

Abstract
IMPORTANCE
Since the implementation of universal newborn screening (NBS) for cystic fibrosis (CF), the timing and magnitude of growth deficiency or its association with correlates of disease among infants with CF who underwent NBS has not been well described.
OBJECTIVE
To examine incremental weight gain, linear growth, and clinical features in the first year of life among infants with CF who underwent NBS.
DESIGN, SETTING, AND PARTICIPANTS
The Baby Observational and Nutrition Study (BONUS), a multicenter, longitudinal, observational cohort study, was conducted during regular CF clinic visits in the first 12 months of life at 28 US Cystic Fibrosis Foundation–accredited Care Centers from January 7, 2012, through May 31, 2015. Participants included 231 infants younger than 3.5 months who underwent NBS and had confirmed CF, with a gestational age of at least 35 weeks, birth weight of at least 2.5 kg, and toleration of full oral feeds. Of these, 222 infants (96.1%) had follow-up beyond 6 months of age and 215 (93.1%) completed 12 months of follow-up.
EXPOSURE
Cystic fibrosis.
MAIN OUTCOME AND MEASURES
Attained weight and length for age and World Health Organization normative z scores at ages 1 to 6 and 8, 10, and 12 months (defined a priori).
RESULTS
Of the 231 infants enrolled, 110 infants (47.6%) were female and 121 (52.4%) were male, with a mean (SD) age of 2.58 (0.69) months. BONUS infants had lower than mean birth weights (mean z score, −0.15; 95% CI, −0.27 to −0.04) and higher birth lengths (mean z score, 0.44; 95% CI, 0.26 to 0.62). They achieved normal weight by 12 months, a significant improvement over a prescreening cohort of newborns with CF from 20 years before the contemporary cohort (mean z score increase, 0.57; 95% CI, 0.37–0.77). However, length was lower than the mean at 12 months (mean z score, −0.56; 95% CI, −0.70 to −0.42). Only 30 infants (13.6%) were at less than the 10th percentile of weight for age, whereas 53 (23.9%) were at less than the 10th percentile of length for age at more than half their visits. Male sex, pancreatic insufficiency, meconium ileus, histamine blocker use, and respiratory Pseudomonas aeruginosa infection were associated with lower weight or length during the first year. Insulinlike growth factor 1 levels were significantly lower among low-length infants. Persistently low-weight infants consumed significantly more calories, and weight and length z scores were negatively correlated with caloric intake.
CONCLUSIONS AND RELEVANCE
Since initiation of universal NBS for CF, significant improvement has occurred in nutritional status, with normalization of weight in the first year of life. However, length stunting remains common.
Before newborn screening (NBS), the diagnosis of cystic fibrosis (CF) was delayed until onset of nutritional failure and many patients demonstrated persistent growth and nutritional deficiencies.1 The advent of NBS for CF has contributed to improved clinical outcomes, including weight and stature.2–5 Improvements in growth have been steady, with median World Health Organization (WHO) weight percentiles of US infants with CF younger than 2 years increasing from less than 20% in 1993 to 40% by 2014.6 However, despite identification of CF in the youngest patients and emphasis on early nutritional supplementation and prompt implementation of pancreatic enzyme replacement therapy (PERT),7 growth in children with CF continues to lag significantly behind that of healthy children. These highly vulnerable infants with CF have not previously undergone evaluation in a prospective geographically diverse study powered to evaluate growth variables and potential contributing factors. Identification of these patterns is an important first step in targeting at-risk patients and could be used to measure response to novel CF therapeutics,8,9 because their use extends into this age group.
The Baby Observational and Nutrition Study (BONUS) was a prospective multicenter study of infants with CF in which the primary aim was to examine the current state of weight gain and linear growth in the first year of life, framed within the context of historic growth patterns in CF.10 The secondary aim of BONUS was to prospectively explore concurrent nutritional, metabolic, respiratory, infectious, and inflammatory characteristics associated with early CF anthropometric measurements.
Methods
Patients and Study Design
BONUS was conducted at 28 US Cystic Fibrosis Foundation–accredited Care Centers in the CF Foundation Therapeutic Development Network. Infants younger than 3.5 months were enrolled if they had a sweat chloride level of at least 60 mEq/L (to convert to millimoles per liter, multiply by 1) by using a quantitative pilocarpine iontophoresis test or 2 well-characterized CF transmembrane conductance regulator gene (CFTR [NCBI Entrez Gene 1080]) mutations. Infants with a gestational age of less than 35 weeks, with a birth weight of less than 2.5 kg, unable to take full oral feeds, or with any serious condition other than pancreatic insufficiency (PI) contributing to malabsorption or interfering with normal growth were excluded. Study visits coincided with guideline-recommended CF clinic visits (monthly until 6 months and at 8, 10, and 12 months of age).7 Anthropometric measures (length, weight, and occipital frontal circumference) were systematically performed at each visit by certified staff.11 Participant guardians completed 3-day diet, stool,12 modified pain,13 and cough14 diaries for the infant before each study visit (eMethods 1 in the Supplement). Breastfeeding frequency and duration, salt intake, vitamin and formula supplementation, and PERT dosages were included in the diet diary. Caloric and nutritional analyses were performed, and the mean was calculated for the 3-day collection. Measurement of fecal elastase levels, respiratory tract cultures, and chest radiography were performed according to Cystic Fibrosis Foundation care guidelines,7 and the 12-month chest radiograph was scored15 centrally by 2 independent readers. Clinical hematologic levels, chemistry, and serum vitamin levels (A, D, and E) were collected at enrollment and 6 and 12 months. Additional details can be found in eMethods 2 in the Supplement. A complete list of cites is found in the list of BONUS investigators at the end of the article. Written informed consent was obtained from all participating parents or guardians, and all participating sites received approval from their institutional review boards.
Data Presentation
We calculated z scores for attained weight, length, and occipital frontal circumference for age by using World Health Organization (WHO) standard growth curves for healthy newborns or healthy infants.16,17 A historic Cystic Fibrosis Foundation Patient Registry birth cohort from 1994–1995 (when only Colorado, Wyoming, and Wisconsin were conducting NBS) was compared with the contemporary BONUS birth cohort for attained weight and length. No standard definition of poor growth in infants with CF exists. For this study, persistently low weight or length were each defined as a z score of less than −1.28 (10th percentile) for more than half the yearly measurements, with at least 1 of these low measurements occurring between 6 and 12 months of age. Infants were classified as having PI if they exhibited fecal elastase levels of 200 μg/g or less or if they had 2 PI-causing CFTR mutations.18 If the fecal elastase value was unavailable (n = 28), PERT use at study termination was used for the purposes of classification (27 of 28 infants) (eTable 1 in the Supplement).
