

Abstract
Background
Cognitive impairment is one of the core features of progressive supranuclear palsy. This study aimed to clarify the profile of cognitive impairment and its underlying pathology in progressive supranuclear palsy.
Methods
We retrospectively reviewed medical records to evaluate the pattern and severity of cognitive impairment in 121 autopsy-confirmed progressive supranuclear palsy patients. A subset of 37 patients underwent neuropsychological evaluation as part of their clinical work-up. The burden of progressive supranuclear palsy-related tau pathology (neurofibrillary tangles/pretangles, coiled bodies, tufted astrocytes, and threads) was semi-quantitatively scored in 20 vulnerable brain regions. Concurrent pathologies potentially associated with cognitive impairment, such as Alzheimer-type pathology, were also assessed. To evaluate possible genetic risk factors for cognitive impairment, genetic analysis for APOE and MAPT was performed.
Results
Ninety patients (74%) had documented cognitive impairment based on neurologic evaluation. In a subgroup with neuropsychological testing (N = 37), executive functioning was the most severely impaired cognitive domain. A global cognitive impairment index (Spearman’s rho −0.49, P = 0.005) and executive functioning were negatively correlated with total tau burden (Spearman’s rho −0.51, P = 0.003), but not correlated with the Alzheimer-type pathology. APOE ε4 carriers had more severe amyloid pathology, but total tau burden and overall test battery mean was not different from APOE ε4 non-carriers.
Conclusion
Cognitive impairment in progressive supranuclear palsy, most notably executive dysfunction, is associated with severity of progressive supranuclear palsy-related tau pathology.
Keywords: progressive supranuclear palsy, Alzheimer’s disease, tau, neuropathology, neuropsychology
Introduction
Progressive supranuclear palsy (PSP) is an atypical parkinsonian disorder associated with supranuclear gaze palsy, postural instability and falls, and cognitive impairment (CI).1–3 Recently, the Movement Disorders Society criteria for clinical diagnosis of PSP included cognitive dysfunction as one of four core features, which also consist of ocular motor dysfunction, postural instability, and akinesia.3 Thus, it is increasingly important to obtain a detailed assessment of the clinical features of CI in PSP. CI in PSP helped give rise to the term “subcortical dementia”, which is characterized by deficits in attention, processing speed, executive function, and verbal fluency.4, 5 The largest prospective study has shown that 40–62% of PSP patients developed CI primarily in frontal-executive dysfunction;6 however, the underlying pathology of CI in PSP still remains unclear.
Pathologic hallmark of PSP is tau accumulation, which can be detected in both neurons and glial cells: neurofibrillary tangles (NFTs), neuropil threads, tufted astrocytes, and coiled bodies.7, 8 These tau pathologies accompanied by neuronal loss and gliosis affect predominantly the globus pallidus, subthalamic nucleus, substantia nigra, and cerebellar dentate nucleus.7, 8 Pathological heterogeneity of PSP has been reported, but attempts to correlate this with clinical symptoms have infrequently found definitive correlations.9–11
The aim of the present study was to investigate the profile of CI and its underlying pathology in PSP. We hypothesized that the burden of PSP-related tau pathology would correlate with the severity of CI. To address this hypothesis, we retrospectively reviewed medical records and neuropsychological evaluations, semi-quantitatively assessed the burden of tau pathology, and analyzed the correlation between neuropsychological test scores and PSP-related tau burden in autopsy-confirmed PSP. To evaluate possible genetic risk factors for CI in PSP, genetic analysis for APOE and MAPT was performed.
Materials and Methods
Subjects
All brain tissue samples used in this study were from the Mayo Clinic Brain Bank for Neurodegenerative Diseases. We selected 121 consecutive autopsy-confirmed PSP cases with available medical records that included clinical assessments by a movement disorder specialist at Mayo Clinic between 1998 and 2016. These cases were received from the following sources: CurePSP (58 cases), Udall Center of Excellence for Parkinson’s disease (47 cases), Mayo Clinic Alzheimer’s Disease (AD) Research Center (13 cases), and State of Florida AD Initiative (3 cases). Some data on these cases have been presented in previous articles.12, 13 All brain autopsies were performed with the consent of the legal next-of-kin or an individual with power-of-attorney. Studies using these autopsy samples were considered exempt from human subject research by the Mayo Clinic Institutional Review Board.
Clinical Assessment
A neurologist (S.K.) and a psychiatrist (K.K.) abstracted the following information from medical records collected throughout the course of disease as previously conducted:12–15 demographic information, clinical symptoms, neurological signs, and results from cognitive screening measures and neuropsychological assessments. Clinical phenotypes of PSP (i.e. Richardson syndrome, PSP-corticobasal syndrome (PSP-CBS), PSP-parkinsonism, PSP-frontotemporal dementia, PSP-speech/language disorder, and PSP with predominant cerebellar ataxia) were classified.3, 12 The determination of the presence of CI was performed in two steps. First, medical records for the main cohort (N = 121) were reviewed for patient symptoms report and diagnostic impressions of the evaluating clinicians for indicators of CI. Examples of these indicators included, but were not limited to “memory loss,” “distractibility,” “difficulty concentrating,” “word finding difficulty,” “difficulty with naming,” “slowed thinking,” “bradyphrenia,” “executive dysfunction,” “apraxia/dyspraxia,” and “visuospatial or perceptual deficits.” This review identified 90 patients with suspected CI. Of these patients, test data from a subgroup of 37 patients who underwent neuropsychological assessment at a point in their neurologic work-up were analyzed by a neuropsychologist (A.P.).15, 16 Additionally, patients were considered to have depression if a diagnosis of depression was documented and the patient was prescribed an antidepressant medication as a primary treatment for their depressive symptoms. Of the 31 patients not classified as suspected CI, none underwent neuropsychological assessment.
