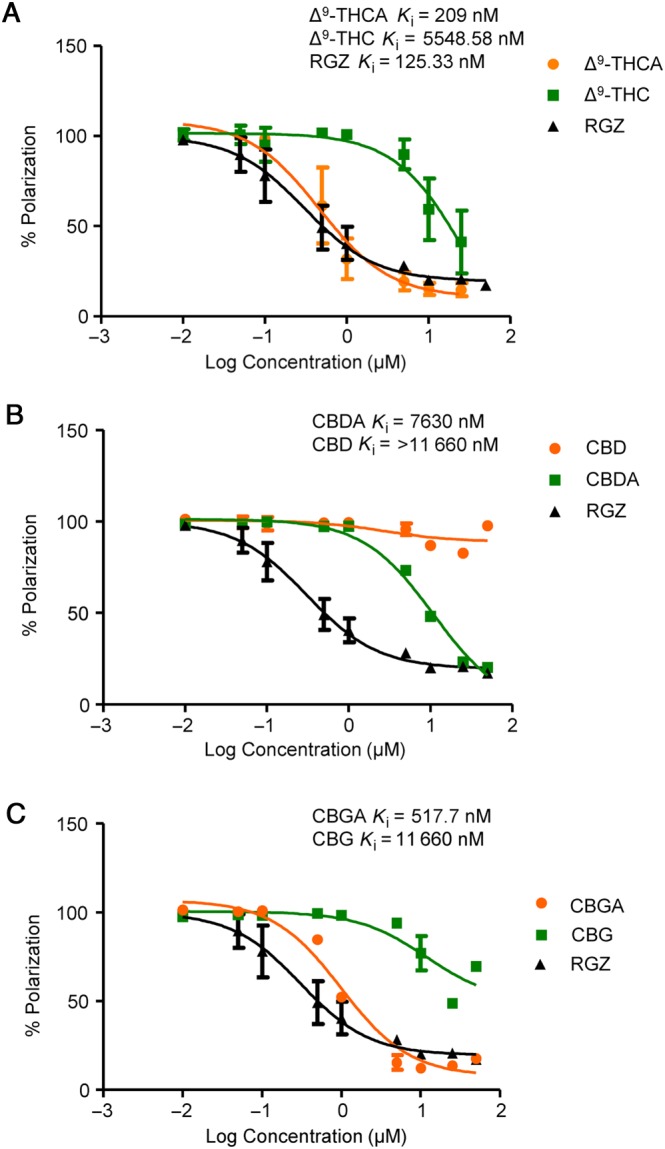
The carboxylic acid group of phytocannabinoids is critical for enhanced PPARγ binding. Cannabinoid binding affinities were tested at the indicated concentrations and compared with the binding affinity of rosiglitazone (RGZ). Data were transformed to a logarithmic function, and the K i values were calculated and are shown in the Figure (n = 5).
Figure 2.
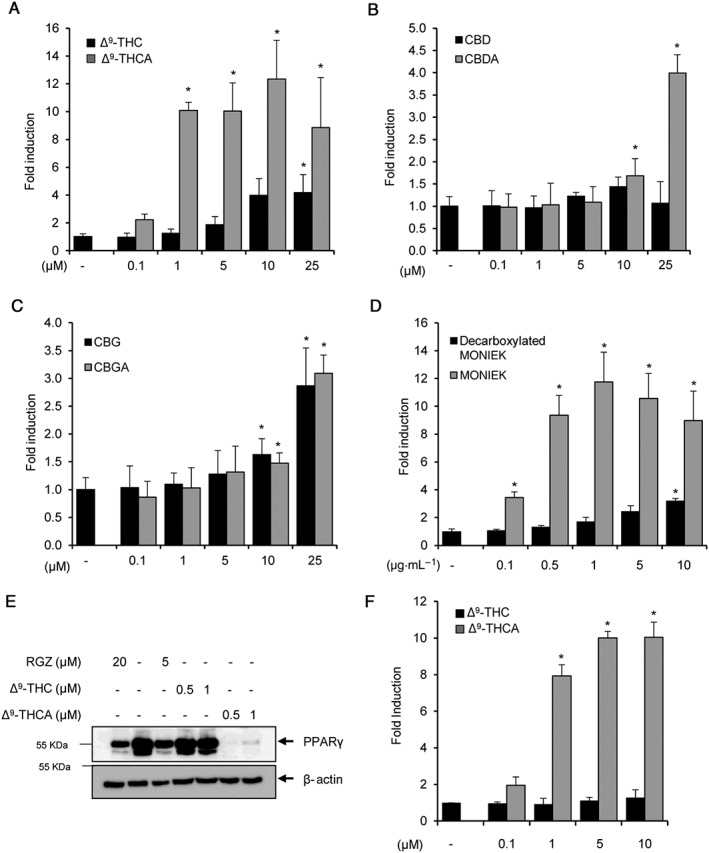
Cannabinoid acids induce PPARγ transcriptional activity and PPARγ degradation. (A–C) HEK‐293T cells were transiently transfected with PPARγ‐GAL4 plus GAL4‐luc and incubated with increasing concentrations of the indicated neutral and cannabinoid acids for 6 h (n = 5). (D) Transfected HEK‐293T cells were stimulated with two phytoextracts derived from the Cannabis variety MONIEK before and after decarboxylation (n = 5). (E) STHdh Q7/Q7 cells were treated with Δ9‐THCA, Δ9‐THC and rosiglitazone (RGZ) for 6 h, and the steady‐state levels of endogenous PPARγ and β‐actin detected by Western blots (n = 5). (F) STHdh Q7/Q7 cells were transiently transfected with PPARγ‐GAL4 plus GAL4‐luc and incubated with increasing concentrations of Δ9‐THCA or Δ9‐THC for 6 h (n = 5). * P < 0.05, significantly different from untreated cells.
PPARγ is known to suffer ligand‐induced degradation in the proteasome (Hauser et al., 2000; Kim et al., 2014). As shown in Figure 2E, Δ9‐THCA and rosiglitazone, but not Δ9‐THC, induced PPARγ degradation in STHdh striatal cells, and a similar effect was found with CBGA and CBDA (Supporting Information Figure S1), demonstrating that the cannabinoid acids also target endogenous PPARγ. We also confirmed that Δ9‐THCA induced PPARγ transcriptional activity in STHdh striatal cells (Figure 2F). Δ9‐THCA and rosiglitazone also induced PPARγ degradation in HEK293T cells showing that the effect of Δ9‐THCA on PPARγ is not cell‐type dependent (Supporting Information Figure S2).