Statistical Analysis
Differences were calculated and comparisons were made with 1- and 2-sample t tests, the Fisher exact test for categorical data, or the Pearson correlation coefficient. Linear mixed-effects models were used to estimate overall change and differences in anthropometric curves across the entire year, accounting for within-patient correlation (eMethods 2 in the Supplement). Risk ratios and 95% CIs were calculated.
Results
From January 7, 2012, through May 31, 2014, 231 infants with CF were enrolled in BONUS, and 215 (93.1%) completed follow-up through 12 months of age (eFigure 1 in the Supplement). Overall, 110 infants (47.6%) were female, 121 (52.4%) were male, 132 (57.1%) were Phe508del homozygous, 211 (91.3%) had PI, and 27 (11.7%) had documented meconium ileus. Mean (SD) age at initial examination was 2.58 (0.69) months. Both CFTR alleles were classes I to III (associated with a severe phenotype19) in 195 infants (84.4%); 16 (6.9%) had 1 class I to III mutation and 1 unknown mutation, 19 (8.2%) had at least 1 class IV to V mutation (associated with a milder phenotype), and 1 (0.4%) had 2 unknown CFTR mutations, consistent with the overall population with CF6 (eTable 1 in the Supplement).
Anthropometrics
As extracted from medical records, birth weights of BONUS infants were statistically lower than those of WHO healthy newborns (mean z score, −0.15; 95% CI, −0.27 to −0.04), with a median birth weight of 3.2 (range, 2.5–4.4) kg. By 12 months of age, BONUS infants demonstrated increased weight gain, and weight for age was not different from that of WHO healthy infants (mean z score, −0.04; 95% CI, −0.17 to 0.09) (Figure 1A). Compared with the 1994–1995 birth cohort, an increase in weight for age was seen at 6 months (mean z score increase, 1.47; 95% CI, 1.24–1.69) and 12 months (mean z score increase, 0.57; 95% CI, 0.37–0.77) (Figure 2A).
Figure 1. Distribution of Baby Observational and Nutritional Study (BONUS) Infant z Scores for Growth at 3, 6, and 12 Months of Age.
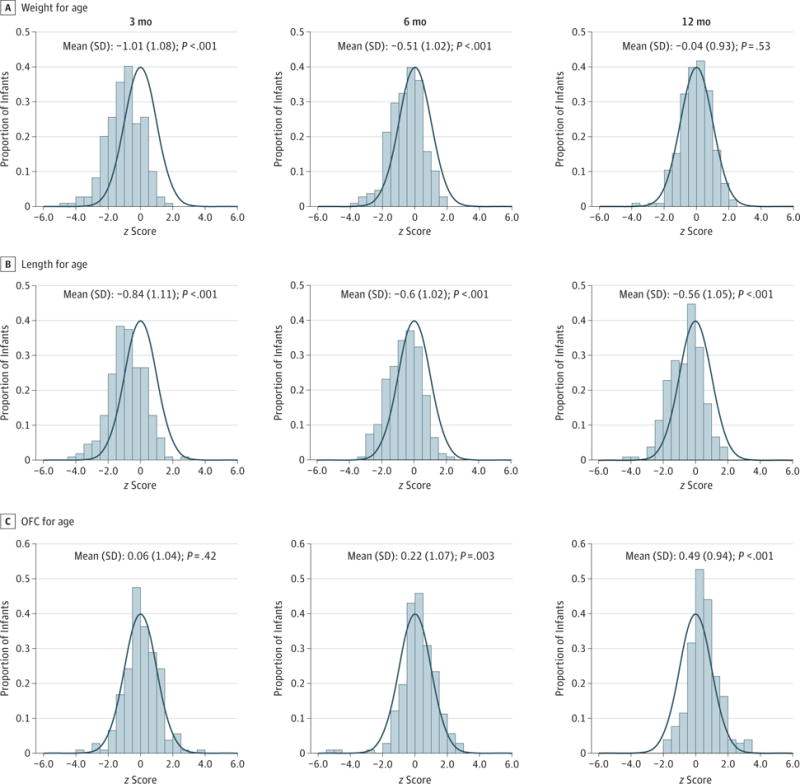
Curves indicate World Health Organization (WHO) standard growth curves for healthy infants16; we calculated z scores for attained weight, length, and occipital frontal circumference (OFC) for age of infants in the BONUS cohort using the WHO standard growth curves.
Figure 2. Baby Observational and Nutritional Study (BONUS) Cohort and Historic Infant Cohort z Scores for Growth During the First Year of Life.
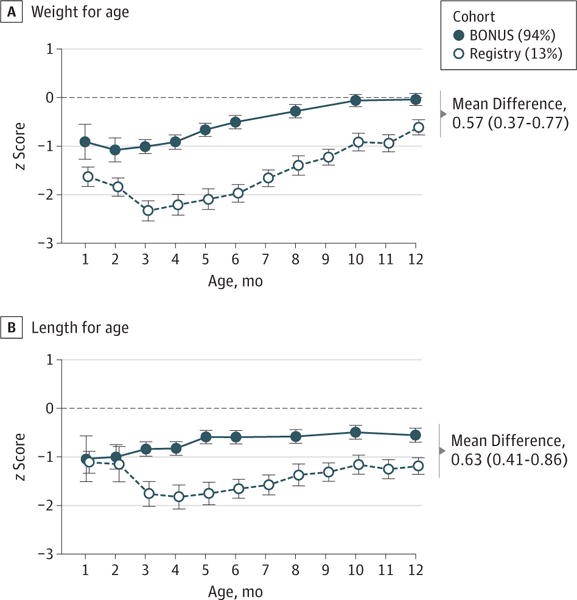
We calculated z scores for mean attained weight and length for age of infants using the World Health Organization standard growth curves.16 The historic cohort includes infants in the 1994–1995 Cystic Fibrosis Foundation Patient Registry (Registry) birth cohort. Error bars indicate 95% CIs. Percentages indicate the percentages of the cohort undergoing newborn or prenatal screening.
Birth lengths of BONUS infants were higher than those of WHO healthy newborns (mean z score, 0.44; 95% CI, 0.26–0.62), with a median birth length of 50.8 (range, 40.1–57.3) cm. However, length for age in BONUS infants measured per study protocol was lower than that in WHO healthy infants (Figure 1B) at 3, 6, and 12 months of age (12-month mean z score, −0.56; 95% CI, −0.70 to −0.42). Midparental height was available in the Cystic Fibrosis Foundation Patient Registry for 52 parent pairs, and as a group, their heights were not significantly lower than US Centers for Disease Control and Prevention 2007–2010 adult white normative values.20 Length was improved compared with the historic cohort at 6 months (z score increase, 1.06; 95% CI, 0.82–1.30) and at 12 months (z score increase, 0.63; 95% CI, 0.41–0.86) (Figure 2B). Occipital frontal circumference was normal or above normal at all ages (Figure 1C).
