

ABSTRACT
Vinexin is a SH3 domain-containing adaptor protein that has diverse roles in cell adhesion, signal transduction, gene regulation and stress granule assembly. In this study, we found that vinexin localizes at the midbody during cell division and facilitates cytokinesis. Knockdown of vinexin in HeLa cells delayed the mitotic cell cycle progression and increased the time of cell abscission and the failure to resolve the cytoplasmic bridge. Midbody-localized vinexin is essential for recruiting rhotekin to this structure for cytokinesis because overexpression of a vinexin mutant without a rhotekin-binding motif or knockdown of rhotekin also impaired cytokinetic abscission and increased the number of cells arrested at the midbody stage. Aberrant expression of vinexin and rhotekin in various cancers has been implicated to promote metastasis because of their functions in cell adhesion and signaling. Our findings reveal a novel role of vinexin and rhotekin in cytokinetic abscission and provide another perspective of how both molecules may affect oncogenic transformation via this fundamental cell cycle process.
KEYWORDS: cell abscission, cytokinesis, midbody, rhotekin, vinexin
Introduction
Adaptor proteins are involved in various cellular processes including signal transduction, cell adhesion, cytoskeletal organization, proliferation and survival.1,2 Vinexin, also known as sorbin and Src homology 3 (SH3) domain-containing protein 3 (SORBS3), has α, β and γ variants encoded from alternatively spliced transcripts. All vinexin isoforms contain a common C-terminus with 3 SH3 motifs that interact with various cytoskeletal proteins and signaling molecules, such as Wiskott-Aldrich syndrome family verprolin-homologous protein (WAVE), vinculin, Sos and rhotekin.3-6 In contrast, the extra N-terminal sequences in α and γ vinexin contain the SoHo domain to associate with membrane protein flotillin for anchoring at lipid rafts.7 The expression of vinexin α and γ is more restricted to skeletal muscle, heart and gonads,4,8,9 whereas vinexin β is most widely expressed in various tissues and cells. Moreover, nuclear vinexin β binds to retinoic acid receptor γ (RARγ) to downregulate RARγ's transcriptional activity.10 Vinexin associates with vinculin to localize at focal adhesions (FAs), where it promotes cell adhesion and spreading.4 Previously, we found that vinexin is translocated from FAs to stress granules (SGs) under arsenite- or heat shock-induced stress. Vinexin is assembled into SGs via its interaction with the RNA-binding protein cytoplasmic polyadenylation element binding protein 4 (CPEB4) and affects SG formation and cell survival.11 In this study, we unexpectedly observed that vinexin β could localize at the midbody during cell division.
Mitosis involves sequential steps including prometaphase, metaphase, anaphase, telophase and cytokinesis. The mitotic progression requires a tight spatio-temporal coordination of many signaling molecules to remodel cytoskeleton components (i.e., tubulin and actin), which leads to equal division of DNA and cytoplasmic contents in 2 daughter cells.12,13 The final stage of mitosis is cytokinesis initiating after segregation of sister chromatins at anaphase. Equatorial activation of Ras homolog gene family member A (RhoA) induces the cleavage furrow ingression by constricting the actomyosin contractile ring, which is composed of myosin and actin filaments. After further constriction, the spindle midzone, a bipolar microtubule array between separating chromosomes, becomes the electron-dense structure called the midbody appearing at the center region of the intercellular bridge.14
Mass spectrometry analysis of biochemically isolated midbodies uncovered the presence of cytoskeleton- and vesicle trafficking-related proteins.15 Proteomic analysis and/or immunostaining revealed several focal adhesion components, such as vinculin, paxillin and talin, at the midbody.15-17 Because hundreds of proteins are identified in this structure, how their assembly is coordinated to regulate cell abscission is unclear. Adaptor proteins such as vinexin with multiple protein-interacting SH3 domains may facilitate the assembly of this structural and signaling complex, but no study has reported the presence of vinexin at the midbody.
In this study, we characterized vinexin in the cell cycle and revealed that vinexin and its binding partner rhotekin play an essential role in cytokinesis. Rhotekin, named for its interaction with GTP bound Rho family proteins,18 is recruited to the midbody by binding to vinexin. Knockdown of either protein impaired cytokinesis in most cells by doubling abscission time. Our results reveal a novel location and function of vinexin and rhotekin in mammalian cytokinesis.
Results
Vinexin is enriched at the midbody bulge during late cytokinesis
Previously, we used live imaging to monitor the redistribution of enhanced green fluorescent protein-tagged vinexin β (EGFP-Vxn β) from FAs to SGs in arsenite-treated HeLa cells11 and unexpectedly observed the presence of an EGFP-Vxn β signal at the midbody during cell division. To follow up this observation, we examined the subcellular distribution of EGFP-Vxn β in mitotic HeLa cells that also expressed mCherry red fluorescent protein-tagged histone 2B (RFP-H2B) to outline the chromosomal architecture. The live images from one dividing cell showed midbody localization of EGFP-Vxn β during telophase and cytokinesis (Fig. 1A, denoted by arrows). However, control EGFP-expressing HeLa cells showed a green fluorescence signal at the midbody with an intensity less than in EGFP-Vxn β-expressing cells (data not shown), which suggests that any overexpressed protein may be non-specifically trapped at the midbody structure.
Figure 1.
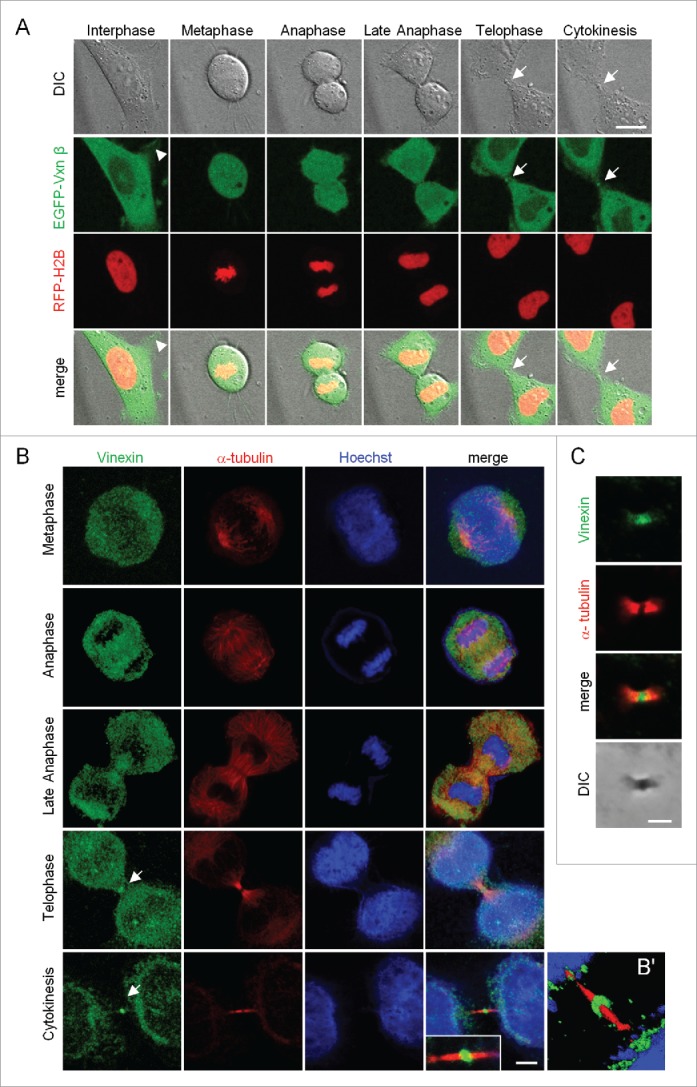
Localization of vinexin at the midbody during mitosis. (A) Live imaging of asynchronized HeLa cells overexpressing EGFP-vinexin β (Vxnβ) and RFP-Histone 2B (H2B). Midbody dense structure formed during cytokinesis observed by differential interference contrast (DIC) microscopy. Arrows and arrowhead mark midbody and focal adhesion, respectively. (B) After nocodazole release, endogenous vinexin was distributed in the cytosol from metaphase to anaphase but enriched at the midbody during cytokinesis. The mitotic stages were defined by the patterns of α-tubulin and Hoechst staining. (B′) A 3D-image reconstructed from confocal z stacks to show the entire midbody structure. (C) Isolated midbodies from Chinese hamster ovary (CHO) cells immunostained for vinexin and α-tubulin. Scales, 10 µm.