To further analyse the effects of Δ9‐THCA at this nuclear receptor, we studied the behaviour of this compound in the presence of rosiglitazone, a full agonist of PPARγ (Lehmann et al., 1995). To achieve this, GAL4‐PPARγ/GAL4‐luc‐transfected HEK293 cells were pre‐incubated with increasing concentrations of Δ9‐THCA and then treated with 1 μM rosiglitazone. Under these conditions, Δ9‐THCA decreased the rosiglitazone‐induced PPARγ transactivation (Figure 3A), suggesting that Δ9‐THCA and rosiglitazone may bind to the same binding site on PPARγ. Next, to investigate the binding characteristics of Δ9‐THCA to PPARγ, the induction of PPARγ activity was studied in washout experiments where Δ9‐THCA was removed from the cell culture solution by washing the cells with PBS after 1 h of treatment and PPARγ activity was measured after 5 h cell culture in the absence of the compound. The results showed that activation of PPARγ by Δ9‐THCA was greatly reduced 5 h after removal of Δ9‐THCA from the cell medium, suggesting that binding of Δ9‐THCA binds to PPARγ in a reversible manner (Figure 3B).
Figure 3.
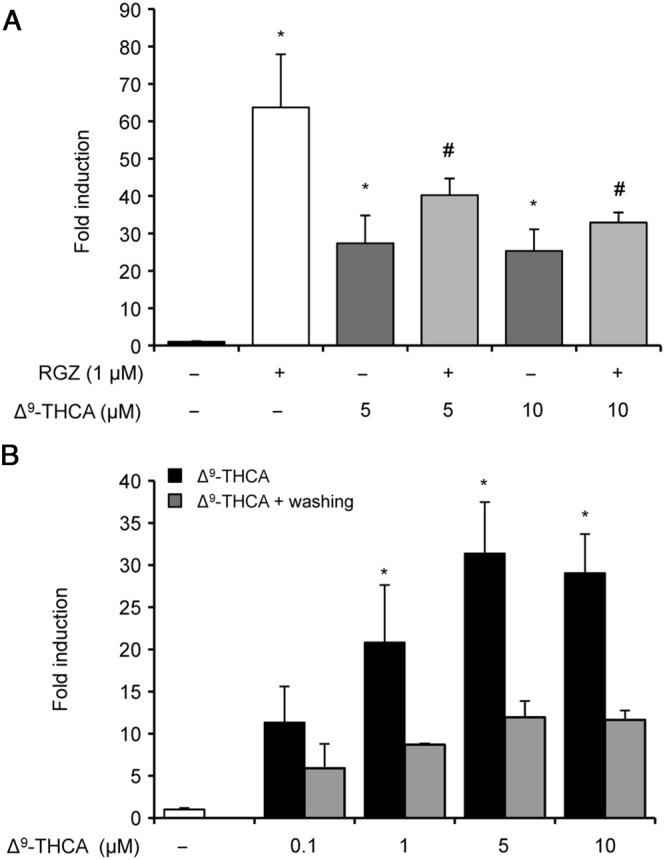
Δ9‐THCA competes with rosiglitazone and activates PPARγ in a reversible manner. HEK‐293T cells were transiently transfected with PPARγ‐GAL4 plus GAL4‐luc. (A) Cells were pretreated with Δ9‐THCA for 1 h and then incubated for 6 h in the presence or absence of rosiglitazone (RGZ) (n = 5). (B) Cells were pretreated with Δ9‐THCA for 1 h and then washed or not with PBS and incubated in complete medium for 6 h (n = 5). Cells were lysed and tested for luciferase activity. * P < 0.05, significantly different from untreated cells # P < 0.05, significantly different from rosiglitazone‐treated cells.
Effects of Δ9‐THCA on mitochondrial biogenesis and PGC‐1α expression
Ligands for PPARγ, such as rosiglitazone, increase mitochondrial biogenesis in neuronal cells (Chiang et al., 2015). Therefore, we carried out experiments to determine if Δ9‐THCA and Δ9‐THC could increase mitochondrial biogenesis. For these studies, N2a cells were incubated with either Δ9‐THCA, Δ9‐THC or rosiglitazone at the indicated concentrations for 72 h and then loaded with Mitotracker Green, which is a probe used to determine mitochondrial mass. The fluorescent intensity changes were recorded and analysed using the IncuCyte® ZOOM Software. Δ9‐THCA treatment induced a significant increase in mitochondrial mass levels, comparable with that induced by rosiglitazone (Figure 4A, B). In addition, Δ9‐THCA and rosiglitazone, but not Δ9‐THC, were able to up‐regulate the expression of PGC‐1α, a PPARγ‐interacting protein and potential HD target that plays a key role in mitochondrial biogenesis (Johri et al., 2013). Interestingly, Δ9‐THCA was more potent than rosiglitazone in inducing PGC‐1α expression (Figure 4C).
Figure 4.
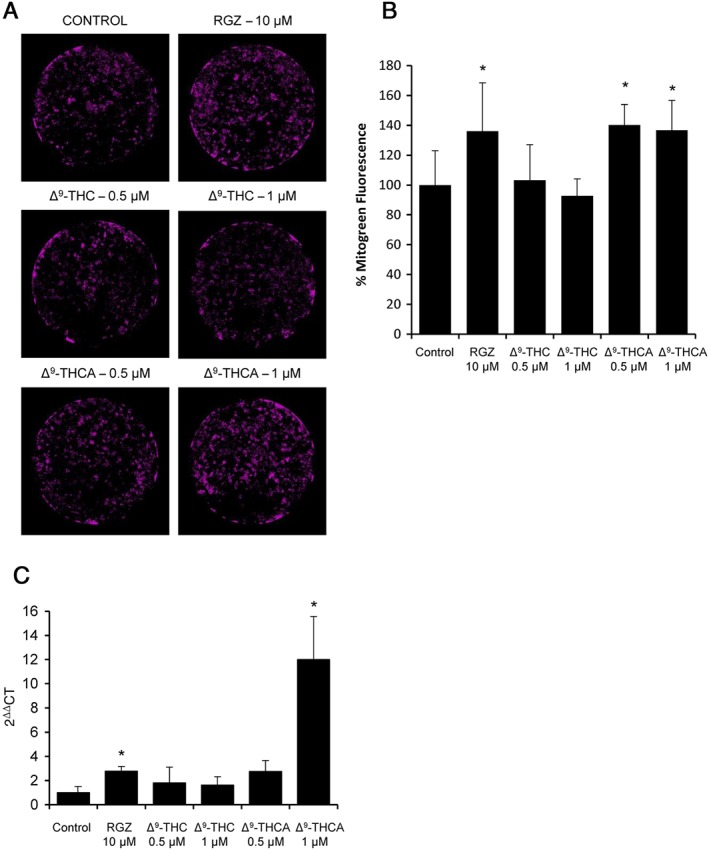
Δ9‐THCA increases mitochondrial biogenesis in N2a cells. (A) The cells were treated with Δ9‐THCA, Δ9‐THC and rosiglitazone (RGZ) for 72 h, and mitochondria stained with the Mitotracker Green dye (n = 5). (B) Quantification of fluorescence intensity (100% = control untreated cells). (C) Δ9‐THCA up‐regulated the expression of PGC‐1α. N2a cells were stimulated with rosiglitazone, Δ9‐THCA or Δ9‐THC, and the levels of PGC‐1α mRNA were analysed by qPCR (n = 5). * P < 0.05, significantly different from control.
