

Summary
The discovery of ubistatins, small molecules that impair proteasomal degradation of proteins by directly binding to polyubiquitin, makes ubiquitin itself a potential therapeutic target. Although ubistatins have the potential for drug development and clinical applications, the lack of structural details of ubiquitin-ubistatin interactions has impeded their development. Here, we characterized a panel of new ubistatin-derivatives using functional and binding assays. The structures of ubiquitin complexes with ubistatin-B and hemi-ubistatin revealed direct interactions with ubiquitin's hydrophobic surface-patch and the basic/polar residues surrounding it. Ubistatin-B binds ubiquitin and diubiquitin tighter than a high-affinity ubiquitin-receptor and shows strong preference for K48-linkages over K11 and K63. Furthermore, ubistatin-B shields ubiquitin conjugates from disassembly by a range of deubiquitinases and by the 26S-proteasome. Finally, ubistatin-B penetrates cancer cells and alters the cellular ubiquitin landscape. These findings highlight versatile properties of ubistatins and have implications for their future development and use in targeting ubiquitin-signaling pathways.
Graphical abstract
characterize a panel of ubistatin derivatives and show that ubistatins inhibit ubiquitination and shield ubiquitin-conjugates from disassembly by a range of deubiquitinases and by the 26S proteasome. Ubistatin B penetrates cancer cells and alters cellular ubiquitin landscape. The structures of ubiquitin complexes with ubistatin B and hemi-ubistatin revealed that hydrophobic and charge/polar interactions are critical for ubistatin:ubiquitin binding.
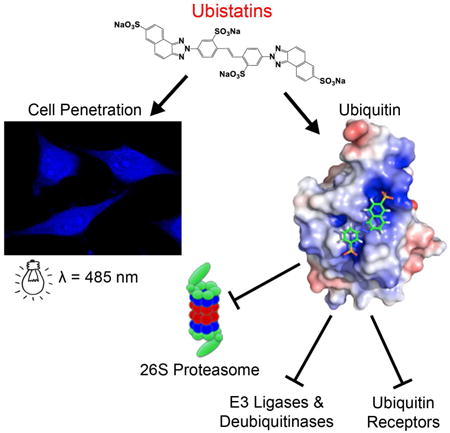
Introduction
The ubiquitin-proteasome system (UPS) is the principal negative regulatory mechanism for short-lived proteins in eukaryotes (Hershko and Ciechanover, 1998). Proteins destined for degradation are post-translationally tagged with ubiquitin (Ub) or a chain of Ub molecules (polyUb) attached to a lysine side chain on a protein through the action of the E1, E2, E3 enzyme cascade (Pickart, 1997). Provided the protein is appropriately modified with Ub, shuttling factors facilitate its transport to the 26S proteasome where dedicated receptors initiate its degradation (Finley et al., 2012; Pickart, 1997).
In eukaryotic organisms deficiencies in components of the UPS – such as E3 Ub ligases, essential subunits of the 26S proteasome, and even mutations in the Ub molecule itself – have severe consequences, including cell death and the onset of disease(Finley et al., 2012; Pickart and VanDemark, 2000; Roscoe et al., 2013). There is currently an intensive effort underway to develop therapeutics for targets upstream of the proteasome, including ubiquitin-selective chaperone p97/VCP, E3 ubiquitin ligases and their regulators, and deubiquitinases (DUBs) (Deshaies, 2014; Skaar et al., 2014). Nevertheless, clinically approved E3 ligase and DUB inhibitors are lacking. Thus far, peptide-based proteasome inhibitors (such as bortezomib and carfilzomib) which bind to the proteolytic β-subunits in the 20S core are the only clinically approved therapeutics targeting the UPS (Cvek, 2012). Because malignancies such as multiple myeloma require enhanced UPS function, these cells are particularly susceptible to proteasome inhibitors (Goldberg, 2012). However, through mechanisms that are not clear, some patients relapse and become refractory to proteasome inhibitors(Ruschak et al., 2011). Unlike the enzymatic components of the UPS (proteasomal subunits, E3 Ub ligases, DUBs), Ub is significantly less likely to tolerate mutations and therefore is potentially a more robust target for therapeutics.
Ubistatins are a family of small molecules that inhibit degradation of polyubiquitinated proteins by the UPS (Verma et al., 2004). By directly binding to polyUb, ubistatins act upstream of the proteasome, preventing recognition by downstream polyUb receptors. Specifically, ubistatins A and B outcompete the proteasomal polyUb receptor Rpn10 and the shuttle protein Rad23 for binding to polyUb chains. Direct binding of ubistatin A to K48-linked di-Ub (K48-Ub2) was detected and mapped to hydrophobic-patch residues (L8, I44, V70) on the surface of Ub(Verma et al., 2004). A subsequent study examined how receptor:Ub interactions were altered by ubistatin in spliceosome assembly assays (Bellare et al., 2008). Although it was established that ubistatin A binds polyUb, the actual mechanism of interactions of ubistatin A and other ubistatin variants with (poly)Ub remained unclear. The lack of high-resolution structural data for the ubistatin:Ub interaction was a significant impediment to our understanding of the mechanism of inhibition and to further development of ubistatins for potential clinical applications.
To this end, a panel of ubistatin B derivatives was synthesized and characterized using functional and NMR-based binding assays. We discovered that the most active compounds contain strongly acidic groups. These efforts led us to focus on ubistatin B and its corresponding hemi-ubistatin. Unexpectedly, ubistatin B binds two Ub molecules simultaneously, while hemi-ubistatin binds Ub significantly weaker and in a 1:1 stoichiometry. We next determined the structure of hemi-ubistatin:Ub complex by NMR, revealing important contributions from key charged groups in Ub. Combined with data from small-angle neutron scattering (SANS), we generated a model of the ubistatin B:Ub complex. Further, we demonstrated that ubistatins inhibit a range of deubiquitinases, including those embedded within the 26S proteasome. Finally, we found that ubistatin B penetrates cell membranes and alters the cellular ubiquitin landscape. These data provide a gateway for future development of more potent pre-clinical candidates to treat cancer cells addicted to an elevated requirement for the UPS.
Results
Synthesis and initial functional screening of ubistatin derivatives
Ubistatin B (ubiB) derivatives (1-8) were synthesized to assess the contribution of substituent groups to Ub binding (Fig 1A). External and internal sulfonic acid groups were either removed or replaced with a less acidic carboxylic acid group. To address the contribution of the two symmetric ring systems to Ub binding, we synthesized “hemi”-ubistatin compounds, 9 (h-ubiB) and 10, containing only a single ring system, as well as compound 11 containing a central bis-sulfonic acid motif (Figs 1B).
Figure 1.

Chemical structures of various synthesized ubistatin derivatives and the development of a structure-activity relationship. (A) Full-ubistatin compounds and (B) hemi-ubistatins. (C) The effect of ubistatin variants on the DUB activity of Rpn11 in purified proteasome: percent inhibition measured at 10 μM (left) and IC50 (right). Data represent the mean and standard deviation (error bars) determined from multiple measurements. See also Table S1. (D) In vitro CFTR ubiquitination reactions in the presence of the indicated ubistatin compounds. Ubiquitinated CFTR (top) is quantified relative to the amount of HA-tagged CFTR protein present in the immunoprecipitate (middle). The results are tabulated in the graph (bottom) and Table S2. (E) Dose-dependent effect of ubistatin B (ubiB) on CFTR ubiquitination. In vitro CFTR ubiquitination reactions were performed as in (D) with the final concentrations of ubiB as shown. The reactions with 0.01 μM ubiB were set to 100% and the extent of CFTR ubiquitination was equivalent to that of a DMSO control (not shown). Data represent the mean and standard deviation (error bars) of at least 3 determinations. See also Table S1.
Ubistatins A and B were shown to inhibit Ub-dependent substrate proteolysis and deubiquitination by 26S proteasomes (Verma et al., 2004). To examine if the isopeptidase activity of the proteasome is impaired, we screened the panel of ubistatin compounds for their ability to inhibit disassembly of a model polyubiquitinated substrate by purified mammalian proteasome. The results indicate that ubistatins can efficiently block DUB activity of proteasome-embedded Rpn11 (Fig 1C, Table S1). Of all the analogs, ubiB had the strongest effect, with an IC50=1.1 μM, while hemi-ubistatins were ineffective at inhibiting Rpn11 activity.
Given that ubistatins block recognition of (poly)Ub by receptors, we set out to test whether ubistatins have any effect on ubiquitination. Toward this goal, we screened the ubistatin compounds for their ability to inhibit ubiquitination of the cystic fibrosis transmembrane conductance regulator (CFTR) in vitro. Previous work established that Ub conjugation is required to select this protein for ER associated degradation (ERAD), and even the wild type form of CFTR is a robust substrate for the ERAD pathway (Cheng et al., 1990). Other work established that the acquisition of Ub in this in vitro assay mirrored the selection of ERAD substrates for degradation in yeast cells (Nakatsukasa et al., 2008). Under the conditions tested, hemi-ubistatins had a minimal effect on CFTR ubiquitination, while the full-ubistatins, ubiB and 2, allowed only 18±2% and 29±13% of the protein to be modified, respectively (Fig 1D, Table S2). Of all the compounds tested, ubiB was clearly the most efficient at inhibiting ubiquitination (Fig 1D), and a titration of ubiB revealed that it inhibited CFTR ubiquitination with IC50 of ≈10 μM (Fig 1E). Based on the ability of ubistatins to interact directly with Ub (Verma et al., 2004), these results reflect the ability of the tested compounds to inhibit the cellular ubiquitination machinery, and suggest that different substituent groups alter the affinities for Ub.
Collectively, these assays expose inhibition of ubiquitination and DUB activity as inherent properties of ubistatins. Furthermore, the findings suggest that substrate degradation via the UPS can be inhibited either upstream by preventing initial ubiquitination, or at the end of the pathway by impairing DUB activity at the proteasome.
Ubistatins recognize a common surface on ubiquitin
Next, we wished to gain insight into the mode of ubistatin binding to Ub. Select full-ubistatins (ubiB, 1, 2, 3, 4, 5) and hemi-ubistatins (h-ubiB and 10) were titrated and screened for direct binding to 15N-enriched Ub by monitoring changes in 1H-15N NMR spectra. Ubistatins capable of binding Ub produced residue-specific chemical shift perturbations (CSPs) highlighting the binding surface in Ub and the residues mediating key contacts (Fig 2A-F). The pattern of residue-specific CSPs for all ubistatins that exhibited binding was consistent with binding to Ub's hydrophobic patch centered on L8, I44, and V70 (Fig 2B,C,E,F). Notably, although ubiB, 1, 2, 3, 4, and 5 all contain the same carbon/nitrogen backbone and only differ in their substituent groups, they exhibited varying capacities to bind Ub. Both ubiB and 2 possess internal and external sulfonates and produced CSPs in and around Ub's hydrophobic patch (Fig 2B, E), while 1 containing external carboxylates and internal sulfonates showed essentially no binding to Ub (Fig 2D). Titration with 3, which contains only external sulfonates also revealed a lack of binding (Fig 2A). The inability to bind Ub was also observed for 4, which contains only internal sulfonates, as well as for 5, which contains both internal and external carboxylates. These results are in strong agreement with both the ubiquitination and proteasome DUB assays and suggest that critical substituent groups, namely acidic sulfonic acid groups, are a major factor that governs Ub binding.
Figure 2.
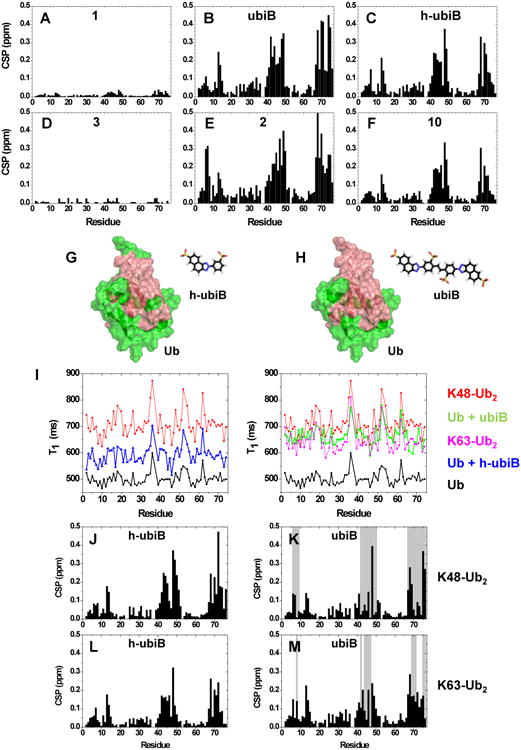
Screening of ubistatins for direct binding to Ub or Ub2 using NMR titration assays. (A-F) Residue-specific amide CSPs in Ub at the endpoint of titration with the indicated ubistatins ([ubistatin]:[Ub]=2 except for 3 where [ubistatin]:[Ub]=1). The starting Ub concentration was 1 mM. (G-H) Residues exhibiting above average CSPs at the endpoint of titration with (G) h-ubiB or (H) ubiB are mapped (painted red) onto the surface of Ub (PDB: 1D3Z); these are residues with CSP >0.082 ppm in (G) and >0.11 ppm in (H). (I) 15N T1 values as a function of residue number in: free Ub (black), Ub at the endpoint in titration with h-ubiB (blue, left panel) or ubiB (green, right), as well as for distal Ub in unbound K48-Ub2 (red) and K63-Ub2 (magenta, right). (J-M) NMR mapping of the interactions of ubiB or h-ubiB with 15N-labeled distal Ub in K48-Ub2 or K63-Ub2. Residue-specific perturbations in 1H-15N NMR spectra of (J) K48-Ub2 with h-ubiB, (K) K48-Ub2 with ubiB, (L) K63-Ub2 with h-ubiB, and (M) K63-Ub2 with ubiB. The titration continued until the molar ratio [ubistatin]:[Ub2]≈4. Black bars represent the magnitude of amide CSPs at the endpoint of titration, while grey bars indicate residues exhibiting strong signal attenuations reflecting severe line broadening caused by intermediate or slow exchange. See also Figs S1-S2.
Unexpectedly, two hemi-ubistatin variants of ubiB, h-ubiB and 10, which were inactive in the functional assays, each produced large residue-specific CSPs (Fig 2C, F). We mapped above average CSPs caused by ubiB and h-ubiB to a nearly identical surface on Ub (Fig 2G, H). The ability of h-ubiB to bind Ub in the same manner as ubiB suggests that h-ubiB, i.e. half of a full ubistatin B, presents an independent Ub-binding element. To exclude the possibility that the observed CSPs reflect a general property of Ub's hydrophobic patch to form non-specific interactions with small hydrophobic molecules, Ub titration with ANS (8-anilinonaphthalene-1-sulfonic acid) resulted in only small CSPs (<0.1 ppm, not shown), supporting the conclusion that hemi-ubistatins represent a new class of specific Ub-binding molecules.