Thus, to evaluate whether endogenous vinexin indeed localizes at the midbody, we released HeLa cells synchronized at G2/M by nocodazole treatment of 1 to 2 h before DNA labeling and immunostaining of vinexin and α-tubulin. Because vinexin β is the major form expressed in HeLa and various cells,4,9 the immunostaining pattern of endogenous vinexin (Fig. 1B) was similar to the distribution of EGFP-Vxn β (Fig. 1A). Vinexin was mostly detected in the cytosol but also at the midbody during telophase and cytokinesis (Fig. 1B). Notably, vinexin was predominantly localized within the intercellular bridge and concentrated at the midbody bulge (Fig. 1B′), a dense structure derived from the midzone after furrow ingression and that could be isolated biochemically and observed by differential interference contrast (DIC) microscopy (Fig. 1C). Immunostaining of purified midbodies also confirmed the presence of vinexin (Fig. 1C).
Knockdown of vinexin reduced cell proliferation and increased the number of cells arrested at the midbody stage
To examine whether vinexin β regulates cell cycle progression, HeLa cells were infected with a lentivirus expressing siCtrl or one of 2 shRNAs, siVxn-1 and siVxn-2, against human vinexin. After puromycin selection, the infected siCtrl, siVxn-1 and siVxn-2 HeLa cells were used for western blot analysis (Fig. 2A) and PrestoBlue cell viability assay (Fig. 2B). Both siVxn constructs efficiently inhibited vinexin β expression (Fig. 2A). Vinexin β deficiency impaired cell proliferation (Fig. 2B). Midbody localization of vinexin suggested that it may control abscission to separate 2 dividing cells. Thus, siCtrl and siVxn cells arrested at G1/S by a thymidine block were released for 12 h and then processed to score the proportion of cells containing the midbody structure. Typically, most synchronized HeLa cells after 12 h release from G1/S should progress through mitosis and complete cell abscission (Fig. S1), so those dividing cells connected with intercellular bridge were arrested at the midbody stage (Fig. 2C, arrows). The number of siVxn cells at the midbody stage was approximately 2-fold that of siCtrl cells (Fig. 2C).
Figure 2.
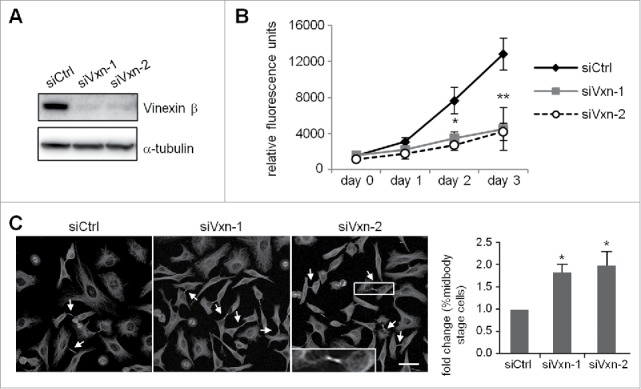
Depletion of vinexin reduces cell proliferation by delaying mitotic progression. (A) HeLa cells infected with a lentivirus expressing control (siCtrl) or shRNA against human vinexin (siVxn-1 or siVxn2) were harvested for immunoblotting assay of vinexin β and α-tubulin. (B) The proliferation rates of siCtrl and siVxn cells were monitored daily by Prestoblue assay. (C) Double thymidine-arrested siCtrl and siVxn cells were released to fresh medium for 12 h, then underwent α-tubulin immunostaining and Hoechst labeling. The midbody stage is when 2 daughter cells are still connected by the intercellular bridge, which is detected by α-tubulin immunostaining (arrows, a magnified example image is shown). Scale, 50 μm. The distribution of siCtrl or siVxn cells at the midbody stage and interphase expressed as mean ± SEM relative ratios with the ratio in siCtrl cells arbitrarily set to 1. Total cell number for each siRNA group from 3 independent experiments is > 1000. ** P < 0.01, * P < 0.05 by Student t test.
Knockdown of vinexin delayed mitotic cell cycle progression and increased abscission time and bi-nucleated cell population
Both shRNAs against vinexin conferred the same cell cycle defects (Fig. 2), which were unlikely due to off-target effects, so we performed the following experiments with siVxn-1 shRNA (hereafter referred to as siVxn) cells. To examine the distribution of siCtrl and siVxn cells in the cell cycle, we used flow cytometry to analyze cellular DNA content and define their stages. Asynchronized siVxn cells showed slightly increased cell population at the G2 phase (Fig. 3A, siCtrl vs siVxn, 13 ± 2% vs 18 ± 3%). Moreover, siVxn cells released from nocodazole-arrested G2/M phase progressed more slowly to G1 than did siCtrl cells (Fig. 3A). At the end of 6 h release, twice more siVxn cells remained in the G2 phase as compared with siCtrl cells (Fig. 3A, 34 ± 2% vs 17 ± 1%, P < 0.01). Nevertheless, the levels of the G2/M checkpoint regulator cyclin B (CCNB1) and its phosphorylated substrate histone 3 (p-H3), which decreased from metaphase to telophase, were comparable in siCtrl and siVxn cells after mitotic release (Fig. S2). These results support that the mitotic defect in siVxn cells likely occurs at a late mitotic stage such as cytokinesis.
Figure 3.
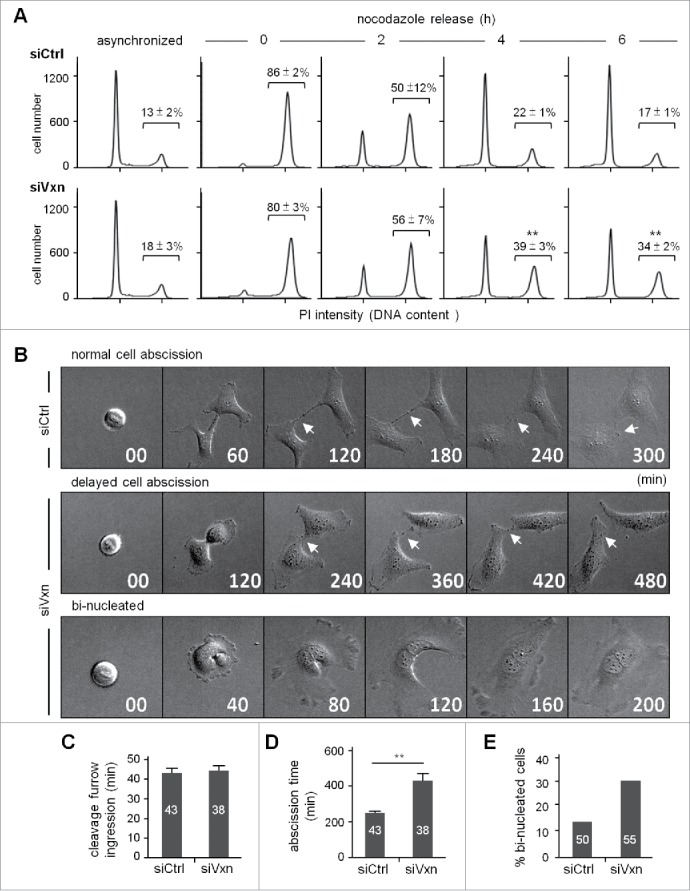
Knockdown of vinexin delays cell abscission. (A) SiCtrl and siVxn HeLa cells were synchronized at G2/M by nocodazole, then released for the indicated time before flow cytometry. Asynchronized siCtrl and siVxn cells were also included. The proportion of siCtrl and siVxn cells at G2 are expressed as mean ± SEM from 3 independent experiments. ** P < 0.01 comparing siCtrl and siVxn groups at each time by Student t test. (B) Time-lapse comparison of cytokinesis in a representative siCtrl cell (top) and 2 siVxn cells (middle and bottom). G1/S synchronized siCtrl and siVxn cells after release from the thymidine block. Midbodies remained at the center of intercellular bridge and midbody remnants inherited by a daughter cell after abscission are denoted by arrows and arrowheads, respectively. An example siVxn cell with incomplete cytokinesis eventually formed a bi-nucleated cell (bottom). The first image of a cell rounding up was considered time 0. For recorded cells with complete cytokinesis, the times required to reach (C) cleave furrow ingression and (D) cell abscission are expressed as mean ± SEM. (E) The percentage of recorded siCtrl and siVxn cells forming bi-nucleated cells. The cell numbers in each group used for analysis are indicated inside the bars. ** P < 0.01 by Student t test.
