

Abstract
Pheochromocytoma is a rare tumor originating from the embryonic neural crest and secreting high levels of catecholamines. The average tumor size is approximately 7 cm, and the average weight is approximately 200 g in the previous publications. Sometimes these tumors may be bigger. In this report, a case of multicystic malignant pheochromocytoma with a huge size is presented, which is seldom reported in the past. A 37-year-old male patient was referred to our hospital for etiological investigation of his recently diagnosed hypertension. Contrast-enhanced computed tomography (CT) examination was performed for further evaluation of the lesion and surrounding tissues. The lesion was 18×8×13 cm in size. It had lobulated margins, large cystic components, and peripheral and septal contrast enhancement. The levels of metanephrine, normetanephrine, adrenaline, noradrenaline, vanilmandelic acid, and dopamine were significantly elevated. The patient was prepared for surgery. In the pathological evaluation, the mass weighed 1018 g and was 18×8×13 cm in size. He was diagnosed with malignant pheochromocytoma. After eight months, a CT examination showed a recurrent mass, liver metastasis, and distant metastasis. The patient received chemotherapy and radiotherapy. Multicystic malignant pheochromocytoma may reach huge sizes without causing any symptoms.
Keywords: Giant, malignant, pheochromocytoma
INTRODUCTION
Pheochromocytoma is a rare tumor originating from the embryonic neural crest and secreting high levels of catecholamines (1). Though it may remain silent, it usually causes clinical symptoms such as headache, palpitation, and hypertension due to these secretions. Asymptomatic cases may have a late diagnosis. The average tumor size is approximately 7 cm, and the average weight is approximately 200 g in the previous publications (2). Sometimes these tumors may be bigger (1, 3–8). In this report, a case of multicystic malignant pheochromocytoma with a huge size is presented, which is seldom reported in the past.
CASE PRESENTATION
A 37-year-old male patient was referred to our hospital for etiological investigation of his recently diagnosed hypertension. In his physical examination, a bulky mass was palpated in the left upper quadrant. Blood electrolyte and glucose levels, haemogram, and other biochemical parameters were unremarkable. Ultrasound examination showed a huge multicystic mass lesion in the left adrenal gland, next to the left kidney. Contrast-enhanced computed tomography (CT) was performed for further evaluation of the lesion and surrounding tissues. The lesion was 18 cm×8 cm×13 cm in size. It had lobulated margins, large cystic components and peripheral and septal contrast enhancement. The tail of the pancreas and spleen was anteriorly displaced, and the left kidney was inferiorly displaced. The left adrenal gland was not visible apart from this lesion. Minimally abdominal free fluid was noted; the right adrenal gland was normal, and there was no finding of distant metastasis. There was no evidence of invasion and lymphadenopathy (Figure 1).
Figure 1.
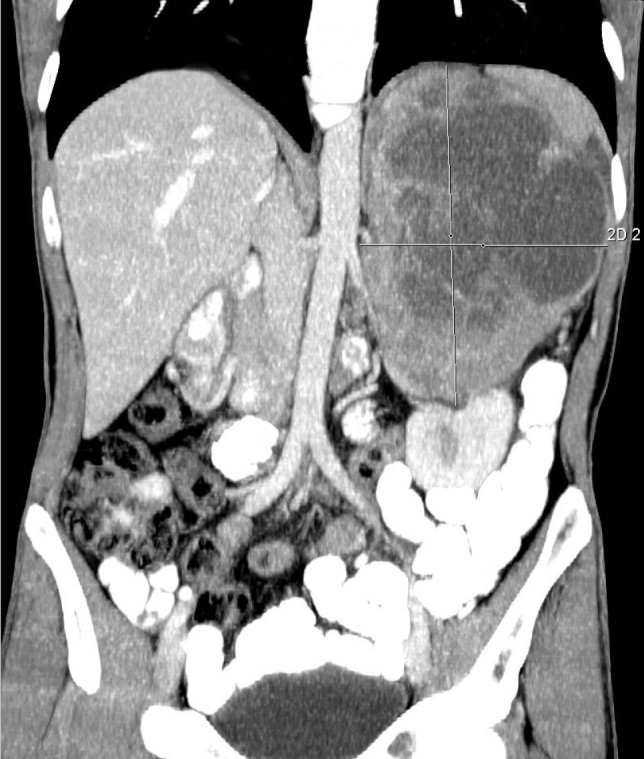
Large pheochromocytoma on computed tomography (coronal section)
Twenty-four hour urine catecholamine levels were studied. The levels of metanephrine, normetanephrine, adrenaline, noradrenaline, vanilmandelic acid, and dopamine were significantly elevated. The 24 h urine levels of metanephrine, normetanephrine, vanilmandelic acid, homovanillate, epinephrine, norepinephrine, and dopamine were 2195 mg/24 h (0–320 mg/24 h), 3707 mg/24 h (0–390 mg/24 h), 33 mg/24 h (1.7–6.5 mg/24 h), 23 mg/24 h (2–7.4 mg/24 h), 65 mg/24 h (1–27 mg/24 h), 154 mg/24 h (1–97 mg/24 h), and 1719 mg/24 h (1–500 mg/24 h), respectively. Blood cortisol level was normal. Blood parathyroid hormone and calcitonin levels were normal. Family history was negative with regards to multiple endocrine neoplasia (MEN) syndrome. The diagnosis of a possibly malignant pheochromocytoma was made. The patient was prepared for surgery; median incision was performed for exploration. The lesion had a close relation to the left lobe of the