Δ9‐THCA attenuates mHtt‐induced cytotoxicity in vitro
The mHtt protein bearing 111 glutamines in the N‐terminal domain (Q111/Q111) induces cytotoxicity depending on the cell type and culture conditions. In Figure 5A, we show that Q111/Q111 induced cytotoxicity under serum‐deprived conditions while Q7/Q7 cells were resistant to serum deprivation. Therefore, we analysed the effects of Δ9‐THCA and Δ9‐THC in STHdh Q111/Q111. We found that neuronal viability after serum deprivation was improved by Δ9‐THCA in STHdh Q111/Q111 cells, and this activity was attenuated in the presence of the PPARγ antagonist GW9662 (Figure 5B). To confirm these results in another cell model, we infected N2a cells with an adenovirus carrying human huntingtin containing 94 polyQ repeats (mHtt‐q94). We found that cytotoxicity induced by mHtt‐q94 was also significantly attenuated by treatment with Δ9‐THCA or Δ9‐THC (Supporting Information Figure S4).
Figure 5.
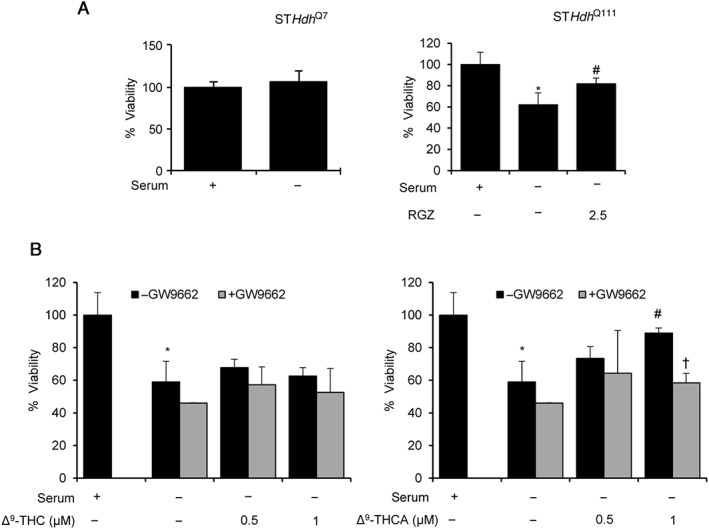
Δ9‐THCA prevents mutated huntingtin‐induced cytotoxicity via PPARγ. (A) Serum deprivation induces neuronal death in STHdh Q111/Q111 but not in STHdh Q7/Q7 cells. Cellular viability was measured by the MTT method (n = 5). (B) Δ9‐THCA and rosiglitazone (RGZ) prevent cell death induced by serum deprivation. STHdh Q111/Q111 cells were cultured under serum deprivation conditions and treated with Δ9‐THCA in the absence or the presence of the PPARγ antagonist GW9662 (5 μM). Cell viability was calculated using the MTT method and referred to control cells (n = 5). * P < 0.05, significantly different from untreated cells; # P < 0.05, significantly different from serum‐starved cells; † P < 0.05, significantly different from serum‐starved cells treated with Δ9‐THCA.
Treatment with Δ9‐THCA protects against striatal neurodegeneration in mice
Systemic administration of 3‐NPA, an irreversible inhibitor of respiratory chain complex II, leads to downstream processes of striatal neurodegeneration that mimic some clinical and pathological effects observed in the human disease (Borlongan et al., 1997). Administration of 3‐NPA results in motor deficits assessed as higher score in hindlimb clasping, hindlimb dystonia, locomotor activity and kyphosis tests compared with control mice. However, treatment with Δ9‐THCA during the development of the neurodegeneration with 3‐NPA, resulted in significant improvement of behavioural symptomatology including hindlimb dystonia, locomotor activity and kyphosis evaluation and a slight amelioration in the hindlimb clasping test (Figure 6A). In contrast, when mice were treated with a combination of Δ9‐THCA and the PPARγ antagonist T0070907, the beneficial effects of Δ9‐THCA were significantly inhibited. No differences in behavioural activity were observed after Δ9‐THCA treatment of control mice, receiving PBS instead of 3‐NPA.
Figure 6.
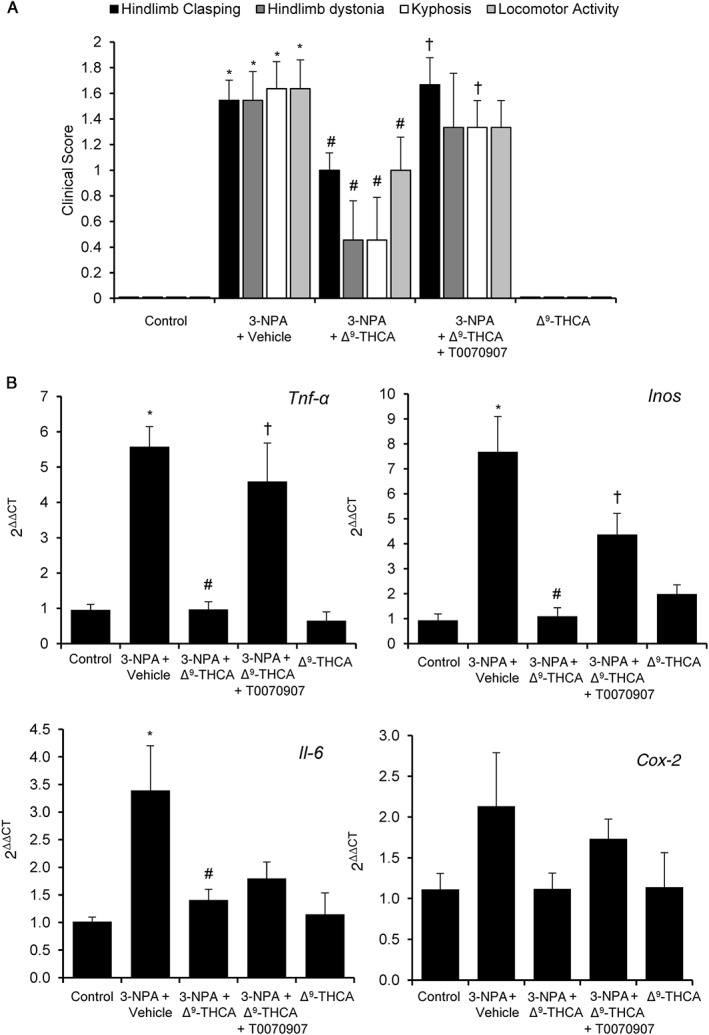
Δ9‐THCA is neuroprotective in 3‐NPA‐treated mice. (A) Behavioural score was determined 12 h after 3‐NPA injections. Mice were treated with Δ9‐THCA (20 mg·kg−1). Hindlimb clasping, general locomotor activity, hindlimb dystonia and kyphosis were measured, and values are expressed as means ± SEM (n = 9). (B) Δ9‐THCA down‐regulates the expression of inflammatory genes in mice brain. RNA was isolated from the striatum, retrotranscribed and analysed by real‐time PCR. Tnf‐α, Inos, Il‐6 and Cox‐2 gene were studied. * P < 0.05, significantly different from control group; # P < 0.05, significantly different from 3‐NPA only group; † P < 0.05, significantly different from 3‐NPA plus Δ9‐THCA group (n = 9 animals per group).