Neuropsychological Assessment
Scores for the following test were available for most patients: Dementia Rating Scale-Second Edition (DRS-2),17 Wechsler Adult Intelligence Scale (WAIS),18 Digit Span and Block Design subtests, Trail Making Test (TMT),19 Wechsler Memory Scale (WMS) Logical Memory,20 Boston Naming Test,21 and Semantic Fluency (Animals).22 Raw test scores were converted to age-corrected standardized scores (T-score, M = 50, SD = 10) based upon procedures using published manuals and widely accepted normative samples.17, 18, 20, 22 Two indices of global cognitive functioning were obtained: DRS-2 Total score and Overall Test Battery Mean (OTBM). The OTBM was calculated as the average standardized score across all neuropsychological domains (with the exception of the DRS-2 Total score) and is a well-established metric for capturing global cognitive functioning.23–25 Individual test scores were categorized into five separate cognitive domains: attention/processing speed, executive functioning, episodic memory, language, and visuospatial/construction (see Supplementary Table 1 for the tests that comprise each cognitive domain).
A subset of patients also completed a depression symptom checklist as a part of their neuropsychological assessment, including Beck Depression Inventory,26 Geriatric Depression Scale,27 and Patient Health Questionnaire-9.28 The severity of depression was classified as minimal, mild, moderate, or severe based upon the published scoring procedures for each checklist. Patients who scored in the minimal symptom severity range were classified as patients without depression, whereas patients with clinically significant depressive symptoms in the mild or greater severity range were considered patients with depression.
Neuropathological Assessment: PSP pathology
Immunohistochemistry for phospho-tau (CP13, 1:1000, from Dr. Peter Davies, Feinstein Institute, North Shore Hospital, NY) was used to establish neuropathological diagnosis of PSP.7 The severity of four tau lesion types, including pretangles/NFTs, coiled bodies, tufted astrocytes, and tau-positive threads, was graded semi-quantitatively on a four-point scale (0, absent; 1, sparse; 2, moderate; 3, frequent)12 by an experienced neuropathologist (D.W.D), blinded to cognitive data, in 20 brain regions: the temporal cortex, motor cortex, caudate/putamen, globus pallidus, basal nucleus, hypothalamus, ventral thalamus, subthalamic nucleus, thalamic fasciculus, red nucleus, substantia nigra, oculomotor complex, midbrain tectum, locus coeruleus, pontine tegmentum, pontine base, medullary tegmentum, inferior olive, dentate nucleus, and cerebellar white matter (Supplementary Figure 1A). Scores from the hippocampus and amygdala were excluded to remove the influence of Alzheimer-type pathology on the analysis. The regional PSP-related tau burden was defined as the sum of scores for all lesion types in each brain region (range: 0–12). The total PSP-related tau burden was calculated as an average of regional PSP-related tau burden across all 20 brain regions.29
To validate the reliability of semi-quantitative assessment of tau burden, we performed quantitative imaging analysis and analyzed the correlation between semi-quantitative and quantitative tau burden. Sections of the motor cortex from 114 PSP cases (sections were unavailable in 7 cases) were scanned on the ScanScopeXT (Aperio Technologies, Vista, CA). The grey matter was annotated using ImageScope-11.2 (Aperio Technologies) and analyzed in Spectrum-11.2 (Aperio Technologies) using a custom-designed color deconvolution algorithm to detect only CP13-positive pathology (Supplementary Figure 1B).30 Total tau burden was expressed as a percent ratio of the area of immunoreactive pixels to the total area of the annotated region. The Spearman rank correlation test showed strong correlation between semi-quantitative and quantitative tau burden (Spearman’s rho 0.86, P = 2 × 10−7) (Supplementary Figure 1C).
Neuropathological Assessment: concurrent pathology
A Braak NFT stage and Thal amyloid phase were assigned to each case with thioflavin S fluorescent microscopy.31, 32 Numbers of senile plaques and NFTs are counted in nine brain regions: mid-frontal, superior temporal, inferior parietal, motor cortex, visual cortex, endplate, CA2/3, CA1, and subiculum. Neuropathological diagnosis of AD was based on the consensus criteria for the neuropathologic diagnosis of AD.33 In this study, both high and intermediate likelihood cases were diagnosed with AD. Neuropathological diagnosis of argyrophilic grain disease, Lewy-related pathology, hippocampal sclerosis and cerebrovascular pathology were established previously described.13 TDP-43 pathology was not included in this study because our previous study showed that TDP-43 pathology was present in 7% of PSP, but it was not associated with CI.13
Genetic Analysis
We performed genetic analysis in cases with available frozen brain tissue (N = 118). For genotyping, genomic DNA was extracted from cerebellum of frozen brain tissue using standard procedures. Genotyping for APOE alleles (SNP rs429358 C/T and rs7412 C/T) and MAPT H1/H2 (SNP rs1052553 A/G, A = H1, G = H2) was assessed with TaqMan SNP genotyping assays (Applied Biosystems, Foster City, CA) as previously reported.12 Genotype calls were obtained with SDS v2.2.2 software (Applied Biosystems).