liver, left kidney, and tail of the pancreas, but there was no invasion. The mass was totally resected. Perioperative or postoperative complication did not develop. The patient was discharged from the hospital on the seventh postoperative day. In the pathological evaluation, the mass weighted 1018 g and was 18 × 8 × 13 cm in size. It showed a high mitotic index and capsular and vascular invasion, and the abdominal fluid sample showed atypical cells leading to the diagnosis of malignant pheochromocytoma. Postoperative 131-I-metaiodobenzylguanidine (MIBG) was administered for treatment. After eight months, a CT examination showed a recurrent mass, which was 12 cm, in the left adrenal gland. There were two liver metastases; one was 9 cm and the other was 1.5 cm in size. Additionally, he had multiple para-aortic, perirenal, and perisplenic enlarged lymph nodes and a small metastatic nodule in the right adrenal gland. There were distant metastases in both lungs and multiple bones, including the lumbosacral vertebrae and right iliac bone on CT. The patient received chemotherapy and radiotherapy. Our patient died eleven months after surgery.
DISCUSSION
Most pheochromocytomas are benign and approximately 10% have malignant behavior (1). With increasing size, the probability of malignancy becomes higher; in lesions over 6 cm, the ratio of benign-to-malignant tumors is 1:8 (9). The average tumor size is approximately 7 cm (2). There are only few reports of huge multicystic malignant pheochromocytoma above 18 cm, and to our knowledge, the pheochromocytoma in this case is the largest one reported from our country (1, 3–8). Seven giant pheochromocytomas larger than 16 cm in current literature have been documented (Table 1). Classical symptoms of pheochromocytoma include headache, palpitation, and hypertension. Some cases may not show any symptoms. The sole reason for the admission of our patient was hypertension. In our case, the lack of prominent symptoms may have resulted in the tumor reaching huge sizes and the late diagnosis.
Table 1.
Pheochromocytomas reported to be larger than 16 cm
| Author/ year | Sex/ age | Country | Size (cm) | Lap/ open | Recurrence | Survival |
|---|---|---|---|---|---|---|
| Grissom et al. (4) | F/54 | USA | 45×25 | Open | Unknown | Unknown |
| Costa et al. (5) | M/46 | Brazil | 30 | Open | No | Died |
| Suga et al. (3) | M/48 | Japan | 21×13 | Open | No | Yes |
| Melegh et al. (6) | M/55 | Hungary | 20 | Open | Unknown | Unknown |
| Ambati et al. (1) | F/77 | Canada | 19×12×18 | Open | No | Yes |
| Pan et al. (7) | M/46 | USA | 18×14×13 | Open | No | Yes |
| Current case | M/37 | Turkey | 18×13 | Open | Yes | Died |
| Costa et al. (5) | F/43 | Brazil | 18 | Open | Yes | Yes |
| Daughtry et al. (8) | M/53 | USA | 17 | Open | Unknown | Unknown |
F: female; M: male; Lap: laparoscopic
Histopathologically, it is not always possible to distinguish benign lesions from malignant ones that do not have an accompanying metastatic lesion or invasion. The presence of capsular and vascular invasion, distant metastasis, high mitotic index and proliferative activity, intra-abdominal malignant-free fluid, and postopererative local recurrence are features of malignancy. Immunohistochemical analysis may be necessary to support the diagnosis. The role of the laparoscopic approach for large tumors is still controversial. In a study, lesion sizes of 12 cm to 14 cm have been cited as the upper limit for laparoscopic adrenalectomy (10). In our case, considering the large size of the tumor and possible malignancy with local invasion, we preferred traditional open surgery. There are not enough studies on the prognosis and survival of malignant pheochromocytoma because of its lower incidence. Malignant pheochromocytoma usually has a poor prognosis and resistance to chemotherapy and radiotherapy. The administration of 131-I-MIBG and long-acting octreotide in patients who are somatostatin receptor positive can be used ın the treatment of malignant pheochromocytoma. In patients with metastatic pheochromocytoma, long-term survival is possible; the overall five-year survival; however, is less than 50%.