Throughout titration with ubiB, Ub spectra exhibited a global decrease in signal intensity (not shown), reflecting an increase in size (slower tumbling) as a consequence of complex formation. This prompted us to address the stoichiometry of h-ubiB and ubiB complexes with Ub. We detected a noticeable increase, from 497±14 ms to 660±25 ms (Table 1), in the 15N longitudinal relaxation time (T1) of Ub upon binding to ubiB, indicating a significant size increase of the tumbling moiety (Fig 2I, right panel). The 15N T1 values for Ub in the presence of ubiB were between those for K48-Ub2 and K63-Ub2, indicating that two Ubs were bound to ubiB simultaneously. By contrast, h-ubiB binding resulted in lower Ub 15N T1 values (590±21 ms; Fig 2I, left panel) which fall between those for free Ub and K63-Ub2, suggesting the presence of 1:1 and 2:1 Ub:h-ubiB complexes. These observations were further corroborated by the dose-dependent decrease in the translational diffusion coefficient of Ub upon addition of ubiB (Fig S1).
Table 1.
15N T1 values (in ms) for Ub and Ub chains free and in the presence of ubistatins, see also Figures 2 and S4, S6, S8. Shown are mean values and standard deviations (in the parentheses) averaged over residues in structured regions of Ub.
| Ub | Ub + h-ubiB |
Ub + ubiB |
Ub72 | Ub72 + ubiB |
UbR74A + ubiB |
UbR74E + ubiB |
UbR42E,R72E + ubiB |
K48-Ub2 | K48-Ub2 + h-ubiB |
K48-Ub2 + ubiB |
K63-Ub2 | K63-Ub2 + h-ubiB |
K63-Ub2 + ubiB |
K48-Ub4 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 497 (14) | 590 (21) | 660 (24) | 446 (19) | 482 (28) | 544 (26) | 556 (28) | 493 (27) | 701 (28) | 810 (34) | 942 (66) | 632 (24) | 848 (31) | 1248 (86) | 1124a (63) |
Data taken from (Varadan et al., 2005).
That a single surface on Ub appeared to bind the ubistatins led us to examine whether the same surface mediated ubistatins' binding to polyUb. Applying the same methodology, we titrated h-ubiB and ubiB separately into solutions of K48-Ub2 or K63-Ub2 in which the distal Ub unit was 15N labeled. The patterns of residue-specific CSPs for all four titrations (Figs 2J-M, S2A) strongly resembled those for monomeric Ub, indicating that both ubiB and h-ubiB employ a conserved binding mechanism for interaction with Ub and with polyUb. Notably, because the hydrophobic patches of the two Ub units in K48-Ub2 are oriented entirely differently from those in K63-Ub2 (Pickart and Fushman, 2004), our results suggest that both ubiB and h-ubiB recognize this binding surface regardless of how it is presented. Furthermore, this result corroborates the conclusion that hemi-ubistatin is the fundamental Ub-binding unit in the full ubistatin molecule. The greater number of residue-specific NMR signal attenuations caused by ubiB in K48-Ub2 compared to K63-Ub2 (Figs 2K,M) also suggests that ubiB has a greater affinity for K48-linkages, in agreement with previous observations (Verma et al., 2004). In addition, the lack of signal attenuations for both linkage types in the presence of h-ubiB (Figs 2J,L, S2A) is in line with our conclusion from the functional in vitro assays (Fig 1C,D) that hemi-ubistatins have lower affinity for (poly)Ub than the full ubistatin. We also observed a significant increase in 15N T1 values for both K48-Ub2 and K63-Ub2 upon addition of the ubistatins, and even a greater difference (judging by the 15N T1 values) between h-ubiB and ubiB in the apparent size of the resulting complexes with Ub2s (Fig S2B).
Structure of the ubiquitin:hemi-ubistatin complex
Our NMR titration data established that both hemi- and full-ubistatins bind the hydrophobic patch in Ub regardless of Ub's polymeric state or linkage composition, provided that the correct substituent groups in the compound are present. We next proceeded to determine the structure for the Ub:h-ubiB complex, in order to reveal structural details of these interactions.
NOESY spectra showed strong cross-peaks between h-ubiB protons and methyl protons of Ub (Fig S3A), which unambiguously positioned h-ubiB in direct contact with I44 and V70 of Ub (Fig S3B). However, these short-range (< 5 Å) distances were insufficient to distinguish between multiple orientations of h-ubiB on Ub surface. Hence, we employed site-directed paramagnetic labeling with nitroxide spin label, MTSL, to obtain long-range distance restraints. The optimal sites for MTSL attachment to Ub were chosen by simulating the paramagnetic effect on each proton in h-ubiB caused by placing MTSL on any residue in Ub, based on NOE-derived models of the Ub:h-ubiB complex (Fig S4A). With this insight, we carried out several experiments attaching MTSL to Ub residues 36, 48, or 63 (cysteine mutants UbI36C, UbK48C, or UbK63C, respectively). MTSL-labeled UbI36C and UbK48C yielded long-range distance restraints subsequently used for structure calculation (Table S3). As a negative control, UbK63C, which positions MTSL far from h-ubiB resulted in a negligible paramagnetic effect (not shown).
The final structure of the Ub:h-ubiB complex (Fig 3A,B) was obtained using the HADDOCK program (de Vries et al., 2010); the statistics are in Table S4 (see also Fig S4C-F). Consistent with intermolecular NOEs, the naphthotriazole moiety of h-ubiB exhibits several hydrophobic contacts with V70 and I44 in Ub (Fig 3B,C). A detailed analysis of the structure suggests that hydrophobic contacts are further stabilized by electrostatic and polar interactions between the sulfonate groups of h-ubiB and several residues in Ub surrounding the hydrophobic patch. Specifically, Ub residues R42 and R72 are in close proximity to one of the two sulfonates of h-ubiB while H68 and G47 are within hydrogen-bonding distance of the other (Fig 3B,C). These contacts are in excellent agreement with the large amide CSPs observed for these residues (Fig 2C).
Figure 3.
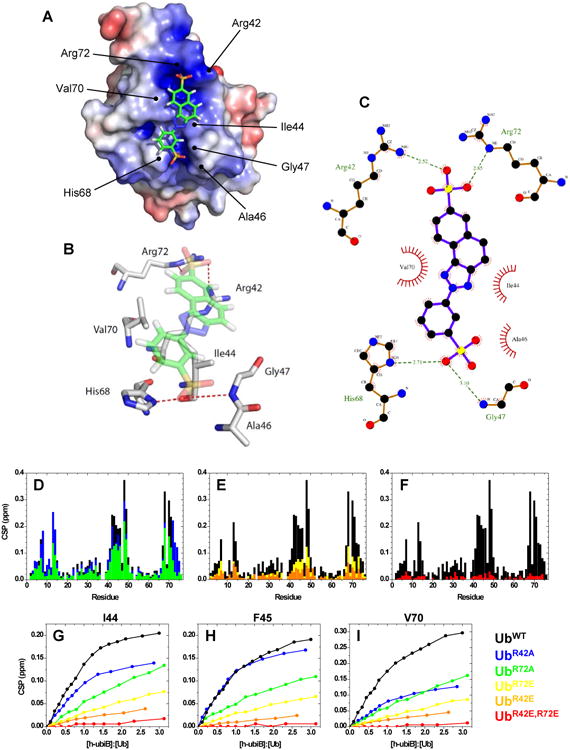
Structure of the Ub:h-ubiB complex. (A) Ub is represented by its electrostatic surface, (equipotential contours from -3kT/e to +3kT/e, positive potential is painted blue and negative is red), while h-ubiB is shown in stick representation. Key Ub residues involved in the interaction are labeled. (B) 3D representation of the contacts between h-ubiB and the interacting groups in Ub. Red dashed lines represent hydrogen bonds. (C) LIGPLOT representation of the hydrophobic contacts (red spoked arcs) and hydrogen bonds (green dashed lines) between h-ubiB and Ub. (D-I) Validation of the structure of Ub:h-ubiB complex by site-directed mutagenesis. Residue-specific CSPs from titration with h-ubiB of UbWT (black) and (D) single-Ala mutants, UbR42A (green) and UbR72A (blue), (E) single-Glu mutants, UbR42E (yellow) and UbR72E (orange), and (F) double-Glu mutant, UbR42E,R72E, red. Representative titration curves for Ub residues (G) I44, (H) F45, and (I) V70, colored according to the indicated mutants. See also Figs S4-S6 and Tables S3, S4.
Electrostatic interactions facilitate ubistatin-ubiquitin binding
To validate the contribution of the electrostatic interactions inferred from our Ub:h-ubiB structure, we systematically mutated cationic residues R42 and R72 one-by-one to neutral (Ala) or oppositely charged (Glu) residues. The electrostatic surface potential of each mutant suggests that the ubistatin binding site undergoes a dramatic change in polarity (Fig S5A). We assessed the ability of h-ubiB to bind several single-site mutants: UbR42A, UbR42E, UbR72A, UbR72E, as well as the double mutant UbR42E,R72E using the same NMR assay as for UbWT (Fig 3D-F). The results clearly show that the charge state at residues 42 and 72 has a dramatic impact on the ability of Ub to bind h-ubiB (Fig 3D-F). By contrast, changing the bulk charge of Ub with the K63D mutation had little effect on h-ubiB binding (Fig S5B,C), verifying that the observed effect of the R42 and R72 mutations was due to changes in specific electrostatic/polar interactions between Ub and h-ubiB. The spectral similarity to UbWT (Fig S5D-E) confirmed that the structure of Ub remained intact for the mutants analyzed.
The impact each Ub mutation has on the strength of h-ubiB interaction is best illustrated by comparing the magnitudes of the residue-specific CSPs at the titration endpoint to those for UbWT, as well as residue-specific titration curves (Fig 3G-I). Notably, both single-site “neutral” mutants (UbR42A and UbR72A) exhibited only a slight decrease in the CSPs compared to UbWT (Fig 3D), suggesting that the remaining interactions (including the hydrophobic effect and the electrostatic attraction to the remaining arginine) are sufficient for binding. Single-site “negative” mutations, UbR42E and UbR72E, significantly reduced the binding, emphasizing the role of the electrostatic contacts and demonstrating that the hydrophobic interaction between Ub and h-ubiB can be overpowered by the point-charge repulsion (Fig 3E). To underscore this point, the double mutant, UbR42E,R72E, showed virtually no detectable CSPs in the presence of h-ubiB, indicating complete abolition of binding (Fig 3F). Following our observation that h-ubiB and ubiB bind to the same surface on Ub, we also tested whether ubiB binds UbR42E,R72E. In line with the h-ubiB results, ubiB produced only negligible CSPs when titrated into UbR42E,R72E and no detectable increase in the 15N T1 (Fig S5G,H, Table 1). These results clearly demonstrate that R42 and R72 are required for ubistatin binding. Furthermore, the importance of the external sulfonic acid groups in ubistatins for efficient Ub binding was corroborated by observations that compounds 4 (lacking external sulfonates) and 1 (external sulfonates replaced with carboxylates) failed to bind Ub (Fig 2D).
To explore the selectivity of ubistatins for Ub we titrated h-ubiB into yeast Rub1 (orthologue of human Nedd8), a Ub-like (UBL) protein highly homologous to Ub. Like Ub, Rub1 contains the L8, I44, V70 hydrophobic surface patch surrounded by basic residues (K6, R42, R74), although R72 is replaced by a threonine (Singh et al., 2012). Interestingly, only minimal CSPs were observed in Rub1, mostly clustered around I44 and V70 (Fig S6). This suggests that residues beyond the hydrophobic patch are major contributors to ubistatin binding, rendering h-ubiB highly specific for Ub.
The hemi- and full ubistatins differ in binding affinity
We next set out to characterize the binding affinities of h-ubiB and ubiB. The NMR titration curves of 15N-labeled Ub with h-ubiB exhibited a characteristic hyperbolic shape (Fig 4A), but did not reach saturation even nearing a 3-fold molar excess of h-ubiB ([h-ubiB]=2.6 mM). From these data we obtained a Kd of 595±200 μM (averaged over 10 residues). By contrast, titration curves for ubiB at the same conditions were consistent with “stoichiometric” binding (not shown), indicating that the Kd was significantly below the concentration of Ub (0.9-1 mM). With this in mind, we performed ubiB titrations starting with Ub concentration of 20 μM (Figs 4B, S7B), which yielded a Kd value of 11.4±2.2 μM (Table 2). To verify this result independently, we took advantage of the native fluorescence of ubiB and measured the change in fluorescence anisotropy upon titration with Ub (Fig 4B). The resulting Kd=14.0±1.6 μM was in agreement with our NMR data.
Figure 4.
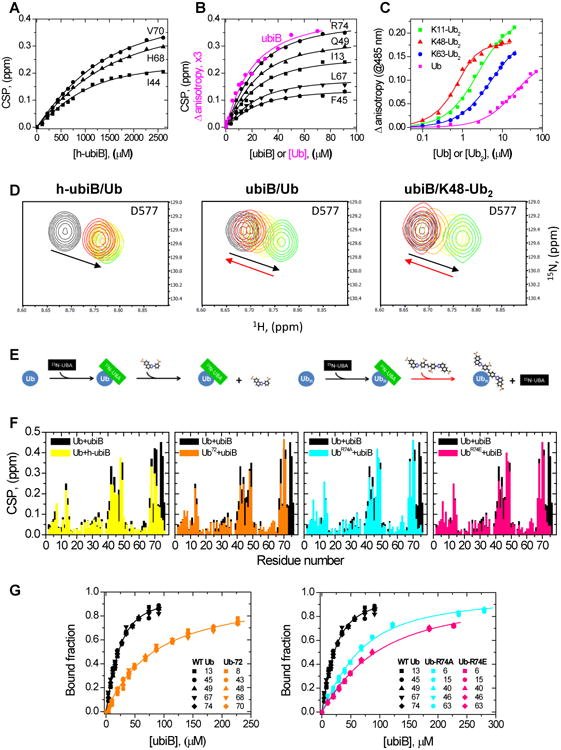
Titration analysis of ubistatin binding to Ub and differently linked Ub2 molecules. (A,B) Representative NMR binding curves for the indicated residues in Ub upon titration with (A) h-ubiB and (B) ubiB. The lines represent the results of a global fit to a 1:1 binding model. Shown in B in magenta, for comparison, are fluorescence data for a reverse titration of ubiB with Ub (from C). (C) Change in ubiB fluorescence anisotropy, monitored at 485 nm, upon titration with Ub (magenta), K11-Ub2 (green), K48-Ub2 (red), or K63-Ub2 (blue). The lines represent the results of fit to a 1:1 binding model. (D) Competition between h-ubiB or ubiB against ubiquilin-1 UBA domain: (left) h-ubiB and Ub, (middle) ubiB and Ub, and (right) ubiB and K48-Ub2. 1H-15N SOFAST-HMQC signals of UBA residue D577: unbound (black contours) and Ub-bound state (green). Signals upon increasing addition of ubistatin are colored yellow (1:1 molar ratio), orange (2:1), and red (3:1). Note that the UBA signals return back to their unbound positions upon addition of ubiB (red arrow), indicating a displacement of UBA from Ub or Ub2 by ubiB. (E) Schematic illustration of the results of the competition assay. (F) Comparison of the residue-specific CSPs in Ub at the endpoint of titration with ubiB and h-ubiB (left) or in WT Ub and the indicated Ub variants upon titration with ubiB. (G) Comparison of the normalized NMR titration curves for select residues in WT Ub, Ub72, UbR74A, and UbR74E. The lines represent the results of global fit of the data to a 1:1 binding model (Table 2). Data coloring is the same as in F. See also Fig S7.
Table 2.