Statistical Analysis
All statistical analyses were performed in SigmaPlot 12.3 (Systat Software, San Jose, CA) and SPSS Statistics 19 (IBM, Chicago, IL). A chi-square or Fisher’s exact test was performed for group comparisons of categorical data as appropriate. Mann-Whitney rank sum test or t-test was used for group comparison analysis of continuous variables as appropriate. The Spearman rank correlation test was used to assess the correlation between each test score or demographic information and PSP-related tau burden. Bonferroni corrections were utilized to adjust for multiple testing separately for some analyses. Significance levels for P values were mentioned in each Table or Figure legend. Hierarchical regressions were conducted to determine whether total tau burden predicted neuropsychological performance after controlling for co-variates.
Results
Comparison between PSP with and without CI
The total cohort included 121 patients with autopsy-confirmed PSP. Of those, 90 (74%) patients were documented with CI based on the record review. The frequencies of CI in each clinical phenotype were 75% in Richardson syndrome (67/89), 57% in PSP-CBS (8/14) and PSP-parkinsonism (4/7), 100% in PSP-frontotemporal dementia (4/4), PSP-speech/language (1/1), and PSP with predominant cerebellar ataxia (1/1), and 80% in unclassified cases because of insufficient clinical information (4/5). Table 1 compares the demographic information and pathologic features between PSP with CI (PSP-CI) and PSP without CI (PSP-NC). The age at onset and death, disease duration, sex ratio, frequency of family history of dementia and parkinsonism, and frequency of clinical diagnosis of PSP did not differ between groups. Of note, depression was more frequently seen in PSP-CI than in PSP-NC (59% vs 35%, P = 0.04). Although the average brain weight was less in PSP-CI than in PSP-NC, other neuropathological features, including Braak neurofibrillary tangle stage, Thal amyloid phase, and frequency of concurrent pathologies of dementia were not different between the two groups.
Table 1.
Comparison of demographic, clinical, and pathologic features between PSP-CI and PSP-NC
| Features | Total (N = 121) |
PSP-CI (N = 90) |
PSP-NC (N = 31) |
P value |
|---|---|---|---|---|
| Male, No. (%) | 74 (61%) | 55 (61%) | 19 (61%) | 0.85 |
| Age at onset, years | 66 ± 8 | 66 ± 8 | 66 ± 9 | 0.74 |
| Age at death, years | 74 ± 8 | 74 ± 8 | 74 ± 9 | 0.98 |
| Disease duration, years | 7 (5, 9) | 7 (5, 9) | 7 (5, 12) | 0.73 |
| Family history of dementia | 28 (23%) | 23 (26%) | 5 (16%) | 0.41 |
| Family history of Parkinsonism | 21 (17%) | 16 (18%) | 6 (19%) | 0.94 |
| Having clinical diagnosis of PSP | 91 (75%) | 70 (78%) | 21 (68%) | 0.38 |
| Pathology | ||||
| Brain weight, grams | 1180 ± 140 | 1160 ± 140 | 1230 ± 150 | 0.02 |
| Braak NFT stage | II (II, III) | II (II, III) | II (II, III) | 0.59 |
| Thal amyloid phase | 0 (0, 3) | 0 (0, 3) | 1 (0, 3) | 0.20 |
| Alzheimer’s disease | 15 (12%) | 9 (10%) | 6 (19%) | 1.00 |
| Argyrophilic grain disease | 31 (26%) | 23 (26%) | 8 (26%) | 1.00 |
| Hippocampal sclerosis | 1 (1%) | 1 (1%) | 0 (0%) | 1.00 |
| Cerebrovascular pathology | 14 (12%) | 10 (11%) | 4 (13%) | 1.00 |
| Lewy-related pathology | 9 (7%) | 8 (9%) | 1 (3%) | 1.00 |
| Total tau burden | 6 (4, 8) | 6 (5, 8) | 6 (4, 8) | <0.001 |
Values are n (%), mean ± SD, and median (25th, 75th %-tile). Abbreviations: NFT, neurofibrillary tangle; PSP-CI, progressive supranuclear palsy with cognitive impairment; PSP-NC, progressive supranuclear palsy without cognitive impairment.
To address the hypothesis that PSP-related tau affects frequency and severity of CI, we compared total tau burden and regional tau burden between PSP-CI and PSP-NC. Total tau burden (Table 1) and regional tau burden in the pontine base and cerebella white matter (Supplementary Figure 2) were significantly higher in PSP-CI than in PSP-NC. In contrast, the number of senile plaques and NFTs in nine brain regions were not significantly different between the two groups (Supplementary Figure 2). These results suggest that PSP-related tau burden, but not concurrent pathologies of dementia, is associated with CI in PSP.
Profile of CI in PSP
To characterize the profile and severity of CI, we analyzed neuropsychological records of 37 patients (57% men, 97% Caucasian). Table 2 shows demographic information and results of neuropsychological tests. For the two global CI indices, the DRS-2 Total Score was more impaired relative to the OTBM. The most impaired domain was executive functioning, although none of the cognitive domain mean scores fell within the clinically impaired range (i.e., 1.5 SD below the mean, T-score < 35). Length of disease duration and duration of interval between testing and death were not significantly correlated with any of the global cognitive index or domain mean scores. Neither the OTBM nor the domain mean scores were significantly different when compared among the PSP clinical phenotypes (Supplementary Table 2).
Table 2.