Nutritional Intake and Fat-Soluble Vitamins
Infants with PI initiated PERT at a mean age of 2.0 months (range, 0.0–4.3 months) with a mean (SD) dose of 1879 (623) U/kg of lipase per meal in the first 6 months, consistent with Cystic Fibrosis Foundation care guidelines.7 Type of feeding at 3 months was as follows: 57 (25.0%) were exclusively breastfed; 127 (55.7%), exclusively formula fed; and 44 (19.3%), both. On the basis of 3-day diet diaries for each participant who did not receive breast milk, median caloric intake in the first year was 107 kcal/kg/d (range, 86–133 kcal/kg/d) (eTable 2 in the Supplement). Total caloric intake could not be calculated for infants who received breast milk. Among exclusively formula-fed infants, 51 (40.2%) received a high-calorie formula (≥24 kcal/30 mL) at 3 months; 77 (52.4%), at 6 months; and 72 (49.0%), at 12 months. Intake of fat (median, 5.0 g/d; 25th-75th percentile, 3.8–6.5 g/d), protein (median, 2.5 g/d; 25th-75th percentile, 1.9–3.2 g/d), and carbohydrates (median, 13.0 g/d; 25th-75th percentile, 10.4–16.2 g/d) were consistent during the first year of life. By 12 months, 37 infants (17.1%) were not receiving formula or breast milk. Exclusive breastfeeding was recorded in patient diaries in 57 infants (25.0%) at 3 months, 35 (15.6%) at 6 months, and 22 (10.0%) at 12 months. These rates are lower than breastfeeding rates in healthy infants born in 2012 (43% at 3 months and 22% at 6 months).21 Exclusive breastfeeding was less common among infants with pancreatic sufficiency vs those with PI at 3 (1 [5.3%] vs 56 [26.8%]), 6 (1 [5.3%] vs 34 [16.6%]), and 12 (1 [5.3%] vs 21 [10.5%]) months. Exclusively breastfed infants weighed more than formula-fed infants or those fed a combination at 3 months of age (mean z score difference, 0.54; 95% CI, 0.22–0.87) (Figure 3A) but not at 6 or 12 months. Feeding type was not associated with infant length at any time point (Figure 3B). Growth z scores had a significant and negative correlation with caloric intake among those who were not breastfed (r = −0.48 for weight; r = −0.31 for length).
Figure 3. Feeding Type and Caloric Intake in the Growth of Baby Observational and Nutritional Study (BONUS) Cohort During the First Year of Life.
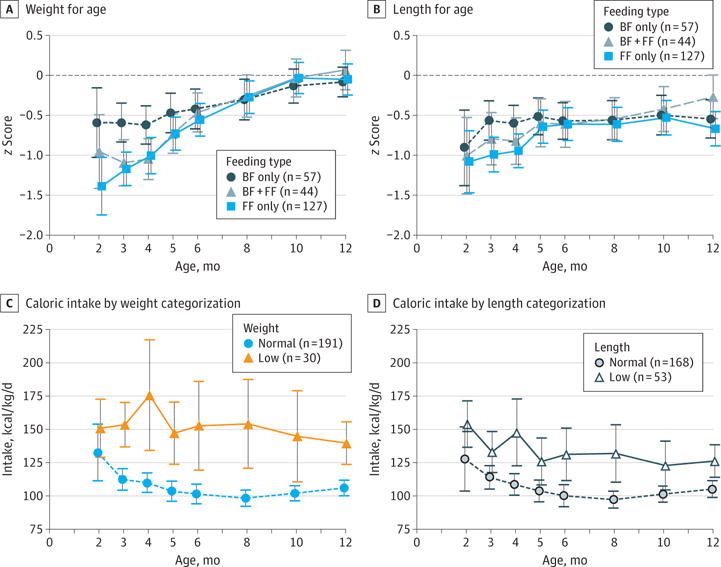
We calculated z scores for mean attained weight and length for age of infants using the the World Health Organization (WHO) standard growth curves16 and plotted these by breastfed (BF) only, formula fed (FF) only, and both. Caloric intake by weight categorization and length categorization was calculated among exclusively FF or solid food–fed BONUS infants. Low categorization indicates that more than 50% of measurements are less than the 10th percentile of the WHO standard curve, with at least 1 such measurement occurring after 6 months of age. Error bars indicate 95% CIs.
With use of cumulative anthropometrics through the first year, participants were categorized into low-weight and low-length groups. Among the 222 BONUS infants with follow-up beyond 6 months, 30 (13.6%) met the study definition of low weight; 53 (23.9%), low length; and 24 (10.8%), both categories. Twenty-eight of 53 low-length infants (53%) were not categorized as low weight. Low-weight infants consumed significantly more calories than did normal-weight infants at 6 months (153 vs 101 kcal/kg/d). Low-length infants also consumed significantly more calories (131 vs 100 kcal/kg/d) (Figures 3C and D).
Most BONUS infants received fat-soluble vitamin (225 [97.8%]) and sodium (226 [98.3%]) supplementation. Median vitamin intake at 12 months among exclusively formula-fed infants was as follows: vitamin A, 8704 IU/d (25th-75th percentile, 845–34 922 IU/d); vitamin E (α-tocopherol), 51.4 IU/d (25th-75th percentile, 0.2–114.2 IU/d); and 25-hydroxyvitamin D, 754 IU/d (25th-75th percentile, 90–1755 IU/d). Serum deficiency of vitamin A (<20 μg/dL; to convert to micromoles per liter, multiply by 0.0349) occurred in 1 of 135 infants (0.7%), and serum deficiency of α-tocopherol (<3.5 mg/L; to convert to micromoles per liter, multiply by 1.33) in 5 of 117 (4.3%).22 Serum levels of total 25-hydroxyvitamin D were less than 30 ng/mL (to convert to nanomoles per liter, multiply by 2.496) at 12 months of age in 104 patients (50.7%) and less than 20 ng/mL in 28 (13.7%).
