

Abstract
Objective:
To provide new insights into the interpretation of genetic variants in a rare neurologic disorder, CDKL5 deficiency, in the contexts of population sequencing data and an updated characterization of the CDKL5 gene.
Methods:
We analyzed all known potentially pathogenic CDKL5 variants by combining data from large-scale population sequencing studies with CDKL5 variants from new and all available clinical cohorts and combined this with computational methods to predict pathogenicity.
Results:
The study has identified several variants that can be reclassified as benign or likely benign. With the addition of novel CDKL5 variants, we confirm that pathogenic missense variants cluster in the catalytic domain of CDKL5 and reclassify a purported missense variant as having a splicing consequence. We provide further evidence that missense variants in the final 3 exons are likely to be benign and not important to disease pathology. We also describe benign splicing and nonsense variants within these exons, suggesting that isoform hCDKL5_5 is likely to have little or no neurologic significance. We also use the available data to make a preliminary estimate of minimum incidence of CDKL5 deficiency.
Conclusions:
These findings have implications for genetic diagnosis, providing evidence for the reclassification of specific variants previously thought to result in CDKL5 deficiency. Together, these analyses support the view that the predominant brain isoform in humans (hCDKL5_1) is crucial for normal neurodevelopment and that the catalytic domain is the primary functional domain.
The phenotype associated with CDKL5 deficiency (MIM: 300203) has become increasingly well defined over the last decade. Its cardinal features are early-onset seizures, often presenting as infantile spasms and usually occurring within the first 3 months of life, global developmental delay, and severely impaired gross motor function.1 CDKL5 deficiency is caused by dominantly acting loss-of-function variants in the X-linked gene CDKL5 (cyclin-dependent kinase-like 5), which plays a crucial role in brain development.2–6 The epidemiology of CDKL5 deficiency has not been studied, and no incidence or prevalence data are available. Nevertheless, the frequency of patients diagnosed with CDKL5 deficiency is increasing due to growing awareness of the disorder and the inclusion of CDKL5 in routine genetic testing of early-onset epileptic encephalopathies.7,8
For rare Mendelian diseases caused by de novo variants, such as CDKL5 deficiency, data from large-scale screening of patient and population samples can be mined to enhance clarification of clinically relevant variants. The Exome Aggregation Consortium (ExAC)9 analyzed high-quality exome DNA sequence data of 60,706 individuals of diverse ancestries, providing opportunities to refine the clinical interpretation of CDKL5 variants. Here, we analyze all known variants observed in CDKL5 patients to date, including novel variants described in this study, along with data from ExAC, the 1000 Genomes Project (1000G),10 and the Single Nucleotide Polymorphism database (dbSNP).11 All variants are analyzed in the context of the updated characterization of the CDKL5 gene12 to provide new insights into the clinical interpretation of variants in CDKL5 deficiency.
METHODS
Standard protocol approvals, registrations, and patient consents.
Written informed consent was obtained from all individuals who participated in this study, and the study was approved by the ethics committees of the respective institutions: University of Western Australia Human Research Ethics Committee (reference # RA/4/1/5024); the Institute of Medical Genetics, University Hospital of Wales (Cardiff, UK) as part of the British Isles Rett Syndrome Survey (REC reference # 15/WA/029); Children's Hospital Colorado (Aurora, CO) (COMIRB 13-2020); established protocols to access clinical data of Sant Joan de Déu Children's Hospital (Barcelona, Spain).
CDKL5 variant data collection.
CDKL5 variants in clinical cohorts (see above) were analyzed by combining data from the CDKL5 variation database at Rett Syndrome Database (RettBASE),13 Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources,14 and all published reports of CDKL5 variants. Population data on CDKL5 variants were sourced from ExAC,9 1000G,10 and dbSNP.11
Computational analysis.
The effects of missense and splicing variants in CDKL5 were predicted using several algorithms (missense: SIFT, PolyPhen, MutationTaster, and PROVEAN; splicing variants: MaxEntScan and dbscSNV), provided within the Ensembl Variant Effect Predictor (VEP).15
Assignment criteria.
The assignment of pathogenicity was based on guidelines for the interpretation of sequence variants.16 The following criteria were used: pathogenic—the same amino acid change as an established pathogenic variant; likely pathogenic—the allele is absent in population data sets and the patient's phenotype is highly specific for CDKL5 disorder, and computational evidence supports a deleterious effect on the gene; uncertain significance—evidence for benign and pathogenic classification is contradictory; likely benign—the variant is detected in a healthy mother and/or sister, and computational evidence suggests no effect on the gene; and benign—the variant is detected in a healthy father and/or brother, or the allele frequency is detected in population data sets at levels too high to explain the prevalence of a rare disorder.
Minigene splicing assay.
DNA fragments of wild-type and mutant CDKL5 exon 14 with flanking intron sequence were synthesized (IDT, Coralville, IA) and cloned into the exon trap vector pET01 (MoBiTec, Göttingen, Germany). Minigenes were transfected into HEK293T cells using jetPRIME (PEQLAB, Erlangen, Germany) according to the manufacturer's instructions. After 24 hours, total RNA was extracted using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). Total RNA was generated, and reverse transcription PCR (RT-PCR) experiments were performed as described previously.17 Further details of these experiments are described in e-Methods.
RESULTS
CDKL5 variants in the population.