Relationship between neuropsychological variables and total tau burden
| Features | N | PSP | Spearman’s rho | P value |
|---|---|---|---|---|
| Age at Testing, years | 37 | 71 ± 8 | −0.49 | 0.002 |
| Age at Disease Onset, years | 36 | 67 ± 8 | −0.49 | 0.008 |
| Age at Death, years | 37 | 75 ± 8 | −0.44 | 0.006 |
| Education Level, years | 37 | 15 ± 3 | −0.12 | 0.49 |
| Global Cognitive Functioning | ||||
| Overall Test Battery Mean | 32 | 40 (34, 48) | −0.49 | 0.005* |
| DRS-2 Total Score | 33 | 33 (23, 47) | −0.47 | 0.006* |
| Attention/Processing Speed | 32 | 45 (37, 49) | −0.42 | 0.02 |
| Executive Functioning | 32 | 37 (28, 46) | −0.51 | 0.003* |
| Episodic Memory | 32 | 44 (35, 54) | −0.46 | 0.008 |
| Language | 32 | 43 (37, 47) | −0.31 | 0.09 |
| Visuospatial/Construction | 32 | 43 (35, 47) | −0.033 | 0.86 |
All test scores are shown as standardized age-corrected T-scores (M = 50, SD = 10). Spearman’s rho is calculated as a correlation between each score and total tau burden.
indicates statistical significance after applying a Bonferroni correction for multiple testing in neuropsychological scores (P < 0.0071).
Twenty-six of the 37 patients were classified as having depression based on a depression screening instrument given at the time of their neuropsychological assessment. Of those, 12 patients (46%) reported clinically significant depressive symptoms. Group difference analysis indicated that there was no significant difference in cognition between patients with and without depression (OTBM: 42 ± 8 vs 40 ± 8, P = 0.88).
Total tau burden negatively correlates with neuropsychological performance
To elucidate the association between tau burden and severity of CI, we performed Spearman correlation analysis between each neuropsychological test and total tau burden (Table 2). The OTBM was negatively correlated with total tau burden (Spearman’s rho −0.49, P = 0.005), but not with the number of senile plaques (Spearman’s rho 0.03, P = 0.87) or NFTs (Spearman’s rho −0.09, P = 0.62) (Figure 1). These results suggest that PSP-related tau pathology is associated with the severity of CI in PSP, but Alzheimer-type pathology is not.
Figure 1.
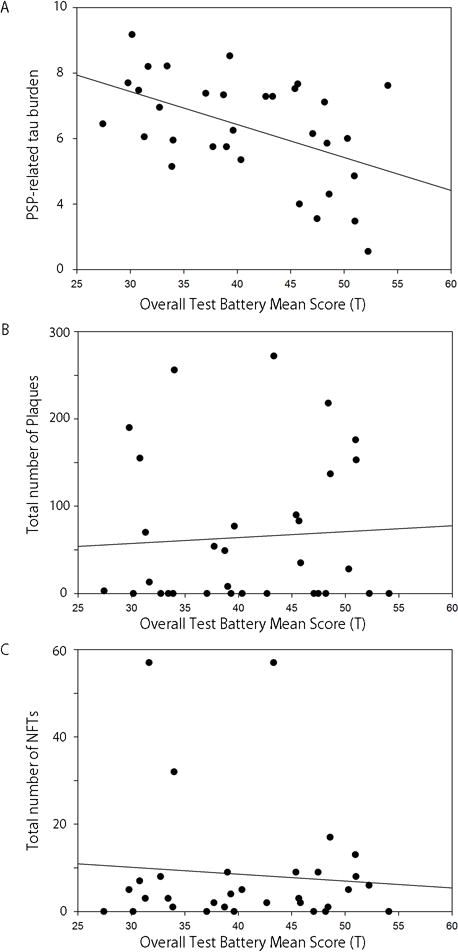
Spearman correlation analyses show that overall test battery mean score is negatively correlated with PSP-related tau burden (A, Spearman’s rho −0.49, P = 0.005), but not with the number of senile plaques (B, Spearman’s rho 0.03, P = 0.87) or neurofibrillary tangles (C, Spearman’s rho −0.09, P = 0.62).
Next, we examined the extent to which each cognitive domain and neuropsychological test score was correlated with total tau burden (Table 2). Deficits in executive functioning (Spearman’s rho −0.51, P = 0.003) were negatively correlated with the total tau burden. Conversely, Visuospatial/Construction was not significantly correlated with total tau burden. Tau burden in some specific regions were negatively correlated with executive functioning (e.g. motor cortex: Spearman’s rho −0.51, P = 0.005; globus pallidus: Spearman’s rho −0.52, P = 0.004), but these were not statistically significant after Bonferroni corrections.
Hierarchical regressions were conducted to determine the contribution that total tau burden predicted CI after controlling for age at testing, age at death, and disease duration (Supplementary Table 3). The first regression included age at testing, age at death, and disease duration in the first model; the second model added total tau burden to determine its contribution to OTBM considering the covariates. The first model accounted for 9.8% of the total variance in OTBM, but was not statistically significant. The second model was statistically significant and accounted for 24.3% of OTBM variance [F (1, 27) = 6.381, P = 0.018]. Of all the variables in the second model, only total tau burden (β = −0.524, P = 0.018) was a statistically significant predictor. The second regression included the age and disease duration variables as in the first regression, but the executive functioning domain mean score was used as the dependent variable. The final model included total tau burden and accounted for 31.8% of the total variance in executive functioning [F (3, 28) = 4.607, P = 0.006]. The change in R2 of .236 was statistically significant [FΔ (1, 28) = 10.723, P = 0.003]. No other neuropsychological domain means were significantly predicted from the age variables, disease duration, or total tau burden.