To determine whether vinexin deficiency specifically affects cell abscission to slow the mitotic progression, we used live imaging to monitor the dynamics of cytokinesis in siCtrl and siVxn cells (representative images in Fig. 3B). To avoid possible artificial effects caused by the microtubule-depolymerizing reagent nocodazole in the abscission process, we used a double thymidine block protocol to arrest cells at the G1/S phase and then imaged cells for 24 h after the release of cells into fresh medium. Cells with a round-up shape before furrow ingression were considered to be at metaphase and the beginning of mitosis (time 0). The time required for mitotic progression from chromosome condensation, chromosome separation to furrow ingression between siCtrl and siVxn cells was similar (Fig. 3C). However, siVxn cells took more time to divide than siCtrl cells (Fig. 3D), which suggests that the mitotic delay mainly occurs at the midbody stage. As an example of dividing siCtrl cells, the intercellular bridge was severed and the midbody remnant was engulfed into one of the daughter cells at about 300 min after metaphase (Fig. 3B, upper panel). The midbody remnant then disappeared, presumably degraded by autophagy (images at the later times not shown). In contrast, siVxn cells took double the time to dissolve the connection between 2 cells (Fig. 3B, middle panel). Some siCtrl and siVxn dividing cells showed a furrow regression phenotype and eventually formed bi-nucleated cells because of abscission failure (Fig. 3B, lower panel). Also, more bi-nucleated cells were formed in dividing siVxn than siCtrl cells (Fig. 3E).
Vinexin regulates cell abscission via its third SH3 domain
The midbody structure is divided into 3 parts: the bulge, dark zone, and flanking zone. The center of the midbody, the bulge, contains centralspindlin and its interacting partners, centrosomal protein CEP55, centriolin, epithelial cell transforming 2 (Ect2) and anillin.19 A narrow region next to the budge refers to the dark zone because of undetectable α-tubulin staining at this center part of microtubule spindles. The microtubule-bundling protein regulator of cytokinesis 1 (PRC1) and its associated motor protein, Kinesin family member 4A (KIF4), are enriched here. The flanking zones are 2 broader bands on microtubules outside the dark zone, where inner centromere protein (INCENP) and Aurora B are accumulated.19
To analyze a possible structural abnormality of midbody in vinexin-knockdown (KD) cells, we examined the distribution of several key molecules including anillin, Aurora B and INCENP and found no significant difference between siCtrl and siVxn cells in midbody-localized immunostaining signals of these proteins (Fig. S3). Thus, to dissect how vinexin affects cell abscission, we first analyzed the midbody localization of EGFP-tagged wild-type (WT) and SH3 motif-deleted vinexin mutants (Fig. 4A) because vinexin binds to its partners through the first 2 SH3 domains or the third SH3 domain. It has been reported that EGFP-tagged midbody-localized proteins, such as EGFP-KIF4 and EGFP-PRC1, tend to distribute aberrantly under high-level overexpression,19 so we only analyzed cells expressing low level of WT or mutant EGFP-Vxn to avoid any artifacts caused by mislocalization of overexpressed proteins. Nevertheless, we could still observe notable fluorescence signal at the midbody even in cells expressing low level of EGFP. Thus, to obtain real midbody-localized signal, the fluorescence intensity of EGFP or EGFP-Vxn in the selected midbody region was subtracted with that in the equivalent nuclear area (Fig. 4B). The images shown here were threshold adjusted after arbitrarily subtracting nuclear EGFP signal (Fig. 4B). We considered the WT and Δ(SH3)3 mutant but not Δ(SH3)1–2 mutant, with fluorescence intensity higher than that of the EGFP control, to be midbody-localized (Fig. 4B). The first 2 SH3 domains of vinexin β interact with cytoskeletal proteins, such as WAVE and vinculin,4,5 and the proteomic study identified vinculin but not WAVE in the isolated midbody structure.15 We confirmed that vinculin was indeed present at the midbody (Fig. 4C) and colocalized with vinexin (Fig. 4D) on immunostaining, so vinexin likely localizes at the midbody by binding to vinexin. Indeed, using lentivirus-introduced shRNA to knock down vinculin (Fig. 4E), we found a significant reduction of vinexin signal at the midbody (Fig. 4F).
Figure 4.
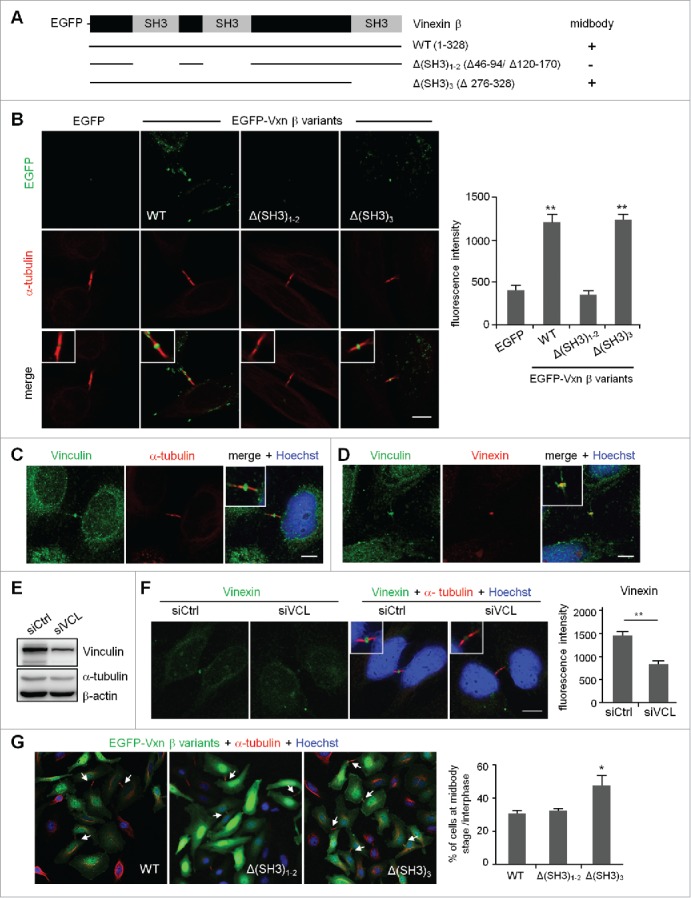
Expression of a SH3 domain-deleted mutant of vinexin increases the population of cells at midbody stage. (A) Illustration of various deletion mutants of vinexin β. (B) Thymidine-arrested cells were transfected with the plasmid expressing EGFP or EGFP-vinexin wild type (WT) or ΔSH3 mutants and then released for 14 h before α-tubulin immunostaining and Hoechst labeling. Quantification of green fluorescence intensity at the midbody stage from 20 cells expressing each EGFP construct. (C, D) Immunostaining of vinculin with (C) α-tubulin or (D) Vinexin at the midbody of HeLa cells. (E) HeLa cells infected with a lentivirus expressing control (siCtrl) or a shRNA against vinculin (siVCL) were harvested for immunoblotting. (F) Thymidine-arrested siCtrl and siVCL cells were released to fresh medium for 12 h, followed by immunostaining of vinexin and α-tubulin. The fluorescence intensity of vinexin at the midbody of HeLa cells was quantified from 20 cells per group. (G) Similar to (B), the images from 3 independent experiments were used to calculate the distribution of cells at the midbody stage and interphase. Arrows denote intercellular bridges. Scales, 10 μm. Data are mean ± SEM. ** P < 0.01, * P < 0.05 by Student t test.