The number of expected and observed variants in CDKL5 from the recent large-scale analysis of genetic variation by ExAC is described in table 1.9 Synonymous variants in CDKL5 are reported to occur at approximately the expected frequency and are found throughout the coding region of CDKL5 (figure 1). By contrast, the number of missense variants observed (n = 157) is lower than expected (table 1). The population sample analyzed in the ExAC study was devoid of severe pediatric disease; therefore, none of these 157 missense variants is, on its own, pathogenic and causative for CDKL5 deficiency. Only 15 (∼10%) of these missense variants are located in the catalytic domain of CDKL5, a 286-amino acid region (∼30% of protein length) crucial for protein function2 (figures 1 and 2). No frameshift or splicing variants were identified in ExAC; however, 2 nonsense variants (described as stop-gain in table 1) were identified: p.Arg952Ter and p.Arg970Ter. Both nonsense variants are found in exon 21 and therefore affect only the hCDKL5_5 isoform, which is expressed almost exclusively in the testis.12,18
Table 1.
Expected and observed CDKL5 variants in ExAC
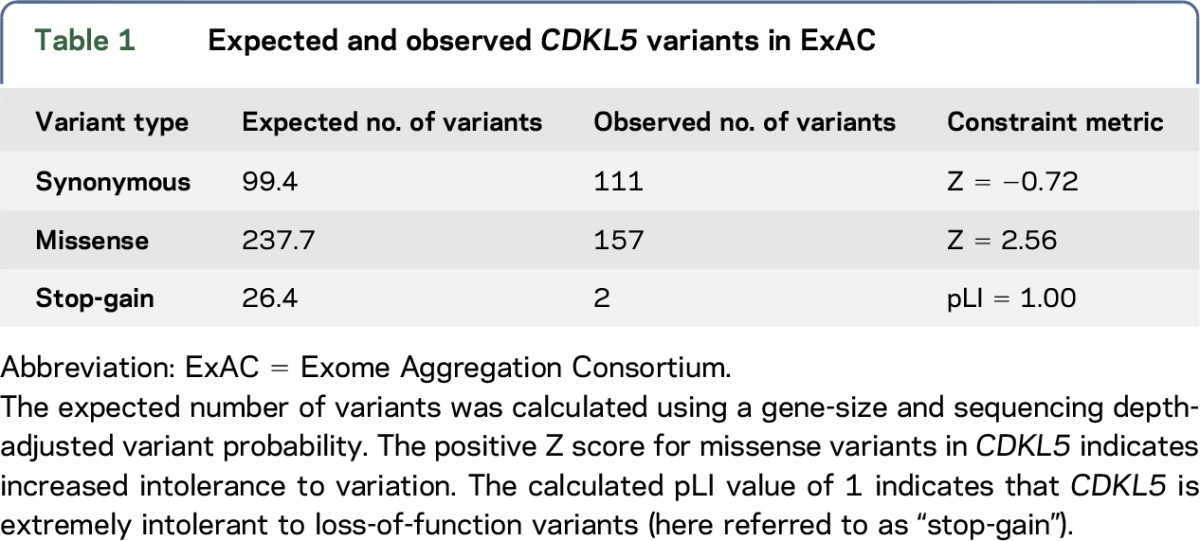
Figure 1. Distribution of exonic CDKL5 variants.

A cartoon of the gene structure is given at the top, with exons of hCDKL5_1, the dominant brain transcript isoform, colored blue-green (coding regions) and black (UTRs). Introns are not drawn to scale. In the diagram beneath, variant types are grouped together, and individual variants are plotted according to their location in the gene. Red indicates pathogenic or likely pathogenic variants; green indicates benign or likely benign variants; and amber indicates variants of uncertain significance.
Figure 2. Pathogenic CDKL5 missense variants cluster in the catalytic domain.

Functional domains in the CDKL5 protein are color coded. Numbers refer to the positions of amino acids. Variants in red (upper) are pathogenic or likely pathogenic. Variants in black (lower) are benign or likely benign. aVariant with a splicing consequence. bVariant of uncertain significance. NES = putative nuclear export signal; NLS = putative nuclear localization signal; ST = serine-threonine kinase active site; TEY = conserved Thr-Glu-Tyr motif.
Some known exonic coding and untranslated regions (UTRs) of CDKL5, recently identified and present only in minor transcript isoforms,12 are not included in the exonic regions covered by ExAC. For example, exon 17 (originally termed 16b19) is present in the hCDKL5_2 isoform and is expressed only at low levels in the developing and adult brain.12 Only 2 exon 17 variants have been identified, both in dbSNP (rs289269000 and rs181987256), neither of which is associated with a disease phenotype.
CDKL5 missense variants in patients.
Missense variants reported in patients are found throughout the portions of CDKL5 that encode the major brain transcript isoform, hCDKL5_1 (figure 1). Only 1 of the 15 missense variants identified within the catalytic domain in ExAC has been reported in a patient with CDKL5 deficiency: c.719G>C (p.Ser240Thr; figure 2). This exon 9 variant has recently been reported as the first familial case of CDKL5-related disease20 in a heterozygous female, who displayed global psychomotor delay and autistic disturbances but no epilepsy. The same variant was detected in her asymptomatic mother, and a skewed X-chromosome inactivation (XCI) ratio was thought to account for this phenotypic difference. We further identified 2 individuals with this variant (1 in ExAC and 1 in 1000G), neither individual having displayed signs of a neurodevelopmental disorder phenotype, consistent with the inclusion criteria in those studies. It is unlikely that 3 asymptomatic carriers of this variant would all have similarly skewed XCI, suggesting that this variant may not in fact be causative. Current evidence suggests that the p.Ser240Thr should be classified as being of “uncertain significance.”