PSP with coexisting AD
We have shown that Alzheimer-type pathology was not related to the presence or severity of CI in PSP. To further support this finding, we compared pathological and clinical features between PSP with AD (PSP/AD) and PSP. As shown in Table 1, 15 out of 121 patients had a concurrent pathological diagnosis of AD. As expected, age at death, Braak neurofibrillary tangle stage and Thal amyloid phase were higher in patients with PSP/AD (Table 3). Interestingly, PSP/AD had lower total tau burden compared to PSP, although the frequency of CI absent of neuropsychological test data was not different between the two groups. Of the 37 patients with neuropsychological data, six patients were PSP/AD. The OTBM was not statistically different between PSP and PSP/AD, which is consistent with the finding that Alzheimer-type pathology did not correlate with the severity of CI. Taken together, coexisting AD did not significantly impact the frequency or severity of CI in PSP.
Table 3.
Clinical and pathological features compared between PSP/AD and PSP
| Features | PSP/AD | PSP | P value |
|---|---|---|---|
| Main cohort | N = 15 | N = 106 | |
| Disease duration, years | 9 (6, 13) | 7 (5, 9) | 0.10 |
| Age at death, years | 81 ± 6 | 73 ± 8 | <0.001 |
| Braak neurofibrillary tangle stage | IV (IV, V) | II (I, III) | <0.001 |
| Thal amyloid phase | 4 (3, 4) | 0 (0, 2) | <0.001 |
| Total tau burden | 1.4 ± 0.4 | 1.6 ± 0.3 | 0.03 |
| Cognitive impairment | 9 (60%) | 81 (76%) | 1.00 |
| Subgroup patients | N = 6 | N = 31 | |
| Disease duration | 8 (6, 10) | 7 (5, 9) | 0.62 |
| Age at death | 79 ± 7 | 74 ± 7 | 0.13 |
| Braak neurofibrillary tangle stage | IV (IV, V) | II (II, III) | <0.001 |
| Thal amyloid phase | 4 (3, 4) | 0 (0, 3) | <0.001 |
| Total tau burden | 1.5 ± 0.4 | 1.6 ± 0.4 | 0.47 |
| Overall Test Battery Mean | 43 (32, 51) | 40 (33, 47) | 0.72 |
| Attention/Processing Speed | 47 (35, 48) | 44 (37, 50) | 0.84 |
| Executive Functioning | 46 (26, 47) | 37 (28, 45) | 0.88 |
| Episodic Memory | 32 (28, 59) | 46 (35, 54) | 0.48 |
| Language | 42 (35, 51) | 43 (36, 47) | 1.00 |
| Visuospatial/Construction | 44 (35, 48) | 43 (35, 47) | 0.98 |
Overall Test Battery Mean and domain mean score are available for 5 patients in PSP/AD and 27 patients in PSP. Abbreviation: PSP, progressive supranuclear palsy; PSP/AD, PSP with Alzheimer’s disease.
Genetic analysis
Genetic analysis revealed no significant difference for either APOE ε4 frequency or MAPT H1/H1 genotype between PSP-CI and PSP-NC. Compared to APOE ε4 non-carriers, APOE ε4 carriers had higher Thal amyloid phase, but unexpectedly, the frequency of CI was lower in APOE ε4 carriers (Table 4). The OTBM and total tau burden were not different between the two groups (Table 4). These results suggest that APOE ε4 allele does not associate with CI in PSP, although it is associated with more severe amyloid pathology. MAPT is a gene of interest because MAPT H1/H1 genotype is a risk factor of PSP; however, there were no difference in clinical and pathological features between H1/H1and H1/H2 genotype (Table 4). The association between genotype and regional tau burden was also analyzed, but there was no significant difference in any regional tau burden between ApoE genotypes or MAPT haplotypes.
Table 4.
Cognitive and pathological features in each APOE4 or MAPT haplotype
| Features |
APOE4 + (N = 28) |
APOE4 − (N = 90) |
P value |
MAPT H1/H1 (N = 103) |
MAPT H1/H2 (N = 15) |
P value |
|---|---|---|---|---|---|---|
| Male, No. (%) | 19 (68%) | 53 (59%) | 0.53 | 61 (59%) | 11 (73%) | 0.45 |
| Age at death, years | 73 ± 9 | 74 ± 8 | 0.66 | 73 ± 9 | 74 ± 8 | 0.66 |
| Cognitive impairment | 14 (56%) | 74 (80%) | 0.002 | 76 (74%) | 12 (80%) | 1.00 |
| Overall Test Battery Mean | 41 ± 9 | 41 ± 8 | 0.89 | 40 ± 8 | 44 ± 8 | 0.24 |
| Braak neurofibrillary tangle stage | III (II, III) | II (II, III) | 0.61 | II (II, III) | II (II, IV) | 0.68 |
| Thal amyloid phase | 3 (1, 4) | 0 (0, 3) | <0.001 | 0 (0, 3) | 1 (0, 3) | 0.71 |
| Alzheimer’s disease | 6 (21%) | 9 (10%) | 1.00 | 12 (12%) | 3 (20%) | 1.00 |
| Lewy-related pathology | 0 (0%) | 8 (9%) | 1.00 | 4 (4%) | 4 (27%) | 1.00 |
| Total tau burden | 7 (6, 7) | 7 (4, 8) | 0.93 | 7 (6, 7) | 7 (4, 8) | 0.93 |
Values are n (%), mean ± SD, and median (25th, 75th %-tile).
Discussion
The main findings of this retrospective clinicopathological study are (1) at least 74% of PSP patients had CI, primarily in executive functioning and (2) the total PSP-related tau burden, but not Alzheimer-type pathology, correlated with severity of CI in PSP. The first finding supports evidence from the literature that were mostly based on clinically-diagnosed PSP patients.5, 6 Our second finding is the first pathological evidence that PSP-related tau pathology is associated with cognitive function in PSP.