To further dissect how midbody recruitment of vinexin affects cell abscission, we analyzed HeLa cells expressing the EGFP-Vxn β WT or ΔSH3-mutant. Cells arrested at G1/S were released for 14 h, then immunostained for α-tubulin to analyze the number of cells partitioning at midbody stage and interphase. Overexpression of the Δ(SH3)3 mutant increased the number of cells at midbody stage as compared with the WT and Δ(SH3)1–2 mutant (Fig. 4G), so the third SH3 motif of vinexin likely recruits additional factors to control cell abscission.
Rhotekin is recruited to the midbody via Vinexin at the late stage of cytokinesis
The third SH3 motif of vinexin interacts with neuronal Wiskott-Aldrich syndrome protein (N-WASP) and rhotekin.5,6 N-WASP and rhotekin are the effectors of small GTPases, Cdc42 and RhoA, respectively.18,20 Both Cdc42 and RhoA are important for remodeling actin cytoskeleton. Rho rearranges pre-existing actin filaments and Cdc42 promotes new actin polymerization by stimulating de novo nucleation of actin or severing actin filaments.21,22 Moreover, active RhoA but not Cdc42 around the cleavage furrow is important for cytokinesis.23,24 Immunostaining of nocodazole-released mitotic cells showed that the distribution of active RhoA changes sequentially from the equatorial cortex, ingressed furrow and then midbody ring to drive mitotic progression.25 Although rhotekin can bind to active RhoA,26 we found the distribution of rhotekin enriched only at the midbody during late cytokinesis (Fig. 5A and S4). Similar to vinexin β distribution pattern (Fig. 1B), the rhotekin-immunoreactive signal was mostly detected in the cytosol and later appeared at the midbody during telophase and cytokinesis (Fig. S4). Moreover, ectopically expressed myc-rhotekin (myc-RTKN) colocalized with vinexin at the midbody (Fig. 5B).
Figure 5.
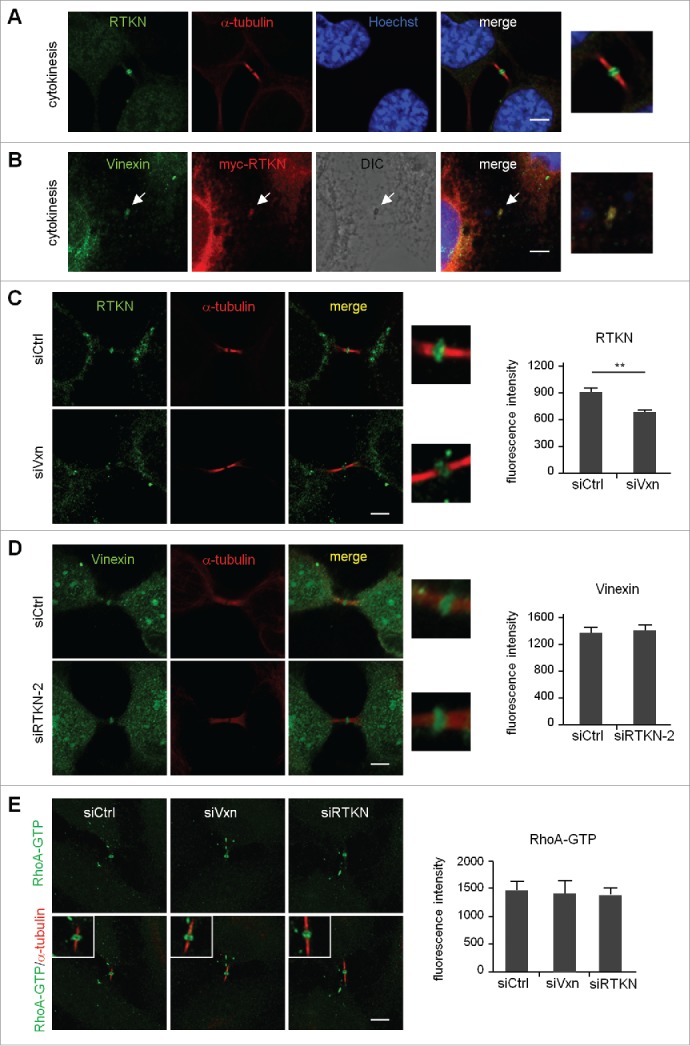
Midbody localization of rhotekin depends on vinexin. (A) Immunostaining of rhotekin (RTKN) and α-tubulin at the midbody of HeLa cells. (B) HeLa cells expressing myc-tagged rhotekin (myc-RTKN) were immunostained with vinexin and myc antibodies. Midbody dense structure observed by DIC microscopy. (C-E) siCtrl, siVxn and siRTKN-2 cells released from mitotic arrest were immunolabeled with the denoted antibodies. The fluorescence intensity of rhotekin, vinexin or RhoA-GTP at the midbody of HeLa cells was quantified from 20 cells per group. Data are mean ± SEM. ** P < 0.01 by Student t test.
Because overexpression of Δ(SH3)3 mutant vinexin increased the number of cells at the midbody stage (Fig. 4G), the midbody localization of rhotekin might depend on its interaction with vinexin. Similar to the generation of siVxn cells, HeLa cells were infected with the lentivirus expressing one of the 2 shRNAs, siRTKN-1 and siRTKN-2, against human rhotekin. After puromycin selection, we used infected HeLa cells for experiments. Because both vinexin and rhotekin antibodies were raised in rabbits, we performed a control experiment showing a significant decrease in the midbody signal of vinexin and rhotekin in siVxn (Fig. S5A) and siRTKN-2 (Fig. S5B) cells, respectively. The rhotekin signal at the midbody was significantly reduced in siVxn cells (Fig. 5C), whereas rhotekin-KD did not affect the amount of midbody-localized vinexin (Fig. 5D). Thus, rhotekin localizes at the midbody in part via its interaction with vinexin. A biochemical study showed that the binding of rhotekin to RhoA-GTP could inhibit its GTPase activity and elevate GTP-bound active RhoA,26 so we then investigated the midbody level of active RhoA and found no change under vinexin- or rhotekin-KD (Fig. 5E).
Knockdown of rhotekin reduced cell proliferation by impairing cytokinesis
Using siRTKN-1 and siRTKN2 cells (Fig. 6A), we then investigated the role of rhotekin in the cell cycle. Similar to siVxn cells (Fig. 2), rhotekin-KD reduced cell proliferation (Fig. 6B) and increased the number of cells arrested at the midbody stage (Fig. 6C, arrows). Moreover, the live images acquired from the release of double thymidine-arrested siCtrl and siRTKN cells showed rhotekin-KD cells with normal furrow ingression but impaired cytokinesis (Fig. 6D) with prolonged abscission time (Fig. 6E). Different from vinexin-KD cells, some RTKN-KD cells entered mitosis normally but underwent apoptosis before furrow ingression. This phenotype is considered to be cytokinesis failure (Fig. 6F). Thus, midbody-localized vinexin and rhotekin appear to cooperate together to regulate cytokinesis.
Figure 6.