Combining all known CDKL5 variant data, there are 59 missense variants in the catalytic domain, which we consider pathogenic or likely pathogenic for CDKL5 deficiency based on our assignment criteria (figure 2). This includes 11 novel missense variants identified in the clinical cohorts in this study (table 2). In each case, the patient received a clinical diagnosis of CDKL5 deficiency, and, in most cases, VEP analysis supported the prediction of a pathogenic variant.
Table 2.
Novel CDKL5 variants in our clinical cohorts
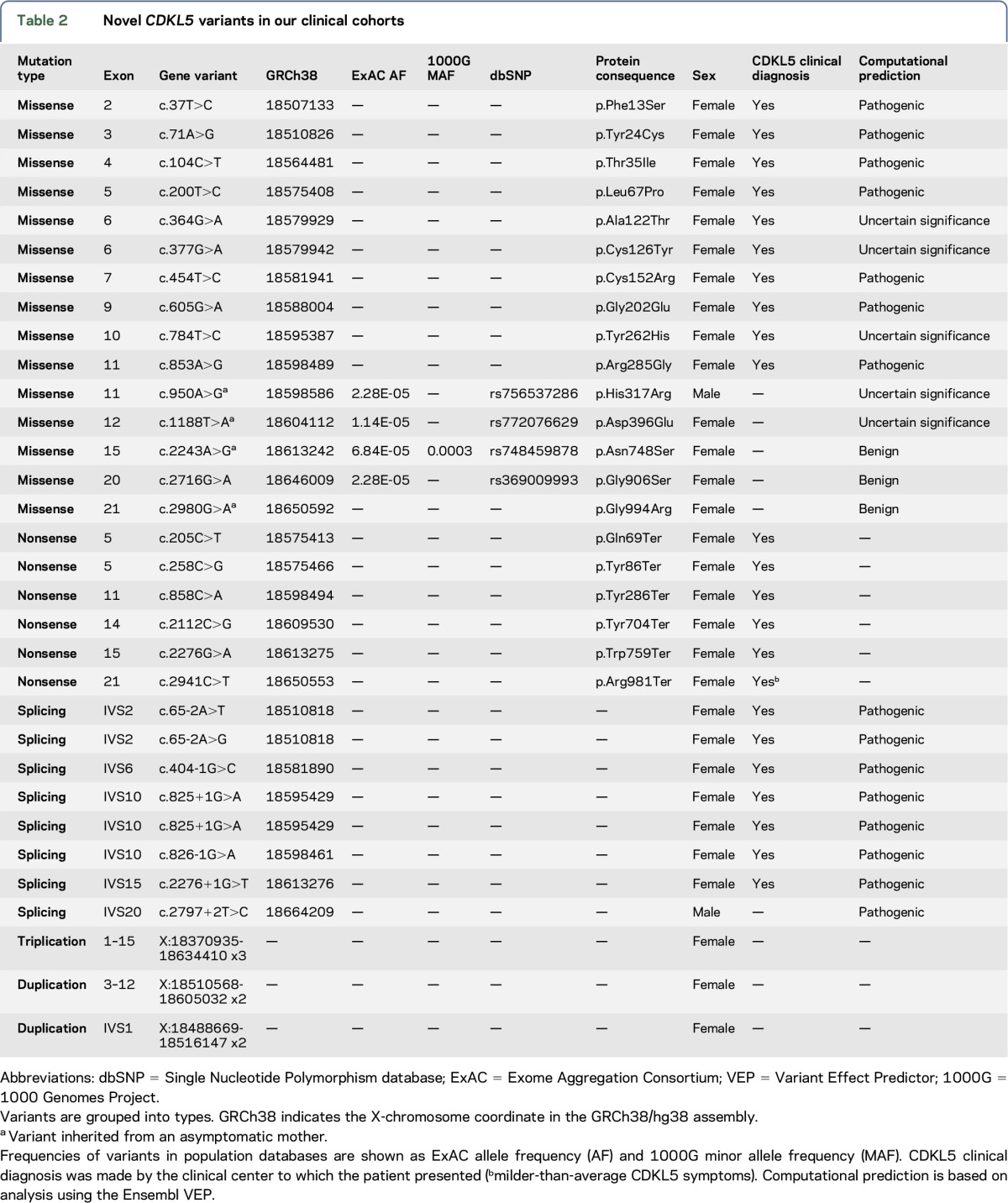
Outside the catalytic domain, there are 179 different missense variants (figure 1). Upstream of the catalytic domain, 3 variants (p.Ile3Phe, p.Asn5Asp, and p.Ile6Thr) are known to be present in ExAC and 1000G, not associated with CDKL5 patients and predicted to be benign by VEP analysis. Fifty-one different missense variants have been found in the last 3 exons (20, 21, and 22). Forty of these are present in ExAC, often in hemizygous males. c.2995G>A (p.Val999Met), previously thought to be pathogenic, is found at notable frequency in ExAC (0.01178) and should therefore be classed as a benign polymorphism. A further 10 are present in dbSNP; none of these variants appear to be responsible for a CDKL5-like phenotype, and VEP analysis predicts these variants to be benign. This is consistent with these exons being absent in the primary brain transcript isoform. In our clinical cohorts, a variant in exon 21, c.2980G>A (p.Gly994Arg), was identified in a patient with developmental delay, but was not clinically diagnosed with CDKL5 deficiency (table 2). The variant was inherited from her asymptomatic mother and, combined with VEP analysis, should therefore be classified as benign.