The results of our study support previous studies on the profile of CI in PSP; multiple cognitive domains are affected, especially executive function.5, 6 We also analyzed the possible influence of depression on CI, since patients with depression are often thought to “masquerade” as CI involving primarily slowed processing speed and inattention due to the depressive symptoms.34, 35 Depression was more frequently seen in PSP-CI compared to PSP-NC; however, when a subset of patients with self-reported depression was compared, no differences in test data were identified. These results suggest that CI in PSP is less affected by psychopathology, such as depression, which is consistent with a previous investigation.36 Nevertheless, it is worth noting that about half of the patients in our PSP cohort had depression. Although the reported frequency of depression in PSP varies 18% to 42%,37, 38 our results suggest that depression may be more common than previously thought; therefore, it is important to assess depressive symptoms in PSP, and if CI is suspected, a thorough objective cognitive evaluation may be necessary to better determine the relationship between depression and cognition.
Pathological analysis confirmed our hypothesis that burden of PSP-related-tau pathology would correlate with the severity of CI in PSP. Total tau burden and regional tau burden in two brain regions (i.e. pontine base, and cerebellar white matter) were significantly higher in PSP-CI than in PSP-NC. Moreover, total tau burden significantly predicted neuropsychological functioning (OTBM and executive functioning) after accounting for age at testing, age at death, and disease duration. Nevertheless, the regions responsible for CI in PSP remain ambiguous. Regional tau burden was nominally higher in several brain regions, but the differences were not statistically significant. We assume that the total tau burden may reflect overall disease severity and that PSP patients in more advanced stages exhibit more severe CI. This is consistent with a previous study that showed CI was related to disease severity whether measured by Clinician Global Impression, Hoehn and Yahr stage, or motor disability.6 As hypothesized by Fiorenzato et al., who examined CI in multiple system atrophy, pathology in circuits between cortical and subcortical regions would affect cognitive functioning rather than in a specific region.39
To examine whether concurrent pathologies affect CI in PSP, we compared clinical and pathological features between PSP/AD and PSP. Although the number of patients with PSP/AD was small and Alzheimer-type pathology was relatively mild, we found no significant difference in frequency and severity of CI in PSP compared to PSP/AD. This is consistent with literature reporting CI in corticobasal degeneration; 59% of corticobasal degeneration had Alzheimer-type pathology, but it had minimal effect on the rate of dementia progression and dementia duration.40 Although it remains inconclusive, there was not enough evidence to suggest that Alzheimer-type pathology affected cognition in PSP-CI.
Genetic analysis for APOE also supported the idea that Alzheimer-type pathology was not associated with CI in PSP. APOE ε4 allele is the strongest genetic risk factor of AD.41 APOE has an important role in Aβ metabolism, Aβ deposition in senile plaques, and amyloid angiopathy.42, 43 In our study, APOE ε4 carriers had more severe amyloid pathology than non-carriers; however, unexpectedly, the frequency of CI was lower in ε4 carriers. Although the reason of low frequency of CI in ε4 carriers was unclear, given the total tau burden was not different, this different frequency of CI might be fortuitous due to the small size. This result suggests that Alzheimer-type pathology driven by APOE ε4 does not increase the frequency of CI in PSP.
A limitation of our study is its retrospective nature and the fact that only a subset of patients (41% with CI) had formal neuropsychological assessment. Furthermore, due to the retrospective nature of this study, the evaluations were performed in the clinical context between multiple clinicians. Also, because assessment of CI in the main cohort was based on physician’s impression and patients’ subjective complaints documented in medical records, the number of PSP patients with CI may be underestimated. Only six patients had PSP/AD in the subgroup; therefore, the detection of different patterns of CI between patients with and without AD was under-powered. Another limitation is that motor symptoms in PSP may cloud the interpretation of cognitive tests, especially with tests that tend to rely on motor speed (i.e., processing speed), as bradykinesia is one of the most frequent motor signs in PSP.16, 44 Cognitive tests relying less on motor-dependent tests should be prioritized to overcome this potential confound. Also, the presence of executive dysfunction could impact learning efficiency in some patients thus attenuating delayed episodic memory scores. The impact of this effect on overall memory performance was unaccounted for in our study. Finally, some brain regions that are typically considered to be related to CI (i.e. the frontal and parietal lobe, hippocampus, and amygdala) were not assessed in diagnostic semi-quantitative assessment of tau pathology in PSP. Given that these data independently collected from the current study, the data shown here were completely unbiased in this sense. A final strength of our study is that the diagnoses of PSP were pathologically confirmed, while most of the existing literature on CI in PSP is based on clinical diagnosis.
Concluding, the results of our study showed that a majority of PSP patients developed CI, primarily involving executive functioning, and that PSP-related tau burden, rather than Alzheimer-type pathology, was correlated with the severity of CI. A comprehensive neuropsychological evaluation may be helpful for identifying CI in patients with PSP as well as assisting in establishing a clinical diagnosis of PSP based on the pattern of CI and the presence or absence of executive dysfunction.
Supplementary Material
Supplementary Figure 1: (A) Semi-quantitative score on a four-point scale of PSP-related tau pathology. Representative images of immunohistochemistry for tau on sections of the globus pallidus (neurofibrillary tangles/pretangles and threads), putamen (tufted astrocytes), and cerebellar white matter (coiled bodies). The scores indicate 1 = sparse, 2 = moderate, and 3 = frequent. Bar = 100 μm. (B) Quantitative assessment of tau burden. The upper image shows immunohistochemistry for tau (CP13) in the grey matter of motor cortex and the lower image shows the custom-designed color deconvolution algorithm to highlight tau deposits (shown in red). Pretangles, tufted astrocytes, threads, and coiled bodies are observed and quantified. (C) The Spearman rank correlation test shows strong correlation between semi-quantitative tau burden and quantitative tau burden (Spearman’s rho 0.86, P = 2 × 10−7).