In addition, 3‐NPA‐lesioned mice showed an up‐regulation of the proinflammatory markers Tnf‐α, Inos and Il‐6 mRNAs in the striatum that was prevented by treatment with Δ9‐THCA. The effect of Δ9‐THCA on Tnf‐α and Inos mRNA expression was abolished in the presence of T0070907, but the PPARγ antagonist did not reverse the inhibitory effect of Δ9‐THCA on Il‐6 mRNA expression. On the other hand, Cox‐2 was not strongly induced by 3‐NPA, but nevertheless, this increased expression was also prevented by Δ9‐THCA (Figure 6B).
Finally, we investigated the effect of Δ9‐THCA in neuronal loss and gliosis induced by 3‐NPA. Histological examination by Nissl staining revealed that treatment with Δ9‐THCA significantly prevented striatal degeneration induced by 3‐NPA (Figure 7). GFAP immunostaining identifies reactive gliosis, an early marker of CNS damage in HD (Hedreen and Folstein, 1995), and we found that 3‐NPA induced a marked astrogliosis determined by GFAP staining and a less severe microgliosis revealed by Iba‐1 staining, which were prevented by Δ9‐THCA treatment (Figure 7B). Altogether, our results demonstrated that Δ9‐THCA reduced the neuroinflammatory status induced by with the injections of 3‐NPA.
Figure 7.
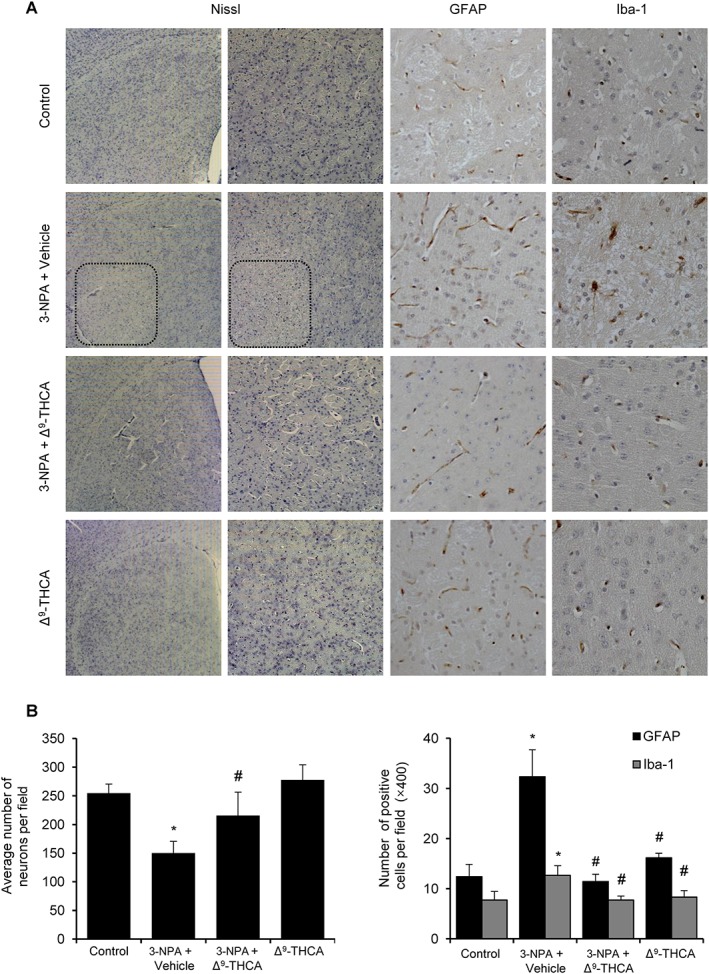
Δ9‐THCA prevents neuronal loss, microgliosis and astrogliosis induced by 3‐NPA administration. (A) Representative images of Nissl, Iba‐1 and GAFP staining performed on coronal striatal brain sections (original magnification 20×). (B) Quantification of the different markers was performed with ImageJ software. Total average number of neurons (Nissl), microglia (Iba‐1+) and astrocytes (GFAP+) is shown. Values are expressed as means ± SEM (n = 6). * P < 0.05, significantly different from control group; # P < 0.05, significantly different from 3‐NPA group (n = 9 animals per group).
Discussion
Over the past years, interest for the medical use of marijuana has grown exponentially, partly fuelled, however, by anecdotal information. To investigate the real potential of medicinal marijuana, preclinical and clinical studies have been launched, with positive results for some neurological conditions like multiple sclerosis and some genetic juvenile forms of epilepsy. All these studies have used the neutral cannabinoids, especially Δ9‐THC and CBD, with little attention to the genuine phytocannabinoids of the plant, namely, their acidic forms. We provide evidence that these compounds hold significant pharmacological potential, with Δ9‐THCA being a potent PPARγ agonist and showing neuroprotective and neuroinflammatory activity in an animal model of HD. Interestingly, other cannabinoid acids (CBDA and CBGA) also outperformed their corresponding neutral cannabinoids in terms of PPARγ binding, and a non‐decarboxylated botanical preparation of Cannabis also showed more potent PPARγ transcriptional activity than its decarboxylated version.