Associations Between Clinical Features and Growth
The Table presents mean differences in attained weight and length z scores for age across the entire year by demographic, diagnostic, and clinical features. Infants with 2 class I to III mutations had significantly lower z scores for length (−0.51; 95% CI, −0.96 to −0.07) but not for weight (−0.17; 95% CI, −0.60 to 0.25). Infants with PI and meconium ileus at birth were more deficient in z scores for weight (−0.77; 95% CI, −1.36 to −0.17) and length (−1.03; 95% CI, −1.71 to −0.35) compared with infants with pancreatic sufficiency (Table). Female BONUS infants had consistently higher z scores for weight (mean, 0.30; 95% CI, 0.06–0.54) than did male infants; of note, 72 boys (59.5%) were homozygous for Phe508del compared with 60 girls (54.5%). Infants who had been prescribed H2 blockers alone or with proton pump inhibitors had lower weight and length. Patients prescribed proton pump inhibitors alone did not have significantly lower z scores for weight (−0.16; 95% CI, −0.47 to 0.16) or height (−0.02; 95% CI, −0.36 to 0.32). Infants with at least 1 Pseudomonas aeruginosa–positive oropharyngeal swab result had lower z scores for weight (−0.38; 95% CI, −0.65 to −0.10); infants with wheezing had a mean lower z score for length (–0.43; 95% CI, −0.76 to −0.10) than did those who did not (Table). Other pulmonary, infectious, and inflammatory factors were not associated with differential growth in infants with CF.
Table.
Growth of BONUS Infants by Demographic, Diagnostic, Nutritional and Gastrointestinal Tract, Respiratory Tract, and Inflammatory Features
| Characteristic | Mean (95% CI) Difference in z Scorea | |
|---|---|---|
| Weight for Age | Length for Age | |
| Demographic and diagnostic | ||
| Female vs male | 0.30 (0.06 to 0.54) | 0.20 (−0.05 to 0.44) |
| PI only vs pancreatic sufficiency | −0.43 (−0.84 to −0.01) | −0.61 (−1.03 to −0.19) |
| PI + meconium ileus vs pancreatic sufficiency | −0.77 (−1.36 to −0.17) | −1.03 (−1.71 to −0.35) |
| CFTR classes I–III vs classes IV–V | −0.17 (−0.60 to 0.25) | −0.51 (−0.96 to −0.07) |
| Nutritional and gastrointestinal tract | ||
| Formula fed + breastfed vs exclusively formula fed at 3 mo | 0.05 (−0.29 to 0.38) | 0.11 (−0.24 to 0.45) |
| Exclusively breastfed vs exclusively formula fed at 3mo | 0.13 (−0.17 to 0.44) | 0.17 (−0.14 to 0.48) |
| Formula fed + breastfed vs exclusively formula fed at 6 mo | 0.19 (−0.14 to 0.52) | 0.18 (−0.16 to 0.52) |
| Exclusively breastfed vs exclusively formula fed at 6 mo | 0.02 (−0.33 to 0.38) | 0.13 (−0.24 to 0.49) |
| H2 blocker use vs no H2 blocker or proton pump inhibitor use | −0.37 (−0.65 to −0.10) | −0.35 (−0.64 to −0.05) |
| Proton pump inhibitor use vs no H2 blocker or proton pump inhibitor use | −0.16 (−0.47 to 0.16) | −0.02 (−0.36 to 0.32) |
| H2 blocker + proton pump inhibitor use vs no H2 blocker or proton pump inhibitor use | −0.85 (−1.16 to −0.53) | −0.64 (−0.97 to −0.32) |
| Respiratory | ||
| Pseudomonas aeruginosa in year vs none | −0.38 (−0.65 to −0.10) | −0.24 (−0.53 to 0.04) |
| Staphylococcus aureus in year vs none | −0.05 (−0.29 to 0.19) | 0.01 (−0.23 to 0.26) |
| Crackles in year vs none | −0.12 (−0.62 to 0.38) | −0.37 (−0.88 to 0.15) |
| Wheezing in year vs none | −0.25 (−0.57 to 0.07) | −0.43 (−0.76 to −0.10) |
| Respiratory-related hospitalization in year vs none | −0.16 (−0.47 to 0.16) | −0.30 (−0.62 to 0.02) |
| Chest radiograph Wisconsin score >5 vs ≤5b | −0.06 (−0.41 to 0.30) | −0.07 (−0.47 to 0.33) |
| Chest radiograph Wisconsin score unknown vs score ≤5b | 0.18 (−0.08 to 0.43) | 0.20 (−0.07 to 0.46) |
| Inflammatory | ||
| Serum WBC>14 000/μL vs <14 000/μL | −0.09 (−0.35 to 0.17) | −0.07 (−0.33 to 0.20) |
| Serum neutrophil level >34% vs ≤34% | −0.16 (−0.41 to 0.10) | −0.14 (−0.40 to 0.12) |
Abbreviations: BONUS, Baby Observational and Nutrition Study; CFTR, CF transmembrane conductance regulator;
H2, histamine; PI, pancreatic insufficiency; WBC, white blood cell count.
SI conversion factors: To convert neutrophils to a proportion of 1.0, multiply by 0.01; WBC to ×109 per liter, multiply by 0.001.
Calculated as the mean difference in World Health Organization z scores. Estimates and 95% CIs are calculated from longitudinal model estimates from the entire year. Positive mean difference estimates represent a higher mean z score for the row label vs the comparison group (row minus comparison group).
Scores range from 0 to 100, with higher scores indicating greater severity of lung disease.
The same clinical features were examined as potential risk factors for low weight and length categorization in the BONUS cohort (eTable 3 in the Supplement). Use of H2 blockers alone demonstrated a relative risk of 2.11 (95% CI, 0.74–5.97) for low weight and 2.78 (95% CI, 1.38–5.61) for low length. Use of H2 blockers in combination with proton pump inhibitors had a relative risk of 4.24 (95% CI, 1.62–11.11) for low weight and 2.86 (95% CI, 1.38–5.93) for low length compared with no H2 blocker or proton pump inhibitor use. Wheezing was associated with a 1.73 times increased risk for low length (95% CI, 1.05–2.85) and a 2.06 times increased risk for low weight (95% CI, 1.03–4.15), whereas P aeruginosa infection in the first year of life was associated with low weight (relative risk, 2.31; 95% CI, 1.20–4.44) but not length (eTable 3 in the Supplement). Fecal calprotectin and blood measures of inflammation were not significantly associated with low weight or length (eFigures 2–6 and eTable 3 in the Supplement).
Serum insulinlike growth factor 1 (IGF-1) levels were lower at 6 months in the BONUS cohort (mean, 51.4 ng/mL; 95% CI, 48.2–54.7 ng/mL [to convert to nanomoles per liter, multiply by 0.131) than in published norms (100 ng/mL)23 and were even lower among infants classified as low weight (mean difference from other BONUS infants, −6.9 ng/mL; 95% CI, −14.0 to 0.3 ng/mL) and low length (mean difference compared with other infants with CF, −8.6 ng/mL; 95% CI, −14.2 to −3.1 ng/ mL) (Figure 4A and B). Serum IGF binding protein 3 levels among BONUS infants (mean, 2262.9 ng/mL; 95% CI, 2178.3–2347.5) were higher than among healthy infants at 6 months of age (1720 ng/mL),23 but levels were lower among low-weight (mean difference, −311 ng/mL; 95% CI, −480 to −141 ng/ mL) and low-length (mean difference, −358 ng/mL; 95% CI, −490 to −226 ng/mL) infants compared with their BONUS peers (Figure 4C and D). Other demographic, diagnostic, feeding, and respiratory tract features were not associated with low weight or length categorization in the BONUS cohort.