The remaining missense variants are distributed between exons 11 and 19 (figure 1). Ninety-seven of these are found in ExAC, many occurring in hemizygotes. In some cases, such as p.Ile508Thr and p.Thr734Ala, the ExAC allele frequency and the presence of these variants in hemizygotes allow them to be reclassified as likely benign. Additional variants present in dbSNP (but not in ExAC) are not associated with any clinical phenotype. In our clinical cohorts, we identified further missense variants in patients, where a clinical diagnosis of CDKL5 deficiency was not given (table 2). p.His317Arg, p.Asp396Glu, p.Asn748Ser, and p.Asp797Asn are present in ExAC/1000G, and all variants were identified as being inherited from a healthy mother or father (table 2), suggesting that they are all benign variants. In our analysis, there are 7 missense variants outside the catalytic domain that should, at present, be classified as “of uncertain significance” pending more information (figure 2). p.Val351Ile is recorded in dbSNP (rs587783150) and p.Thr538Ala is recorded in the RettBASE,13 for which no clinical or screening information is available. p.Leu302Phe and p.Asp422Glu are unique variants that have been associated with seizure phenotypes, although the phenotype of these cases is atypical in comparison to other CDKL5 patients reported in these studies.21,22 p.Asn399Thr and p.Val793Ala have also been associated with epilepsy phenotypes4,23; these variants have also been observed as singletons in ExAC.
The most compelling evidence of a pathogenic missense variant outside the catalytic domain concerns the c.2152G>A (p.Val718Met) variant. This variant has been identified in 3 unrelated CDKL5 patients in 3 independent studies.24–26 In each case, the patient presented with symptoms consistent with CDKL5 deficiency and harbored no known variants in other epilepsy-related genes. This variant is not present in the 1000G or the ExAC population databases. VEP analysis of c.2152G>A, which affects the last base of exon 14, predicted a detrimental effect on splicing (MaxEntScan diff: 2.89). We assessed this variant in vitro using a minigene splicing assay, showing that splicing is disrupted, resulting in exon 14 being omitted from the transcript (figure 3). We therefore suggest that the c.2152G>A variant be categorized as a splicing rather than a missense variant. The consequence (r.2047_2152del) is a frameshift variant that would generate a premature stop codon in exon 16. This finding removes evidence supporting the existence of pathogenic missense variants outside the catalytic domain of CDKL5, and we therefore conclude that caution should be applied in interpretations of pathogenicity whenever an apparent missense variant is found outside the catalytic domain.
Figure 3. CDKL5 variant c.2152G>A causes skipping of exon 14 in HEK293T cells.

(A) Schematic representation of the minigene constructs used in the in vitro splicing assay (not to scale). The pET01 vector contains 5′ and 3′ exons separated by an intron sequence. Minigenes contain wild-type (wt) or mutant (mut; c.2152G>A) CDKL5 exon 14 sequences flanked by portions of their natural introns (thick lines). Primers used for RT-PCR experiments are indicated by arrows. Splicing events, indicated by dashed lines, would result in a 348-bp or a 242-bp product depending on inclusion or otherwise of CDKL5 exon 14. (B) Agarose gel showing RT-PCR results from the splicing assay using pET01 vector and minigenes of wild-type or mutated CDKL5 exon 14. Upper bands (348 bp) indicate the presence of exon 14, whereas lower bands (242 bp) indicate exclusion of exon 14. Marker indicates 100 bp ladder; −RT indicates a negative control without reverse transcriptase.
Nonsense, frameshift, and splicing variants.
Pathogenic nonsense, frameshift, and splicing variants are found throughout the coding region of hCDKL5_1, the predominant brain transcript isoform (figure 1). Efforts have been made to establish genotype-phenotype relationships based on the position of these variants in the gene, by predicting structural and functional consequences.8 However, the levels of functional CDKL5 in patients with truncating variants at different points throughout the gene are still not known. Novel nonsense and splicing variants identified in the clinical cohorts in this study are described in table 2, and novel frameshift variants in table e-1, http://links.lww.com/NXG/A2.
In exon 19, the last exon utilized by the hCDKL5_1 transcript, a variant described in dbSNP (rs863225289) has been identified in a patient with early infantile epileptic encephalopathy 2 (MIM: 300672). This variant lies downstream of the internal splice donor site in exon 19 and so would not be expected to affect the protein product made from the testis-specific hCDKL5_5 transcript isoform. However, in relation to hCDKL5_1, the major brain transcript isoform, this is a nonsense variant (c.2176C>T, p.Gln906Ter, with respect to hCDKL5_1). This may trigger nonsense-mediated decay (NMD; due to the presence of a long 3′-UTR27) and result in typical CDKL5 deficiency. Additional observations of this variant would be required to validate this conjecture, but it is consistent with the idea that truncating variants in exon 19 may be pathogenic.
