

Abstract
The C-terminal selective electrophilic activation of polypeptides is essential for site-specific peptide modification and conjugation techniques such as Native Chemical Ligation (NCL). Peptide C-terminal α-thioesters are particularly valuable precursors for NCL, due to their hydrolytic stability in aqueous buffers and reactivity toward thiol nucleophiles. The synthesis of peptide α-thioesters, however, requires harsh acidic conditions or complex chemical manipulations, which ultimately limits their functional group compatibility and broad utility. Herein, we report a readily accessible N-mercaptoethoxyglycinamide (MEGA) solid-phase linker for the facile synthesis of latent peptide α-thioesters. Incubating peptide-MEGA sequences with 2-mercaptoethanesulfonic acid at mildly acidic pH yielded α-thioesters that were directly used in NCL without purification. The MEGA linker yielded robust access to peptide α-thioesters ranging in length from 4 to 35 amino acids, and greatly simplified the synthesis of cyclic peptides. Finally, the high utility of MEGA was demonstrated by the one-pot synthesis of a functional analog of the Sunflower Trypsin Inhibitor 1.
Graphical Abstract
Native chemical ligation (NCL) is a powerful tool for the total synthesis and semisynthesis of full-length proteins with site-specific post-translational modifications.1 Successful applications of NCL require access to an N-terminal Cys-containing peptide fragment and a peptide C-terminal α-thioester. After an initial transthioesterification, whereby the Cys side-chain displaces the thiol from the C-terminal thioester fragment, a spontaneous S-to-N acyl shift leads to the thermodynamically stable amide bond (Figure 1).
Figure 1.
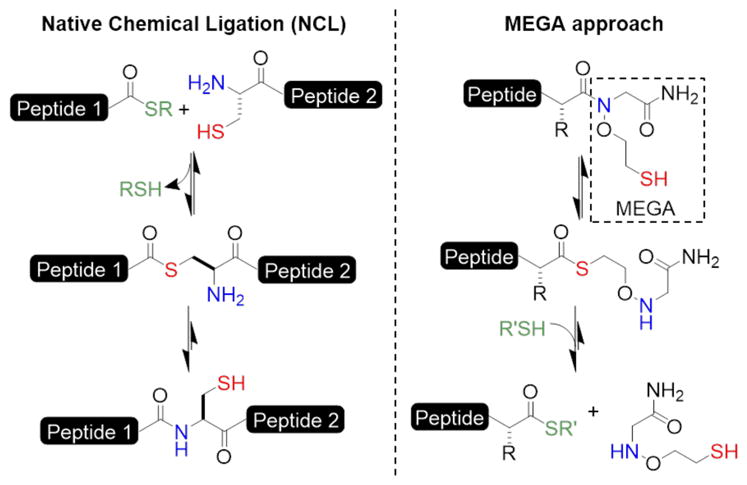
The principle of Native Chemical Ligation (NCL) and the N-mercaptoethoxyglycinamide (MEGA) approach to peptide thioesterification.
Peptide fragments bearing an N-terminal Cys are readily obtained by solid phase peptide synthesis (SPPS) using a 9-fluorenylmethoxycarbonyl (Fmoc-) a-amine protecting group strategy,2–4 or by heterologous expression in Escherichia coli.5 In contrast, the direct synthesis of peptide α-thioesters by Fmoc-chemistry is limited by their inherent lability toward the organic bases employed for Fmoc-deprotection. Peptide α-thioesters may indeed be synthesized with a C-terminal thiol resin-linker using the tert-butyloxycarbonyl (Boc-) a-amine protecting group strategy.6–7 However, applications of Boc-chemistry have several limitations, including incompatibility with specific phosphorylated8 and glycosylated amino acids9 and the use of HF gas for peptide cleavage from the solid-phase.10 Recently, Kent and coworkers reported trifluoromethanesulfonic acid (TFMSA) as an alternative to HF, but the broad utility and functional group compatibility of TFMSA is currently unknown.11
Most common strategies to generate peptide a-thioesters use Fmoc-chemistry in conjunction with multi-step manipulation after SPPS,12–13 or modification of the solid-phase linker prior to peptide assembly,14–15 each with its inherent limitations and synthetic challenges.16 Several thioesterification strategies utilize modified C-terminal amino acids,14 or strongly acidic conditions and elevated temperatures with Cys17 to favor intramolecular N-to-S acyl shift of the backbone amide bond, followed by transthioesterification with external thiols. Functionalized resins containing alkyl thiols that are suitably poised for nucleophilic attack at the C-terminal amide bond,18 also known as crypto-thioesters,19 hold promise due to the minimal chemical manipulation required post-SPPS.20 The complex chemistry required to install crypto-thioesters, however, limits their accessibility to a handful of laboratories. Therefore, efforts to expand the utility of NCL would benefit from facile and high-yielding Fmoc-based strategies to access peptide α-thioesters.