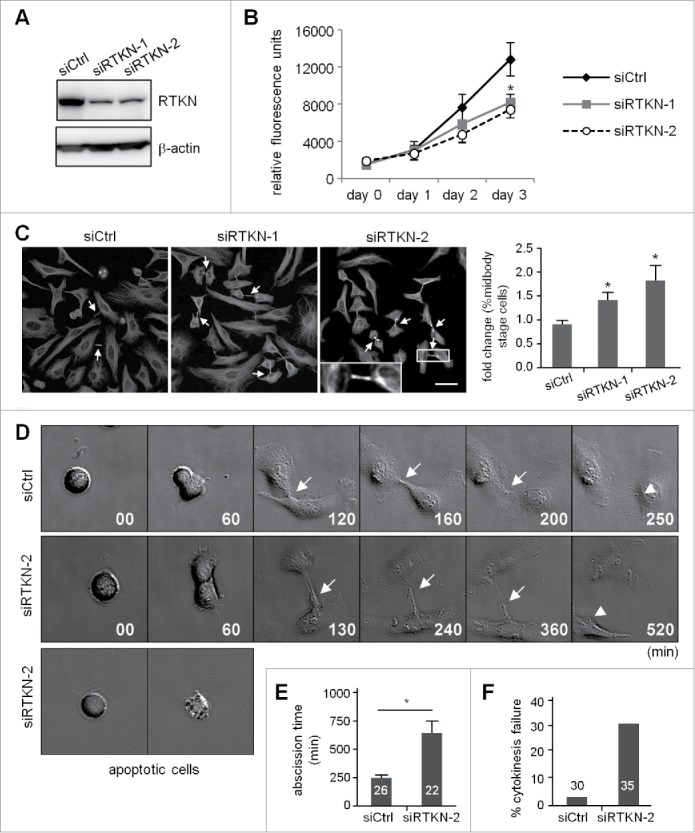
Knockdown of rhotekin impairs cell proliferation by cytokinectic delay and failure. (A) HeLa cells infected with a lentivirus expressing control (siCtrl) or shRNAs against human rhotekin (siRTKN-1, siRTKN-2) were harvested for immunoblotting of rhotekin and β-actin. (B) The proliferation rate of siCtrl and siRTKN cells monitored by Prestoblue assay. (C) Double thymidine-arrested siCtrl and siRTKN cells were released to fresh medium for 12 h, then underwent α-tubulin immunostaining and Hoechst labeling. The midbody stage is defined when 2 daughter cells are still connected by an intercellular bridge (arrows). Scale, 50 μm. The distribution of siCtrl or siVxn cells at the midbody stage and interphase is expressed as relative ratios with the ratio in siCtrl cells arbitrarily set to 1. Total cell number for each siRNA group from 3 independent experiments is ∼1000. (D) Time-lapse comparison of cytokinesis in a representative siCtrl cell (top) and siRTKN cell (middle). G1/S-synchronized siCtrl and siRTKN cells after release from the thymidine block were time-lapse recorded. Midbodies remained at the center of intercellular bridge and midbody remnants inherited by a daughter cell after abscission are denoted by arrows and arrowheads, respectively. Some siRTKN cells underwent apoptosis (bottom). The first image of a cell rounding up was considered time 0. (E) For recorded cells with complete cytokinesis, the time required to complete cell abscission is expressed as mean ± SEM. (F) The percentage of recorded siCtrl and siVxn cells with incomplete cytokinesis eventually forming a bi-nucleated cell. The cell numbers in each group used for analysis are labeled inside the bars. * P < 0.05 by Student t test.
Discussion
Vinexin modulates various cellular pathways including signal transduction, gene expression, cytoskeletal organization and stress granule assembly by interacting with distinct binding proteins. Although rhotekin was identified as a binding partner of vinexin by a yeast 2-hybrid screen,6 the physiologic function of such an interaction remains unclear. In this study, we identified a novel subcellular location for vinexin and rhotekin, whereby the 2 proteins coordinate at the midbody stage to promote cell abscission during late cytokinesis.
Assembly of the contractile ring during anaphase initiates cytokinesis in mammalian cells. Similar to vinexin, RhoA also affects the formation of a focal adhesion complex and actin stress fibers in cultured cells.27,28 Nevertheless, RhoA-knockout (KO) mouse embryonic fibroblasts (MEFs) possess normal stress fibers and focal adhesions unless in conjunction with RhoB and RhoC deficiency.29 In contrast, RhoA- but not RhoB- or RhoC-KO MEFs show defective cytokinesis and chromosome segregation.29 Thus, RhoA plays an indispensable role to orchestrate dynamic remodeling of cytoskeleton for cell division. Several guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs), such as Ect2, GEF-H1, MgcRacGAP and ARHGAP19, regulate the dynamic cycling of RhoA between the active GTP-bound and inactive GDP-bound conformations during cytokinesis.23,30-33 During cytokinesis, the mitotic spindle directs the formation of a contractile ring at the equatorial cell cortex, which then contracts to cause cleavage furrow ingression. RhoA accumulates at the equatorial cortex and continues to concentrate at the cleavage furrow during cytokinesis.23,25,32 RhoA is critical to control actin filament assembly and enhance actomyosin interaction via binding to downstream effectors, such as mammalian homolog of Drosophila Diaphanous (mDia) and Rho-associated kinase ROCK.34
In this study, we identified that another RhoA effector, rhotekin, participates in cytokinesis. Knockdown of vinexin or rhotekin reduced the level of midbody-localized rhotekin (Fig. 5C and S5B) and delayed cell abscission after furrow ingression (Fig. 3 and Fig. 6). However, how rhotekin affects mitotic cell division is not clear. Although phosphorylation of rhotekin by protein kinase D can increase active RhoA and stress fibers,26 we found that active RhoA level at the midbody was not affected by reducing the amount of rhotekin at the midbody in vinexin-KD or rhotekin-KD cells (Fig. 5E). In addition to a prolin-rich domain binding to the third SH3 motif of vinexin and a Rho-binding domain interacting with Rho proteins, rhotekin also contains a C-terminal SPV-motif binding to the PDZ domain35 and a pleckstrin-homology domain, which can interact with proteins and phosphatidylinositol lipids within biologic membranes.36,37 Thus, other domains or sequences in rhotekin may recruit unidentified factors to promote cell abscission. Alternatively, rhotekin may facilitate interactions of components in the midbody complex without affecting their localization.
Because the cytokinetic defect in the majority of (∼70%) vinexin-KD or rhotekin-KD cells occurs after furrow ingression to double abscission time (Fig. 3D and Fig. 6E), the midbody localization of vinexin and rhotekin likely recruits factors involved in late cytokinesis, such as the exocyst complex.38 Exocyst and endosomal sorting complex required for transport (ESCRT) complexes are required for vesicle anchorage and cleavage, respectively, at the scission site. Cells depleted of components in these 2 complexes show delayed abscission and furrow regression.39,40 In contrast, RhoA and Aurora B play roles in both furrow ingression and cell abscission.41-44 Thus, cytokinesis involves multiple inter-coordinated steps and requires different midbody components to complete these processes. Notably, approximately 30% of cells are binucleated when knocking down vinexin (Fig. 3E) but exhibit cytokinesis failure and apoptosis at metaphase when knocking down rhotekin (Fig. 6F). Given that both vinexin and rhotekin interact with several other proteins, the distinct phenotypes in the 30% of knockdown cells indicate that the 2 proteins may bind to other partners and play additional roles before furrow ingression besides working together at the midbody. As well, the post-translational modification of vinexin declines with the progression of mitosis (Fig. S2), but what the nature of this modification is and whether such a change is important to modulate vinexin function await further investigation.
The temporal and spatial assembly of midbody components and their interactions remain largely unknown. Several focal adhesion components such as Talin and Paxillin.16,17 also play a role in cytokinesis. Previous proteomic study identified 160 proteins but not vinexin and rhotekin in this structure in Chinese hamster ovary (CHO) cells.15 Because vinexin was also detected in the isolated midbodies from CHO cells (Fig. 1C), the role of vinexin and rhotekin in cytokinesis is unlikely to be cell type-specific and we expected more midbody components than those reported.15 Carcinogenesis is often associated with an increase in metastatic potential caused by the loss of cell adhesion structures.45,46 Several cancer tissues show aberrant expression of vinexin.47,48 Moreover, in NIH3T3 fibroblasts, v-Src-mediated transformation suppresses vinexin β expression to increase cell migration,47,48 and upregulation of vinexin β also decreases anchorage-independent growth of LNCaP prostatic cancer cells (Mizutani et al, 2007), so vinexin may affect oncogenic transformation via its role in cell adhesion. Our findings here reveal a novel function of vinexin and rhotekin in cytokinetic abscission. Because the improper separation of genetic materials caused by defective mitosis or aberrant furrow regression results in polyploidy and tumorigenesis,49,50 delayed cell abscission in vinexin- or rhotekin-deficient cells may increase the population of cells carrying aberrant genetic material and lead to tumorigenesis. This study provides a novel mechanism of how vinexin and rhotekin may affect oncogenic transformation via this fundamental cell cycle process.