Dissociation constants (in mM) for h-ubiB and ubiB binding to Ub, Ub2, and Ub mutants, see Figures 4 and S7. The numbers in the parentheses represent standard deviations. Data marked with an asterisk were obtained from ubiB fluorescence anisotropy measurements; the rest of the Kd values were obtained from NMR titrations of 15N-labeled Ub or Ub2 with ubiB or h-ubiB.
| Ub + h-ubiB | Ub + ubiB | Ub + ubiB* | Ub72 + ubiB | UbR74A + ubiB | UbR74E + ubiB | UbR42E + ubiB | UbR42E,R72E + ubiB | K48-Ub2 + ubiB* | K11-Ub2 + ubiB* | K63-Ub2 + ubiB* |
|---|---|---|---|---|---|---|---|---|---|---|
| 595 (200) | 11.35 (2.2) | 14.0* (1.6) | 73.1 (7.3) | 40.3 (9.1) | 67.6 (15.0) | 396 (77) | >1000 | 0.264* (0.023) | 1.83* (0.05) | 4.88* (0.22) |
Finally, to quantify the strength of ubiB binding to di-Ub chains, we measured the change in fluorescence anisotropy of ubiB upon titration with K11-, K48-, or K63-linked Ub2 (Fig 4C). K48-Ub2 exhibited the tightest binding (Kd=264±23 nM) supporting previous reports (Verma et al., 2004); however, other Ub2s also bound ubiB quite tightly, with Kd values of 1.83±0.05 μM (K11-Ub2) and 4.88±0.22 μM (K63-Ub2).
To further verify our results, we devised a direct NMR competition assay (Fig 4D,E), which tested the ability of ubiB and h-ubiB to compete for Ub binding against the Ub-associated (UBA) domain of ubiquilin-1, one of the strongest Ub binders (Kd∼20 μM) among known Ub-binding domains (Raasi et al., 2005; Zhang et al., 2008). In this assay, unlabeled Ub was added to 200 mM 15N-labeled UBA at a 1:1 molar ratio, causing the UBA signals to shift to the Ub-bound state. Then, either h-ubiB or ubiB was gradually added. The addition of ubiB caused the UBA signals to return to their positions in the unbound state (Fig 4D, center), indicating that ubiB outcompetes UBA for binding to Ub. Furthermore, ubiB outcompeted UBA for binding to K48-Ub2 (Fig 4D, right). By contrast, h-ubiB was unable to outcompete UBA, even at 3-fold molar excess (Fig 4D, left). These results indicate that the relative affinity of ubiB for Ub and K48-linked polyUb is greater than that of the UBA (Fig 4E), while the affinity of h-ubiB is drastically lower, in agreement with our NMR and fluorescence titration data (Table 2).
Several reasons may account for the stronger binding of Ub to ubiB compared to h-ubiB. First, ubiB contains twice the number of negatively charged (SO3-) groups which could strengthen the interaction with the positively-charged residues surrounding the hydrophobic patch of Ub. Second, ubiB presents a larger contiguous Ub-binding surface spanning two h-ubiB molecules, which doubles the effective local concentration of the Ub-binding sites (ubiB vs h-ubiB) and provides an increased likelihood for Ub to re-associate with the same ubiB molecule after dissociation. Furthermore, the extended binding surface could facilitate bidentate and possibly avid binding of the two halves of ubiB to Ub or polyUb. Last but not least, Ub:ubiB binding could be strengthened by additional interatomic contacts that are not present or weaker in the Ub:h-ubiB complex. It is also possible that the resulting ubiB:2xUb complex is further strengthened by the interactions between the two Ubs bound to the same ubiB molecule.
To shed light on possible differences in Ub's contacts with h-ubiB and ubiB, we compared shifts in the NMR signals of each residue in Ub upon titration with these two compounds. A detailed analysis revealed that despite strong similarity of the magnitudes and the directions of the signal shifts caused by ubiB and h-ubiB for most of Ub residues, there is a striking difference in the behavior of the C-terminal residues(Figs 4F, S7A,B). Specifically, R74, G75, and even G76 exhibited substantial CSPs upon addition of ubiB whereas their signals barely shifted upon h-ubiB binding. To examine possible contributions of these residues to stronger ubiB binding, we deleted the C-terminal tail of Ub (residues 73-76); this variant will be referred to as Ub72. This resulted in a 6-fold decrease in the binding affinity (Kd=73.1 μM, Table 2). Furthermore, there was only a minor increase in 15N T1 of Ub72 upon ubiB binding, suggesting a 1:1 stoichiometry of the resulting complex (Table 1). These results clearly indicate that Ub's C-terminus plays a role in stronger ubiB binding and Ub dimerization. Notably, the directions of the signal shifts and the CSPs for the rest of Ub residues remained essentially unchanged (Figs 4F, S7A-C), suggesting that the nature of the local interactions between Ub72 and ubiB remained similar to Ub:h-ubiB interactions. Since Ub's C-terminus contains a basic residue (R74) that may contribute to ubiB binding through electrostatic interactions, we examined the effect of R74 substitution with Ala (UbR74A) or Glu (UbR74E) (Fig S7D-G). These mutations weakened ubiB binding by 3.5 to 6-fold, respectively, (Table 2, Fig 4G) and the 15N T1 at the end of the titration was markedly lower than for Ub:ubiB complex and even slightly less than for Ub:h-ubiB complex (Table 1, Fig S7H). Notably, no CSPs were observed for the C-terminal residues (Figs 4F, S7F), and, as in the case of Ub72, the directions of the signal shifts and the CSPs for the rest of Ub residues were essentially the same as for h-ubiB:Ub binding (Fig S7D-E). Collectively, these results point to the role of Ub's C-terminal residues, especially R74, in strengthening ubiB:Ub binding and promoting the formation of the ternary ubiB:2xUb complex.
Structural modeling of ubistatin B interactions with Ub
Thus far, our data demonstrated that ubiB binds to Ub's hydrophobic patch and the resulting complex includes two Ub molecules. Furthermore, the strong similarity of both the magnitudes (CSPs, Fig 2B,C) and the directions of the NMR signal shifts (Fig S7A,B) upon Ub binding to ubiB and h-ubiB indicates similarity of the interatomic contacts between Ub and these compounds. In order to gain additional structural insights into these interactions, we performed SANS measurements for Ub and K48-Ub2 in complex with ubiB. A drastic change in the SANS profiles of both Ub and K48-Ub2 upon addition of ubiB (Fig 5A,B) indicates that the overall shape (hence structural arrangement) of these proteins in complex with ubiB is different from their free states. In fact, the analysis showed a significant increase in the radius of gyration (Rg) for both Ub and Ub2 (Table 3). Specifically, ubiB binding to Ub caused a two-fold increase in I(q≈0) and resulted in the Rg value close to that of unbound K48-Ub2. These results clearly corroborate the stoichiometry of the ubiB:2xUb complex inferred from our NMR data. Furthermore, the overall similarity between the SANS profiles, as well as the pair distribution functions for the Ub+ubiB and free K48-Ub2 samples suggests that structural arrangement of the two Ubs in complex with ubiB resembles that of K48-Ub2.
Figure 5.
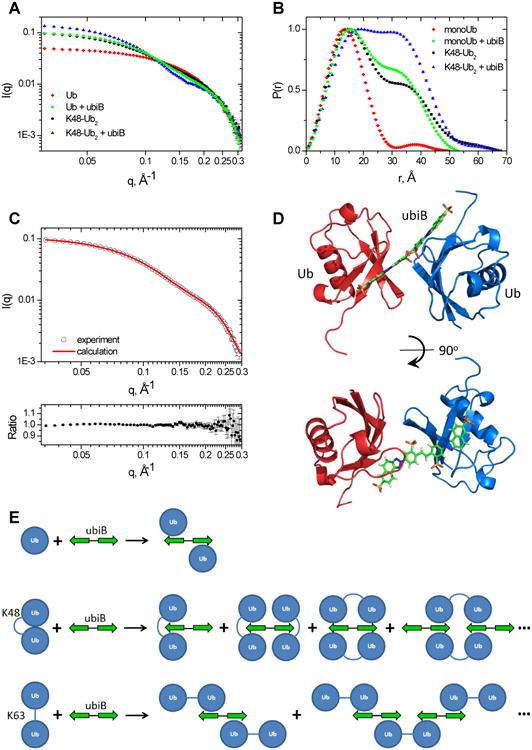
Characterization of the complexes of Ub and K48-Ub2 with ubiB using SANS. (A) SANS intensity and (B) pair distribution function for Ub or K48-Ub2 alone and in the presence of an equimolar amount of ubiB. (C) Agreement between the experimental SANS intensity (I(q)exp, open circles) and the I(q)calc calculated for the structure shown in (D). Top: overlay of the I(q) values; bottom: the ratio of I(q)exp to I(q)calc. The error bars in (A) and (C) represent standard errors in the experimental I(q) values, and in (B) standard errors in the derived P(r). (D) Structural model of the Ub:ubiB:Ub complex obtained by rigid-body docking. Ubs are shown as ribbons colored blue and red, ubiB is in stick representation. (E) Schematic illustration of the Ub:ubiB:Ub complex and possible structural arrangements of K48-Ub2 and K63-Ub2 in complex with ubiB, see also Figs S8-S9.
Table 3.
Radius of gyration (Rg, in Å) derived from the SANS data for various samples studied. The numbers in the parentheses represent standard deviations.
| Sample | Ub | Ub + ubiB | K48-Ub2 | K48-Ub2 + ubiB |
|---|---|---|---|---|
| Rg | 11.3 (0.1) | 17.4 (0.1) | 18.4 (0.1) | 20.1 (0.1) |
Inspired by these observations, we built a model of the ubiB:2xUb complex based on a combination of the NMR and SANS data and using HADDOCK (de Vries et al., 2010) supplemented with molecular dynamics (MD) simulations. The generated structure of the complex that agrees best with the SANS data (Fig 5C) is shown in Figure 5D; the convergence of the top 10 structures is illustrated in Figure S8A.
The structure of the ternary complex, Ub:ubiB:Ub, features a sandwich-like arrangement, with two Ubs encompassing the ubiB molecule situated between them (Fig 5D). The Ubs are positioned one on each side of the compound and oriented such that they contact the ligand through their β-sheet surface containing the hydrophobic patch. This arrangement allows the ubiB molecule to interact with two Ubs simultaneously by utilizing both halves (i.e., the naphthotriazole moieties), one for each Ub. The interface between each Ub and the corresponding half of ubiB is similar to that between Ub and h-ubiB, and the flexible C-terminus of Ub is positioned such that it can interact with ubiB's sulfonates (see above). It is worth mentioning that the NMR restraints-driven docking also produced a different arrangement, with both Ubs positioned on the same face of ubiB while still contacting different halves of ubiB (Fig S8B). However, that structure was in poor agreement with the SANS data (cf. Fig 5C). It is noteworthy that the SANS data can clearly distinguish between the structural arrangement in which the two Ubs are positioned on the opposite faces (sides) of ubiB and the one where both Ubs are bound to the same face.
The structure of the Ub:ubiB:Ub complex (Fig 5D) was then used as the starting structure for a 2 μs MD simulation. Although both ubiB and Ubs moved and rearranged during the simulation, the ternary complex remained intact. Interestingly, some of the MD snapshots featured the C-terminal G76 of one Ub in close proximity to K48 of the other Ub (Fig S8C) and resembled the structural arrangement in the closed conformation of K48-Ub2 (Cook et al., 1992; Varadan et al., 2002), thus suggesting that ubiB can intercalate into K48-Ub2.
The SANS and P(r) profiles for K48-Ub2 in the ubiB-bound state are drastically different from those of both the ternary Ub:ubiB:Ub complex and K48-Ub2 alone (Fig 5A,B). The analysis revealed a larger Rg compared to K48-Ub2 (Table 3) and a marked increase in the fraction of atoms with distances >20 Å from each other (Fig 5B), suggesting a substantial increase in the apparent size of Ub2 upon ubiB binding. Combined with the higher I(q≈0) value at approximately the same Ub2 concentration (Fig 5A) and significantly longer 15N T1 than for free Ub2 (Fig S2B, Table 1), these results point to the presence of more than one Ub2 molecule in the complex (as illustrated in Fig 5E). Based on the observed dimerization of Ub upon ubiB binding one might expect that Ub2 would follow a similar trend and form a dimer in the ubiB-bound state. In fact, both the Rg and the pair distribution function measured for Ub2+ubiB match predictions for the closed conformation of K48-linked tetra-Ub (PDB: 2O6V) (Fig S9A). These results suggest that the ubiB:K48-Ub2 interaction involves two Ub2 molecules and that the shape of the complex is similar to the closed form of K48-Ub4 (packed as a dimer of Ub2s). The actual structural arrangement in such complexes is unclear at this point, but several possible scenarios are sketched in Figure 5E (also Fig S9B).
The architecture of polyUb complexes with ubistatins would depend on the ability of the Ub chain to adopt specific conformations, and could be linkage dependent. The compact packing of K48-Ub2s in the ubiB-bound state might reflect the ability of K48-linked chains to adopt conformations that enable close contacts between the hydrophobic patches of the interacting Ub moieties (Fushman and Walker, 2010; Pickart and Fushman, 2004; Varadan et al., 2002). By contrast, K63-linked chains, which adopt more extended conformations (Pickart and Fushman, 2004; Varadan et al., 2004), might form extended complexes with ubiB (e.g., as illustrated in Figure 5E). In fact, the stark increase in 15N T1 for K63-Ub2 in the ubiB-bound state compared to ubiB-bound K48-Ub2 and even free K48-Ub4 (Table 2, Fig S2B) points to this possibility.
Ubistatins shield ubiquitin conjugates from deubiquitinases
In light of the observation that select ubistatins impair activity of the proteasomal DUB Rpn11 (Fig 1C, Table S1), we set out to examine their effect on other DUBs. We found that both ubiB and h-ubiB exhibited a concentration-dependent effect on the ability of another proteasome-associated DUB, Ubp6, to disassemble K48-Ub2 (Fig 6A) and K63-Ub2 (not shown). Preservation of K48-Ub2 was observed at or above stoichiometric amounts of ubistatin to Ub. With both linkage types, ubiB exhibited a stronger ability than h-ubiB to block deubiquitination by Ubp6.
Figure 6.
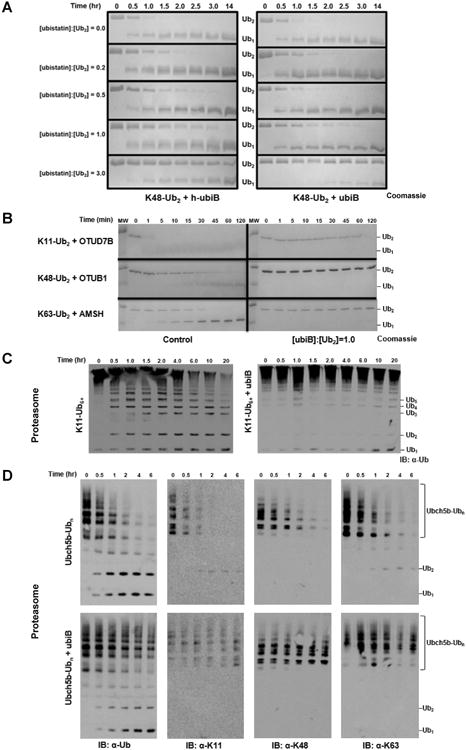
Ubistatins shield polyUb from disassembly by DUBs. (A) Disassembly of K48-Ub2 by Ubp6 over the indicated time course in the presence of varying concentrations of h-ubiB (left) or ubiB (right). (B) Linkage-selective DUBs rapidly disassemble their respective linkage type (left), while stoichiometric amounts of ubiB prevent any cleavage within the time course (right). (C) Disassembly of K11-Ub6+ by yeast 26S proteasome in the absence (left) and presence (right) of ubiB. (D) Processing of Ubch5b-Ubn substrate by the proteasome in the absence (top row) or presence (bottom row) of ubiB. The change in the Ub profile over time was followed with the indicated antibody.