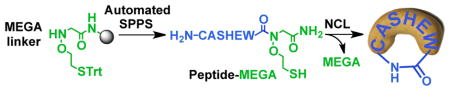
Herein we report a novel and versatile N-mercaptoethoxyglycinamide (MEGA) linker approach for the facile synthesis of peptide α-thioesters. The MEGA approach was inspired by our serendipitous discovery that NCL products containing the ligation auxiliary, 2-(aminooxy)ethanethiol, may undergo thiolysis at room temperature to regenerate the initial reactive α-thioester fragment (Figure 1). We demonstrate that peptides synthesized with the C-terminal MEGA linker are readily converted to their α-thioester form under mildly acidic conditions and can be directly applied toward NCL without further purification. Importantly, the MEGA linker strategy is compatible with a wide-range of C-terminal amino acids, including sterically demanding β-branched amino acids. Finally, MEGA is particularly useful for the one-pot synthesis of cyclic peptides, as shown by our synthesis of a functional analog of the Sunflower Trypsin Inhibitor-1 (SFT-1).21
The MEGA linker was assembled in two steps prior to peptide synthesis. Briefly, commercially available Rink amide resin was condensed with bromoacetic acid, and the bromoacetylated resin incubated with S-trityl protected 2-(aminooxy)ethanethiol (1) to obtain the MEGA linked resin (Table 1). Compound 1 was synthesized in multi-gram quantities in three high-yielding steps with a single column purification.22 Next, we explored conditions for coupling the first amino acid to the secondary amine of MEGA. DIC-mediated coupling with the additive ethyl (hydroxy-imino)cyanoacetate (Oxyma) sufficed for sterically unhindered amino acids-Gly and Ala. Consistent with previously reported conditions for coupling onto secondary amines, coupling reagents such as HATU or bis(trichloromethyl) carbonate (BTC) were required for other amino acids.23 However, this did not result in significant racemization during resin loading, as seen from the high purity of crude peptides released from the resin (Supplementary Figure S1).
Table 1.
Synthesis of AWKX-MEGA peptides
| |||||
|---|---|---|---|---|---|
| Entry | X | Crude Puritya [%] | Isolated Yieldb [%] | Calcd. MW | Obsd. MWc |
| 1 | K | 85 | 51 | 664.8 | 664.6 |
| 2 | Q | 85 | 47 | 664.8 | 664.8 |
| 3 | V | 75 | 47 | 635.8 | 635.5 |
| 4 | G | 71 | 45 | 593.7 | 593.4 |
| 5 | A | 77 | 44 | 607.7 | 607.4 |
| 6 | L | 79 | 43 | 649.8 | 649.5 |
| 7 | S | 72 | 42 | 623.7 | 623.6 |
| 8 | F | 89 | 33 | 683.8 | 683.6 |
| 9 | C | 44 | 31 | 639.8 | 639.5 |
| 10 | R | 40 | 20 | 692.9 | 692.5 |
| 11 | T | 79 | 19 | 637.8 | 637.5 |
| 12 | D | 62 | 18 | 651.8 | 651.4 |
| 13 | D-A | 82 | 56 | 607.7 | 607.4 |
| 14 | D-C | 58 | 43 | 639.8 | 639.5 |
Purity of peptide based on RP-HPLC peak integration at 280 nm.
Isolated yield based on 0.05 mmol scale synthesis.
ESI-MS [M+H]+ ion.