Supplementary Figure 2: Median total tau burden (A), number of senile plaques (B), and number of NFTs (C) in PSP-CI (left bars) and PSP-NC (right bars). * indicates statistical significance after applying a Bonferroni correction for multiple testing in neuropsychological scores (P < 0.0025). Abbreviations: PSP-CI, progressive supranuclear palsy with cognitive impairment; PSP-NC, progressive supranuclear palsy without cognitive impairment.
Acknowledgments
We would like to thank the patients and their families who donated brains to help further the scientific understanding of neurodegeneration. The authors would also like to acknowledge Linda Rousseau and Virginia Phillips (Mayo Clinic, Jacksonville) for histologic support, Monica Castanedes-Casey (Mayo Clinic, Jacksonville) for immunohistochemistry support, Drs. William P. Cheshire, Kevin B. Boylan (Mayo Clinic, Jacksonville), Erika Driver-Dunckley (Mayo Clinic, Scottsdale), and David S. Knopman (Mayo Clinic, Rochester) for contributing patients, and Drs. Jane Cerhan, Robert Ivnik, Mary Machulda, Jeff Smigielski, Glenn Smith, Max Trenerry (Mayo Clinic, Rochester), Tanis Ferman, John Lucas, Otto Pedraza, Beth Rush, Mark Schwartz (Mayo Clinic, Jacksonville), and Dona Locke (Mayo Clinic, Scottsdale) for neuropsychological testing.
Funding
This work is supported by NIH grants P50 NS072187 and R35 NS097261 and a Jaye F. and Betty F. Dyer Foundation Fellowship in progressive supranuclear palsy research.
List of abbreviations
- CI
cognitive impairment
- DRS
Dementia Rating Scale
- OTBM
overall test battery mean
- PSP
progressive supranuclear palsy
- PSP/AD
PSP with coexisting Alzheimer’s disease
- PSP-CI
PSP with CI
- PSP-NC
PSP without CI
- TMT
Trail Making Test
- WAIS
Wechsler Adult Intelligence Scale
- WMS
Wechsler Memory Scale.
Footnotes
Financial Disclosures of all authors
Dr. Koga reports no disclosures.
Dr. Parks reports no disclosures.
Dr. Kasanuki reports no disclosures.
Dr. Sanchez-Contreras reports no disclosures.
Mr. Matthew C. Baker reports no disclosures.
Dr. Josephs receives research support from the NIH (R01-DC010367, R01-DC012519 & R01-AG037491) and the Alzheimer’s Association. Dr. Josephs is an editorial board member of Acta Neuropathologica, Journal of Neurology and Parkinsonism and Related Disorders.
Dr. Ahlskog reports no disclosures.
Dr. Uitti receives research support by the NIH (P50-NS072187, and from Advanced Neuromodulation Systems, Inc./St. Jude Medical, Dr. Uitti is an Associate Editor of Neurology and an editorial board member of Parkinsonism & Related Disorders.
Dr. Graff-Radford reports no disclosures.
Dr. van Gerpen receives research funds from the Mayo Clinic CR program and NIH (P50-NS072187). This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Dr. Wszolek is partially supported by the NIH/NINDS P50 NS072187, NIH/NIA (primary) and NIH/NINDS (secondary) 1U01AG045390-01A1, Mayo Clinic Center for Regenerative Medicine, Mayo Clinic Center for Individualized Medicine, Mayo Clinic Neuroscience Focused Research Team (Cecilia and Dan Carmichael Family Foundation, and the James C. and Sarah K. Kennedy Fund for Neurodegenerative Disease Research at Mayo Clinic in Florida), the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch, and The Sol Goldman Charitable Trust. Dr. Wszolek serves as Co-Editor-in-Chief of Parkinsonism and Related Disorders, Associate Editor of the European Journal of Neurology, and on the editorial boards of Neurologia i Neurochirurgia Polska, the Medical Journal of the Rzeszow University, and Clinical and Experimental Medical Letters; holds and has contractual rights for receipt of future royalty payments from patents re: A novel polynucleotide involved in heritable Parkinson’s disease; receives royalties from editing Parkinsonism and Related Disorders (Elsevier, 2015, 2016) and the European Journal of Neurology (Wiley- Blackwell, 2015, 2016).
Dr. Rademakers receives research support from the NIH (P50-NS072187, R01-NS076471 and R01-NS080882).
Dr. Dickson receives support from the NIH (P50-NS072187). Dr. Dickson is an editorial board member of Acta Neuropathologica, Annals of Neurology, Brain, Brain Pathology, and Neuropathology, and he is editor in chief of American Journal of Neurodegenerative Disease, and International Journal of Clinical and Experimental Pathology.