These observations encouraged us to test if ubiB had a similar effect on other DUBs cleaving a variety of Ub conjugates. Highlighting the ability of ubiB to bind diverse linkage types, ubiB prevented the dimeric Ub substrates K11-Ub2, K48-Ub2, and K63-Ub2 from being disassembled by their respective linkage-specific DUBs: Cezanne (OTUD7B), OTUB1, and AMSH (Fig 6B).
We then examined if ubiB affects DUB activity of the 26S proteasome, which is essential for cellular viability (Goldberg, 2012). Recent work revealed that K11-linked chains of six or more Ub units signal substrates for rapid degradation by the proteasome (Meyer and Rape, 2014) and that proteasomal DUBs rapidly disassemble K11 linkages (Mansour et al., 2015). Using K11-Ub6+ as a substrate, in the absence of ubiB the proteasome produced numerous cleavage intermediates after just 30 minutes, and virtually all of the starting material was depleted at the end of the 20 hr time course (Fig 6C). However, in the presence of ubiB, the high molecular weight K11-Ub6+ corresponding to the starting product remained constant and only trace amounts of cleavage intermediates appeared, likely in concentrations approaching the detection limit of the antibody (Fig 6C). Finally, we utilized Ubch5b-Ubn, an autoubiquitinated E2 carrying all possible Ub linkages (Mansour et al., 2015), in order to determine if and how ubiB alters the ability of the proteasome to deubiquitinate a complex substrate, independent of degradation. In the absence of ubiB, the 26S proteasome removed virtually all high molecular weight forms of Ubch5b-Ubn within 2 hrs. By contrast, significant amount of processing intermediates still remained in the presence of ubiB (Fig 6D). To understand which linkage types were processed, we then analyzed each time point using three linkage-specific antibodies (for K11, K48, or K63). In the absence of ubiB the respective blots showed complete removal of K11, K48, or K63 linkages by the proteasome at the latest time points (Fig 6D). By contrast, the addition of ubiB preserved all three linkage types in the Ubch5b-Ubn substrate, and eliminated low molecular weight intermediates. The results of this experiment indicate that ubiB protects the K11, K48, and K63 linkages from disassembly by the proteasome when Ubch5b-Ubn is used as ubiquitinated substrate.
Ubistatin B penetrates human cancer cells and perturbs the ubiquitin landscape
The in vivo effects of ubistatins have been previously demonstrated in cell extracts, Xenopus eggs, and through microinjection (Bellare et al., 2008; Verma et al., 2004). During our preliminary testing of ubiB on cancerous human cell lines we consistently observed that ubiB associated with cells in the absence of chemical permeabilization. To determine if this effect was solely due to ubiB interactions with the plasma membrane or via internalization, we incubated HeLa and RCC4 cells with 10 μM of ubiB for 6 hrs. We observed distinct ubiB fluorescence at and within the boundaries of the fluorescently labeled plasma membrane in each cell type (Fig S10A). Notably, the morphology of the treated cells (Fig S10B) indicated that a 10 μM dose of ubiB was not toxic, at least within this time period. Given the relatively high affinity of ubiB for polyUb, we further speculated that ubiB directly interacts with polyUb within cells. Next, we conducted immunofluorescence assays with anti-Ub, as well as three linkage-specific antibodies on fixed, permeabilized HeLa cells pretreated with ubiB. Signals from both ubiB and all four antibodies were distributed broadly within the cell, including the nucleus (Fig S10E).
We then set to investigate changes to the cellular polyUb landscape following ubiB treatment. Using the standard lysis method with RIPA buffer, we observed signal from ubiB in harvested cells, which was retained in both pelleted cell debris and supernatants following lysis (Fig S10C). Analysis by Western blot indicated that treatment with ubiB increases the amount of Ub conjugates, including those with K48 and K63 linkages, in a dose-dependent manner (Fig S10D). To further examine this effect, HeLa cells treated with ubiB or proteasome inhibitor MG132 were directly lysed by boiling in Laemmli buffer and analyzed with a Ub-specific antibody (Fig 7). Both ubiB and MG132 treatments resulted in marked changes to the Ub landscape, notably, an increase in high molecular weight Ub conjugates. On a closer examination, Ub conjugates below 70 kDa are greatly diminished in the ubiB-treated cells, in contrast with MG132 and the control. Both MG132 and ubiB impair substrate degradation by the proteasome, albeit through distinct mechanisms. While MG132 acts directly on the proteolytic sites in the 20S core particle, ubiB's binding to polyUb chains shields them from recognition by proteasomal Ub receptors and shuttling factors (Verma et al., 2004), as well as a spectrum of DUBs (Fig 6). The latter properties are a likely reason for the increase in high molecular weight Ub conjugates upon ubiB treatment.
Figure 7.
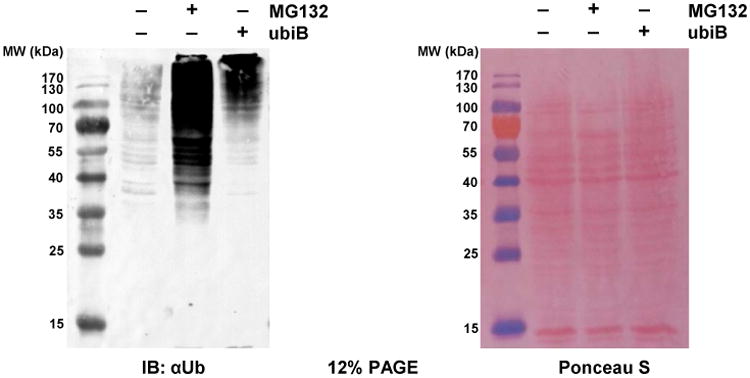
The impact of ubiB internalization on the Ub landscape in human cancer cells. HeLa cells were treated with ubiB or MG132 for 8 hrs prior to lysis. Anti-Ub is used to probe the Ub landscape of HeLa cells lysed in SDS. Despite the dramatic difference in high molecular weight Ub conjugates, Ponceau S staining (right panel) shows equal loading of proteins across all lanes. See Fig S10 for cell images and additional analysis of the Ub landscape.
Note that the observed accumulation of Ub conjugates upon ubiB treatment does not contradict the ability of ubiB to block substrate ubiquitination (Fig 1D-E), because the effects of ubiB on deubiquitination and ubiquitination occur at different thresholds (IC50 of 1 μM and 10 μM, respectively), with deubiquitination being the more sensitive. For example, at 1 μM ubiB there is at most 5-10% inhibition of ubiquitination (Fig 1E), but ∼50% inhibition of deubiquitination (Table S1). Achieving complete inhibition of ubiquitination would require high ubiB concentrations (≥100 μM) that might not be attainable in cells. Therefore, the inhibitory effect of ubiB on deubiquitination predominates.
To summarize, we detected ubiB uptake using imaging of either live cells or after fixation. These results are consistent with the observation that ubiB perturbs the cellular pool of Ub conjugates. Further work will be required to determine if ubiB binds primarily to Ub chains within cells and whether it can serve as a fluorescent marker for cellular Ub.
Discussion
This study provides a detailed characterization of how ubistatins interact with Ub and polyUb chains, describing previously unknown functional consequences of these interactions. Based on the screening of ubistatin derivatives in CFTR ubiquitination, DUB shielding, and (poly)Ub binding assays, we demonstrate that ubistatin B (ubiB) has the greatest ability to interact with Ub conjugates. By dissecting the binding interaction of a hemi-ubistatin (h-ubiB) with Ub, we found that ubiB contains two Ub-binding entities. Furthermore, confirming that ubiB can simultaneously bind multiple Ub units, we detected the formation of higher-order complexes of ubiB with Ub and with polyUb. Notably, all active ubistatin variants recognized a conserved binding surface on Ub, regardless of whether Ub was present as a monomer or polymers of different linkage types. The structure of the Ub:h-ubiB complex and SANS-derived models of the Ub:ubiB:Ub complex provide atomic-level details of how ubistatins interact with Ub. In particular, this study revealed critical role of electrostatic/polar interactions between ubistatin's acidic groups and the basic residues surrounding the surface hydrophobic patch of Ub. The precise orientation of these critical residues on the surface of Ub narrowly restricts ubistatin binding, such that ubiB, for example, is specific for Ub yet reasonably inert to its closest homologue Rub1. That ubistatins can associate with more than one Ub simultaneously means that they can intercalate into polyUb and thus shield Ub-conjugates or polyUb chains from a range of DUBs (including those associated with the proteasome). The ability of ubistatins to penetrate mammalian cells now allows for a broad range of potential applications that merit additional investigation into this class of compounds.
In the cell, diverse classes of Ub-binding domains (UBDs) are found across several protein families that enable the broad spectrum of polyUb signals to be interpreted (Komander and Rape, 2012). Our discovery of synthetic molecules capable of binding Ub as well or better than natural UBDs provides for numerous potential future applications. Thus far, two distinct nano-scale systems, gold or silver nanoparticles(Calzolai et al., 2010; Mangini et al., 2014) and fullerenol (Zanzoni et al., 2015) have been reported to form specific interactions with Ub. Several metal ions (Cd, Cu, Hg, Pt, Zn) have also been found to form adducts with Ub in which H68 coordinates at least one ion (e.g., (Arena et al., 2013; Falini et al., 2008)). Ongoing efforts will undoubtedly uncover other small molecules capable of binding Ub. However, ubistatins already present many advantages over other existing synthetic Ub-binding molecules: the ubistatins are highly specific for Ub, bind Ub tightly, penetrate cells, have well characterized in vitro and in vivo outcomes, and provide a fluorescent readout.
From a historical perspective, ubistatin A was the first small molecule shown to bind K48-linked polyUb (Verma et al., 2004). Here, we demonstrate that ubistatin derivatives bind both Ub and polyUb. The Ub:ubistatin interaction is unique in that ubistatins share no resemblance to any known Ub-binding molecules; however, these compounds can now serve as a starting material to isolate or design more effective preclinical candidates. In fact, our characterization of the ubistatin derivatives suggests that substituent groups and positions can be chemically modified without dramatically reducing Ub binding. The similarities carry over to polyUb, as the changes in NMR spectra of K48-Ub2 exhibit nearly identical patterns for ubistatin B (ubiB) (Fig 2K) and ubistatin A (Verma et al., 2004). Combined NMR and SANS analyses also showed that the interaction of ubiB with polyUb chains of different linkages (e.g., K48- and K63-linked) can result in different spatial arrangements and stoichiometries. It is tempting to speculate that related small molecules can be identified or designed that specifically bind interaction surfaces on the numerous UBL domains found in the cell. For instance, minor derivatization of ubistatins may improve binding to Rub1 (Nedd8 in humans), which contains the same hydrophobic patch as Ub but differs in the surrounding residues. This approach could be extended to other UBL family members (e.g., FAT10, ISG15, SUMO) to interfere with their own unique signaling pathways or to modulate the interactions of a UBL within multidomain systems, such as Parkin or USP7.
The development of agents to target various aspects of the UPS has been underway for over a decade and has already yielded important outcomes (Buckley and Crews, 2014). In this regard, we propose that Ub itself may represent an alternative and potentially more robust therapeutic target compared to the enzymatic components of the UPS. The highly conserved amino acid sequence of Ub suggests that it is unlikely that a treated cell could tolerate drug refractory mutations in Ub. To highlight this point, mutation of several ubistatin-interacting residues in Ub, especially R42 and R72, are lethal in yeast (Roscoe et al., 2013), effectively making Ub a static target. However, the chemical properties of ubistatins have raised some concerns regarding their use as drugs (Bellows and Tyers, 2004). To mitigate some of these concerns here we show that ubistatins can penetrate cells and have measurable effects on the Ub landscape. The use of ubistatins in cell-based and animal experiments or as therapeutics would be enhanced by the development of pro-drug forms of the parent molecule that shield the sulfonate groups by reversible covalent bonds or non-covalent complexes. Even if some other factors limit ubistatins as therapeutics, the intrinsic fluorescence and the ability of ubistatins to accommodate modular substituent groups will allow them to serve as powerful reagents in biochemical studies(Bellare et al., 2008).
The mechanism of action of ubistatins is reminiscent of that for endogenously expressed UBDs (Sims et al., 2012; Yoshida et al., 2015). In each case, the cell experiences a buildup of polyUb, but notably this is not lethal over short time periods. The shielding by a given UBD likely limits DUB activity, further contributing to polyUb buildup. More generally, molecules like ubistatins and select other proteins allow for the controlled accumulation of polyUb linkage types: ubiB has the greatest effect on K48 linkages, the triple-UIM RAP80 derivative (Rx3-A7) is highly selective for K63 linkages (Sims et al., 2012), and TR-TUBEs cause an increase in all Ub-Ub linkage types (Yoshida et al., 2015). Yet, the chemical properties of ubistatins combined with resistance to proteases offer advantages compared to the protein-based UBDs. Ubistatins may also have applications in modulating cellular signaling pathways, such as those triggered by CXCR4 receptors (Majetschak, 2011). Finally, given that ubistatins have been shown to arrest the cell cycle, producing effects similar to proteasome inhibitors (Verma et al., 2004), and combined with the data presented in this study, we believe that the ubiquitin signal is a plausible candidate for therapeutic intervention in the ubiquitin-proteasome pathway.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for reagents should be directed to the leading author: David Fushman (fushman@umd.edu).
Experimental Model and Subject Details
HeLa (female) and RCC4 (male) human cancer cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in a humidified incubator set at 37°C with 5% CO2.
Method Details
Certain commercial equipment, instruments, or materials are identified in this paper to foster understanding. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
Molecular Biology
Human wild-type Ub in pET3a was used a template for all Ub mutants. Mutagenesis was carried out using KOD Hot Start Master Mix and corresponding primer pairs. Followed by DpnI digestion, PCR reactions were transformed into DH5α cells and selected against LB-ampicillin agar. Individual colonies were verified by DNA sequencing from the T7 promoter.
Preparation of proteins
Unlabeled and 15N-enriched recombinant Ub and Ub variants were obtained using bacterial expression (in E.coli) and purification as described (Varadan et al., 2002) and verified using SDS-PAGE and 1H-15N NMR spectra. Ub dimers were assembled using linkage-specific E2 enzymes as detailed elsewhere (Varadan et al., 2005; Varadan et al., 2004; Varadan et al., 2002). Ub variants UbK48R, UbK63R, UbD77, or Ub74 containing chain-terminating mutations were used to provide with full control of chain length and the location of the isotope-labeled Ub unit. DUBs were expressed and purified following established protocols. Yeast proteasome and recombinant Ubp6 were purified as described (Mansour et al., 2015).