Materials and methods
Antibodies and chemicals
Antibodies used were for β-actin (A5441), α-tubulin (T5168), myc (M4439), Vinculin (V9264) from Sigma-Aldrich; RhoA (26C4) from Santa Cruz Biotechnology; Aurora B (BD611082) from BD Biosciences; Anillin (GTX107742), Vinexin (GTX115362) and Rhotekin (GTX114161) from GeneTex; GFP (#29779) from AnaSpec; INCENP (ab36453) from Abcam; Cyclin B1 (#4135), p-Histone H3 (Ser10 residue, #9701) from Cell Signaling Technology; AlexaFluor 488 and 594-conjugated secondary antibodies and Hoechst 33342 (H3570) from Invitrogen. All chemicals were obtained from Sigma-Aldrich.
Plasmid construction and shRNA constructs
The shRNA clones, TRCN0000123145 (siVxn-1, GAAGGGTGACATTGTCTACAT), TRCN0000123148 (siVxn-2, CGGAACGTTCCCTGGAAATTA), TRCN0000116752 (siVCL, CCCAGGCAAATCAGTTACTAA), TRCN0000048659 (siRTKN-1, CCTAAGCATCAGTAACCAGTA) and TRCN0000048660 (siRTKN-2, GACCTGAATATGCTCTACATT) targeted to human vinexin, vinculin and rhotekin were from the RNAi Core Facility (Academia Sinica). The vinexin β DNA fragments were amplified from mouse testis cDNA and cloned in the enhanced green fluorescent protein (EGFP) plasmid as described.11 SH3 domain-deleted vinexin mutants were generated by use of the QuikChange site-directed mutagenesis kit (Stratagene).
Cell culture, lentiviral infection and transfection
HEK-293T (CRL-11268, ATCC), HeLa (CCL−2, ATCC), and CHO-K1 (CCL−61, ATCC) cells were cultured in Dulbecco's modified essential medium supplemented with 10% fetal bovine serum. HEK-293T cells were used to produce lentiviruses expressing siVxn or siRTKN shRNA as described.51 Transfection of HEK-293T and HeLa cells involved use of Lipofectamin 2000 (Invitrogen). To generate vinexin- or rhotekin-knockdown (KD) cells, HeLa cells were infected with the lentivirus and with 1 µg/ml polybrene overnight (day 1), replaced with new medium next day (day 2), and selected with 1 µg/ml puromycin for another day (day 3) before subculture for experiments.
Immunofluorescence staining
For mitotic arrest, HeLa cells grown on coverslips were treated with 2.5 mM thymidine for 16 h and released for 2 h in fresh medium containing 250 μM nocodazole. The mitotic arrested cells were released for 30 min in fresh medium to proceed with the cell cycle from metaphase to anaphase or for 2 h to enrich cells at cytokinesis. Cells were washed twice with phosphate buffered saline (PBS), fixed with 4% formaldehyde for 20 min, permeabilized with 0.1% Triton X-100 for 5 min, blocked with 3% bovine serum albumin for 30 min, then immunostained with indicated antibodies. For active RhoA staining, cells were fixed in prechilled 10% trichloroacetic acid on ice for 15 min as described.25
Image acquisition and quantification
To quantify fluorescence intensities of designated targets at the midbody stage, images from 20 immunostained cells in control or vinexin-KD condition were obtained under the same exposure setting that maximized the dynamic range of pixel intensity and then analyzed by using MetaMorph 4.5. The fluorescence images were acquired under a LSM700 confocal microscope (Carl Zeiss) with a Plan-Apochromat 63X oil objective lens. Because the fluorescence signals were not saturated (12 bit image, maximal pixel intensity < 4095), they reflect the relative amount of designated proteins present at the midbody.
Time lapse phase-contrast imaging
To synchronize cells at G1/S, HeLa cells were treated with double thymindine block and released into fresh medium.52 The culture dish was then placed on a 37°C preheated stage and cell images were recorded every 10 min for 24 h by use of an inverted Leica DMI 6000B microscope with a 40x objective. The round-shape cells before furrow ingression were considered the beginning of metaphase. The time required for a monitored metaphase cell to complete cytokinesis (i.e., cleavage of intercellular bridge followed by entrance of a midbody remnant into a daughter cell) was considered the abscission time. Images were analyzed by using MetaMorph 4.5 (Universal Imaging).
Midbody isolation
Midbody isolation was performed as described15,53 with some modification. Briefly, CHO cells synchronized with 2.5 mM thymidine for 16 h were washed and released to nocodazole-containing medium for 10 h. The shaken-off mitotic cells were replaced with fresh medium for 1 h with agitation in a CO2 incubator before the addition of 5 μg/ml taxol and phalloidin for 3 min to stabilize spindles and the contractile ring, respectively. Cells were pelleted, washed once with H2O without disturbing the cell pellet and then resuspended in a hypotonic buffer (20 μg/ml taxol, 0.25% Triton X-100, 2 mM PIPES, pH 6.9) with 30-s vortexing to disrupt cells and release midbodies. After centrifugation at 600xg to remove nuclei, midbodies in the supernatant were pelleted at 2000 xg for 20 min, resuspended in PBS and spun onto poly-L-Lysine–coated coverslips placed in a 12-well culture plate. Coverslips were used for immunostaining.
Flow cytometry of cell cycle profile
To obtain the cell cycle profile of HeLa cells, cells synchronized by using a double thymidine block and release protocol54 were harvested at the indicated times after release. To arrest cells at G2/M, lentivirus-infected HeLa cells were synchronized with 250 µM nocodazole for 12 h and released into fresh medium for various times before harvesting. The collected cells were fixed with 70% ethanol on ice for 10 min, washed twice with PBS, then incubated in 50 μg/ml propidium iodide, 100 μg/ml RNase A and 0.1% Tween in PBS for 30 min at room temperature. The DNA-staining profile was analyzed by use of FACS Calibur (BD Biosciences) with a 488-nm argon laser and Flowjo software. Dead cells and debris defined from the forward- and side-scatter intensity profile were excluded from the analysis.
PrestoBlue assay of cell proliferation
HeLa cells after control, vinexin or rhotekin KD were seeded at 5×104 cells per well on a 24-well plate. After 6 h incubation, cells were incubated with 10-fold–diluted PrestoBlue reagent (Invitrogen) in complete medium for 1 h at 37°C, then fluorescence was measured at 585 nm by use of the Gemeni EM microplate reader (Molecular Devices). Cells were replaced with fresh medium and incubated for 24 h before repeating the PrestoBlue assay for 3 consecutive days.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Tang Tang for mCherry-H2B plasmid, INCENP and Aurora B antibodies and Ruey-Hwa Chen for RhoA antibody. They thank Joel Richter for HEK-293T cells, Te-Chang Lee for CHO-K1 cells and Sheau-Yann Shieh for HeLa cells. They appreciate Tzu-Wen Tai in the IBMS core facility for helping with FACS analyses and the RNAi Core Facility at Academia Sinica for the shRNA clones.
Funding
This work was supported by grants from the Ministry of Science and Technology [MoST105–2311-B-001–078 -MY3, MoST104–2321-B-001-064-] and Academia Sinica [AS-103-TP-B05] in Taiwan. The RNAi Core Facility was supported by the National Core Facility Program for Biotechnology [MoST 104–2319-B-001–001].