Three nonsense variants have been detected in exon 21, which is specific to isoform hCDKL5_5: c.2854C>T, p.Arg952Ter; c.2908C>T, p.Arg970Ter; and c.2941C>T, p.Arg981Ter. Both p.Arg952Ter and p.Arg970Ter variants have been described in patients with CDKL5-like symptoms.28,29 However, in both cases, delay in seizure onset and an absence of other features of CDKL5-associated pathology were reported. p.Arg952Ter was identified in the patient's mother, grandmother, and half-sister, and in 6 individuals in a control population.28 A further 12 instances of this allele are present in ExAC, including 3 hemizygotes (allele frequency 0.0001367). A recent case study also identified this variant in an asymptomatic hemizygous male.30 p.Arg970Ter occurs in a single heterozygous female in ExAC (in the case study, the variant was not detected in the mother and the father was deceased29). Together, these findings suggest that variants in these late exons do not cause a CDKL5 deficiency phenotype.
A c.2941C>T, p.Arg981Ter variant was identified in our clinical cohorts (table 2). A girl presented with a phenotype consistent with CDKL5 deficiency—profound cognitive impairment, mild dysmorphic appearance, stereotypical hand wringing, and epilepsy (from age 6 months). Both parents were deceased; therefore, it is unknown whether this variant was inherited or occurred de novo. This variant is not present in ExAC or in 1000G, and the clinical evidence would suggest defining this as a pathogenic variant. However, this variant lies immediately downstream of the 2 nonsense variants described above, both of which have considerable evidence suggesting that they should be classified as benign. We conclude that, at present, p.Arg981Ter should be classified as being of “uncertain significance.”
It is important that no pathogenic frameshift or splicing variants were found affecting exons 20, 21, and 22 (figure 1). A novel splicing variant was identified in our clinical cohorts (table 2): c.2797+2T>C. The patient, a boy, was not clinically diagnosed with CDKL5 deficiency and was found to have inherited the variant from his asymptomatic mother. VEP analysis of this variant predicts the abolition of the splice donor site of exon 20, which would be likely to result in a premature stop codon.
Copy number variations and CDKL5 duplication.
Several studies have reported duplications of Xp22 associated with intellectual disability and autism phenotypes,31–34 but the duplicated regions reported (spanning 8–21 Mb) included as many as 80 genes, and interpretation of gene-specific overexpression effects in such circumstances is problematic. A more recent study described 3 unrelated families with more compact duplication regions incorporating CDKL5.35 Four different duplicated regions were described, ranging from 540 to 935 kb in size. In the case of the 683-kb duplication harbored by three of the patients, the duplicated region does not include exon 1 of CDKL5, so the predominant adult brain isoform, hCDKL5_1, would not be expected to be overexpressed in these individuals. The authors point out that the alternative hCDKL5_5 transcript may be expressed and overexpression effects during fetal development may result in the observed phenotype. Three patients also harbored additional duplicated regions on other chromosomes, and parents harboring these same CDKL5-containing duplications were either phenotypically unaffected or displayed mild intellectual disability.35 Therefore, the pathway leading to the phenotypes in these individuals remains somewhat unclear, as experimental confirmation of effects on CDKL5 transcript and protein levels is not yet available. Here, we report 2 duplications and 1 triplication in our clinical cohorts (table 2). However, in each of these copy number variations, only a part of the gene is duplicated; therefore, it is extremely unlikely that an increased level of CDKL5 would be present in these individuals.
Variants in regulatory and UTRs.
The recent identification of novel exons and transcription start sites at the 5′ end of the gene suggests the presence of multiple promoter regions.12 However, most brain transcripts are driven by a putative promoter upstream of exon 1. Several variants in the 5′-UTR have been reported, but so far only those associated with the deletion of exon 1 or 2, or the disruption of splicing from exon 1 to exon 2 (c.-162-2A>G36), have been shown to be pathogenic. Only 2 variants in the putative promoter region, c.-440G>T and c.-189C>T, have so far been found in patients with a CDKL5-like phenotype,3 but at present their significance remains uncertain. The large 6.6 kb 3′-UTR of hCDKL5_1 has only recently been defined,12 so the region has not been analyzed in patients either by targeted sequencing or by exome analysis, and we found no evidence for the presence of clinically relevant variants in the 1000G data.
Incidence of CDKL5 deficiency.
The incidence of CDKL5 deficiency is unknown. We have analyzed data from the International CDKL5 Disorder Database8 to provide a lower estimate of birth prevalence in Australia of 0.21 cases per 100,000 live births (95% CI 0.12–0.33) for the years 1982–2014. Although a birth prevalence in this range would indicate that CDKL5 deficiency is an ultra rare disorder, it is likely that this figure will increase, as targeted next-generation sequencing for investigation of early-onset epileptic encephalopathy becomes more common.7,25
In our analysis, the most frequent CDKL5 deficiency–causing variants are 2 nonsense variants in exon 12: c.1648C>T (p.Arg550Ter) and c.1675C>T (p.Arg559Ter). Each of these variants affects only 3% of all patients, highlighting the high degree of allelic heterogeneity in CDKL5 deficiency.