Administration of Δ9‐THCA and activation of cannabinoid CB1 receptors have already been shown to exert a neuroprotective action in different models of CNS diseases including HD (Blazquez et al., 2011; Fernández‐Ruiz et al., 2015; Basavargiappa et al., 2017). However, in other transgenic models of HD, treatment with CB1 receptor ligands, including Δ9‐THC, did not improve the progression of the pathology (Dowie et al., 2010). Moreover, a botanical preparation containing Δ9‐THC/CBD (1:1 ratio) failed to improve symptomatology in a recent Phase II clinical trial with HD patients (López‐Sendon Moreno et al., 2016). HD progression occurs concomitantly with an early decline of presynaptic CB1 receptors (McCaw et al., 2004), and therefore, targeting CB1 receptors may be a plausible therapeutic strategy in the initial stages of HD, to be later replaced by anti‐inflammatory drugs. In this regard, drugs targeting PPARγ, the nuclear receptor for some cannabinoids, have been shown to be beneficial by attenuating microglia inflammation and by modulating the peripheral adaptive immune response (Kim et al., 2015). In addition, preclinical evidence suggests that PPARγ ligands many exert beneficial effects in many CNS diseases such as amyotrophic lateral sclerosis, Parkinson's disease, Alzheimer's disease, HD, multiple sclerosis and stroke (Katsouri et al., 2012).
One important dysregulated gene in HD is that for PGC‐1α, a transcriptional co‐activator protein involved in energy homeostasis and adaptive thermogenesis. PGC‐1α mRNA levels are decreased in autopsy samples of human HD striatum, and striatal cell death in HD may be due to the altered energy metabolism and excitotoxicity induced by the aggregation of expanded Htt (Browne and Beal, 2004). Here, we have shown that Δ9‐THCA up‐regulated PGC‐1α mRNA expression and prevented mHtt‐induced cell death in two different cellular models. Thus, the protective effect of Δ9‐THCA could be mediated by restoring the energy metabolism in the target cells and by down‐regulating the expression of proinflammatory mediators that are commonly associated with HD (Rocha et al., 2016).
Δ9‐THCA is a non‐psychotropic cannabinoid, but its binding to CB1 receptors is still debated. While some authors showed that Δ9‐THCA binds CB1 receptors with a K i value of 23.5 nM (Rosenthaler et al., 2014), others had found a negligible activity (Ahmed et al., 2008). A possible explanation for these contrasting results could be the occurrence of decarboxylation during storage of the compound or under some experimental conditions. A recent study showed that freshly prepared and highly pure Δ9‐THCA (98%) has a low binding affinity for CB1 and CB2 receptors (McPartland et al., 2017). Thus, it seems that the biological activities of Δ9‐THCA are not mediated by interaction with these classical membrane receptors. In this context , Δ9‐THCA exhibited anti‐emetic and immunomodulatory activities through CB1‐dependent and CB1‐independent mechanisms respectively (Verhoeckx et al., 2006; Rock et al., 2013). The sample of Δ9‐THCA used in this study was 97% pure (Supporting Information Figure S4), and as its PPARγ binding and transcriptional activities were 20‐fold higher than those of Δ9‐THC, we suggest that PPARγ assays could be used to monitor Δ9‐THCA decarboxylation during storage, as an alternative to the chemical analysis.
We also showed that T0070907 prevented the neuroprotective effect of Δ9‐THCA in 3‐NPA‐lesioned mice. We suggest that Δ9‐THCA enters the CNS and that PPARγ is the major target responsible for the neuroprotective and anti‐inflammatory activity for this cannabinoid. Δ9‐THCA also showed neuroprotective activities in vitro (Moldzio et al., 2012), and there is anecdotal evidence that tinctures of Δ9‐THCA may have anti‐seizure activity (https://tokesignals.com/parents‐thca‐tincture‐works‐just‐as‐well‐as‐cbd‐for‐pediatric‐seizures‐heres‐how‐to‐make‐it/). PPARγ signalling has a role in neuroinflammation and epilepsy, and it is possible that the potential anti‐seizure activities of Δ9‐THCA and tinctures containing this cannabinoid, could be mediated by the modulation of this nuclear receptor (Chuang et al., 2012; Wong et al., 2015). Interestingly, non‐decarboxylated botanical preparations of medicinal marijuana may contain also high levels of other non‐psychotropic cannabinoid acids such as CBDA and CBGA that also target PPARγ.
The effect of cannabinoid acids on PPARγ is not without precedent. Δ9‐THC is metabolized in the body to produce the major, non‐psychotropic metabolite, THC‐11‐oic acid. Interestingly, ajulemic acid, a synthetic analogue of THC‐11‐oic acid, is a potent PPARγ agonist, suggesting that a COOH group is critical for the activation of the PPARγ pathway (Ambrosio et al., 2007). Furthermore, the formation of THC‐11‐oic acid after Δ9‐THC treatment could underlie the in vivo biological effects of Δ9‐THC mediated by the PPARγ pathway (Vara et al., 2013; Fishbein‐Kaminietsky et al., 2014).
PPARγ ligands include a wide array of natural and synthetic molecules among which the best characterized are the glitazones, a group of thiazolidinediones that have been extensively used in patients with Type 2 diabetes. The glitazones bind to the canonical ligand‐binding pocket (LPB) located within the nuclear receptor ligand‐binding domain of PPARγ and act as full agonists (Hughes et al., 2012). However, PPARγ ligands of this type have undesirable side effects like weight gain, oedema, liver injury, cancer and an increased risk of heart failure (Rosen, 2010). Furthermore, reduced bone mass and increased risk of peripheral fractures in thiazolidinedione‐treated patients are the results of inhibition of bone marrow osteoblastogenesis (Grey et al., 2007). Therefore, considerable research efforts have recently been modulators (SPPARMs), compounds that improve glucose homeostasis but elicit reduced side effects, because they are partial agonists at PPARγ, as shown by selective receptor–cofactor interactions and target gene regulation. Plant‐derived compounds represent a good source of SPPARMs that bind to the canonical ligand‐binding site and act as partial agonists, and Δ9‐THCA is another compound to add to this list of natural compounds (Wang et al., 2014). We have shown that Δ9‐THCA binds to purified PPARγ (K i = 209 nM), activates chimeric Gal4‐PPARγ‐dependent reporter gene expression as a partial agonist (with a maximal efficacy sixfold lower than rosiglitazone) and antagonizes the effect of rosiglitazone upon co‐treatment. Moreover, some of the activities of Δ9‐THCA are blocked by GW9662 and T0070907, which are synthetic irreversible PPARγ antagonists that covalently attach to Cys285 located within the LBP of PPARγ. Altogether, these data suggest that Δ9‐THCA binds to the canonical LPB in a reversible manner. However, a second functional binding site in the PPARγ LBP has been identified, and functional PPARγ agonists targeting this second site are not affected by GW9662, and it has been suggested that ligands targeting different binding sites mediate distinct biological responses (Hughes et al., 2014). We found that T0070907 did not prevent the effect of Δ9‐THCA on Il‐6 mRNA expression in vivo, and so, it is possible that Δ9‐THCA may also bind to alternative binding sites. In addition, Δ9‐THCA outperforms rosiglitazone in inducing PPARγ degradation, a mechanism that involves ubiquitination and degradation by the proteasome and serves to limit the PPARγ response to specific ligands (Hauser et al., 2000; Kim et al., 2014). Although the exact mechanism of action of Δ9‐THCA on the PPARγ pathway remains to be fully elucidated, this compound may qualify as a potentially safe SPPARM.