After coupling the first amino acid, a series of 4-mer peptides with the sequence AWKX-MEGA was synthesized to assess the C-terminal amino acid compatibility of MEGA (Table 1, Supplementary Figure S2). Acidolytic cleavage from the resin and purification by RP-HPLC yielded good quantities of 14 different AWKX-MEGA peptides. Importantly, the high purity of each crude peptide indicated that the N-O bond was stable throughout manual peptide synthesis and cleavage from the solid support (Table 1, Supplementary Figure S1). AWKX-MEGA peptides were subsequently tested for the production of isolable α-thioesters. We envisioned that thioester formation from the N-oxyamide 2 (Figure 2A) may proceed by an N-to-S acyl shift to form the rearranged thioester 3. Collapse of the tetrahedral intermediate provides the initial thioester 3 and addition of an excess of external thiol promotes thiol exchange to generate a stable, isolable thioester 4 (Figure 2A). Although thioesterification proceeds through a 6-membered cyclic intermediate, rather than the 5-membered intermediate for N-alkylated Cys14 or bis-sulfanylethylamide-based strategies,15 the good aminooxy leaving group enabled thioesterification at mildly acidic pH and room temperature for the AWKG-MEGA peptide (Supplementary Figure S3A–B). This is in contrast with pH ~1 required for thioesterification from N-alkylated Cys.14
Figure 2.
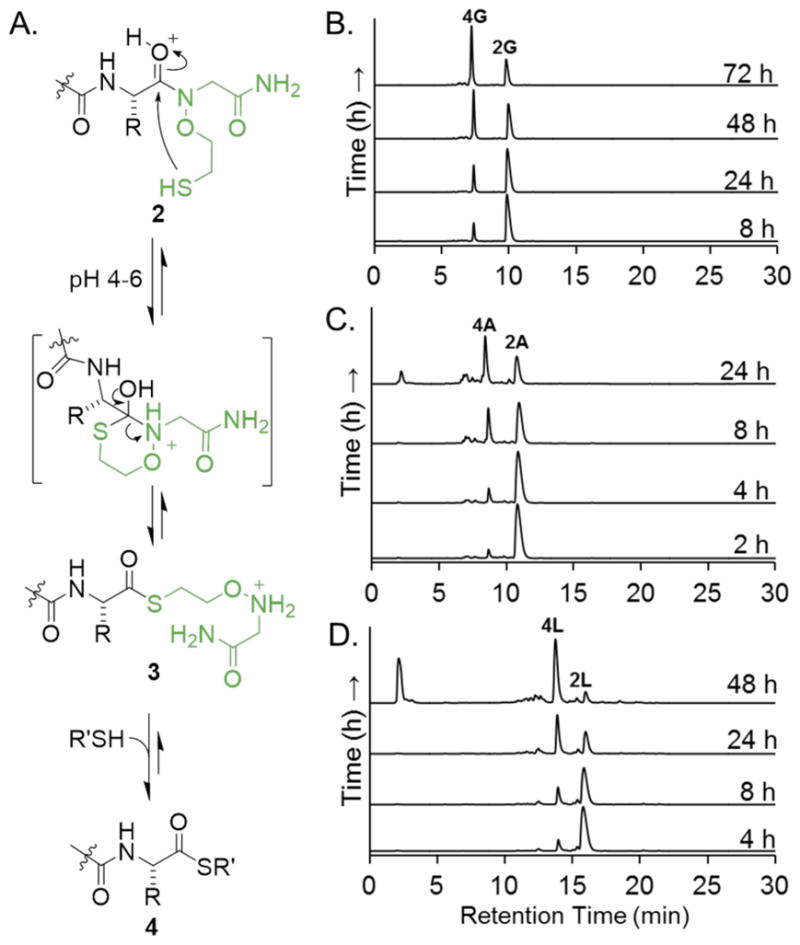
Generation of AWKX-MESNa thioesters. A. Proposed mechanism of thioester formation from peptide-MEGA. B-D. C18 RP-HPLC time-course of thioester formation for AWKG-MEGA (B), AWKA-MEGA (C) and AWKL-MEGA (D) peptides. Buffer: 200–400 mM MESNa, 100 mM NaH2PO4, 25–50 mM TCEP. RP-HPLC: 10–60% CH3CN in water, 30 min gradient.