Figure 4. Plasma Hormonal Growth Factors by Weight and Length at Enrollment and 6 and 12 Months of Age in the Baby Observational and Nutritional Study (BONUS) Cohort.
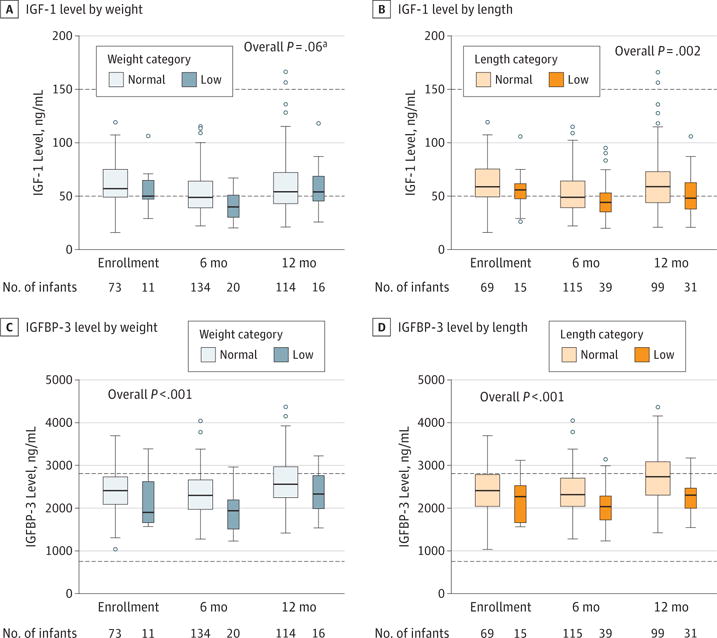
Distributions of insulinlike growth factor 1 (IGF-1) and IGF binding protein 3 (IGFBP-3) are shown by normal vs low weight and length categorizations. Boxes indicate 25th, 50th, and 75th percentiles; dashed horizontal lines, reference level23; open circles, outliers; and whiskers, 1.5 times the 25th to 75th percentile. Low categorization indicates that more than 50% of measurements are less than the 10th percentile of the World Health Organization standard curve,16 with at least 1 measurement occurring after 6 months of age. To convert IGF to nanomoles per liter, multiply by 0.131.
aCalculated as comparison of plasma hormonal levels between low and normal growth categorizations across all 3 points.
Discussion
We herein report, to our knowledge, the largest multicenter prospective study examining the growth and nutritional status of a cohort of infants with CF diagnosed by NBS. Although z scores for weight for age lagged behind WHO normative values at birth and during the first 6 months of age, BONUS infant z scores for weight increased to those of WHO healthy infants by 12 months. Of note, head circumference was normal at birth and throughout the first year of life. Correction in weight gain is a striking improvement compared with a 1994–1995 historic cohort, before universal NBS for CF in the United States (Figure 2). Of interest, the observed improvement in weight occurred regardless of feeding type (formula vs breastfeeding), consistent with prior observations.24 Furthermore, historic markers of nutritional status, caloric and macronutrient intake, or vitamin supplementation were often higher in low-weight and low-length infants and did not predict normal growth. One could speculate that key factors in successful correction of weight include early implementation of PERT not possible before universal NBS in the United States and current aggressive approaches to nutritional supplementation.
Despite significant progress in improved weight among infants with CF, a subgroup of low-weight infants continued to falter. In this group, increased caloric intake and addition of H2 blockers to improve enzyme efficacy did not correlate with better growth outcomes. Additional caloric and supplement intake very likely prevented more severe failure to thrive; thus, we caution that these results not be interpreted to suggest that caloric supplementation in infants with CF is not necessary. However, this subpopulation was unable to overcome growth deficits, suggesting that other factors contributed to poor weight gain and linear growth. We examined an extensive list of pulmonary, nutritional, hormonal, infectious, and inflammatory factors that might be associated with poor growth. Colonization with P aeruginosa and wheezing were associated with the highest risk for low weight or length, even in this very young and healthy population, consistent with infant CF studies in Aus-tralia.25,26 Despite the absence of identifiable associated inflammatory measures in this study, poor weight gain and eventual poor lung function may have a common etiology rather than a causal relationship.
In contrast to our observations with weight improvement, we found persistent delay at 1 year in linear growth in most BONUS infants. Given the rigor taken in certifying BONUS study dieticians and research coordinators,11 we do not believe these findings to be biased toward shorter length. Stunting was not seen in infants with pancreatic sufficiency, and pancreatic phenotype is more strongly associated with genotype than are other clinical manifestations.27–29 In general, patients with pancreatic sufficiency tend to have milder CFTR mutations (classes IV and V).28 Although the dramatic difference in linear growth may be attributable to changes in absorption of critical micronutrients despite supplementation, poor growth may ultimately be a consequence of more severe CFTR dysfunction.
Of note, the BONUS cohort had lower serum IGF-1 levels than published values in healthy infants,23 and infants with low length also had lower serum IGF-1 and IGF binding protein 3 levels than the rest of the BONUS cohort. Insulinlike growth factor 1, which drives anabolism and linear growth and influences immune cell function,30 is known to be variable in the first year of life23 and decreased in CF.31 An ex vivo cell model suggests that IGF-1 augments CFTR function.32 Furthermore, reduced serum IGF-1 levels have been documented in newborn pigs with transgenic CF, suggesting a direct linkage to CFTR dysfunction.33 Of interest, newborns with CF are known to have reduced IGF-1 concentrations compared with healthy control infants, even before development of significant malabsorption, lung infections, or respiratory compromise.33 Growth during infancy is the outcome of a variety of complex anabolic, catabolic, and inflammatory interactions. One such delicate interaction is the effect of sleep disorders on the growth hormone–IGF-1 axis34 and restoration of linear growth and normalization of inflammatory markers after correction of obstructive sleep apnea.35 Might subtle abnormalities such as disrupted sleep tip an infant with CF into a more catabolic state, and could growth hormone somehow modulate CFTR for an additive effect on weight and height?36 Chronic inflammation is also associated with disruption of the growth hormone–IGF-1 axis in CF, potentially contributing to growth failure.37 Although the mechanism behind reduced IGF-1 levels in infants with CF is unknown, the effect of increased severity of CFTR mutations on height in BONUS and the 2014 CF Foundation annual data report6 implies an intrinsic relationship. Recent reports of improved growth in children with CF who have a G551D mutation and were treated with ivacaftor (a CFTR potentiator) indicate a direct effect of CFTR on growth factors.