The limited shelf life of Δ9‐THCA will undoubtedly complicate its development, but the compound enjoys a remarkable stability in human fluids, even serving as a marker to distinguish the recreational use of marijuana from the medical use of the semi‐synthetic version of Δ9‐THC (Marinol) (Raikos et al., 2014). Such degradation of Δ9‐THCA might be prevented by binding to plasma proteins like albumin, and this could provide a clue in the development of stable formulations, as done, incidentally, also for of Δ9‐THC itself, a highly unstable compound in the pure state. Alternatively, Δ9‐THCA may serve as a scaffold to develop more stable analogues that retain its PPARγ agonist activity but are devoid of narcotic properties.
Taken together, the results of our study show that cannabinoid acids are more potent PPARγ agonists and transcriptional activators than their decarboxylated analogues. These data would strongly suggest Δ9‐THCA as a lead structure for the development of novel drugs for the management of HD and, possibly, other neurodegenerative and inflammatory diseases.
Author contributions
X.N., G.M., S.M., G.A. and E.M. contributed to the conception and design of the study. X.N., S.C., C.F.V. and C.S.C. performed the isolation of cannabinoids and phytoextracts. C.D.R., I.C., C.N., B.P. and M.L.B. performed the in vitro and in vivo experiments. X.N., C.D.R., G.A. and E.M. wrote the manuscript. All the authors contributed to the analysis and interpretation of data, critically reviewed and approved the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Cannabinoid acids induce higher PPARγ degradation compared to their decarboxylated forms. STHdh Q7/Q7 cells were treated with Δ9‐THCA‐A, CBG, CBGA, CBD or CBDA for 6 hours and the steady state levels of endogenous PPARγ and β‐actin detected by Western blots.
Figure S2 HEK‐293T cells were seeded at 1 x 104 in 60 mm dishes and 24 h later treated with RGZ or Δ9‐THCA for 6 hours and the steady state levels of endogenous PPARγ and β‐actin detected by Western blot.
Figure S3 Δ9‐THCA prevents mHtt‐CFP‐induced cell toxicity. A) N2a cells were seeded at 3.5 x103 in poly‐d‐lysine treated 96‐well plates. After 24 hours, cells were infected for 5 h with 1 μl/ml of adenovirus expressing CFP‐tagged human huntingtin exon 1 harboring a pathogenic polyQ tract of 94 CAG repeats. Then, medium was replaced by fresh medium and the cells treated with Δ9‐THCA or Δ9‐THC at the indicated doses and images were taken after 72 hours. A) Cell viability was determined using the IncuCyte HD software and non‐infected cells were considered as 100 % of cell viability. B) DAPI stained nuclei and expressions of CFP‐tagged mHttq94 protein were analyzed by confocal microscopy.
Figure S4 Isolated Δ9‐THCA analyzed by GC‐MS. Δ9‐THCA was diluted in 3 mL of hexane. An intermediate solution was prepared and 50 μL were taken and mixed with 20 uL of IS (CBD‐d3) and dried together in a concentrator. The dry residue was reconstituted in 15 μL of pyridine and 135 μL of derivative mixture BSTFA: TMCS (98:2 v/v) and incubated at 37 C for 1 hour. Crimp vials were used in order to avoid leakage. Δ9‐THCA purity was 97%.
Acknowledgements
This work was partially supported by the MINECO grant RTC‐2015‐3364 cofounded by the European Development Regional Fund in the Framework of the Operative Program ‘Reinforcement of research, technological development and innovation’ and by ICEX, España Exportación e Inversiones, programme INVEST IN SPAIN grant 201503487 to Phytoplant Research S.L. E.M. was also supported by the MINECO grant SAF2014‐53763‐P. B.P. was supported by the iPFIS programme fellowship (MINECO). We thank Dr José J. Lucas (Severo Ochoa Molecular Biology Center, Madrid, Spain) and Dr Andrea Ruiz (University Complutense of Madrid, Spain) for providing with adenovirus carrying mHtt‐q94.
Nadal, X. , del Río, C. , Casano, S. , Palomares, B. , Ferreiro‐Vera, C. , Navarrete, C. , Sánchez‐Carnerero, C. , Cantarero, I. , Bellido, M. L. , Meyer, S. , Morello, G. , Appendino, G. , and Muñoz, E. (2017) Tetrahydrocannabinolic acid is a potent PPARγ agonist with neuroprotective activity. British Journal of Pharmacology, 174: 4263–4276. doi: 10.1111/bph.14019.