Thioesterification conditions were further optimized for various AWKX-MEGA peptides. We observed that the nature of the C-terminal amino acid, thiol nucleophile, pH, and reaction temperature significantly influenced thioester yields (Figure 2B–D and Table 2). Transthioesterification was particularly sensitive to the external thiol employed. While the sodium salt of 2-mercaptoethanesulfonic acid (MESNa) led to the highest yields for all amino acids tested, other commonly employed thiols such as 3-mercaptopropionic acid and 2,2,2-trifluoroethanethiol did not yield significant quantities of the corresponding α-thioesters (Supplemental Figures S3C–S3D). Thioesterification yields were best under mildly acidic conditions (pH 4.0–6.0), and no reaction was observed at pH < 4. Mildly basic reaction conditions, pH 7.0–8.0, yielded a mixture of thioester and hydrolyzed products at elevated temperatures (Supplemental Figure S3E). Consistent with previous reports with N-alkylated Cys,14 AWKX-MEGA peptides exhibited slow thioesterification kinetics at room temperature when X was not Gly. However, elevated temperatures improved yields for all peptide thioesters (Table 2 and Supplementary Figure S4). Even for the AWKG-MEGA peptide, increasing the reaction temperature from 25 °C to 37 °C led to a concomitant increase in thioester yield from 29% to 60% (Supplementary Figure S5).
Table 2.
Optimized thioesterification conditions for AWKX-MEGA peptides.
| Entry | X | Temperature (°C) | pH | Reaction Time (h) | Yield (%)a |
|---|---|---|---|---|---|
| 1 | L | 70 | 4.5 | 48 | 67.8 |
| 2 | K | 70 | 4.5 | 48 | 67.5 |
| 3 | F | 70 | 4.5 | 48 | 65.8 |
| 4 | G | 37 | 5.6 | 72 | 60.5 |
| 5 | D-A | 70 | 5.6 | 24 | 57.1 |
| 6 | Q | 50 | 4.5 | 72 | 48.8 |
| 7 | A | 70 | 5.6 | 24 | 45.2 |
| 8 | T | 70 | 4.5 | 48 | 43.1 |
| 9 | S | 50 | 4.5 | 48 | 42.8 |
| 10 | R | 70 | 5.6 | 72 | 41.1 |
| 11 | D-C | 50 | 4.5 | 48 | 38.3 |
| 12 | D | 50 | 5.6 | 8 | 32.5 |
| 13 | C | 50 | 4.5 | 48 | 32.5 |
| 14 | V | 70 | 5.6 | 72 | 26.3 |
Based on RP-HPLC peak integration at 280 nm.
Steric hindrance in β-branched C-terminal amino acids drastically reduces reaction rates during NCL.7 Hence, we tested AWKV-MEGA and AWKT-MEGA in thioesterification reactions. While Val produced only a modest 26% yield, Thr generated significantly more of the corresponding α-thioester (Table 2, entries 8 and 14). We surmise that hydrogen bonding with the hydroxyl group of Thr may limit conformational freedom in the MEGA side-chain and enhance its reactivity. The lack of hydrogen bonding with the Val side-chain precludes such conformational stabilization and results in lower yields. Overall, β-branching in the C-terminal amino acid proved a greater hindrance toward thioesterification than absolute bulk of the side-chain, as seen by the good yields for AWKF and AWKL thioesters in comparison with AWKV (Table 2, entries 1, 3 and 14).
Surprisingly, although the AWKD-MEGA peptide showed good initial kinetics of thioesterification, we observed a steep drop in thioester product recovered after >8 h (Supplementary Figure S6). MS characterization of the reaction products at 24 h suggested the formation of a C-terminal aspartic anhydride, followed by its hydrolysis to aspartate (Supplementary Figure S7). This suggests that some C-terminal amino acids with nucleophilic side-chains may cyclize upon incubation at elevated temperatures for extended periods of time. Gratifyingly, however, a peptide with C-terminal Lys produced the corresponding thioester in good yield (Table 2, entry 2). Finally, we also addressed the potential for C-terminal epimerization during thioesterification. The D-epimers of AWKA-MEGA and AWKC-MEGA were synthesized and converted to their respective thioesters (Table 1 and 2, and Supplementary Figure S8). Cys was chosen as it is particularly prone to racemization in its activated form.24 All diastereomeric thioesters displayed different RP-HPLC retention times upon co-injection, and to our delight, an average of <1% epimerization was observed throughout the time-course of thioesterification. Thus, the MEGA linker approach leads to minimal epimerization for Ala and Cys, and hence good overall thioesterification yields.