DISCUSSION
Large-scale sequencing studies are powerful tools for the analysis of genetic variants in rare diseases, such as CDKL5 deficiency. Analysis of CDKL5 variants in this study reinforces the work of recent studies that propose hCDKL5_1 as being the predominant functional isoform required for normal neurodevelopment and brain function. We have shown that missense variants outside the catalytic domain are unlikely to be pathogenic. We have also highlighted specific cases that should currently be classified as being of “uncertain significance” and have functionally reclassified a pathogenic missense variant as a splicing variant. Furthermore, we saw no evidence that missense variants outside the catalytic domain were overrepresented in patient populations compared with their allele frequencies in population databases. However, we cannot rule out the possibility that this lack of association is the result of some missense variants having reduced penetrance and thus existing in unaffected individuals in population cohorts. An analysis of male and female variants separately (in a compact subset of 15 missense variants in the catalytic domain) was suggestive of no significant sex differences in allele frequency (not shown). However, further analysis of all missense variants would be recommended in a future study.
There is a lack of evidence for pathogenic variants in exons 20, 21, and 22. Early studies of the CDKL5 gene had deemed hCDKL5_5 (formerly known as hCDKL5115) to be the predominant CDKL5 transcript and protein isoform. However, we now know that this isoform utilizing exons 20–22 is expressed almost exclusively in the testis and found only at extremely low levels in the adult brain.2,12,18 A recent analysis of missense variants in these exons concluded that genetic variation in this C-terminus was likely to have little or no significance to a CDKL5 disorder phenotype.37 We find further evidence of this in our study. Furthermore, the identification of nonsense variants in these exons in the population is an important observation, consistent with a previous study in which patients with a 136-kb deletion lost only these 3′ CDKL5 exons and the overlapping RS1 gene.38 All patients in the study showed only a retinoschisis phenotype, consistent with RS1 deficiency. Together, the data reinforces the view that variants identified at the 3′ end of the CDKL5 gene should be interpreted with caution.
Although there is substantial evidence that hCDKL5_1 is the predominant brain isoform, hCDKL5_5 continues to be cited as a reference sequence. Consequently, exon 17 and full-length exon 19 are not always routinely sequenced in targeted gene panels containing CDKL5 and are often excluded from exome analysis (as in ExAC). This has potentially important consequences for molecular diagnosis, and it is possible that pathogenic variants in these exon regions are therefore underreported. It is possible that the presence of isoform hCDKL5_2 (which contains exon 17) is not crucial for normal CDKL5 function, given that the levels of this isoform are only 10% (or less) of hCDKL5_1, at all stages of development.12 It may be critical to analyze full-length exon 19, as nonsense and frameshift variants even at the 3′ end of the gene are likely to be pathogenic. The presence of long 3′-UTRs is known to be a major factor in triggering the NMD process,27 and the recent identification of 6.6 kb and 9.9 kb 3′-UTRs in CDKL5 brain isoforms12 suggests that NMD could play an important role in the downregulation of mRNA in the event of a premature termination codon, even in the last coding exon (exon 19 in all CDKL5 brain isoforms). Therefore, we suggest that exon 17 and the full-length form of exon 19 should be included in all molecular diagnostic screens for CDKL5 variants, whether by targeted gene panels or by exome sequencing.
The pathogenicity of CDKL5 variants can ultimately be tested in animal models or engineered human cell lines.39 However, this study provides evidence for the reclassification of specific CDKL5 variants and insights for genetic diagnosis. Although pathogenic CDKL5 variants are found across the majority of the coding regions of the gene, missense variants clearly cluster in the N-terminal catalytic domain. Missense variants outside this domain and all variants in exons 20, 21, and 22 are likely to be benign. In contrast to the well-described duplication syndromes involving closely related genes MECP2 and FOXG1,40,41 we believe that more evidence is required to conclude that there is a well-defined CDKL5 duplication syndrome. Continued evaluation of cases investigating both genotypic and phenotypic expressions as well as diagnoses of copy number variations involving CDKL5 may help to elucidate this aspect of CDKL5 biology.
GLOSSARY
- dbSNP
Single Nucleotide Polymorphism database
- ExAC
Exome Aggregation Consortium
- NMD
nonsense-mediated decay
- RettBASE
Rett Syndrome Database
- RT
reverse transcription
- UTR
untranslated region
- VEP
Variant Effect Predictor
- XCI
X-chromosome inactivation
- 1000G
1000 Genomes Project
AUTHOR CONTRIBUTIONS
Ralph D. Hector: study concept and design, analysis and interpretation of data, and drafting of the manuscript. Vera M. Kalscheuer and Friederike Hennig: analysis and interpretation of data, in vitro experiments, and critical revision of the manuscript for intellectual content. Helen Leonard Jenny Downs, Angus Clarke, Tim A. Benke, Judith Armstrong, and Mercedes Pineda: acquisition of patient data, analysis and interpretation of data, and critical revision of the manuscript for intellectual content. Mark E.S. Bailey and Stuart R. Cobb: analysis and interpretation of data and drafting of the manuscript.
STUDY FUNDING
The study was funded by CDKL5 UK.