In addition to IGF-1 deficiency, 104 BONUS infants (50.7%) had 25-hydroxyvitamin D levels lower than the CF standard of 30 ng/mL,38 and 28 (13.7%) had levels lower than the Institute of Medicine reference level of 20 ng/mL,39 despite a median supplement intake of 750 IU/d, nearly 3-fold the recommended daily intake for healthy infants. Vitamin D deficiency is associated with reduced bone density in adolescents and adults with CF.40
Limitations
Limitations of this study include its observational nature and possible indication bias. For example, the highest caloric intake and use of H2 blockers was seen in infants with the lowest weight gain, likely a caregiver’s or a clinician’s response to poor growth. Although we captured notable and persistent length deficiencies, we did not have complete information about genetic potential based on midparental height (only 52 of 231 parental pairs). A comprehensive comparison of the BONUS cohort with the 1994–1995 birth cohort was not conducted; therefore, baseline clinical features may have confounded growth findings. However, annual Cystic Fibrosis Foundation Patient Registry reports indicate that genetic disposition has been relatively constant.6 Newborn screening likely led to the identification of more mild cases of CF in infancy; thus, this may have contributed to the observed improvements in growth. A small number of participants did not complete the study (16 [6.9%]), and 41 patients did not contribute blood specimens at all points; this may have biased results in an unknown way. Monitoring of early pulmonary disease was not as extensive as in prior studies, where lower airway culture and high-resolution computed tomography findings were prospectively collected.25,26 Thus, we may not have been able to detect the effects of early lower airway inflammation or bacterial infections. We also acknowledge that, in a subset of participants without fecal elastase values (n = 28), their determination of PI was post hoc and based on data available from clinical encounters; this likely overestimated PI and did not quantitate malabsorption. Last, as an observational study, no adjustments for multiple comparisons were performed; thus, results must be interpreted with care.
Conclusions
BONUS demonstrated that NBS for CF in the United States has likely contributed to correction of malnutrition and suboptimal growth in infants with CF. Weight improvement appears to be more responsive than linear growth to early initiation of enzyme replacement and nutritional supplementation. More extensive examination of PERT and gastrointestinal tract symptoms in this cohort may inform prevention of weight and length faltering during infancy in patients with CF. The association between low length and lower IGF-1 and IGF binding protein 3 concentrations—although often not considered as clinically significant—raises questions of CFTR’s potential role in ensuring functional IGF levels and merits further study. As CFTR modulators become available, infant length, rather than just weight, may ultimately be a surrogate measure of treatment success and benefit. This large cohort and its biorepository have set the stage for investigating new approaches to management of infant malnutrition in CF and preventing its later consequences.
Supplementary Material
Key Points.
Question
What is the current state of growth deficiencies among infants with cystic fibrosis after implementation of the universal newborn screen?
Findings
In this study of 231 infants from 28 US Cystic Fibrosis Foundation–accredited Care Centers followed up for the first year of life, normal weight was achieved by 12 months of age (comparable to the World Health Organization cohort); however, linear growth continued to lag behind.
Meaning
Since initiation of universal newborn screening for cystic fibrosis, significant improvement in nutritional status during infancy has occurred, but stunting remains common.
Acknowledgments
Funding/Support: This study was supported by grants BONUS11KO from Cystic Fiborosis Foundation Therapeutics; grants R01DK095738, P30DK089507, UL1TR000423, and P30 DK072482 from the NIH; and grant LEUNG14GE0 from the CFF.
Role of the Funder/Sponsor: The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Author Contributions: Dr Heltshe had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Leung, Heltshe, Borowitz, Gelfond, Heubi, Ramsey.
Acquisition, analysis, or interpretation of data: All authors.
Drafting of the manuscript: Leung, Heltshe, Borowitz, Gelfond, Kloster, Stalvey, Ramsey.
Critical revision of the manuscript for important intellectual content: All authors.
Statistical analysis: Leung, Heltshe, Kloster, Stalvey.
Obtained funding: Heltshe, Borowitz, Ramsey.
Administrative, technical, or material support: Leung, Heltshe, Borowitz.
Study supervision: Leung, Heltshe, Borowitz, Heubi, Stalvey, Ramsey.
Conflict of Interest Disclosures: Dr Leung reports consulting for Vertex and research/grant support from Bristol-Meyers Squibb, Gilead, Abbvie, and Roche Pharmaceuticals outside the submitted work. Dr Stalvey reports serving on the Education Advisory Board for Vertex and as a medical consultant for Versartis, Inc, and research/grant support from Versartis, Inc, Genentech, Inc, and the National Institutes of Health (NIH; grants DK072482 and K08DK094784) outside the submitted work. Dr Ramsey reports during the past 3 years grant support from the Cystic Fibrosis Foundation (CFF) and the NIH and serving as the principal investigator on contracts between Seattle Children’s Hospital and Aridis Pharmaceuticals, LLC, Celtaxsys, Kalobios, Flatley Discovery Labs, LLV, Vertex Pharmaceuticals, Inc, Laurent Therapeutics, Inc, Nilvalis Therapeutics, Inc, and Synedgen, Inc. No other disclosures were reported.
Group Information: The following principal investigators and study site coordinators participated in the Baby Observational and Nutrition Study (BONUS): A. Stecenko, M. Schechter, and E. Hunter (Emory University, Atlanta, Georgia); W. Hoover, H. Hathorne, and K. Brand (University of Alabama at Birmingham); A. Filbrun and M. Linn (University of Michigan, Ann Arbor); D. Borowitz and N. Caci (Women and Children’s Hospital of Buffalo, Buffalo, New York); B. McWilliams and I. Brazil (Children’s Medical Center of Central Texas, Austin); J. Heubi and M Bushman (Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio); S. McColley and A. Bowen (Chicago Lurie Children’s Hospital, Chicago, Illinois); K. McCoy and K. Sakellaris (Nationwide Children’s Hospital, Columbus, Ohio); P. Sharma, C. Cannon, and A. Hebert (University of Texas Southwestern Medical Center, Dallas); S. Sagel and M. Anthony (Children’s Hospital Colorado, Denver); M. Dyson and H. Urbanek (Cook Children’s Medical Center, Fort Worth, Texas); S. Millard and C. Gile (Henen DeVos Women and Children’s Center, Grand Rapids, Michigan); G. Graff and L. Allwein (Hershey Medical Center, Hershey, Pennsylvania); P. Hiatt and N. Schaap (Baylor College of Medicine, Houston, Texas); N. Krupp and L. Bendy (Riley Hospital for Children, Indianapolis, Indiana); R. Ahrens and M. Teresi (University of Iowa, Iowa City); A. Berlinski and L. L. Ramsey (Arkansas Children’s Hospital, Little Rock); J. McNamara and M. Johnson (Minnesota Children’s Hospital and Clinics, Minneapolis); R. Brown and P. Berry (Vanderbilt Children’s Hospital, Nashville, Tennessee); N. Mehdi, J. Royall, and D. Thomas (Children’s Hospital of Oklahoma, Oklahoma City); R. Rubenstein and E. Donnelly (Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania); D. Weiner, S. Hurban, and E. Hartigan (Children’s Hospital of Pittsburgh and University of Pittsburgh, Pittsburgh, Pennsylvania); M. Powers and K. Simmons (Oregon Health & Sciences University, Portland); B. Chatfield and H. Oldroyd (University of Utah, Salt Lake City); L. Hoffman and S. McNamara (Seattle Children’s Hospital, Seattle, Washington); J. Wooldridge and A. Cooper (Cardinal Glennon Children’s Hospital and St Louis University, St Louis, Missouri); A. Faro and D. Rodgers (St Louis Children’s Hospital, St Louis, Missouri); C. Fortner and V. Suttmore (SUNY Upstate Medical University, Syracuse); J. Hocevar-Trnka, M. Kloster, M. Skalland, and A. Fowler (Cystic Fibrosis Foundation Therapeutics Development Network Coordinating Center, Seattle Children’s Research Institute, Seattle, Washington).