References
- [1].Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science 1997; 278:2075-80; PMID:9405336; http://dx.doi.org/ 10.1126/science.278.5346.2075 [DOI] [PubMed] [Google Scholar]
- [2].Kioka N, Ueda K, Amachi T. Vinexin, CAP/ponsin, ArgBP2: a novel adaptor protein family regulating cytoskeletal organization and signal transduction. Cell Structure Function 2002; 27:1-7; PMID:11937713; http://dx.doi.org/ 10.1247/csf.27.1 [DOI] [PubMed] [Google Scholar]
- [3].Akamatsu M, Aota S, Suwa A, Ueda K, Amachi T, Yamada KM, Akiyama SK, Kioka N. Vinexin forms a signaling complex with Sos and modulates epidermal growth factor-induced c-Jun N-terminal kinase/stress-activated protein kinase activities. J Biol Chem 1999; 274:35933-7; PMID:10585480; http://dx.doi.org/ 10.1074/jbc.274.50.35933 [DOI] [PubMed] [Google Scholar]
- [4].Kioka N, Sakata S, Kawauchi T, Amachi T, Akiyama SK, Okazaki K, Yaen C, Yamada KM, Aota S. Vinexin: a novel vinculin-binding protein with multiple SH3 domains enhances actin cytoskeletal organization. J Cell Biol 1999; 144:59-69; PMID:9885244; http://dx.doi.org/ 10.1083/jcb.144.1.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mitsushima M, Sezaki T, Akahane R, Ueda K, Suetsugu S, Takenawa T, Kioka N. Protein kinase A-dependent increase in WAVE2 expression induced by the focal adhesion protein vinexin. Genes Cells: Devoted Mol Cell Mechan 2006; 11:281-92; http://dx.doi.org/ 10.1111/j.1365-2443.2006.00932.x [DOI] [PubMed] [Google Scholar]
- [6].Nagata K, Ito H, Iwamoto I, Morishita R, Asano T. Interaction of a multi-domain adaptor protein, vinexin, with a Rho-effector, Rhotekin. Med Mol Morphology 2009; 42:9-15; PMID:19294487; http://dx.doi.org/ 10.1007/s00795-008-0433-8 [DOI] [PubMed] [Google Scholar]
- [7].Kimura A, Baumann CA, Chiang SH, Saltiel AR. The sorbin homology domain: a motif for the targeting of proteins to lipid rafts. Proc Natl Acad Sci U S A 2001; 98:9098-103; PMID:11481476; http://dx.doi.org/ 10.1073/pnas.151252898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kawauchi T, Ikeya M, Takada S, Ueda K, Shirai M, Takihara Y, Kioka N, Amachi T. Expression of vinexin alpha in the dorsal half of the eye and in the cardiac outflow tract and atrioventricular canal. Mechanisms Dev 2001; 106:147-50; PMID:11472845; http://dx.doi.org/ 10.1016/S0925-4773(01)00421-X [DOI] [PubMed] [Google Scholar]
- [9].Matsuyama M, Mizusaki H, Shimono A, Mukai T, Okumura K, Abe K, Shimada K, Morohashi K. A novel isoform of Vinexin, Vinexin gamma, regulates Sox9 gene expression through activation of MAPK cascade in mouse fetal gonad. Genes Cells 2005; 10:421-34; http://dx.doi.org/ 10.1111/j.1365-2443.2005.00844.x [DOI] [PubMed] [Google Scholar]
- [10].Bour G, Plassat JL, Bauer A, Lalevee S, Rochette-Egly C. Vinexin beta interacts with the non-phosphorylated AF-1 domain of retinoid receptor gamma (RARgamma) and represses RARgamma-mediated transcription. J Biol Chem 2005; 280:17027-37; PMID:15734736; http://dx.doi.org/ 10.1074/jbc.M501344200 [DOI] [PubMed] [Google Scholar]
- [11].Chang YW, Huang YS. Arsenite-activated JNK signaling enhances CPEB4-Vinexin interaction to facilitate stress granule assembly and cell survival. PloS One 2014; 9:e107961; PMID:25237887; http://dx.doi.org/ 10.1371/journal.pone.0107961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Green RA, Paluch E, Oegema K. Cytokinesis in animal cells. Annual Rev Cell Dev Biol 2012; 28:29-58; PMID:22804577; http://dx.doi.org/ 10.1146/annurev-cellbio-101011-155718 [DOI] [PubMed] [Google Scholar]
- [13].Steigemann P, Gerlich DW. Cytokinetic abscission: cellular dynamics at the midbody. Trends Cell Biol 2009; 19:606-16; PMID:19733077; http://dx.doi.org/ 10.1016/j.tcb.2009.07.008 [DOI] [PubMed] [Google Scholar]
- [14].D'Avino PP, Giansanti MG, Petronczki M. Cytokinesis in animal cells. Cold Spring Harbor Perspectives Biol 2015; 7:a015834; PMID:25680833; http://dx.doi.org/ 10.1101/cshperspect.a015834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Skop AR, Liu H, Yates J 3rd, Meyer BJ, Heald R. Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanisms. Science 2004; 305:61-6; PMID:15166316; http://dx.doi.org/ 10.1126/science.1097931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bellissent-Waydelich A, Vanier MT, Albiges-Rizo C, Simon-Assmann P. Talin concentrates to the midbody region during mammalian cell cytokinesis. J Histochem Cytochem 1999; 47:1357-68; PMID:10544209; http://dx.doi.org/ 10.1177/002215549904701102 [DOI] [PubMed] [Google Scholar]
- [17].Shafikhani SH, Mostov K, Engel J. Focal adhesion components are essential for mammalian cell cytokinesis. Cell Cycle 2008; 7:2868-76; PMID:18787414; http://dx.doi.org/ 10.4161/cc.7.18.6674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Reid T, Furuyashiki T, Ishizaki T, Watanabe G, Watanabe N, Fujisawa K, Morii N, Madaule P, Narumiya S. Rhotekin, a new putative target for Rho bearing homology to a serine/threonine kinase, PKN, and rhophilin in the rho-binding domain. J Biol Chem 1996; 271:13556-60; PMID:8662891; http://dx.doi.org/ 10.1074/jbc.271.43.26739 [DOI] [PubMed] [Google Scholar]
- [19].Hu CK, Coughlin M, Mitchison TJ. Midbody assembly and its regulation during cytokinesis. Mol Biol Cell 2012; 23:1024-34; PMID:22278743; http://dx.doi.org/ 10.1091/mbc.E11-08-0721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, Kirschner MW. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 1999; 97:221-31; PMID:10219243; http://dx.doi.org/ 10.1016/S0092-8674(00)80732-1 [DOI] [PubMed] [Google Scholar]
- [21].Hall A. Rho GTPases and the actin cytoskeleton. Science 1998; 279:509-14; PMID:9438836; http://dx.doi.org/ 10.1126/science.279.5350.509 [DOI] [PubMed] [Google Scholar]
- [22].Tapon N, Hall A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol 1997; 9:86-92; PMID:9013670; http://dx.doi.org/ 10.1016/S0955-0674(97)80156-1 [DOI] [PubMed] [Google Scholar]
- [23].Birkenfeld J, Nalbant P, Bohl BP, Pertz O, Hahn KM, Bokoch GM. GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev Cell 2007; 12:699-712; PMID:17488622; http://dx.doi.org/ 10.1016/j.devcel.2007.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yoshizaki H, Ohba Y, Kurokawa K, Itoh RE, Nakamura T, Mochizuki N, Nagashima K, Matsuda M. Activity of Rho-family GTPases during cell division as visualized with FRET-based probes. J Cell Biol 2003; 162:223-32; PMID:12860967; http://dx.doi.org/ 10.1083/jcb.200212049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yonemura S, Hirao-Minakuchi K, Nishimura Y. Rho localization in cells and tissues. Exp Cell Res 2004; 295:300-14; PMID:15093731; http://dx.doi.org/ 10.1016/j.yexcr.2004.01.005 [DOI] [PubMed] [Google Scholar]
- [26].Pusapati GV, Eiseler T, Rykx A, Vandoninck S, Derua R, Waelkens E, Van Lint J, von Wichert G, Seufferlein T. Protein kinase D regulates RhoA activity via rhotekin phosphorylation. J Biol Chem 2012; 287:9473-83; PMID:22228765; http://dx.doi.org/ 10.1074/jbc.M112.339564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ridley AJ. Rho GTPases and cell migration. J Cell Sci 2001; 114:2713-22; PMID:11683406 [DOI] [PubMed] [Google Scholar]