NMR experiments
All NMR experiments were performed at 23° on Bruker Avance III 600 MHz and 800 MHz NMR spectrometers equipped with cryoprobes. The protein samples were prepared in 20 mM (1 M = 1 mol/L) phosphate buffer (pH 6.8) containing 0.02% (w/v) NaN3 and 5% D2O. 15N longitudinal relaxation time (T1) was measured using standard pulse sequences. Each experiment was acquired as a pseudo-3D data set with 1024 points in 1H, 80 increments in 15N, and typically up to five T1-relaxation delays. The data were processed using TopSpin (Bruker Biospin Inc) and analyzed using in-house Matlab software. Residues having overlapping NMR signals were excluded from the T1 analyses.
Chemical shift perturbations mapping
Differences in chemical shifts of amide-group nuclei (1HN, 15N) between two samples (A and B) were quantified as amide chemical shift perturbations (CSPs), defined as follows:
| (1) |
Here δH and δN are chemical shifts of 1H and 15N, respectively, for a given backbone N-H group. The same equation was used to quantify spectral perturbations upon titration; in this case, A refers to the unbound species, and B corresponds to various steps in the titration.
NMR-based titration assays
NMR titration assays were performed by adding increasing amounts of unlabeled ligand (ubistatin) to 15N-labeled protein (e.g., Ub) and monitoring changes (shifts, attenuations) in protein signals in 1H-15N HSQC or SOFAST-HMQC spectra. Signal shifts upon titration were quantified as CSPs (see Equation 1, where in this case A corresponds to the free protein and B to the current step in the titration) and fitted to the following equation:
| (2) |
where fB is the bound fraction of the protein, and CSPmax is the maximum possible value of the signal shift, i.e. in the fully ligand-bound state. The analysis was performed using in-house Matlab program Kdfit (Varadan et al., 2004). Various binding stoichiometry models were considered, as detailed in (Varadan et al., 2004). Specifically, for a 1:1 binding model:
| (3) |
where [Pt] and [Lt] are the total molar concentrations of the protein and the ligand, respectively.
A buffer containing 20 mM sodium phosphate at pH 7.5 greatly improved the solubility of the ubistatins, and there was no detectable effect on Ub, which was in 20 mM sodium phosphate buffer at pH 6.8. Control spectra (not shown) revealed negligible CSPs in Ub, even when the pH changed from 6.8 to 7.5. The ability to dissolve ubistatins at higher concentrations allowed us to add smaller volumes of ubistatin stock solution to the 15N Ub sample.
NMR signal assignment of hemi-ubistatin and Ub
NMR signals of the nine protons in h-ubiB were assigned using 2D 1H- 1H TOCSY and NOESY spectra. Proton signals in unbound h-ubiB are separated except for H4, H5, and H9, which overlap. Because of the large CSPs observed in Ub upon binding of h-ubiB, NMR signal assignment of Ub in the bound state was confirmed through 3D H_CCCONH and C_CCCONH TOCSY spectra collected on 13C/15N-enriched mono-Ub in complex with h-ubiB. The assignment of Ub amide signals from the 1H-15N NMR spectra in the course of the titration was used as a starting point. The nine proton signals from Ub-bound h-ubiB were assigned using 2D 1H, 1H-TOCSY and NOESY spectra with 13C, 15N filtering to eliminate signals from Ub. Having determined the assignments of h-ubiB and Ub in the bound state we then performed 3D 13C-edited, 13C 15N-filtered NOESY-HSQC experiment to detect intermolecular NOEs. To obtain high resolution and greater sensitivity with more scans, 2D versions of the 13C-edited/filtered NOESY-HSQC with either 1H or 13C in the indirect dimension were collected as well, the representative spectra are shown in (Figure S3B). Interproton distances for the eighteen assigned intermolecular NOEs were calibrated using a 2D 1H- 1H NOESY experiment that allows for both inter- and intra-molecular NOEs to appear in the same spectrum (Table S3).
Site-directed spin labeling and paramagnetic relaxation enhancement (PRE) measurements
Site-directed spin labeling was used to obtain long-range intermolecular distance constraints for determining the structure of Ub:h-ubiB complex. The nitroxide paramagnetic spin label, 1-oxyl-2,2,5,5-tetramethyl-3-pyrroline-3-methyl methanesulfonate (MTSL), was attached to a Cys at position 12, 36, 48, or 63 in Ub, introduced via site-directed mutagenesis, as described (Varadan et al., 2005; Varadan et al., 2004). The paramagnetic relaxation enhancement (PRE) effects caused by MTSL were quantified as the ratio of the signal intensities in the NMR spectra of h-ubiB or Ub recorded with MTSL in the oxidized and reduced states. The location of the unpaired electron of MTSL was reconstructed using in-house Matlab program SLfit (Ryabov and Fushman, 2006) based on the ratio of intensities of Ub signals in 1H-15N HSQC spectra recorded with MTSL in oxidized and reduced states, while distances from the electron to individual protons in h-ubiB were determined from signal intensities in 1D 1H-1H NOESY spectra with 15N filtering.
Structure modeling for Ub:h-ubiB complex
The structure of the Ub:h-ubiB complex was obtained using the biomolecular docking program HADDOCK v2.1 (de Vries et al., 2010). As no atomic-resolution structure of h-ubiB exists, the compound was first built in “Marvinsketch” (http://www.chemaxon.com) and then energy minimized to determine the representative conformation; see details in Figure S4C. Among the two possible conformations of the h-ubiB compound, due to electrostatic repulsion of sulfonate groups the trans conformation exhibits lower energy and was chosen for the docking process.
For the starting Ub structure we used the solution NMR optimized coordinates from PDB ID 1D3Z. Ambiguous and unambiguous constraints used to drive and restrain structure calculations of the complex are reported in Table S3. Ambiguous constraints were defined on the basis of the CSPs data collected on 15N Ub or h-ubiB upon addition of its binding partner. To avoid treating the whole ligand as active, we defined ambiguous constraints (AIR) between residues of the Ub protein and a set of H atoms of h-ubiB that show some contacts in the 3D [13C]-edited, [13C,15N]-filtered NOESY-HSQC experiment only. A total of 18 unambiguous distance constraints arising from NOEs were introduced as well as 18 other unambiguous distances derived from PRE data. To accurately describe unambiguous distances arising from PRE data, cysteine residues and MTSL atoms were introduced in silico at positions 36 and 48 on Ub and distances were defined between the oxygen of MTSL and any other proton of h-ubiB (Table S3). During the docking process, the number of steps for the rigid body stage was set to zero and the temperature for the simulated annealing was set to 500 K. A total of 2000 structures were generated at the rigid body stage, the best 200 of them were selected for semi-flexible refinement, and 100 were finely retained for the final refinement in water.
The final solvent-refined structures were subjected to clustering and the ten best structures in the lowest energy cluster were retained for analysis (see Table S4 for results). The resulting structures were clustered with a 1.5 Å cut off and the Ub:h-ubiB interaction contacts were analyzed within the LIGPLOT program (Wallace et al., 1995).
The HADDOCK score used during the final stage was calculated as a weighted contribution of different energetic terms: 1.0×EvdW + 0.2×Eelec + 0.1×EAIR + 1.0×Edesolv. The electrostatic surface of Ub was computed using the adaptive Poisson-Boltzmann solver (APBS).
Small-angle neutron scattering (SANS) measurements and analysis
Scattering measurements were performed on the 30 m SANS instruments at the NIST Center for Neutron Research (NCNR) in Gaithersburg, MD. Samples of free Ub (4.5 mg/mL) or K48-Ub2 (4.29 mg/mL) and equimolar mixtures of ubiB with Ub (4.5 mg/mL) or K48-Ub2 (4.24 mg/mL) were prepared in 20 mM sodium phosphate buffer (pD 6.8) in D2O. The neutron wavelength, λ, was 6 Å, with a wavelength spread, Δλ/λ, of 0.15. Scattered neutrons were detected with a 64 cm×64 cm two-dimensional position-sensitive detector with 128×128 pixels at a resolution of 0.5 cm/pixel. A sample-to-detector distance of 1.5 m was used to cover the range 0.03 Å-1 ≤ q ≤ 0.4 Å-1 , where q = 4π sin(θ)/λ and 2θ is the scattering angle. The data were reduced using the IGOR program with routines developed at the NCNR (Kline, 2006) to produce scattering intensity, I(q). Guinier analysis was carried out using the equation I(q)/I(0) ≈ exp(−q2Rg2/3) to estimate the radius of gyration (Rg) and the forward scattering intensity, I(0), of the samples. Atom pair distribution, P(r), was calculated using GNOM (Semenyuk and Svergun, 1991).
NMR & SANS-based modeling of ubiB:2xUb complex structure
The ternary ubiB:2xUb system consisted of two Ub molecules and the ubiB ligand, which were randomly sampled in space. As a first step, 2000 structures (replicas) of the system were generated by means of rigid-body docking using HADDOCK (de Vries et al., 2010) software where only the first step of the HADDOCK protocol was considered. The strong similarity of both the magnitudes (CSPs, Figure 2B,C) and the directions of the NMR signal shifts (Figure S7A,B) upon Ub binding to ubistatin B and hemi-ubistatin B indicates similarity of the interatomic contacts in the Ub:ubiB and Ub:h-ubiB complexes. Therefore the intermolecular distance constraints between ubiB and each Ub molecule were the same as for Ub:h-ubiB interaction (Table S3) except that each Ub was assumed to interact with a different half of ubiB (i.e., one Ub with the left and the other Ub with the right naphthotriazole moiety). For each generated structure, the corresponding SANS profile was calculated and compared with the experimental I(q). The I(q) and P(r) profiles were calculated for each structure using the Xtal2sas (Curtis et al., 2012; Kline, 2006) module of the SASSIE (Curtis et al., 2012) program. The agreement between the experimental and theoretical SANS profiles was quantified using the target function:
| (4) |
where Np (=77) is the total number of SANS data points, I(qi)exp is the measured scattering intensity, I(qi)calc is the intensity calculated using Xtal2sas for scattering vector qi, and σi is the experimental error. The whole set of structures was sorted according to their corresponding χ2 values, ranging from 47.8 to 1020, while spanning a range of Rg values from 16 Å to 20.4 Å. The HADDOCK calculation then proceeded through the flexible docking step, after which 200 best-score final structures were selected and analyzed. The cluster of the top 10 structures is shown in Figure S8A.
In parallel, the structure that best matched the experimental SANS profile after the rigid-body docking step was subjected to molecular dynamics (MD) to account for the various degrees of freedom that were held fixed in the rigid-body docking and to adequately represent the energetics. For the MD protocol, each Ub was modeled with the AMBER99SB force field and the ligand with the GAFF force field. The model was ionized and solvated with TIP3P water molecules in a 75 Å × 75 Å × 75 Å unit cell. The resulting ∼38,000 atoms system was energy minimized and equilibrated locally with ACEMD (Harvey et al., 2009) for 2 ns under NPT conditions, of 1 atm at 300 K, nonbonded cutoff of 9 Å, rigid bonds, and PME electrostatics. A time step of 4 fs was used, in conjunction with a hydrogen mass repartitioning scheme. During minimization and the first 2 ns of equilibration, the protein's Cα and ligand's heavy atoms were restrained by a harmonic potential with k = 1 kcal·mol-1·Å-2 (1 cal = 4.184 J). No biasing potentials were imposed during MD production runs, i.e. the ligand and the protein were allowed to diffuse freely through the solvent. The complete MD simulation resulted in a 2 μs trajectory, from which 50,000 snapshots were extracted, each snapshot representing 40 ps of the trajectory. The structures were aligned with respect to one of the Ub molecules prior to SANS profile calculation.
The ensemble of structures extracted from the MD trajectory spanned a range of Rg values from 16.1 to 19.6 Å while the range of χ2 values was from 47.9 to 5032. The best-χ2 structure from the MD trajectory is shown in Figure S8C. As a word of caution, the mathematical problem of deriving structures from SAS data is largely underdetermined. Thus, the structure that best matches our experimental data (Figure 5C,D) has to be interpreted as a structure that could undergo fluctuations around its average position.
Fluorescence-based binding assays
Fluorescence measurements were carried out on an ISS PC1 photon counting spectrofluorimeter at 21°C in PBS pH 7.4 buffer. The concentration of ubiB was held constant at 1 μM for each titration (10 μM for Ub titration) and the concentration of the ligand (in this case, Ub or Ub2) was varied. Emission and excitation spectra were acquired in the L-configuration. Anisotropy experiments were performed in T-configuration using 0.5 mm slits, excitation of 386 nm and detection at 485 nm. Data were processed using Vinci software and fit to a single-site binding model (see Equation 3) using in-house Matlab program Kdfit (Varadan et al., 2004). The measured change, Δaniso, in fluorescence anisotropy upon titration was fit to the following equation:
| (5) |
where fB is the bound fraction of ubiB, see Equation 2 (where in this case [Pt] is the molar concentration of ubiB and [Lt] is the molar concentration of Ub or Ub2).
Proteasome/Rpn11 inhibition assay
Proteasomes isolated from human erythrocytes were obtained from (BioMol/Enzo Life Sciences). The Rpn11 substrate, Ub4pep, comprised a His6 tag for purification followed by four consecutive repeats of Ub linked in tail-to-head fashion (i.e., the C-terminal G76 of the upstream Ub fused directly to M1 of the downstream Ub), and with the 4th Ub followed by a 30-mer peptide sequence that contained a unique cysteine for labeling. A DNA sequence encoding the Ub4pep substrate was synthesized and cloned into pJexpress401 vector for E. coli expression. Oregon Green® 488 was covalently coupled to the unique cysteine in Ub4pep. Purified Ub4pep was incubated with the 5× molar excess of the dye in reaction buffer comprising 50 mM sodium phosphate, 200 mM NaCl, 10% Glycerol, and 100 μM TCEP, pH 7.0. The coupling reaction was quenched by the addition of 10 mM DTT. The mixture of fluorescent dye and protein was then applied to a Superdex 75HR column (GE Healthcare) to separate the free label from the labeled protein.
Ubistatin variants were first assayed for inhibition of changes in fluorescence polarization at 10 μM in assay buffer (50 mM Tris, pH 7.4, 0.01% NP-40, 1 mM MgCl2, 1 mM ATP, 1 mM DTT). IC50 values of active analogues were obtained by assaying at a series of concentrations (0, 0.39, 0.78, 1.56, 3.13, 6.25, 12.5, 25, 50, and 100 μM) in quadruplicate.
In Vitro Assay of CFTR Ubiquitination
In vitro CFTR ubiquitination reactions were performed as described (Nakatsukasa et al., 2008). Briefly, microsomes containing HA-tagged CFTR were incubated with yeast cytosol, 125I-ubiquitin, an ATP regenerating system and either 100 μM of the indicated compound or the equivalent volume of DMSO at 26°C for 60 min. When the effects of the ubistatins were examined, the compounds were dissolved in DMSO to concentrations such that adding 1 μL of the stock resulted in the desired concentration in a reaction. The reactions were incubated at 26°C for 60 min and were quenched by the addition of SDS to 1% and N-ethylmaleimide (NEM) to 10mM in 1xTBS. After a further incubation at 37°C for 30 min, TritonX-100 was added to ∼1% (and the SDS diluted to 0.1%) by the addition of 900 μL of 1.1% TritonX-100 in 1xTBS supplemented with NEM and PIC. Immunoprecipitation of CFTR was accomplished by addition of anti-HA monoclonal antibody (clone 12CA5, Roche) and Protein A Sepharose CL4B (GE Healthcare) and incubation at 4°C for 16 hrs. Immunoprecipitates were washed 3 times in 50mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.2% SDS, 5 mM EDTA, 10 mM NEM and PIC, and subjected to SDS-PAGE. Half of each sample was used to detect HA-CFTR by western blot, and the other half was used to detect of 125I-Ubiquitin-conjugated CFTR by phosphorimager analysis. The data were quantified using ImageGauge Ver. 3.45 software (Fuji Life Sciences) and normalized to the amount of HA-tagged CFTR protein present in the immunoprecipitation, as measured by western blot. The extent of CFTR ubiquitination in the DMSO control reactions was set to 100%.