References
- Agarwal S, Yadav A, Chaturvedi RK (2017). Peroxisome proliferator‐activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem Biophys Res Commun 483: 1166–1177. [DOI] [PubMed] [Google Scholar]
- Ahmed SA, Ross SA, Slade D, Radwan MM, Zulfiqar F, Matsumoto RR et al (2008). Cannabinoid ester constituents from high‐potency Cannabis sativa . J Nat Prod 71: 536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosio AL, Dias SM, Poliikarpov I, Zurier RB, Burstein SH, Garratt RC (2007). Ajulemic acid, a synthetic nonpsychoactive cannabinoid acid, bound to the ligand binding domain of the human peroxisome proliferator‐activated receptor gamma. J Biol Chem 282: 18625–18633. [DOI] [PubMed] [Google Scholar]
- Basavargiappa BS, Shivakumar M, Joshi V, Subbanna S (2017). Endocannabinoid system in neurodegenerative disorders. J Neurochem 160 (3): 480–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazquez C, Chiarlone A, Sagredo O, Aguado T, Pazos MR, Resel E et al (2011). Loss of striatal type 1 cannabinoid receptors is a key pathogenic factor in Huntington's disease. Brain 134: 119–136. [DOI] [PubMed] [Google Scholar]
- Borlongan CV, Nishino H, Sanberg PR (1997). Systemic, but not intraparenchymal, administration of 3‐nitropropionic acid mimics the neuropathology of Huntington's disease: a speculative explanation. Neurosci Res 28: 185–189. [DOI] [PubMed] [Google Scholar]
- Browne SE, Beal MF (2004). The energetics of Huntington's disease. Neurochem Res 29: 531–546. [DOI] [PubMed] [Google Scholar]
- Cer RZ, Mudunuri U, Stephens R, Lebeda FJ (2009). IC50‐to‐Ki: a web‐based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res 37: W441–W445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang MC, Cheng YC, Nicol CJ, Lin KH, Yen CH, Chen SJ et al (2015). Rosiglitazone activation of PPARgamma‐dependent signaling is neuroprotective in mutant huntingtin expressing cells. Exp Cell Res 338: 183–193. [DOI] [PubMed] [Google Scholar]
- Chiang MC, Chern Y, Huang RN (2012). PPARgamma rescue of the mitochondrial dysfunction in Huntington's disease. Neurobiol Dis 45: 322–328. [DOI] [PubMed] [Google Scholar]
- Chuang YC, Lin TK, Huang HY, Chang WN, Liou CW, Chen SD et al (2012). Peroxisome proliferator‐activated receptors gamma/mitochondrial uncoupling protein 2 signaling protects against seizure‐induced neuronal cell death in the hippocampus following experimental status epilepticus. J Neuroinflammation 9: 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz‐Alonso J, Paraiso‐Luna J, Navarrete C, del Río C, Cantarero I, Palomares B et al (2016). VCE‐003.2, a novel cannabigerol derivative, enhances neuronal progenitor cell survival and alleviates symptomatology in murine models of Huntington's disease. Sci Rep 6: 29789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowie MJ, Howard ML, Nicholson LF, Faull RL, Hannan AJ, Glass M (2010). Behavioural and molecular consequences of chronic cannabinoid treatment in Huntington's disease transgenic mice. Neuroscience 170: 324–336. [DOI] [PubMed] [Google Scholar]
- Fernagut PO, Diguet E, Stefanova N, Biran M, Wenning GK, Canioni P et al (2002). Subacute systemic 3‐nitropropionic acid intoxication induces a distinct motor disorder in adult C57Bl/6 mice: behavioural and histopathological characterisation. Neuroscience 114: 1005–1017. [DOI] [PubMed] [Google Scholar]
- Fernández‐Ruiz J, Moro MA, Martínez‐Orgado J (2015). Cannabinoids in neurodegenerative disorders and stroke/brain trauma: from preclinical models to clinical applications. Neurotherapeutics 12: 793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbein‐Kaminietsky M, Gafni M, Sarne Y (2014). Ultralow doses of cannabinoid drugs protect the mouse brain from inflammation‐induced cognitive damage. J Neurosci Res 92: 1669–1677. [DOI] [PubMed] [Google Scholar]
- Fuhr L, Rousseau M, Plauth A, Schroeder FC, Sauer S (2015). Amorfrutins are natural PPARgamma agonists with potent anti‐inflammatory properties. J Nat Prod 78: 1160–1164. [DOI] [PubMed] [Google Scholar]
- Grey A, Bolland M, Gamble G, Wattie D, Horne A, Davidson J et al (2007). The peroxisome proliferator‐activated receptor‐gamma agonist rosiglitazone decreases bone formation and bone mineral density in healthy postmenopausal women: a randomized, controlled trial. J Clin Endocrinol Metab 92: 1305–1310. [DOI] [PubMed] [Google Scholar]
- Hauser S, Adelmant G, Sarraf P, Wright HM, Mueller E, Spiegelman BM (2000). Degradation of the peroxisome proliferator‐activated receptor gamma is linked to ligand‐dependent activation. J Biol Chem 275: 18527–18533. [DOI] [PubMed] [Google Scholar]
- Hedreen JC, Folstein SE (1995). Early loss of neostriatal striosome neurons in Huntington's disease. J Neuropathol Exp Neurol 54: 105–120. [DOI] [PubMed] [Google Scholar]
- Hughes TS, Chalmers MJ, Novick S, Kuruvilla DS, Chang MR, Kamenecka TM et al (2012). Ligand and receptor dynamics contribute to the mechanism of graded PPARγ agonism. Structure 20: 139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TS, Giri PK, de Vera IM, Marciano DP, Kuruvilla DS, Shin Y et al (2014). An alternate binding site for PPARγ ligands. Nat Commun 5: 3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Albertz J, Guo Z, Peng Q, Rudow G, Troncoso JC et al (2013). Neuroprotective effects of PPAR‐gamma agonist rosiglitazone in N171‐82Q mouse model of Huntington's disease. J Neurochem 125: 410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johri A, Chandra A, Beal MF (2013). PGC‐1alpha, mitochondrial dysfunction, and Huntington's disease. Free Radic Biol Med 62: 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsouri L, Blondrath K, Sastre M (2012). Peroxisome proliferator‐activated receptor‐gamma cofactors in neurodegeneration. IUBMB Life 64: 958–964. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Park KW, Lee EW, Jang WA, Seo J, Shin S et al (2014). Suppression of PPARgamma through MKRN1‐mediated ubiquitination and degradation prevents adipocyte differentiation. Cell Death Differ 21: 594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Song J, Park KW (2015). The multifaceted factor peroxisome proliferator‐activated receptor gamma (PPARgamma) in metabolism, immunity, and cancer. Arch Pharm Res 38: 302–312. [DOI] [PubMed] [Google Scholar]
- Krejci Z, Santavy F (1955). Isolace dalších látek z listí indického konopí Cannabis sativa L. [Isolation of other substances from the leaves of the Indian hemp (Cannabis sativa L., varietas indica.)]. Acta Univ Palacki Olomuc Fac Med 6: 59–66. [Google Scholar]
- Lehmann JM, Moore LB, Smith‐Oliver TA, Wilkison WO, Willson TM, Kliewer SA (1995). An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator‐activated receptor gamma (PPAR gamma). J Biol Chem 270: 12953–12956. [DOI] [PubMed] [Google Scholar]
- López‐Sendon Moreno JL, García‐Caldentey J, Trigo‐Cubillo P, Ruiz Romero C, García Ribas G, Alonso Arias MA et al (2016). A double‐blind,randomized, cross‐over, placebo‐controlled, pilot trial with Sativex in Huntington's disease. J Neurol 263: 1390–1400. [DOI] [PubMed] [Google Scholar]
- McCaw EA, Hu H, Gomez GT, Hebb AL, Kelly ME, Denovan‐Wright EM (2004). Structure, expression and regulation of the cannabinoid receptor gene (CB1) in Huntington's disease transgenic mice. Eur J Biochem 271: 4909–4920. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPartland JM, McDonald C, Young M, Grant Phillip S, Furkert DP, Glass M (2017). Affinity and efficacy studies of tetrahydrocannabinolic acid A at cannabinoid receptor types one and two. Cannabis Cannabinoid Res 2: 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldzio R, Pacher T, Krewenka C, Kranner B, Novak J, Duvigneau JC et al (2012). Effects of cannabinoids delta(9)‐tetrahydrocannabinol, delta(9)‐tetrahydrocannabinolic acid and cannabidiol in MPP+ affected murine mesencephalic cultures. Phytomedicine 19: 819–824. [DOI] [PubMed] [Google Scholar]
- Nadal X (2016). Methods of purifying cannabinoids, compositions and kits thereof. US 20160214920 A1.