Additional Contributions: We thank the Cystic Fibrosis Foundation Therapeutic Development Network sites, participating patients and families, and the Cystic Fibrosis Foundation Patient Registry for their contributions to this study.
Contributor Information
Daniel H. Leung, Division of Gastroenterology, Hepatology, and Nutrition, Department of Pediatrics, Baylor College of Medicine, Houston, Texas.
Sonya L. Heltshe, Cystic Fibrosis Foundation Therapeutics Development Network Coordinating Center, Seattle Children’s Research Institute, Seattle, Washington; Division of Pulmonary and Sleep Medicine, Department of Pediatrics, University of Washington, Seattle.
Drucy Borowitz, Department of Pediatrics, University of Buffalo, Buffalo, New YorkCystic Fibrosis Foundation, Bethesda, Maryland.
Daniel Gelfond, Division of Gastroenterology/Nutrition, Department of Pediatrics, University of Rochester, Rochester, New York.
Margaret Kloster, Cystic Fibrosis Foundation Therapeutics Development Network Coordinating Center, Seattle Children’s Research Institute, Seattle, Washington.
James E. Heubi, Division of Gastroenterology and Nutrition, Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio; Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, Ohio.
Michael Stalvey, Department of Pediatrics, University of Alabama at Birmingham.
Bonnie W. Ramsey, Cystic Fibrosis Foundation Therapeutics Development Network Coordinating Center, Seattle Children’s Research Institute, Seattle, Washington; Division of Pulmonary and Sleep Medicine, Department of Pediatrics, University of Washington, Seattle.
References
- 1.Farrell PM, Lai HJ, Li Z, et al. Evidence on improved outcomes with early diagnosis of cystic fibrosis through neonatal screening: enough is enough! J Pediatr. 2005;147(3(suppl)):S30–S36. doi: 10.1016/j.jpeds.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 2.Farrell PM, Kosorok MR, Rock MJ, et al. Wisconsin Cystic Fibrosis Neonatal Screening Study Group Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Pediatrics. 2001;107(1):1–13. doi: 10.1542/peds.107.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Dijk FN, McKay K, Barzi F, Gaskin KJ, Fitzgerald DA. Improved survival in cystic fibrosis patients diagnosed by newborn screening compared to a historical cohort from the same centre. Arch Dis Child. 2011;96(12):1118–1123. doi: 10.1136/archdischild-2011-300449. [DOI] [PubMed] [Google Scholar]
- 4.Siret D, Bretaudeau G, Branger B, et al. Comparing the clinical evolution of cystic fibrosis screened neonatally to that of cystic fibrosis diagnosed from clinical symptoms: a 10-year retrospective study in a French region (Brittany) Pediatr Pulmonol. 2003;35(5):342–349. doi: 10.1002/ppul.10259. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, Lindstrom MJ, Farrell PM, Lai HJ, Wisconsin Cystic Fibrosis Neonatal Screening Group Pubertal height growth and adult height in cystic fibrosis after newborn screening. Pediatrics. 2016;137(5):e20152907. doi: 10.1542/peds.2015-2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cystic Fibrosis Foundation Patient Registry. Annual Data Report 2014. Bethesda, MD: Cystic Fibrosis Foundation; 2014. [Google Scholar]
- 7.Borowitz D, Robinson KA, Rosenfeld M, et al. Cystic Fibrosis Foundation Cystic Fibrosis Foudation evidence-based guidelines for management of infants with cystic fibrosis. J Pediatr. 2009;155(6(suppl)):S73–S93. doi: 10.1016/j.jpeds.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramsey BW, Davies J, McElvaney NG, et al. VX08-770-102 Study Group A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wainwright CE, Elborn JS, Ramsey BW, et al. TRAFFIC Study Group; TRANSPORT Study Group Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–231. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.ClinicalTrials.gov. Baby Observational and Nutritional Study (BONUS) NCT01424696. https://clinicaltrials.gov/ct2/show/NCT01424696. Accessed January 31, 2017.