DISCLOSURE
R.D. Hector has received travel funding from the LouLou foundation and has received research support from CDKL5 UK and the LouLou Foundation. V.M. Kalscheuer has received research support from the Max Planck Society, the German Federal Ministry of Education and Research #01EW1408A, and the University of Pennsylvania Orphan Disease Center on behalf of the Loulou Foundation #CDKL5-17-103-01. F. Hennig reports no disclosures. H. Leonard has served on the editorial board of Children; received research support from an NHMRC Senior Research Fellowship #1117105; has received funding for the maintenance of the International Rett syndrome Database (InterRett) from Rettsyndrome.org and the International CDKL5 Disorder Database from the International Foundation for CDKL5 Research, NHMRC Program grant 572742, and an NHMRC Senior Research Fellowship #572568; has received an honorarium and travel funding to attend the International Rett Syndrome Symposium; and has received travel funding from the Italian, Russian, and Australia Rett syndrome parent associations, Newron Pharmaceuticals, and the LouLou Foundation. J. Downs has received travel funding/speaker honoraria from the Rett Syndrome Association of Australia and Biogen; has been an employee of Telethon Kids Institute and Curtin University; has received funding from NHMRC project grants (1103745 and 1103746); and has received research support from Telethon Kids Institute and the International Foundation for CDKL5 Research. A. Clarke has received research support from ESRC, CDKL5 UK, and the UK FOXG1 Support Group; has received travel funding from the Nuffield Council on Bioethics and the European Society of Human Genetics; has served on the editorial boards of Human Genetics, European Journal of Human Genetics, Communication & Medicine, the Journal of Community Genetics, Wiley's Encyclopedia of Life Sciences, and Genome Medicine; is a medical adviser to the (UK) Ectodermal Dysplasia Society and Rett UK; and has been supported by Edimer Pharmaceuticals in relation to a clinical trial of treatment for a form of ectodermal dysplasia. T.A. Benke has served on the scientific advisory boards of AveXis and Marinus; has received research support from Questcor Pharmaceuticals, NIH grants RO1 NS076577, U10NS077277, and U54 HD061222, unrestricted research grant from Mallinckrodt pharmaceuticals (not related to this study); has received research support from the Rett Syndrome Research Trust, the Citizens United for Research in Epilepsy (CURE), and International Foundation for CDKL5 Research (IFCR); and has received institutional support from the University of Colorado School of Medicine, Children's Hospital Colorado, and the Children's Hospital of Colorado Foundation Ponzio Family Chair for Neurology Research. J. Armstrong reports no disclosures. M. Pineda has received consulting fees or honoraria and payment for lectures from Actelion Pharmaceuticals Ltd and BioMarin and has been a consultant of a child neurologist on NPC for Actelion during the last 5 years. M.E.S. Bailey has received research support from RETTCO Inc., CDKL5 UK, the Rett Syndrome Research Trust, the Rett Syndrome Association Scotland, the Rosetrees Trust, the Stoneygate Trust, and the Chief Scientist Office (Scottish Government Health Directorate; grants ETM/334 and CGA/16/3). S.R. Cobb has received research support from CDKL5 UK, the Rett Syndrome Research Trust, the Rett Syndrome Association Scotland, the Chief Scientist Office, the Rosetrees Trust, and the R.S. Macdonald Charitable Trust. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Fehr S, Wilson M, Downs J, et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur J Hum Genet 2013;21:266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalscheuer VM, Tao J, Donnelly A, et al. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am J Hum Genet 2003;72:1401–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans JC, Archer HL, Colley JP, et al. Early onset seizures and Rett-like features associated with mutations in CDKL5. Eur J Hum Genet 2005;13:1113–1120. [DOI] [PubMed] [Google Scholar]
- 4.Archer HL, Evans J, Edwards S, et al. CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. J Med Genet 2006;43:729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tao J, Van Esch H, Hagedorn-Greiwe M, et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet 2004;75:1149–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ricciardi S, Ungaro F, Hambrock M, et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat Cel Biol 2012;14:911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gokben S, Onay H, Yilmaz S, et al. Targeted next generation sequencing: the diagnostic value in early-onset epileptic encephalopathy. Acta Neurol Belg 2017;117:131–138. [DOI] [PubMed] [Google Scholar]
- 8.Fehr S, Wong K, Chin R, et al. Seizure variables and their relationship to genotype and functional abilities in the CDKL5 disorder. Neurology 2016;87:2206–2213. [DOI] [PubMed] [Google Scholar]
- 9.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Genomes Project C, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001;29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hector RD, Dando O, Landsberger N, et al. Characterisation of CDKL5 transcript isoforms in human and mouse. PLoS One 2016;11:e0157758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christodoulou J, Grimm A, Maher T, Bennetts B. RettBASE: the IRSA MECP2 variation database-a new mutation database in evolution. Hum Mutat 2003;21:466–472. [DOI] [PubMed] [Google Scholar]