To test the utility of MEGA for longer sequences, the 35-mer peptide p53(1–35)-MEGA was prepared via microwave-assisted automated SPPS on a Liberty Blue peptide synthesizer (CEM corporation, Matthews, NC) (Supplementary Figure S9A). Initial attempts revealed that 90 °C coupling and deprotection cycles with 20% (v/v) piperidine in DMF led to significant N-O bond cleavage and reduced yields. We addressed this issue by decreasing coupling and deprotection temperatures to 50 °C, and employed 5% (w/v) piperazine and 0.1 M HOBt in DMF as a less basic mixture for Fmoc-deprotection. Under these optimized conditions N-O bond cleavage was completely eliminated from the crude peptide product and pure p53(1–35)-MEGA was obtained in 22% isolated yield (Supplementary Figures S9B–C). The final peptide was subjected to thioesterification and generated the corresponding MESNa thioester in 75% yield (Supplementary Figures S9D-E). The fact that MEGA-linked resin is fully compatible with microwave-assisted SPPS will facilitate the synthesis of otherwise challenging sequences in higher yields and purity.25
With the successful synthesis of thioesters of varying lengths, we next investigated the scope for one-pot NCL between AWKX-MEGA peptides and an N-terminal Cys-containing peptide, CASW (5) (Figure 3A, Supplementary Figure S10). First, AWKX-MEGA thioesterification was undertaken with optimized conditions (Table 2), followed by the addition of 5 at pH 7.5. NCL proceeded rapidly at room temperature to generate the peptide AWKXCASW (6) in 1 h with minimal thioester hydrolysis (Figure 3B–D). The ability to directly use thioesters generated with MEGA in NCL, without intermediate purification, is particularly attractive for higher overall yields of ligation products.
Figure 3.
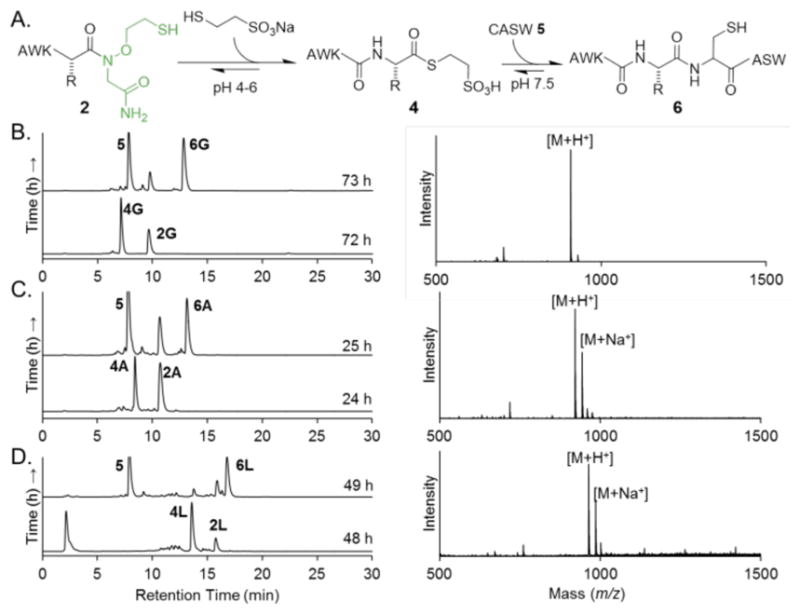
One-pot thioesterification of AWKX-MEGA and native chemical ligation (NCL) with CASW (5). A. Scheme for one-pot NCL of AWKX-MEGA peptides. B. C18 RP-HPLC time-course of NCL for AWKG-MEGA (left) and ESI-MS of isolated AWKGCASW (right). Calcd. [M+H]+ 907.6 Da, obsd. 907.9 Da. C. C18 RP-HPLC time-course of NCL for AWKA-MEGA (left) and ESI-MS of isolated AWKACASW (right). Calcd. [M+H]+ 921.9 Da, obsd. 921.7 Da. D. C18 RP-HPLC time-course of NCL for AWKL-MEGA (left) and ESI-MS of isolated AWKLCASW (right). Calcd. [M+H]+ 963.2 Da, obsd. 963.8 Da. RP-HPLC: 10–60% CH3CN in water, 30 min gradient.