- 11.Coburn-Miller C, Casey S, Luong Q, et al. Standardization of research-quality anthropometric measurement of infants and implementation in a multicenter study. Clin Transl Sci. 2015;8(4):330–333. doi: 10.1111/cts.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chumpitazi BP, Lane MM, Czyzewski DI, Weidler EM, Swank PR, Shulman RJ. Creation and initial evaluation of a Stool Form Scale for children. J Pediatr. 2010;157(4):594–597. doi: 10.1016/j.jpeds.2010.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taddio A, Nulman I, Koren BS, Stevens B, Koren G. A revised measure of acute pain in infants. J Pain Symptom Manage. 1995;10(6):456–463. doi: 10.1016/0885-3924(95)00058-7. [DOI] [PubMed] [Google Scholar]
- 14.West SE, Zeng L, Lee BL, et al. Respiratory infections with Pseudomonas aeruginosa in children with cystic fibrosis: early detection by serology and assessment of risk factors. JAMA. 2002;287(22):2958–2967. doi: 10.1001/jama.287.22.2958. [DOI] [PubMed] [Google Scholar]
- 15.Koscik RE, Kosorok MR, Farrell PM, et al. Wisconsin cystic fibrosis chest radiograph scoring system: validation and standardization for application to longitudinal studies. Pediatr Pulmonol. 2000;29(6):457–467. doi: 10.1002/(sici)1099-0496(200006)29:6<457::aid-ppul8>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 16.WHO Multicentre Growth Reference Study Group. WHO Child Growth Standards: Methods and Development. Geneva, Switzerland: World Health Organization; 2006. [Google Scholar]
- 17.Grummer-Strawn LM, Reinold C, Krebs NF, Centers for Disease Control and Prevention (CDC) Use of World Health Organization and CDC growth charts for children aged 0–59 months in the United States. MMWR Recomm Rep. 2010;59(RR-9):1–15. [PubMed] [Google Scholar]
- 18.O’Sullivan BP, Baker D, Leung KG, Reed G, Baker SS, Borowitz D. Evolution of pancreatic function during the first year in infants with cystic fibrosis. J Pediatr. 2013;162(4):808–812.e1. doi: 10.1016/j.jpeds.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 19.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352(19):1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 20.Fryar CD, Gu Q, Ogden CL. Anthropometric reference data for children and adults: United States, 2007–2010. Vital Health Stat 11. 2012;(252):1–48. [PubMed] [Google Scholar]
- 21.Centers for Disease Control and Prevention. Breastfeeding Among US Children Born 2002–2012, CDC National Immunization Surveys. Atlanta, GA: United States Centers for Disease Control; 2016. [Google Scholar]
- 22.American Academy of Pediatrics Committee on NutritionKleinman RE, editor. Pediatric Nutrition Handbook. 7th. Elk Grove Village, IL: American Academy of Pediatrics; 2014. pp. 634–635. [Google Scholar]
- 23.Juul A, Dalgaard P, Blum WF, et al. Serum levels of insulin-like growth factor (IGF)–binding protein-3 (IGFBP-3) in healthy infants, children, and adolescents: the relation to IGF-I, IGF-II, IGFBP-1, IGFBP-2, age, sex, body mass index, and pubertal maturation. J Clin Endocrinol Metab. 1995;80(8):2534–2542. doi: 10.1210/jcem.80.8.7543116. [DOI] [PubMed] [Google Scholar]
- 24.Holliday KE, Allen JR, Waters DL, Gruca MA, Thompson SM, Gaskin KJ. Growth of human milk-fed and formula-fed infants with cystic fibrosis. J Pediatr. 1991;118(1):77–79. doi: 10.1016/s0022-3476(05)81850-5. [DOI] [PubMed] [Google Scholar]
- 25.Ramsey KA, Ranganathan S, Park J, et al. ARESTCF Early respiratory infection is associated with reduced spirometry in children with cystic fibrosis. Am J Respir Crit Care Med. 2014;190(10):1111–1116. doi: 10.1164/rccm.201407-1277OC. [DOI] [PubMed] [Google Scholar]
- 26.Ranganathan SC, Parsons F, Gangell C, Brennan S, Stick SM, Sly PD, Australian Respiratory Early Surveillance Team for Cystic Fibrosis Evolution of pulmonary inflammation and nutritional status in infants and young children with cystic fibrosis. Thorax. 2011;66(5):408–413. doi: 10.1136/thx.2010.139493. [DOI] [PubMed] [Google Scholar]
- 27.Bartlett JR, Friedman KJ, Ling SC, et al. Gene Modifier Study Group Genetic modifiers of liver disease in cystic fibrosis. JAMA. 2009;302(10):1076–1083. doi: 10.1001/jama.2009.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmed N, Corey M, Forstner G, et al. Molecular consequences of cystic fibrosis transmembrane regulator (CFTR) gene mutations in the exocrine pancreas. Gut. 2003;52(8):1159–1164. doi: 10.1136/gut.52.8.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campbell PW, III, Phillips JA., III The cystic fibrosis gene and relationships to clinical status. Semin Respir Infect. 1992;7(3):150–157. [PubMed] [Google Scholar]
- 30.Gifford AH, Nymon AB, Ashare A. Serum insulin-like growth factor-1 (IGF-1) during CF pulmonary exacerbation: trends and biomarker correlations. Pediatr Pulmonol. 2014;49(4):335–341. doi: 10.1002/ppul.22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laursen EM, Juul A, Lanng S, et al. Diminished concentrations of insulin-like growth factor I in cystic fibrosis. Arch Dis Child. 1995;72(6):494–497. doi: 10.1136/adc.72.6.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee HW, Cheng J, Kovbasnjuk O, Donowitz M, Guggino WB. Insulin-like growth factor 1 (IGF-1) enhances the protein expression of CFTR. PLoS One. 2013;8(3):e59992. doi: 10.1371/journal.pone.0059992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rogan MP, Reznikov LR, Pezzulo AA, et al. Pigs and humans with cystic fibrosis have reduced insulin-like growth factor 1 (IGF1) levels at birth. Proc Natl Acad Sci USA. 2010;107(47):20571–20575. doi: 10.1073/pnas.1015281107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gianotti L, Pivetti S, Lanfranco F, et al. Concomitant impairment of growth hormone secretion and peripheral sensitivity in obese patients with obstructive sleep apnea syndrome. J Clin Endocrinol Metab. 2002;87(11):5052–5057. doi: 10.1210/jc.2001-011441. [DOI] [PubMed] [Google Scholar]
- 35.Nachalon Y, Lowenthal N, Greenberg-Dotan S, Goldbart AD. Inflammation and growth in young children with obstructive sleep apnea syndrome before and after adenotonsillectomy. Mediators Inflamm. 2014;2014:1–7. doi: 10.1155/2014/146893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stalvey MS, Anbar RD, Konstan MW, et al. A multi-center controlled trial of growth hormone treatment in children with cystic fibrosis. Pediatr Pulmonol. 2012;47(3):252–263. doi: 10.1002/ppul.21546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wong SC, Dobie R, Altowati MA, Werther GA, Farquharson C, Ahmed SF. Growth and the growth hormone-insulin like growth factor 1 axis in children with chronic inflammation: current evidence, gaps in knowledge, and future directions. Endocr Rev. 2016;37(1):62–110. doi: 10.1210/er.2015-1026. [DOI] [PubMed] [Google Scholar]
- 38.Tangpricha V, Kelly A, Stephenson A, et al. Cystic Fibrosis Foundation Vitamin D Evidence-Based Review Committee An update on the screening, diagnosis, management, and treatment of vitamin D deficiency in individuals with cystic fibrosis: evidence-based recommendations from the Cystic Fibrosis Foundation. J Clin Endocrinol Metab. 2012;97(4):1082–1093. doi: 10.1210/jc.2011-3050. [DOI] [PubMed] [Google Scholar]
- 39.Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96(1):53–58. doi: 10.1210/jc.2010-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donovan DS, Jr, Papadopoulos A, Staron RB, et al. Bone mass and vitamin D deficiency in adults with advanced cystic fibrosis lung disease. Am J Respir Crit Care Med. 1998;157(6, pt 1):1892–1899. doi: 10.1164/ajrccm.157.6.9712089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.