Gel-based assay for inhibition of proteasome and DUBs
A total of 25 μM K48-Ub2 or K63-Ub2 was incubated with 5 μM Ubp6 in 50 μL of PBS pH 7.4 buffer. For linkage specific DUBs, 20 μM of K11-Ub2, K48-Ub2, or K63-Ub2 was incubated with 150 nM Cezanne (OTUD7B), OTUB1, or AMSH respectively. For proteasome assay 5 μM of K11-Ub6+ or Ubch5b-Ubn was incubated with 50 nM of purified yeast 26S proteasome (containing Ubp6). All reactions were carried out at 30°C. Samples were taken at the indicated time points and stored in 5× loading dye until analysis on 15% SDS-PAGE. Reactions that included h-ubiB or ubiB were allowed to equilibrate for 20 min prior to addition of the DUB. Visualization was achieved by Coomassie staining. Western blot detection was carried out using polyclonal rabbit anti-Ub (Dako z0458) and linkage specific rabbit monoclonal anti-K11 (Millipore 2021885), anti-K48 (Millipore 2197314), and anti-K63 (Millipore 2063204), all at a 1:1,000 dilution. IgG goat anti-rabbit HRP conjugate (Bio-Rad) at a 1:50,000 dilution was used as the secondary antibody for all blots.
Characterization of ubiB internalization in live cells using fluorescence microscopy
HeLa and RCC4 cells were cultured in DMEM supplemented with 10% fetal bovine serum in a glass bottom dish. Cells were treated with 10 μM ubiB for 6 hrs and washed three times with PBS buffer. The plasma membrane was visualized by treating cells with a 1:500 dilution of wheat germ agglutinin, Texas Red® conjugate (Life Technologies) for 15 min. A Zeiss LSM700 inverted confocal microscope was used to image live cells in Z-stack configuration at 1 μM increments with a 63× oil immersion lens. Images were acquired at 512×512 pixels with the 405 nm laser for excitation of ubiB and the 555 nm laser to excite the red plasma membrane probe. All images were processed using Zen software (Zeiss) and analyzed with the Fiji distribution of ImageJ.
HeLa cells grown in media containing 10 μM ubiB for 6 hrs on a glass coverslip were used for immunofluorescence. Following treatment, cells were fixed in ice cold 100% (v/v) methanol for 5 min at room temperature, permeabilized in PBS, 15% (v/v) Triton X-100, and blocked overnight at 4°C in PBST buffer containing 1% (w/v) BSA, 22.52 mg/mL glycine, and 0.1% Tween 20. The same four Ub antibodies (see above) were used with an anti-rabbit-Cy5 as the secondary antibody. Throughout this procedure ubiB remained within the cells. Cover slips were mounted to microscope slide and sealed. Images were acquired and analyzed as above in the live cell assay.
Detecting ubiB-induced perturbations of the Ub landscape
HeLa cells were treated with 10 μM MG132 or 2 μM ubiB for 8 hrs, washed two times in PBS pH 7.4 buffer, and harvested. Equal amounts of HeLa cells representing each condition were combined with 2× protein loading dye and immediately lysed at 95°C for 10 minutes. Samples were loaded onto a 12% SDS gel, transferred to a PVDF membrane, blotted against rabbit anti-Ub (Dako) in a 1:1,000 dilution, and detected with goat anti-rabbit HRP (BioRad) in a 1:50,000 dilution.
In a separate assay, following 6 hrs ubiB treatment, HeLa cells were lysed chemically in RIPA buffer (25 mM Tris pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium doxycholate, 0.1% SDS) in the presence of protease inhibitor cocktail (Millipore 539134). At each stage of lysis, the presence of ubiB was visually detected using a 308 nm UV transilluminator and photographed (Figure S10C). Cell debris were cleared by centrifugation and the protein content of the supernatant was quantified using Bradford assay. Equal amounts of protein were loaded and subjected to Western blot with rabbit anti-Ub as described above.
Synthesis of ubistatin compounds
All ubistatin compounds used for study were designed and synthesized as detailed below.
General Procedures
Starting materials and reagents were purchased from commercial suppliers and used without further purification. All products were characterized by LC/MS and NMR analysis. Analytical LC/MS chromatography was performed on Waters Alliance HT Liquid Chromatograph coupled with Waters SQ Mass Spectrometer featuring electro spray ionization (ESI+ and ESI-) with MS scans from 100 to 1500 m/z and cone voltage 50 V; mobile phase: 0.01% NH4OH in water/acetonitrile; column: 4.6 × 30 mm XBridge C18 3.5 mm column, 3 mL/min, 2.5 min, Gradient: 5-95% acetonitrile. 1H NMR spectra were recorded on a Varian 500 MHz spectrometer or a Bruker 300 MHz spectrometer and are reported in ppm and referenced to residual protons in the NMR solvent. 13C NMR spectra were recorded at 125 MHz on a Varian spectrometer; 13C shifts are reported in ppm and referenced to carbon resonances in the NMR solvent. Data below are reported as chemical shift (d, in ppm), splitting (s = singlet, d = doublet, t = triplet, m = multiplet; br = broad), coupling constant in Hz; integration. Masses are reported in daltons.
Ubistatin B (ubiB)
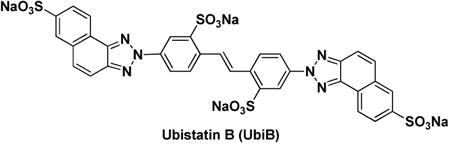
(Dobas et al., 1957): To 925 mg (2.50 mmol) of 4,4′-diamino-2,2′-stilbenedisulfonic acid was added 10 mL of water and 1 mL of conc. HCl and the mixture was cooled in an ice bath. A solution of 345 mg (5.00 mmol) NaNO2 in 2 mL of water was added over 30 min. After stirring an additional 10 min, the paste was added to a solution of 1.11 g (5.00 mmol) of 6-amino-2-naphthalenesulfonic acid, 200 mg of NaOH (5.00 mmol), and 1.23 g (15.0 mmol) of NaOAc in 150 mL of water cooled in an ice bath. A deep red color appears quickly and the solution was stirred overnight, warming to room temperature. The next day Na2CO3 (800 mg, 7.55 mmol) was added and the mixture was stirred 1 h before the addition of 7.5 mL of NH4OH(aq) and 2.50 g of CuSO4·5 H2O (10 mmol) dissolved in 20 mL of water. The mixture was heated at 90 °C for 1 h, the mixture turning green. The reaction was cooled to 70 °C and 7 mL of conc. HCl was added to give a dark solution which was removed from the oil bath. Precipitates form with the addition of 30 g of NaCl. The solids were collected by filtration and dissolved in ca. 40 mL hot water before addition of ca. 200 mL of acetone and cooling. Filtration and rinsing with acetone yielded 916 mg of yellow solid (42%). 1H NMR (500 MHz, CD3OD) δ: 9.11 (s, 2H), 8.73 (d, J = 8.23 Hz, 2H), 8.53 (d, J = 8.21 Hz, 2H), 8.49 (s, 2H), 8.45 (s, 2H), 8.32 (d, J = 13 8.43 Hz, 2H), 8.19 (d, J = 8.20 Hz, 2H), 7.91-7.95 (m, 4H); 13C NMR (125 MHz, D2O) δ: 143.15, 141.82, 141.69, 140.98, 137.24, 135.05, 131.45, 130.09, 129.25, 128.03, 126.57, 124.76, 124.27, 124.02, 121.45, 118.34, 115.87; MS: M-H = 833 (C34H22N6O12S4 = 834). IR Spectrum matched that reported by Bio-Rad Laboratories (BR182452).
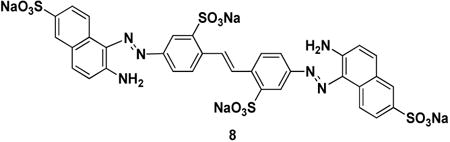
A small amount of the intermediate dye (8) was isolated after the Na2CO3 addition from an aliquot of the reaction mixture by adding NaCl(s) and filtering off the red precipitate. 1H NMR (300 MHz, D2O) δ: 8.66 (d, J = 8.90 Hz, 2H), 8.24 (d, J = 1.88 Hz, 2H), 8.04 (s, 2H), 7.96 (s, 2H), 7.81-7.84 (m, 4H), 7.72 (dd, J = 1.19, 7.93 Hz, 2H), 7.24 (d, J = 8.37 Hz, 2H), 6.38 (d, J = 8.94 Hz, 2H); MS: M-H = 837 (C34H26N6O12S4 = 838).
The following compounds were made using the above procedure on the same scale:
Compound 1
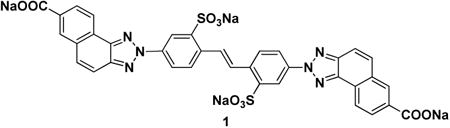
Synthesized from 4,4′-diamino-2,2′-stilbenedisulfonic acid and 2-aminonaphthoic acid, reddish brown solid (45%): 1H NMR(300 MHz, D2O/DMSO-d6) δ: 8.79 (d, J = 2.22 Hz, 2H), 8.50-8.53 (m, 4H), 8.35-8.38 (m, 2H), 8.25 (s, 2H), 8.22-8.25 (m, 2H), 8.03 (d, J = 9.29 Hz, 2H), 7.88-7.97 (m, 4H); MS: M-H = 761 (C36H22N6O10S2 = 762).
Compound 2
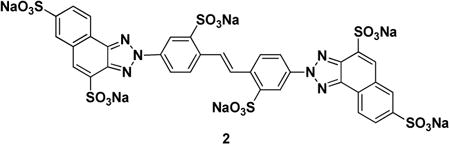
Synthesized from 4,4′-diamino-2,2′-stilbenedisulfonic acid and 2-amino-3,6-naphthalenedisulfonic acid, yellow solid (40%): 1H NMR (500 MHz, CD3OD) δ: 9.11 (d, J = 2.27 Hz, 2H), 8.73 (d, J = 8.35 Hz, 2H), 8.64 (dd, J = 2.26, 8.58 Hz, 2H), 8.54 (s, 2H), 8.42 (s, 2H), 8.36 (s, 2H), 8.29 (d, J = 8.70 Hz, 2H), 8.23 (dd, J = 1.48, 8.35 Hz, 2H); MS: M-H = 993 (C34H22N6O18S6 = 994).
Compound 9 (h-ubiB)
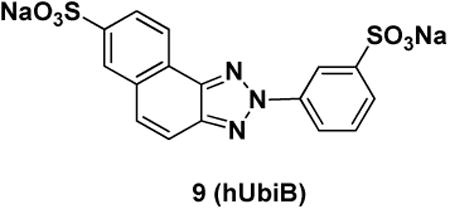
Synthesized from 3-aminobenzenesulfonic acid and 6-amino-2-naphthalene sulfonic acid, off-white solid (44%): 1H NMR (300 MHz, D2O) δ: 8.21 (s, 1H), 8.14 (d. J = 7.70 Hz, 1H), 8.00 (s, 1H), 7.88 (d, J = 7.61 Hz, 1H), 7.71 (d, J = 7.57 Hz, 1H), 7.57 (d, J = 7.73 Hz, 1H), 7.30-7.37 (m, 3H); MS: M-H = 404 (C16H11N3O6S2 = 405).
Compound 10
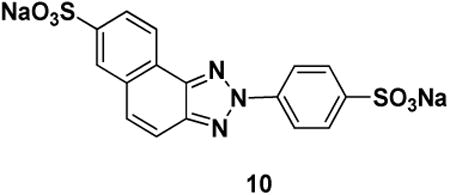
Synthesized from 4-aminobenzenesulfonic acid and 6-amino-2-naphthalene sulfonic acid, off-white solid (68%): 1H NMR (300 MHz, D2O) δ: 8.09 (s, 1H), 8.05 (d. J 8.24 Hz, 1H), 7.86-7.95 (m, 4H), 7.80 (d, J = 7.80 Hz, 1H), 7.40 (d, J = 9.19 Hz, 1H), 7.27 (d, J = 9.21 Hz, 1H); MS: M-H = 404 (C16H11N3O6S2 = 405).
Compound 3
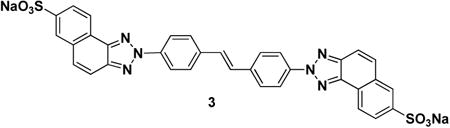
To 354 mg (125 mmol) of 4,4′-diaminostilbene was added 5 mL of water and 0.5 mL of conc. HCl and the mixture was cooled in an ice bath. A solution of 172 mg (2.50 mmol) NaNO2 in 1 mL of water was added over 30 min. After stirring an additional 10 min, the paste was added to a solution of 560 mg (2.50 mmol) of 6-amino-2-naphthalenesulfonic acid, 100 mg of NaOH (2.50 mmol), and 610 g (7.5 mmol) of NaOAc in 50 mL of water cooled in an ice bath. A deep red color appeared quickly and the mixture was stirred overnight, warming to room temperature. The next day Na2CO3, 400 mg (3.25 mmol), was added and the mixture was stirred 1 h before filtering off the red solid. The solids were added to 50 mL of pyridine and 600 mg of copper (II) acetate and heated at 130 °C for 5 h, the mixture turned deep green. After cooling and filtering, 80 mg of solid was dissolved in a 1:1 mixture of MeOH:DMF with a small amount of HCl(aq). The solution was added to a 5 g PEAX (basic) solid phase extraction column and rinsed with MeOH. The product elutes with the addition of acidic (HCl(aq)) DMF solution, concentration yielded 27 mg of dark brown solid (4%): (300 MHz, D2O) δ: 8.38-8.41 (m, 4H), 8.19 (s, 2H), 8.02 (d, J = 7.06 Hz, 2H), 7.88 (d, J = 7.09 Hz, 2H), 7.84 (d, J = 7.30 Hz, 2H), 7.54-7.57 (m, 8H); MS: M-H = 673 (C34H22N6O6S2 = 674).