- 14.Firth HV, Richards SM, Bevan AP, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am J Hum Genet 2009;84:524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLaren W, Gil L, Hunt SE, et al. The Ensembl variant effect predictor. Genome Biol 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Musante L, Kunde SA, Sulistio TO, et al. Common pathological mutations in PQBP1 induce nonsense-mediated mRNA decay and enhance exclusion of the mutant exon. Hum Mutat 2010;31:90–98. [DOI] [PubMed] [Google Scholar]
- 18.Williamson SL, Giudici L, Kilstrup-Nielsen C, et al. A novel transcript of cyclin-dependent kinase-like 5 (CDKL5) has an alternative C-terminus and is the predominant transcript in brain. Hum Genet 2012;131:187–200. [DOI] [PubMed] [Google Scholar]
- 19.Fichou Y, Nectoux J, Bahi-Buisson N, Chelly J, Bienvenu T. An isoform of the severe encephalopathy-related CDKL5 gene, including a novel exon with extremely high sequence conservation, is specifically expressed in brain. J Hum Genet 2011;56:52–57. [DOI] [PubMed] [Google Scholar]
- 20.Allou L, Julia S, Amsallem D, et al. Rett-like phenotypes: expanding the genetic heterogeneity to the KCNA2 gene and first familial case of CDKL5-related disease. Clin Genet 2016;91:431–440. [DOI] [PubMed] [Google Scholar]
- 21.Liang JS, Shimojima K, Takayama R, et al. CDKL5 alterations lead to early epileptic encephalopathy in both genders. Epilepsia 2011;52:1835–1842. [DOI] [PubMed] [Google Scholar]
- 22.Roche Martínez A, Armstrong J, Gerotinab E, Fonsa C, Campistola J, Pineda M. CDKL5 in different atypical Rett syndrome variants: description of the first eight patients from Spain. J Pediatr Epilepsy 2012;1:27–35. [Google Scholar]
- 23.Sprovieri T, Conforti FL, Fiumara A, et al. A novel mutation in the X-linked cyclin-dependent kinase-like 5 (CDKL5) gene associated with a severe Rett phenotype. Am J Med Genet A 2009;149A:722–725. [DOI] [PubMed] [Google Scholar]
- 24.Bahi-Buisson N, Nectoux J, Rosas-Vargas H, et al. Key clinical features to identify girls with CDKL5 mutations. Brain 2008;131:2647–2661. [DOI] [PubMed] [Google Scholar]
- 25.Trump N, McTague A, Brittain H, et al. Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J Med Genet 2016;53:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lilles S, Talvik I, Noormets K, et al. CDKL5 gene-related epileptic encephalopathy in Estonia: four cases, one novel mutation causing severe phenotype in a boy, and overview of the literature. Neuropediatrics 2016;47:631–367. [DOI] [PubMed] [Google Scholar]
- 27.Yepiskoposyan H, Aeschimann F, Nilsson D, Okoniewski M, Muhlemann O. Autoregulation of the nonsense-mediated mRNA decay pathway in human cells. RNA 2011;17:2108–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Intusoma U, Hayeeduereh F, Plong-On O, et al. Mutation screening of the CDKL5 gene in cryptogenic infantile intractable epilepsy and review of clinical sensitivity. Eur J Paediatr Neurol 2011;15:432–438. [DOI] [PubMed] [Google Scholar]
- 29.Psoni S, Willems PJ, Kanavakis E, et al. A novel p.Arg970X mutation in the last exon of the CDKL5 gene resulting in late-onset seizure disorder. Eur J Paediatr Neurol 2010;14:188–191. [DOI] [PubMed] [Google Scholar]
- 30.Wang T, Guo H, Xiong B, et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun 2016;7:13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Froyen G, Van Esch H, Bauters M, et al. Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: important role for increased gene dosage of XLMR genes. Hum Mutat 2007;28:1034–1042. [DOI] [PubMed] [Google Scholar]
- 32.Tzschach A, Chen W, Erdogan F, et al. Characterization of interstitial Xp duplications in two families by tiling path array CGH. Am J Med Genet Part A 2008;146A:197–203. [DOI] [PubMed] [Google Scholar]
- 33.Thorson L, Bryke C, Rice G, et al. Clinical and molecular characterization of overlapping interstitial Xp21-p22 duplications in two unrelated individuals. Am J Med Genet Part A 2010;152A:904–915. [DOI] [PubMed] [Google Scholar]
- 34.Sismani C, Anastasiadou V, Kousoulidou L, et al. 9 Mb familial duplication in chromosome band Xp22.2-22.13 associated with mental retardation, hypotonia and developmental delay, scoliosis, cardiovascular problems and mild dysmorphic facial features. Eur J Med Genet 2011;54:e510–e515. [DOI] [PubMed] [Google Scholar]
- 35.Szafranski P, Golla S, Jin W, et al. Neurodevelopmental and neurobehavioral characteristics in males and females with CDKL5 duplications. Eur J Hum Genet 2015;23:915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nemos C, Lambert L, Giuliano F, et al. Mutational spectrum of CDKL5 in early-onset encephalopathies: a study of a large collection of French patients and review of the literature. Clin Genet 2009;76:357–371. [DOI] [PubMed] [Google Scholar]
- 37.Diebold B, Delepine C, Gataullina S, Delahaye A, Nectoux J, Bienvenu T. Mutations in the C-terminus of CDKL5: proceed with caution. Eur J Hum Genet 2014;22:270–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huopaniemi L, Tyynismaa H, Rantala A, Rosenberg T, Alitalo T. Characterization of two unusual RS1 gene deletions segregating in Danish retinoschisis families. Hum Mutat 2000;16:307–314. [DOI] [PubMed] [Google Scholar]
- 39.Wang IT, Allen M, Goffin D, et al. Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc Natl Acad Sci USA 2012;109:21516–21521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seltzer LE, Sohnee A, Paciorkowski AR, et al. Developmental and epilepsy follow-up of children with duplications of FOXG1 on 14q12. Ann Neurol 2013;74:S161. [Google Scholar]
- 41.Van Esch H, Bauters M, Ignatius J, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet 2005;77:442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]