- [28].Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annual Rev Biochem 1999; 68:459-86; PMID:10872457; http://dx.doi.org/ 10.1146/annurev.biochem.68.1.459 [DOI] [PubMed] [Google Scholar]
- [29].Melendez J, Stengel K, Zhou X, Chauhan BK, Debidda M, Andreassen P, Lang RA, Zheng Y. RhoA GTPase is dispensable for actomyosin regulation but is essential for mitosis in primary mouse embryonic fibroblasts. J Biol Chem 2011; 286:15132-7; PMID:21454503; http://dx.doi.org/ 10.1074/jbc.C111.229336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Minoshima Y, Kawashima T, Hirose K, Tonozuka Y, Kawajiri A, Bao YC, Deng X, Tatsuka M, Narumiya S, May WS Jr, et al.. Phosphorylation by aurora B converts MgcRacGAP to a RhoGAP during cytokinesis. Dev Cell 2003; 4:549-60; PMID:12689593; http://dx.doi.org/ 10.1016/S1534-5807(03)00089-3 [DOI] [PubMed] [Google Scholar]
- [31].David MD, Petit D, Bertoglio J. The RhoGAP ARHGAP19 controls cytokinesis and chromosome segregation in T lymphocytes. J Cell Sci 2014; 127:400-10; PMID:24259668; http://dx.doi.org/ 10.1242/jcs.135079 [DOI] [PubMed] [Google Scholar]
- [32].Nishimura Y, Yonemura S. Centralspindlin regulates ECT2 and RhoA accumulation at the equatorial cortex during cytokinesis. J Cell Sci 2006; 119:104-14; PMID:16352658; http://dx.doi.org/ 10.1242/jcs.02737 [DOI] [PubMed] [Google Scholar]
- [33].Zanin E, Desai A, Poser I, Toyoda Y, Andree C, Moebius C, Bickle M, Conradt B, Piekny A, Oegema K. A conserved RhoGAP limits M phase contractility and coordinates with microtubule asters to confine RhoA during cytokinesis. Dev Cell 2013; 26:496-510; PMID:24012485; http://dx.doi.org/ 10.1016/j.devcel.2013.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nature Cell Biology 1999; 1:136-43; PMID:10559899; http://dx.doi.org/ 10.1038/11056 [DOI] [PubMed] [Google Scholar]
- [35].Ito H, Iwamoto I, Morishita R, Nozawa Y, Asano T, Nagata K. Identification of a PDZ protein, PIST, as a binding partner for Rho effector Rhotekin: biochemical and cell-biological characterization of Rhotekin-PIST interaction. Biochem J 2006; 397:389-98; PMID:16646955; http://dx.doi.org/ 10.1042/BJ20052015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Saraste M, Hyvonen M. Pleckstrin homology domains: a fact file. Curr Opin Structural Biol 1995; 5:403-8; PMID:7583640; http://dx.doi.org/ 10.1016/0959-440X(95)80104-9 [DOI] [PubMed] [Google Scholar]
- [37].Wang DS, Shaw G. The association of the C-terminal region of beta I sigma II spectrin to brain membranes is mediated by a PH domain, does not require membrane proteins, and coincides with a inositol-1,4,5 triphosphate binding site. Biochem Biophys Res Commun 1995; 217:608-15; PMID:7503742; http://dx.doi.org/ 10.1006/bbrc.1995.2818 [DOI] [PubMed] [Google Scholar]
- [38].Agromayor M, Martin-Serrano J. Knowing when to cut and run: mechanisms that control cytokinetic abscission. Trends Cell Biol 2013; 23:433-41; PMID:23706391; http://dx.doi.org/ 10.1016/j.tcb.2013.04.006 [DOI] [PubMed] [Google Scholar]
- [39].Gromley A, Yeaman C, Rosa J, Redick S, Chen CT, Mirabelle S, Guha M, Sillibourne J, Doxsey SJ. Centriolin anchoring of exocyst and SNARE complexes at the midbody is required for secretory-vesicle-mediated abscission. Cell 2005; 123:75-87; PMID:16213214; http://dx.doi.org/ 10.1016/j.cell.2005.07.027 [DOI] [PubMed] [Google Scholar]
- [40].Thoresen SB, Campsteijn C, Vietri M, Schink KO, Liestol K, Andersen JS, Raiborg C, Stenmark H. ANCHR mediates Aurora-B-dependent abscission checkpoint control through retention of VPS4. Nat Cell Biol 2014; 16:550-60; PMID:24814515; http://dx.doi.org/ 10.1038/ncb2959 [DOI] [PubMed] [Google Scholar]
- [41].Gai M, Camera P, Dema A, Bianchi F, Berto G, Scarpa E, Germena G, Di Cunto F. Citron kinase controls abscission through RhoA and anillin. Mol Biol Cell 2011; 22:3768-78; PMID:21849473; http://dx.doi.org/ 10.1091/mbc.E10-12-0952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Morin P, Flors C, Olson MF. Constitutively active RhoA inhibits proliferation by retarding G(1) to S phase cell cycle progression and impairing cytokinesis. European J Cell Biol 2009; 88:495-507; PMID:19515453; http://dx.doi.org/ 10.1016/j.ejcb.2009.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kallio MJ, McCleland ML, Stukenberg PT, Gorbsky GJ. Inhibition of aurora B kinase blocks chromosome segregation, overrides the spindle checkpoint, and perturbs microtubule dynamics in mitosis. Curr Biol 2002; 12:900-5; PMID:12062053; http://dx.doi.org/ 10.1016/S0960-9822(02)00887-4 [DOI] [PubMed] [Google Scholar]
- [44].Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, Gerlich DW. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 2009; 136:473-84; PMID:19203582; http://dx.doi.org/ 10.1016/j.cell.2008.12.020 [DOI] [PubMed] [Google Scholar]
- [45].Frame MC, Fincham VJ, Carragher NO, Wyke JA. v-Src's hold over actin and cell adhesions. Nat Rev Mol Cell Biol 2002; 3:233-45; PMID:11994743; http://dx.doi.org/ 10.1038/nrm779 [DOI] [PubMed] [Google Scholar]
- [46].Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol 2004; 5:816-26; PMID:15459662; http://dx.doi.org/ 10.1038/nrm1490 [DOI] [PubMed] [Google Scholar]
- [47].Mizutani K, Ito H, Iwamoto I, Morishita R, Deguchi T, Nozawa Y, Asano T, Nagata KI. Essential roles of ERK-mediated phosphorylation of vinexin in cell spreading, migration and anchorage-independent growth. Oncogene 2007; 26:7122-31; PMID:17486060; http://dx.doi.org/ 10.1038/sj.onc.1210512 [DOI] [PubMed] [Google Scholar]
- [48].Umemoto T, Inomoto T, Ueda K, Hamaguchi M, Kioka N. v-Src-mediated transformation suppresses the expression of focal adhesion protein vinexin. Cancer Letters 2009; 279:22-9; PMID:19217206; http://dx.doi.org/ 10.1016/j.canlet.2009.01.017 [DOI] [PubMed] [Google Scholar]
- [49].Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005; 437:1043-7; PMID:16222300; http://dx.doi.org/ 10.1038/nature04217 [DOI] [PubMed] [Google Scholar]
- [50].Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genetics Dev 2007; 17:157-62; http://dx.doi.org/ 10.1016/j.gde.2007.02.011 [DOI] [PubMed] [Google Scholar]
- [51].Huang YS, Kan MC, Lin CL, Richter JD. CPEB3 and CPEB4 in neurons: analysis of RNA-binding specificity and translational control of AMPA receptor GluR2 mRNA. EMBO J 2006; 25:4865-76; PMID:17024188; http://dx.doi.org/ 10.1038/sj.emboj.7601322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ott C, Lippincott-Schwartz J. Cytokinetic Abscission: Timing the Separation. Curr Biol 2015; 25:R722-4; PMID:26294187; http://dx.doi.org/ 10.1016/j.cub.2015.06.069 [DOI] [PubMed] [Google Scholar]
- [53].Mullins JM, McIntosh JR. Isolation and initial characterization of the mammalian midbody. J Cell Biol 1982; 94:654-61; PMID:7130277; http://dx.doi.org/ 10.1083/jcb.94.3.654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ma HT, Poon RY. Synchronization of HeLa cells. Methods Mol Biol 2011; 761:151-61; PMID:21755447 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.