Compound 4
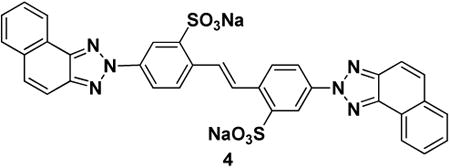
To 1.29 g (3.49 mmol) of 4,4′-diamino-2,2′-stilbenedisulfonic acid was added 20 mL of water and 1.7 mL of conc. HCl and the mixture was cooled in an ice bath. A solution of 480 mg (6.98 mmol) NaNO2 in 5 mL of water was added over 30 min. After stirring an additional 10 min, the paste was added to a solution of 1.00 g (6.98 mmol) of 2-naphthylamine and 1.71 g (20.9 mmol) of NaOAc in 100 mL of MeOH cooled in an ice bath. A red color appeared quickly and the solution was stirred 1 h before addition of Na2CO3, (1.0 g, 8.1 mmol) and the mixture was stirred overnight. Another 100 mL of water and 100 mL of MeOH was added before addition of 11 mL of NH4OH(aq) and a solution of 3.6 g of CuSO4·5 H2O (20 mmol) dissolved in 20 mL of water and the reaction was heated at 80 °C for 24 h. After cooling and filtration, the collected green solids were stirred in acetone for 1 h and filtered. The solid was then treated with 900 mL of refluxing MeOH and filtered, the filtrate was then treated two more times with hot MeOH. The MeOH washings were combined and concentrated giving 1.26 g of a yellow solid (54%): 1H NMR (500 MHz, DMSO-d6) δ: 8.84 (d, J = 2.37 Hz, 2H), 8.63 (d, J = 7.68 Hz, 2H), 8.37 (dd, J = 2.32, 8.48 Hz, 2H), 8.34 (s, 2H), 8.10 (d, J = 7.73 Hz, 2H), 7.93-7.99 (m, 6H), 7.73-7.80 (m, 4H); 13C NMR (125 MHz, DMSO-d6) δ: 147.86, 143.83, 142.96, 138.33, 135.65, 132.75, 130.81, 130.02, 128.80, 128.76, 128.51, 127.83, 124.59, 123.63, 120.61, 119.81, 117.02; MS: M-H = 673 (C34H22N6O6S2 = 674).
1,2-Bis(2-carboxy-4-nitrophenyl)ethane
(Müller et al., 1985). A solution of 2.91 g (16.0 mmol) of 2-methyl-5-nitrobenzoic acid in 10 mL of basic water (NaOH) was warmed to 50 °C. To this was added a solution of 48 mL of 30% (w/v) NaOH in water and 42 mL of 10-12% sodium hypochlorite over 1 h. Precipitate forms and the mixture was stirred at 50 °C overnight. The solid product was filtered and rinsed with cold water. The white solid was then dissolved in a large amount of water, 1.56 g of white solid precipitated out upon addition of 1 N HCl (54%). 1H NMR (300 MHz, DMSO-d6) δ: 8.64 (d, J = 3.52 Hz, 2H), 8.20 (dd, J = 2.55, 8.45 Hz, 2H), 7.50 (d, J = 8.50 Hz, 2H), 3.39 (s, 4H); MS: M-H = 359 (C16H12N2O8 = 360).
1,2-Bis(4-amino-2-carboxyphenyl)ethane
(Hein and Pierce, 1954). Iron powder (7.29 g, 130 mmol) in 50 mL of water and 1 mL conc. HCl was heated to 120 °C, and to this mixture was added a solution of 940 mg of 1,2-bis(2-carboxy-4-nitrophenyl)ethane in 20 mL slightly basic water. The mixture was heated 1.5 h before cooling and addition of 1 N NaOH until the mixture was basic. After filtration, the solution was made acidic with HCl (aq) and the precipitated solids were filtered and rinsed with acetone giving 415 mg of off-white solid (57%). 1H NMR (300 MHz, DMSO-d6) δ: 7.03 (d, J = 2.5, 2H), 6.89 (d, J = 8.2, 2H), 6.63 (dd, J = 2.5, 8.2, 2H), 4.60 (br s, 4H), 2.88 (s, 4H). 13C NMR (75 MHz, DMSO-d6) δ: 169.31, 146.35, 131.01, 130.52, 130.05, 117.21, 115.24, 35.25. MS: M-H = 299 (C16H16N2O4 = 300).
Compound 7
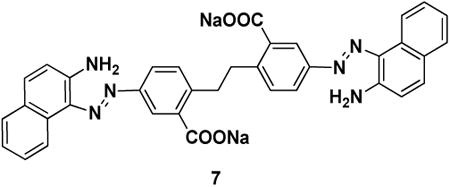
To 415 mg of 1,2-bis(4-amino-2-carboxyphenyl)ethane (1.38 mmol) in 10 mL of water and 0.75 mL of conc. HCl cooled to 0 °C was added a solution of 191 mg (2.76 mmol) of NaNO2 in 2 mL of water over 30 min. The solution was then added to a solution of 395 mg (2.76 mmol) of 2-naphthylamine and 679 mg of NaOAc (8.28 mmol) in 50 mL of MeOH cooled in an ice bath, the solution turning orange quickly. After 20 min, 400 mg (3.25 mmol) of Na2CO3 was added. A small amount of the reaction mixture was filtered and rinsed with MeOH, the orange solid was pure by LC/MS and NMR analysis. 1H NMR (300 MHz, DMSO-d6) δ: 8.64 (d, J = 8.53 Hz, 2H), 8.25 (s, 2H), 7.94 (d, J = 7.23 Hz, 2H), 7.70-7.76 (m, 4H), 7.49-7.54 (m, 2H), 7.37 (d, J = 4.02 Hz, 2H), 7.29-7.35 (m, 2H), 7.08 (d, J = 8.89 Hz, 2H), 3.28 (s, 4H); MS: M-H = 607 (C36H28N6O4 = 608).
Compound 6
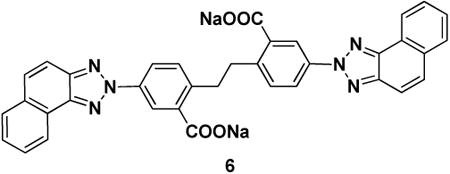
To the mixture above was added 5 mL of NH4OH(aq) and 1.37 g (5.52 mmol) of CuSO4·5 H2O dissolved in 20 mL of water and the reaction was heated at 90 °C overnight. The next day the green mixture with off-white precipitate was cooled and filtered. The filtrate was added to a separatory funnel with 1 N HCl and EtOAc. The water was extracted several times with EtOAc, the EtOAc layers were combined, dried (MgSO4) and concentrated to give 35 mg of dark solid product (4%) which was converted to the bis sodium salt by treatment with NaOH. 1H NMR (300 MHz, DMSO-d6) δ: 8.59 (d, J = 7.18 Hz, 2H), 8.42 (d, J = 2.43 Hz, 2H), 8.04-8.10 (m, 4H), 7.88-7.96 (m, 4H), 7.69-7.79 (m, 4H), 7.46 (d, J = 8.31 Hz, 2H), 3.29 (s, 4H); MS: M-H = 603 (C36H24N6O4 = 604).
Compound 5
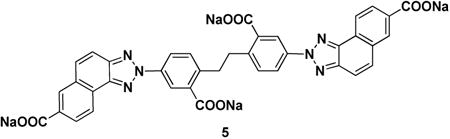
To a suspension of 260 mg (0.87 mmol) of 1,2-bis(4-amino-2-carboxyphenyl)ethane in 2 mL of water cooled in an ice bath was added 0.7 mL conc. HCl. A solution of 120 mg (1.73 mmol) of NaNO2 in 1 mL of water was added dropwise over 20 min. This was then added dropwise to a cold solution of 321 mg (1.73 mmol) of 2-amino-6-naphthoic acid, 70 mg of NaOH, and 430 mg of NaOAc in 30 mL of water and the resulting suspension was stirred overnight warming to room temperature. The next day 284 mg of Na2CO3 was added and the mixture was stirred 1 h before addition of 2.7 mL of NH4OH(aq) and 890 mg of CuSO4·5 H2O in 10 mL. The mixture turns dark and was heated at 90 °C for 1 h before cooling to 70 °C. The addition of a few drops of 1 N HCl dissolves all solids in the basic solution which was cooled to room temperature. The addition of NaCl precipitates out pink solid which was purified by reverse phase chromatography to produce 80 mg of red solid product (12%). 1H NMR (300 MHz, D2O) δ: 8.60 (d, J = 8.3, 2H), 8.56 (d, J = 1.0, 2H), 8.53 (d, J = 2.3, 2H), 8.33 – 8.26 (m, 2H), 8.21 (dd, J = 2.4, 8.3, 2H), 7.85 (q, J = 9.2, 4H), 7.54 (d, J = 8.4, 2H), 3.38 (s, 4H). MS: M-H = 691 (C38H24N6O8 = 692).
Supplementary Material
Key Resources Table.
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal Anti-ubiquitin | Dako | Cat#z0458; RPID: AB_2315524 |
| Rabbit monoclonal K11 Ubiquitin linkage specific, clone 2A3/2E6 | Merck-Millipore | Cat# MABS107-I; RRID:AB_2713901 |
| Rabbit monoclonal K48 Ubiquitin linkage specific, clone Apu2 | Merck-Millipore | Cat# 05-1307; RRID:AB_1587578 |
| Rabbit monoclonal K63 Ubiquitin linkage specific, clone Apu3 | Merck-Millipore | Cat# 05-1308; RRID:AB_1587580 |
| IgG goat anti-rabbit (H L)-HRP conjugate | BioRad | Cat#170-6515; RPID: AB_11125142 |
| IgG H&L Goat anti-rabbit (Cy5®) preadsorbed | Abcam | Cat#ab97077; RPID: AB_10679461 |
| Mouse anti-HA monoclonal unconjugated (clone 12CA5) | Roche | Cat# 11583816001; RRID: AB_514505 |
| Bacterial and Virus Strains | ||
| BL-21(DE3) Rosetta™ Chemically Competent E. coli | Novagen | Cat# 71397 |
| M15 Chemically Competent Cells | Qiagen | N/A |
| MAX Efficiency™ DH5α™ Chemically Competent Cells | ThermoFisher | Cat#18258012 |
| Biological Samples | ||
| Human Proteasomes - erythrocyte isolated | BioMol/Enzo Life Sciences | Cat# BML-PW9310-0050 |
| Yeast (S. cerevisiae) proteasomes | Mansour et al., 2015 | N/A |
| ER-derived microsomes | Nakatsukasa et al., 2008 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2-mercaptoethanol 99% | Sigma-Aldrich | Cat#M3148 |
| Dithiothreitol (DTT) | Gold Bio | Cat#DTT25 |
| Isopropyl-β-D-1-thiogalactoside (IPTG) | Gold Bio | Cat#I2481C50 |
| Complete protease inhibitor cocktail | Roche | Cat#11836145001 |
| Potassium Phosphate dihydrate KH2PO4 | Merck | Cat#529568 |
| Potassium Phosphate dibasic K2HPO4 | Merck | Cat#105104 |
| Sodium Phosphate dihydrate NaH2PO4 | Merck | Cat#567545 |
| Sodium Phosphate dibasic Na2HPO4 | Merck | Cat#567550 |
| Ampicillin sodium salt | Sigma | Cat#A9518 |
| Kanamycin | Sigma | Cat#K4000 |
| Chloroamphenicol | Sigma | Cat#C0378 |
| KOD Hot Start Master Mix | Novagen | Cat#71842 |
| Phenylmethanesulfonyl fluoride (PMSF) | Sigma-Aldrich | Cat#P7626 |
| IGEPAL CA-630 | Sigma-Aldrich | Cat#18896 |
| Adenosine 5′-triphosphate, disodium, trihydrate (ATP) | Fisher Scientific | Cat#10326943 |
| Ammonium Chloride (15N, 99%) | Cambridge Isotope Laboratories, Inc | Cat#NLM-467 |
| D-Glucose (U-13C6, 99%) | Cambridge Isotope Laboratories, Inc | CLM-1396-5 |
| Albumin, from Bovine Serum | Sigma-Aldrich | Cat#A3294 |
| Coomassie R-250 Brilliant Blue | ThermoFisher | Cat#20278 |
| Tris(2-carboxyethyl)phosphine-HCl (TCEP-HCl) | Gold Bio | Cat# TCEP25 |
| MG132 | Calbiochem Merck Millipore | Cat#474790 |
| 1-oxyl-2,2,5,5-tetramethyl-3-pyrroline-3-methyl methanesulfonate (MTSL) | Toronto Research Chemicals | Cat#O875000 |
| Wheat Germ Agglutinin, Texas Red® conjugate | Life Technologies | Cat#W7024 |
| Oregon Green® 488 maleimide | ThermoFisher | Cat#O6034 |
| I125-Ubiquitin | This work | |
| Ub4pep∼Oregon Green® conjugate | This work | |
| Ubistatin B (ubiB) | Verma et al., 2004 | N/A |
| Ubistatin compound 1 | This work | N/A |
| Ubistatin compound 2 | This work | N/A |
| Ubistatin compound 3 | This work | N/A |
| Ubistatin compound 4 | This work | N/A |
| Ubistatin compound 5 | This work | N/A |
| Ubistatin compound 6 | This work | N/A |
| Ubistatin compound 7 | This work | N/A |
| Ubistatin compound 8 | This work | N/A |
| Ubistatin compound 9 / Hemi-ubistatin B (h-ubiB) | This work | N/A |
| Hemi-Ubistatin compound 10 | This work | N/A |
| Related ubistatin compound 11 | This work | N/A |
| 8-Anilino-1-naphthalenesulfonic acid (ANS) | Sigma | Cat#A1028-5G |
| 15N-labeled (distal) dimeric Ub (K48-Ub2) | This work | N/A |
| 15N-labeled (distal) dimeric Ub (K63-Ub2) | This work | N/A |
| 15N-labeled monomeric Ub | This work | N/A |
| 13C/15N-labeled monomeric Ub | This work | N/A |
| 15N-labeled monomeric UbR42A | This work | N/A |
| 15N-labeled monomeric UbR42E | This work | N/A |
| 15N-labeled monomeric UbR72A | This work | N/A |
| 15N-labeled monomeric UbR72E | This work | N/A |
| 15N-labeled monomeric UbR42E,R72E | This work | N/A |
| 15N-labeled monomeric UbR74A | This work | N/A |
| 15N-labeled monomeric UbR74E | This work | N/A |
| 15N-labeled monomeric UbK63D | This work | N/A |
| 15N-labeled monomeric Ub72 (∆73-76) | This work | N/A |
| 15N-labeled monomeric UbT12C | This work | N/A |
| 15N-labeled monomeric UbI36C | This work | N/A |
| 15N-labeled monomeric UbK48C | This work | N/A |
| 15N-labeled monomeric UbK63C | This work | N/A |
| 15N-labeled RUB1 | Singh et al., 2012 | N/A |
| 15N--labeled UBA of UBQLN1 | This work | N/A |
| K11-linked dimeric Ub (K11-Ub2) | This work | N/A |
| K11-linked >hexameric Ub (K11-Ub6+) | This work | N/A |
| K48-linked dimeric Ub (K48-Ub2) | This work | N/A |
| K63-linked dimeric Ub (K63-Ub2) | This work | N/A |
| 6xHis-Ubp6 full length | This work | N/A |
| GST-Cezanne OTUD7B (OTU domain) | This work | N/A |
| 6xHis-OTUB1 (OTU domain) | This work | N/A |
| GST-AMSH (JAMM/MPN+ domain) | This work | N/A |
| UbcH5B-Ubn | This work | N/A |
| Experimental Models: Cell Lines | ||
| HeLa | ATCC | ATCC® CCL-2™ |
| RCC4 | Gift from Allan Weissman (NCI) | N/A |
| Recombinant DNA | ||
| pJexpress401 Ub4pep | This work | N/A |
| pET3a untagged Human wild-type Ubiquitin | Varadan et al., 2002 | N/A |
| pET3a untagged UbR42A | This work | N/A |
| pET3a untagged UbR42E | This work | N/A |
| pET3a untagged UbR72A | This work | N/A |
| pET3a untagged UbR72E | This work | N/A |
| pET3a untagged UbR42E/R72E | This work | N/A |
| pET3a untagged UbR74A | This work | N/A |
| pET3a untagged UbR74E | This work | N/A |
| pET3a untagged UbK63D | This work | N/A |
| pET3a untagged Ub72 | This work | N/A |
| pET3a untagged UbT12C | This work | N/A |
| pET3a untagged UbI36C | This work | N/A |
| pET3a untagged UbK48C | This work | N/A |
| pET3a untagged UbK63C | This work | N/A |
| pTXB1-RUB1 | Singh et al., 2012 | N/A |
| pGEX2T-1 UBQLN1 UBA domain | Raasi et al., 2005 | N/A |
| pQE30 Ubp6 full length | Mansour et al., 2015 | N/A |
| pOPINK Cezanne/OTUD7B 53-446 (OTU domain) | (Mevissen et al., 2013) | Addgene:61581 |
| pProEX-OTUB1 (OTU domain) | (Wang et al., 2009) | Addgene:26959 |
| AMSH | Fushman Lab | N/A |
| pET3a human UbcH5B | Mansour et al., 2015 | N/A |
| pSM1152 (2μ URA3 PPGKCFTR∷HA) | Nakatsukasa et al., 2008 | N/A |
| Software and Algorithms | ||
| ImageGauge version 3.45 | Fuji Life Sciences | www.fujifilm.com |
| Marvinsketch | ChemAxon | http://www.chemaxon.com |
| ORCA package | Max-Planck Institut für Chemische Energiekonversion | https://orcaforum.cec.mpg.de/ |
| PRODRG server | GlycoBioChem Ltd. | http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg |
| LIGPLOT program | EMBL-EBI | https://www.ebi.ac.uk/thornton-srv/software/LIGPLOT/ |
| Vinci software | Vinci Inc. | http://www.iss.com/fluorescence/software/vinci.html |
| Zen Lite | Carl Zeiss | www.zeiss.com |
| Fiji | LOCI - University of Wisconsin-Madison. | https://fiji.sc |
| Bruker Topspin version 3.5 | Bruker Inc. | www.bruker.com |
| Computer aided resonance assignment (CARA) | Wüthrich lab, ETH | Cara.nmr.ch |
| SPARKY 3 | Goddard and Kneller, UCSF | https://www.cgl.ucsf.edu/home/sparky/ |
| HADDOCK v2.1 | Bonvin Lab, University of Utrecht | haddock.science.uu.nl/ |
| SASSIE | NCNR, NIST | https://sassie-web.chem.utk.edu/sassie2/ |
| PyMOL | Schrödinger, LLC | http://www.pymol.org |
| KdFit, Matlab program for analysis of titration data | Fushman Lab | http://gandalf.umd.edu/FushmanLab/pdsw |
| SLfit, Matlab program for analysis of spin-labeling data | Fushman Lab | http://gandalf.umd.edu/FushmanLab/pdsw |
| Other | ||
| 5 mL GST Trap | GE Life Sciences | Cat#17513001 |
| 5 mL His Trap | GE Life Sciences | Cat#17-5248-01 |
| 5 mL SP HP | GE Life Sciences | Cat#17-5054-01 |
| 16/60 Superdex 75 | GE Life Sciences | Cat#28989333 |
| Protein A Sepharose CL4B | GE Life Sciences | Cat#17-0780-01 |
| 4.6 × 30 mm XBridge C18 3.5 μm column | Waters | N/A |
Highlights.