- Quintanilla RA, Utreras E, Cabezas‐Opazo FA (2014). Role of PPAR gamma in the differentiation and function of neurons. PPAR Res 2014: 768594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raikos N, Schmid H, Nussbaumer S, Ambach L, Lanz S, Längin A et al (2014). Determination of Δ9‐tetrahydrocannabinolic acid A (Δ9‐THCA‐A) in whole blood and plasma by LC‐MS/MS and application in authentic samples from drivers suspected of driving under the influence of Cannabis . Forensic Sci Int 243: 130–136. [DOI] [PubMed] [Google Scholar]
- Rocha NP, Ribeiro FM, Furr‐Stimming E, Teixeira AL (2016). Neuroimmunology of Huntington's disease: revisiting evidence from human studies. Mediators Inflamm 2016: 8653132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock EM, Kopstick RL, Limebeer CL, Parker LA (2013). Tetrahydrocannabinolic acid reduces nausea‐induced conditioned gaping in rats and vomiting in Suncus murinus . Br J Pharmacol 170: 641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen CJ (2010). Revisiting the rosiglitazone story – lessons learned. N Engl J Med 363: 803–806. [DOI] [PubMed] [Google Scholar]
- Rosenthaler S, Pohn B, Kolmanz C, Huu CN, Krewenka C, Huber A et al (2014). Differences in receptor binding affinity of several phytocannabinoids do not explain their effects on neural cell cultures. Neurotoxicol Teratol 46: 49–56. [DOI] [PubMed] [Google Scholar]
- Skerrett R, Malm T, Landreth G (2014). Nuclear receptors in neurodegenerative diseases. Neurobiol Dis 72: 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Spiegelman BM (2008). Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem 77: 289–312. [DOI] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch‐Maguire P, Wheeler VC, Sharp AH, Persichetti F et al (2000). Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet 9: 2799–2809. [DOI] [PubMed] [Google Scholar]
- Valdeolivas S, Navarrete C, Cantarero I, Bellido ML, Muñoz E, Sagredo O (2015). Neuroprotective properties of cannabigerol in Huntington's disease: studies in R6/2 mice and 3‐nitropropionate‐lesioned mice. Neurotherapeutics 12: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vara D, Morell C, Rodriguez‐Henche N, Diaz‐Laviada I (2013). Involvement of PPARgamma in the antitumoral action of cannabinoids on hepatocellular carcinoma. Cell Death Dis 4: e618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeckx KC, Korthout HA, Van Meeteren‐Kreikamp AP, Ehlert KA, Wang M, Van der Greef J et al (2006). Unheated Cannabis sativa extracts and its major compound THC‐acid have potential immuno‐modulating properties not mediated by CB1 and CB2 receptor coupled pathways. Int Immunopharmacol 6: 656–665. [DOI] [PubMed] [Google Scholar]
- Wang L, Waltenberger B, Pferschy‐Wenzig EM, Blunder M, Liu X, Malainer C et al (2014). Natural product agonists of peroxisome proliferator‐activated receptor gamma (PPARγ): a review. Biochem Pharmacol 92: 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Wang YH, Avula B, Radwan MM, Wanas AS, Van Antwerp J et al (2016). Decarboxylation study of acidic cannabinoids: a novel approach using ultra‐high‐performance supercritical fluid chromatography/photodiode array‐mass spectrometry. Cannabis Cannabinoids Res 1: 262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodd TB, Spivey WTN, Easterfield TT (1896). The resin of Indian hemp. J Chem Soc 69: 539–546. [Google Scholar]
- Wong SB, Cheng SJ, Hung WC, Lee WT, Min MY (2015). Rosiglitazone suppresses in vitro seizures in hippocampal slice by inhibiting presynaptic glutamate release in a model of temporal lobe epilepsy. PLoS One 10: e0144806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Cannabinoid acids induce higher PPARγ degradation compared to their decarboxylated forms. STHdh Q7/Q7 cells were treated with Δ9‐THCA‐A, CBG, CBGA, CBD or CBDA for 6 hours and the steady state levels of endogenous PPARγ and β‐actin detected by Western blots.
Figure S2 HEK‐293T cells were seeded at 1 x 104 in 60 mm dishes and 24 h later treated with RGZ or Δ9‐THCA for 6 hours and the steady state levels of endogenous PPARγ and β‐actin detected by Western blot.
Figure S3 Δ9‐THCA prevents mHtt‐CFP‐induced cell toxicity. A) N2a cells were seeded at 3.5 x103 in poly‐d‐lysine treated 96‐well plates. After 24 hours, cells were infected for 5 h with 1 μl/ml of adenovirus expressing CFP‐tagged human huntingtin exon 1 harboring a pathogenic polyQ tract of 94 CAG repeats. Then, medium was replaced by fresh medium and the cells treated with Δ9‐THCA or Δ9‐THC at the indicated doses and images were taken after 72 hours. A) Cell viability was determined using the IncuCyte HD software and non‐infected cells were considered as 100 % of cell viability. B) DAPI stained nuclei and expressions of CFP‐tagged mHttq94 protein were analyzed by confocal microscopy.
Figure S4 Isolated Δ9‐THCA analyzed by GC‐MS. Δ9‐THCA was diluted in 3 mL of hexane. An intermediate solution was prepared and 50 μL were taken and mixed with 20 uL of IS (CBD‐d3) and dried together in a concentrator. The dry residue was reconstituted in 15 μL of pyridine and 135 μL of derivative mixture BSTFA: TMCS (98:2 v/v) and incubated at 37 C for 1 hour. Crimp vials were used in order to avoid leakage. Δ9‐THCA purity was 97%.