When the cases in Table 1 were evaluated, the largest pheochromocytoma in the world was reported to be 45 cm. All patients were operated with the open method. Two cases were benign and six were malignant. One case was uncertain. Mortality and recurrence developed in two malignant cases.
CONCLUSION
Multicystic malignant pheochromocytoma may reach huge sizes without causing any symptoms.
Footnotes
Informed Consent: Written informed consent was obtained from patient who participated in this case.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - E.U., T.K., A.O.G., M.A.İ.; Design - E.U., T.K., A.O.G., M.A.İ.; Supervision - E.U., T.K., A.O.G., M.A.İ.; Funding - E.U., T.K.; Materials - E.U., T.K., M.A.İ; Data Collection and/or Processing - E.U., T.K., M.A.İ.; Analysis and/or Interpretation - E.U., T.K., A.O.G., M.A.İ.; Literature Review - E.U., T.K., A.O.G.; Writer - E.U., T.K., M.A.İ.; Critical Review - E.U., T.K., A.O.G.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study has received no financial support.
REFERENCES
- 1.Ambati D, Jana K, Domes T. Largest pheochromocytoma reported in Canada: A case study and literature review. Can Urol Assoc J. 2014;5–6:E374–377. doi: 10.5489/cuaj.1719. http://dx.doi.org/10.5489/cuaj.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26:551–566. doi: 10.1097/00000478-200205000-00002. http://dx.doi.org/10.1097/00000478-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Suga K, Motoyama K, Hara A, Kume N, Ariga M, Matsunaga N. Tc-99m MIBG imaging in a huge clinically silent pheochromocytoma with cystic degeneration and massive hemorrhage. Clin Nucl Med. 2000;25:796–800. doi: 10.1097/00003072-200010000-00009. http://dx.doi.org/10.1097/00003072-200010000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Grissom JR, Yamase HT, Prosser PR. Giant pheochromocytoma with sarcoidosis. South Med J. 1979;72:1605–1607. doi: 10.1097/00007611-197912000-00035. http://dx.doi.org/10.1097/00007611-197912000-00035. [DOI] [PubMed] [Google Scholar]
- 5.Costa SRP, Cabral NM, Abhrão AT, Costa RB, Silva LM, Lupinacci RA. Giant cystic malignant pheochromocytoma invading right hepatic lobe: Report on two cases. Sao Paulo Med J. 2008;26:229–231. doi: 10.1590/S1516-31802008000400008. http://dx.doi.org/10.1590/S1516-31802008000400008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melegh Z, Rényi-Vámos F, Tanyay Z. Giant cystic pheochromocytoma located in the renal hilus. Pathol Res Pract. 2002;198:103–106. doi: 10.1078/0344-0338-00194. http://dx.doi.org/10.1078/0344-0338-00194. [DOI] [PubMed] [Google Scholar]
- 7.Pan Z, Repertinger S, Deng C, Sharma P. A giant cystic pheochromocytoma of the adrenal gland. Endocr Pathol. 2008;19:133–138. doi: 10.1007/s12022-008-9016-4. http://dx.doi.org/10.1007/s12022-008-9016-4. [DOI] [PubMed] [Google Scholar]
- 8.Daughtry JD, Susan LP, Straffon RA, Stewart BH. A case of a giant pheochromocytoma. J Urol. 1977;118:840–842. doi: 10.1016/s0022-5347(17)58215-4. [DOI] [PubMed] [Google Scholar]
- 9.Alexandraki KI, Grossman AB. Adrenal incidentalomas: ‘the rule of four’. Clin Med. 2008;8:201–204. doi: 10.7861/clinmedicine.8-2-201. http://dx.doi.org/10.7861/clinmedicine.8-2-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zografos GN, Kothonidis K, Ageli C, Kopanakis N, Dimitriou K, Papaliodi E, et al. Laparoscopic resection of large adrenal ganglioneuroma. JSLS. 2007;11:487–492. [PMC free article] [PubMed] [Google Scholar]