Ubistatin B binds Ub selectively and prefers K48-linked Ub chains over K11 or K63
Hydrophobic and charge/polar interactions are critical for ubistatin:Ub binding
Ubistatins block disassembly of Ub conjugates by various DUBs and by 26S proteasome
Ubistatin B penetrates cancer cells and alters cellular Ub landscape
Acknowledgments
We thank George H. Lorimer and Arnon Henn for access to their fluorimeters, Ananya Majumdar for help with setting up triple-resonance NMR experiments, Rajesh Singh for Rub1 sample, Allan Weissman for RCC4 cells, and Konstantin Berlin for useful discussions. The project has been funded in part from the NCI Initiative for Chemical Genetics, NIH, under Contract No. N01-CO-12400 to Stuart L. Schreiber, by NIH grants GM065334 to DF and GM095755 to DF and MHG, ISF grant 909-14 to MHG, and by NIH grants GM075061 and DK079307 and by Cystic Fibrosis Foundation Therapeutics grant BRODSK13XXO to JLB, and Fulbright postdoctoral fellowship to MAN, and utilized NMR instrumentation supported in part by NSF grant DBI1040158 and neutron-scattering facilities supported in part by the NSF under Agreement No. DMR-0944772.
Footnotes
Author Contributions: TAL synthesized ubistatin compounds. Rpn11 functional assays were performed by FP, T-FC, RJD; CFTR assays by JLB and JLG-F; NMR measurements by MAN, DZ, CMC, SMB, DF; SANS studies by CAC, SK, DF; structure calculations by OW and MAN; fluorescence measurements by MAN, WM, AT, OH and analyzed by DF; DUB assays by MAN, WM, OH, and cellular assays by MAN, AT, and MHG. All authors contributed to writing the manuscript and revisions.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arena G, Bellia F, Frasca G, Grasso G, Lanza V, Rizzarelli E, Tabbi G, Zito V, Milardi D. Inorganic stressors of ubiquitin. Inorganic chemistry. 2013;52:9567–9573. doi: 10.1021/ic401276x. [DOI] [PubMed] [Google Scholar]
- Bellare P, Small EC, Huang X, Wohlschlegel JA, Staley JP, Sontheimer EJ. A role for ubiquitin in the spliceosome assembly pathway. Nat Struct Mol Biol. 2008;15:444–451. doi: 10.1038/nsmb.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows DS, Tyers M. Cell biology. Chemical genetics hits. Science. 2004;306:67–68. doi: 10.1126/science.1104611. [DOI] [PubMed] [Google Scholar]
- Buckley DL, Crews CM. Small-molecule control of intracellular protein levels through modulation of the ubiquitin proteasome system. Angew Chem Int Ed Engl. 2014;53:2312–2330. doi: 10.1002/anie.201307761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzolai L, Franchini F, Gilliland D, Rossi F. Protein--nanoparticle interaction: identification of the ubiquitin--gold nanoparticle interaction site. Nano letters. 2010;10:3101–3105. doi: 10.1021/nl101746v. [DOI] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Cook WJ, Jeffrey LC, Carson M, Chen Z, Pickart CM. Structure of a diubiquitin conjugate and a model for interaction with ubiquitin conjugating enzyme (E2) J Biol Chem. 1992;267:16467–16471. doi: 10.2210/pdb1aar/pdb. [DOI] [PubMed] [Google Scholar]
- Curtis JE, Raghunandan S, Nanda H, Krueger S. SASSIE: A program to study intrinsically disordered biological molecules and macromolecular ensembles using experimental scattering restraints. Comput Phys Commun. 2012;183:382–389. [Google Scholar]
- Cvek B. Proteasome inhibitors. Prog Mol Biol Transl Sci. 2012;109:161–226. doi: 10.1016/B978-0-12-397863-9.00005-5. [DOI] [PubMed] [Google Scholar]
- de Vries SJ, van Dijk M, Bonvin AM. The HADDOCK web server for data-driven biomolecular docking. Nature protocols. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC biology. 2014;12:94. doi: 10.1186/s12915-014-0094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobas J, Pirkl J, Hanousek V. Fluorescent derivatives of 1,2,3-triazole. II. Sulfonated benzo-and naphthotriazoles derived from 4,4′diaminostilbene-2,2′disulfonic acid. Chemicke Listy pro Vedu a Prumysl. 1957;51:1113–1121. [Google Scholar]
- Falini G, Fermani S, Tosi G, Arnesano F, Natile G. Structural probing of Zn(II), Cd(II) and Hg(II) binding to human ubiquitin. Chem Commun (Camb) 2008:5960–5962. doi: 10.1039/b813463d. [DOI] [PubMed] [Google Scholar]
- Finley D, Ulrich HD, Sommer T, Kaiser P. The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics. 2012;192:319–360. doi: 10.1534/genetics.112.140467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushman D, Walker O. Exploring the linkage dependence of polyubiquitin conformations using molecular modeling. J Mol Biol. 2010;395:803–814. doi: 10.1016/j.jmb.2009.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL. Development of proteasome inhibitors as research tools and cancer drugs. J Cell Biol. 2012;199:583–588. doi: 10.1083/jcb.201210077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey MJ, Giupponi G, De Fabritiis G. ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J Chem Theory Comput. 2009;5:1632–1639. doi: 10.1021/ct9000685. [DOI] [PubMed] [Google Scholar]
- Hein DW, Pierce ES. Optical Bleaching Agents. I. Derivatives of Dichlorodiaminostilbenedisulfonic Acid. J Am Chem Soc. 1954;76:2725–2731. [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Kline SR. Reduction and analysis of SANS and USANS data using IGOR Pro. J Appl Cryst. 2006;39:895–900. [Google Scholar]
- Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Majetschak M. Extra cellular ubiquitin: immune modulator and endogenous opponent of damage-associated molecular pattern molecules. Journal of leukocyte biology. 2011;89:205–219. doi: 10.1189/jlb.0510316. [DOI] [PubMed] [Google Scholar]
- Mangini V, Dell'Aglio M, De Stradis A, De Giacomo A, De Pascale O, Natile G, Arnesano F. Amyloid transition of ubiquitin on silver nanoparticles produced by pulsed laser ablation in liquid as a function of stabilizer and single-point mutations. Chemistry. 2014;20:10745–10751. doi: 10.1002/chem.201402934. [DOI] [PubMed] [Google Scholar]
- Mansour W, Nakasone MA, von Delbrueck M, Yu Z, Krutauz D, Reis N, Kleifeld O, Sommer T, Fushman D, Glickman MH. Disassembly of Lys11- and mixed-linkage polyubiquitin conjugates provide insights into function of proteasomal deubiquitinases Rpn11 and Ubp6. J Biol Chem. 2015;290:4688–4704. doi: 10.1074/jbc.M114.568295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevissen TE, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, Ekkebus R, Kulathu Y, Wauer T, El Oualid F, et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell. 2013;154:169–184. doi: 10.1016/j.cell.2013.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HJ, Rape M. Enhanced protein degradation by branched ubiquitin chains. Cell. 2014;157:910–921. doi: 10.1016/j.cell.2014.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller A, Kuhnert B, Sonntag C, Wünsch KH. Synthese von 2-Styryl-naphtho[1,2-d] thiazolen (Synthesis of 2-Styryl-naptho[1,2-d] thiazoles) Journal für Praktische Chemie. 1985;327:698–704. [Google Scholar]
- Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–112. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM. Targeting of substrates to the 26S proteasome. FASEB J. 1997;11:1055–1066. doi: 10.1096/fasebj.11.13.9367341. [DOI] [PubMed] [Google Scholar]
- Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Pickart CM, VanDemark AP. Opening doors into the proteasome. Nat Struct Biol. 2000;7:999–1001. doi: 10.1038/81018. [DOI] [PubMed] [Google Scholar]
- Raasi S, Varadan R, Fushman D, Pickart CM. Diverse polyubiquitin interaction properties of ubiquitin-associated domains. Nat Struct Mol Biol. 2005;12:708–714. doi: 10.1038/nsmb962. [DOI] [PubMed] [Google Scholar]
- Roscoe BP, Thayer KM, Zeldovich KB, Fushman D, Bolon DN. Analyses of the effects of all ubiquitin point mutants on yeast growth rate. J Mol Biol. 2013;425:1363–1377. doi: 10.1016/j.jmb.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruschak AM, Slassi M, Kay LE, Schimmer AD. Novel proteasome inhibitors to overcome bortezomib resistance. Journal of the National Cancer Institute. 2011;103:1007–1017. doi: 10.1093/jnci/djr160. [DOI] [PubMed] [Google Scholar]
- Ryabov Y, Fushman D. Interdomain mobility in di-ubiquitin revealed by NMR. Proteins. 2006;63:787–796. doi: 10.1002/prot.20917. [DOI] [PubMed] [Google Scholar]
- Semenyuk AV, Svergun DI. GNOM -- a program package for small-angle scattering data processing. J Appl Cryst. 1991;24:537–540. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JJ, Scavone F, Cooper EM, Kane LA, Youle RJ, Boeke JD, Cohen RE. Polyubiquitin-sensor proteins reveal localization and linkage-type dependence of cellular ubiquitin signaling. Nature methods. 2012;9:303–309. doi: 10.1038/nmeth.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, Zerath S, Kleifeld O, Scheffner M, Glickman MH, Fushman D. Recognition and cleavage of related to ubiquitin 1 (Rub1) and Rub1-ubiquitin chains by components of the ubiquitin-proteasome system. Mol Cell Proteomics. 2012;11:1595–1611. doi: 10.1074/mcp.M112.022467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nature reviews Drug discovery. 2014;13:889–903. doi: 10.1038/nrd4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadan R, Assfalg M, Fushman D. Using NMR spectroscopy to monitor ubiquitin chain conformation and interactions with ubiquitin-binding domains. In: Deshaies RJ, editor. Ubiquitin and Protein Degradation, Meth Enzym. part B. Vol. 399. 2005. pp. 177–192. [DOI] [PubMed] [Google Scholar]
- Varadan R, Assfalg M, Haririnia A, Raasi S, Pickart C, Fushman D. Solution conformation of Lys63-linked di-ubiquitin chain provides clues to functional diversity of polyubiquitin signaling. J Biol Chem. 2004;279:7055–7063. doi: 10.1074/jbc.M309184200. [DOI] [PubMed] [Google Scholar]
- Varadan R, Walker O, Pickart C, Fushman D. Structural properties of polyubiquitin chains in solution. J Mol Biol. 2002;324:637–647. doi: 10.1016/s0022-2836(02)01198-1. [DOI] [PubMed] [Google Scholar]
- Verma R, Peters NR, D'Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, Zhang M, Coffino P, Fushman D, Deshaies RJ, King RW. Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science. 2004;306:117–120. doi: 10.1126/science.1100946. [DOI] [PubMed] [Google Scholar]
- Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- Wang T, Yin L, Cooper EM, Lai MY, Dickey S, Pickart CM, Fushman D, Wilkinson KD, Cohen RE, Wolberger C. Evidence for bidentate substrate binding as the basis for the K48 linkage specificity of otubain 1. J Mol Biol. 2009;386:1011–1023. doi: 10.1016/j.jmb.2008.12.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Saeki Y, Murakami A, Kawawaki J, Tsuchiya H, Yoshihara H, Shindo M, Tanaka K. A comprehensive method for detecting ubiquitinated substrates using TR-TUBE. Proc Natl Acad Sci U S A. 2015;112:4630–4635. doi: 10.1073/pnas.1422313112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanzoni S, Ceccon A, Assfalg M, Singh RK, Fushman D, D'Onofrio M. Polyhydroxylated [60]fullerene binds specifically to functional recognition sites on a monomeric and a dimeric ubiquitin. Nanoscale. 2015;7:7197–7205. doi: 10.1039/c5nr00539f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Raasi S, Fushman D. Affinity makes the difference: nonselective interaction of the UBA domain of Ubiquilin-1 with monomeric ubiquitin and polyubiquitin chains. J Mol Biol. 2008;377:162–180. doi: 10.1016/j.jmb.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.