Following successful one-pot intermolecular NCL with MEGA, we envisioned its application for intramolecular NCL to access head-to-tail cyclized peptides. Cyclic peptides are useful scaffolds in therapeutic discovery efforts due to their conformational rigidity, which entropically favors enhanced target binding compared to linear peptides. Additionally, the lack of free N- and C-termini in cyclic peptides confers resistance to exopeptidases and enhances membrane permeation.26 To this end, we synthesized two short peptides, CASHEW-MEGA and CRGD(D-F)-MEGA (Supplementary Figures S11–S12). The cyclic form of CRGD(D-F) binds the integrin αVβ3 receptor with nanomolar affinity.27 Both peptides were prepared by automated SPPS in overall isolated yields of 56% and 45%, respectively (Supplementary Figure 13). The peptides were subjected to one-pot thioesterification and cyclization and both reactions proceeded to completion in < 8 h with no significant side-products detected by HPLC and MS (Supplementary Figure S14). Although the synthesis of cyclic peptides by MEGA requires the presence of an N-terminal thiol for ligation, the ability to desulfurize Cys,28 and other thiol-containing Cys surrogates,29 post-cyclization will provide facile access to a wide range of cyclic peptides that may not have Cys in their primary sequences.
We also assessed the extent of epimerization during peptide cyclization. C-terminally activated His residues are particularly prone to epimerization due to the proximity of the imidazole side-chain to the backbone a-hydrogen.30 Therefore we synthesized two epimeric peptides, CLAS(D-H)-MEGA and CLAS(L-H)-MEGA, and measured their degree of epimerization during one-pot cyclization (Supplementary Figure S15). The diastereomeric cyclic CLASH peptides were well-resolved by RP-HPLC (Supplementary Figure S16A–D) and we found that CLAS(L-H)-MEGA epimerized <2% after 24 h (Supplementary Figure S16E).
Encouraged by our excellent results with short cyclic peptides, we applied the MEGA strategy toward a challenging target-the Sunflower Trypsin Inhibitor-1 (SFT-1). SFT-1 is a 14-mer cyclic peptide that is structurally constrained by three Pro residues, an extensive hydrogen bond network and also contains one internal disulfide.21 SFT-1 is the smallest Bowman-Birk type trypsin inhibitor and analogs of SFT-1 are active against a range of proteases including matriptase and kallikreins (Figure 4A). One potent analog of SFT-1, the I10G mutant, was previously obtained using manual peptide synthesis and Boc-chemistry.31 In order to simplify access to SFT-1(I10G), we synthesized the linear sequence 14-mer peptide CFPDGRCTKSIPPG-MEGA by automated SPPS (Figure 4B and Supplementary Figure 17). The purified peptide was incubated in thioesterification buffer for 24 h at 50 °C to provide the cyclized product in 30% isolated yield after RP-HPLC purification (Supplementary Figure S18). The cyclized peptide was then quantitatively oxidized to the disulfide form by incubation in ammonium bicarbonate buffer overnight (Figure 4C). Finally, we tested SFT-1 (I10G) for inhibitory activity against bovine Trypsin by employing the well-characterized substrate N-(a)-benzoyl-L-arginine-4-nitroanilide (BAPNA). Trypsin hydrolyzes BAPNA to release the yellow colored 4-nitroaniline, which is easily detected by its absorbance at 410 nm. Assays were conducted in 96-well plate format with 100 nM Trypsin, 500 μM BAPNA and varying concentration of SFT-1 (I10G). Consistent with previous reports, we observed a robust dose-response curve with IC50 = 150.2 ± 1.1 nM (Figure 4D). Thus, our Fmoc-compatible and automated approach simplified access to a bioactive cyclic peptide.
Figure 4.
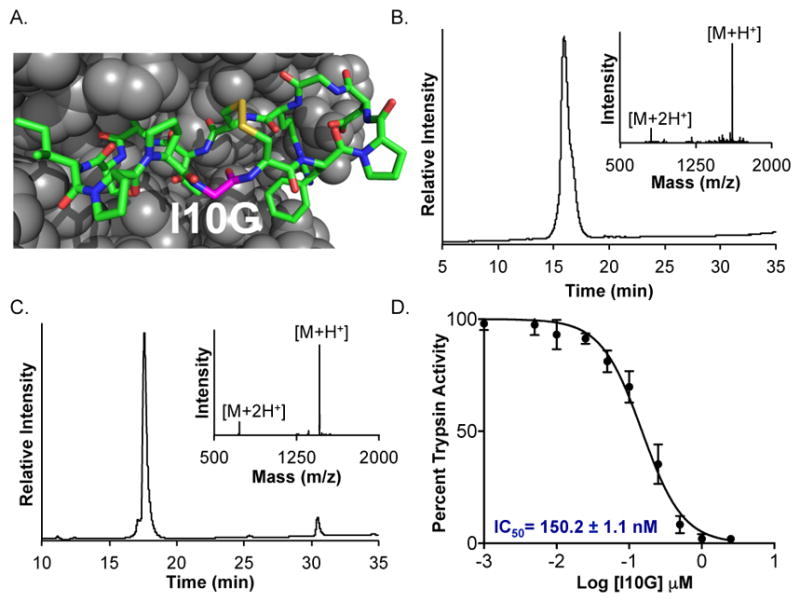
One-pot synthesis and trypsin inhibitory activity of SFT-1 (I10G). A. Model of SFT-1(I10G) (stick representation) based on the X-ray structure of SFT-1 bound to bovine trypsin (gray spheres). PDB code 1SFI. B. C18 RP-HPLC of SFT-1(I10G)-MEGA peptide. Inset is the ESI-MS of SFT-1(I10G)-MEGA; Calcd. [M+H+] 1,609.9 Da, obsd. 1,609.8 ± 0.2 Da. C. C18 RP-HPLC of cyclized and oxidized SFT-1 (I10G) product. Inset is the ESI-MS of purified SFT-1 (I10G); Calcd. [M+H+] 1,458.7 Da, obsd. 1,458.6 ± 0.3 Da. D. Dose-response curve for the inhibition of bovine trypsin activity by SFT-1 (110G). RP-HPLC: 0–73% CH3CN in water, 30 min gradient.
In conclusion, we have applied our serendipitous discovery that NCL products containing the ligation auxiliary 2-(aminooxy)ethanethiol may undergo thiolysis to develop a synthetically facile and robust strategy for accessing peptide α-thioesters. The MEGA linker uses readily accessible starting materials and yields latent peptide α-thioesters directly applicable to one-pot inter- and intramolecular NCL. It is compatible with a wide-range of C-terminal amino acids and peptide lengths, and is particularly useful for the synthesis of cyclic peptides. Compatibility with microwave-assisted automated SPPS, demonstrated by our synthesis of a 35-mer p53 N-terminal peptide, is especially exciting for applications of the MEGA linker in the total chemical synthesis of post-translationally modified proteins.
Supplementary Material
Acknowledgments
We are grateful to the Department of Chemistry at the University of Washington for generous support. C.C. acknowledges the National Institutes of Health, grant 1R01M110430. C.E.W. gratefully acknowledges support from the NSF GRFP (grant number DGH-1256082) and an ARCS foundation fellowship.
Footnotes
Notes
The authors declare no competing financial interests.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Science. 1994;266:776. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 2.Flavell RR, Muir TW. Acc Chem Res. 2009;42:107. doi: 10.1021/ar800129c. [DOI] [PubMed] [Google Scholar]
- 3.Dhall A, Chatterjee C. ACS Chem Biol. 2011;6:987. doi: 10.1021/cb200142c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weller CE, Pilkerton ME, Chatterjee C. Biopolymers. 2014;101:144. doi: 10.1002/bip.22253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erlanson DA, Chytil M, Verdine GL. Chem Biol. 1996;3:981. doi: 10.1016/s1074-5521(96)90165-9. [DOI] [PubMed] [Google Scholar]
- 6.Camarero JA, Cotton GJ, Adeva A, Muir TW. J Pept Res. 1998;51:303. doi: 10.1111/j.1399-3011.1998.tb00428.x. [DOI] [PubMed] [Google Scholar]
- 7.Hackeng TM, Griffin JH, Dawson PE. Proc Natl Acad Sci USA. 1999;96:10068. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Otvos L, Elekes I, Lee VMY. Int J Pept Protein Res. 1989;34:129. doi: 10.1111/j.1399-3011.1989.tb01501.x. [DOI] [PubMed] [Google Scholar]
- 9.Gamblin DP, Scanlan EM, Davis BG. Chem Rev. 2009;109:131. doi: 10.1021/cr078291i. [DOI] [PubMed] [Google Scholar]
- 10.Muttenthaler M, Albericio F, Dawson PE. Nat Protoc. 2015;10:1067. doi: 10.1038/nprot.2015.061. [DOI] [PubMed] [Google Scholar]
- 11.Gates ZP, Dhayalan B, Kent SB. Chem Commun. 2016;52:13979. doi: 10.1039/c6cc07891e. [DOI] [PubMed] [Google Scholar]
- 12.Zheng JS, Tang S, Huang YC, Liu L. Acc Chem Res. 2013;46:2475. doi: 10.1021/ar400012w. [DOI] [PubMed] [Google Scholar]
- 13.Blanco-Canosa JB, Nardone B, Albericio F, Dawson PE. J Am Chem Soc. 2015;137:7197. doi: 10.1021/jacs.5b03504. [DOI] [PubMed] [Google Scholar]
- 14.Erlich LA, Kumar KS, Haj-Yahya M, Dawson PE, Brik A. Org Biomol Chem. 2010;8:2392. doi: 10.1039/c000332h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ollivier N, Dheur J, Mhidia R, Blanpain A, Melnyk O. Org Lett. 2010;12:5238. doi: 10.1021/ol102273u. [DOI] [PubMed] [Google Scholar]
- 16.Mong SK, Vinogradov AA, Simon MD, Pentelute BL. ChemBioChem. 2014;15:721. doi: 10.1002/cbic.201300797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang J, Reynolds NL, Tyrrell C, Dorin JR, Macmillan D. Org Biomol Chem. 2009;7:4918. doi: 10.1039/b913886b. [DOI] [PubMed] [Google Scholar]
- 18.Taichi M, Hemu X, Qiu Y, Tam JP. Org Lett. 2013;15:2620. doi: 10.1021/ol400801k. [DOI] [PubMed] [Google Scholar]
- 19.Sato K, Shigenaga A, Tsuji K, Sumikawa Y, Sakamoto K, Otaka A. ChemBioChem. 2011;12:1840. doi: 10.1002/cbic.201100241. [DOI] [PubMed] [Google Scholar]
- 20.Tailhades J, Patil NA, Hossain MA, Wade JD. J Pept Sci. 2015;21:139. doi: 10.1002/psc.2749. [DOI] [PubMed] [Google Scholar]
- 21.Korsinczky ML, Schirra HJ, Craik DJ. Curr Protein Pept Sci. 2004;5:351. doi: 10.2174/1389203043379594. [DOI] [PubMed] [Google Scholar]
- 22.Weller CE, Huang W, Chatterjee C. ChemBioChem. 2014;15:1263. doi: 10.1002/cbic.201402135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patgiri A, Witten MR, Arora PS. Org Biomol Chem. 2010;8:1773. doi: 10.1039/c000905a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han Y, Albericio F, Barany G. J Org Chem. 1997;62:4307. doi: 10.1021/jo9622744. [DOI] [PubMed] [Google Scholar]
- 25.Bacsa B, Kappe CO. Nat Protoc. 2007;2:2222. doi: 10.1038/nprot.2007.300. [DOI] [PubMed] [Google Scholar]
- 26.Wang CK, Craik DJ. Biopolymers. 2016;106:901. doi: 10.1002/bip.22878. [DOI] [PubMed] [Google Scholar]
- 27.Prante O, Einsiedel J, Haubner R, Gmeiner P, Wester HJ, Kuwert T, Maschauer S. Bioconjug Chem. 2007;18:254. doi: 10.1021/bc060340v. [DOI] [PubMed] [Google Scholar]
- 28.Yan LZ, Dawson PE. J Am Chem Soc. 2001;123:526. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]
- 29.Noisier AFM, Albericio F. Amino Acids Pept Prot. 2014;39:1. [Google Scholar]
- 30.Jones JH, Ramage WI, Witty MJ. Int J Pept Protein Res. 1980;15:301. doi: 10.1111/j.1399-3011.1980.tb02581.x. [DOI] [PubMed] [Google Scholar]
- 31.Quimbar P, Malik U, Sommerhoff CP, Kaas Q, Chan LY, Huang YH, Grundhuber M, Dunse K, Craik DJ, Anderson MA, Daly NL. J Biol Chem. 2013;288:13885. doi: 10.1074/jbc.M113.460030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.