Author Contributions
SK: Conception of the project; Acquisition, analysis and interpretation of data; drafting of manuscript; execution of the statistical analysis; writing of the first draft
AP: Conception of the project; Acquisition, analysis and interpretation of data; execution of the statistical analysis; review and edit the draft
KK: Acquisition, analysis and interpretation of data; review and critique
MSC: Acquisition, analysis and interpretation of data;
MB: Acquisition, analysis and interpretation of data;
KAJ: Review and critique; contribution of patients
JEA: Review and critique; contribution of patients
RJU: Review and critique; contribution of patients
NGR: Review and critique; contribution of patients
JAG: Review and critique; contribution of patients
ZKW: Review and critique; contribution of patients
RR: Review and critique; interpretation of data
DWD: Conception and organization of the project; interpretation of data; review and critique
References
- 1.Steele JC, Richardson JC, Olszewski J. Progressive Supranuclear Palsy. A Heterogeneous Degeneration Involving the Brain Stem, Basal Ganglia and Cerebellum with Vertical Gaze and Pseudobulbar Palsy, Nuchal Dystonia and Dementia. Arch Neurol. 1964;10:333–359. doi: 10.1001/archneur.1964.00460160003001. [DOI] [PubMed] [Google Scholar]
- 2.Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47(1):1–9. doi: 10.1212/wnl.47.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Hoglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord. 2017 doi: 10.1002/mds.26987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albert ML, Feldman RG, Willis AL. The ‘subcortical dementia’ of progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 1974;37(2):121–130. doi: 10.1136/jnnp.37.2.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bak TH, Crawford LM, Hearn VC, Mathuranath PS, Hodges JR. Subcortical dementia revisited: similarities and differences in cognitive function between progressive supranuclear palsy (PSP), corticobasal degeneration (CBD) and multiple system atrophy (MSA) Neurocase. 2005;11(4):268–273. doi: 10.1080/13554790590962997. [DOI] [PubMed] [Google Scholar]
- 6.Brown RG, Lacomblez L, Landwehrmeyer BG, et al. Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy. Brain. 2010;133(Pt 8):2382–2393. doi: 10.1093/brain/awq158. [DOI] [PubMed] [Google Scholar]
- 7.Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy) Neurology. 1994;44(11):2015–2019. doi: 10.1212/wnl.44.11.2015. [DOI] [PubMed] [Google Scholar]
- 8.Litvan I, Hauw JJ, Bartko JJ, et al. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol. 1996;55(1):97–105. doi: 10.1097/00005072-199601000-00010. [DOI] [PubMed] [Google Scholar]
- 9.Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol. 2010;23(4):394–400. doi: 10.1097/WCO.0b013e32833be924. [DOI] [PubMed] [Google Scholar]
- 10.Ahmed Z, Josephs KA, Gonzalez J, DelleDonne A, Dickson DW. Clinical and neuropathologic features of progressive supranuclear palsy with severe pallido-nigro-luysial degeneration and axonal dystrophy. Brain. 2008;131(Pt 2):460–472. doi: 10.1093/brain/awm301. [DOI] [PubMed] [Google Scholar]
- 11.Ishizawa K, Dickson DW. Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. J Neuropathol Exp Neurol. 2001;60(6):647–657. doi: 10.1093/jnen/60.6.647. [DOI] [PubMed] [Google Scholar]
- 12.Koga S, Josephs KA, Ogaki K, et al. Cerebellar ataxia in progressive supranuclear palsy: An autopsy study of PSP-C. Mov Disord. 2016;31(5):653–662. doi: 10.1002/mds.26499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koga S, Sanchez-Contreras M, Josephs KA, et al. Distribution and characteristics of transactive response DNA binding protein 43 kDa pathology in progressive supranuclear palsy. Mov Disord. 2017;32(2):246–255. doi: 10.1002/mds.26809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koga S, Aoki N, Uitti RJ, et al. When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology. 2015;85(5):404–412. doi: 10.1212/WNL.0000000000001807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koga S, Parks A, Uitti RJ, et al. Profile of cognitive impairment and underlying pathology in multiple system atrophy. Mov Disord. 2017;32(3):405–413. doi: 10.1002/mds.26874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koga S, Parks A, Dickson DW. Reply re: “Profile of cognitive impairment and underlying pathology in multiple system atrophy”. Mov Disord. 2017 doi: 10.1002/mds.26874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jurica PJ, Leitten CL, Mattis S. DRS-2 : Dementia rating scale-2 : professional manual. Lutz, FL: Psychological Assessment Resources; 2001. [Google Scholar]
- 18.Wechsler D. Wechsler Adult Intelligence Scale-Fourth Edition (WAIS-IV) San Antonio, TX: Pearson; 2008. [Google Scholar]
- 19.Armitage SG. An analysis of certain psychological tests used for the evaluation of brain injury. Washington: American Psychological Association; 1946. [Google Scholar]
- 20.Wechsler D. Wechsler Memory Scale - Fourth Edition (WMS-IV) San Antonio, TX: Peason; 2009. [Google Scholar]
- 21.Kaplan E, H G, Weintraub S. Boston Naming Test 2. Philadelphia, PA: Lippincott Williams & Wilkins; 2002. [Google Scholar]
- 22.Mitrushina MN. Handbook of normative data for neuropsychological assessment. 2nd. Oxford: Oxford University Press; 2005. [Google Scholar]
- 23.Green P, Rohling ML, Lees-Haley PR, Allen LM., 3rd Effort has a greater effect on test scores than severe brain injury in compensation claimants. Brain Inj. 2001;15(12):1045–1060. doi: 10.1080/02699050110088254. [DOI] [PubMed] [Google Scholar]
- 24.Rohling ML. Generating a linear function for residual impairment for TBI: a comparison of the HRB and a flexible battery approach. Arch Clin Neuropsychol. 2000;15(8):821–822. [Google Scholar]
- 25.Vogel SJ, Banks SJ, Cummings JL, Miller JB. Concordance of the Montreal cognitive assessment with standard neuropsychological measures. Alzheimers Dement (Amst) 2015;1(3):289–294. doi: 10.1016/j.dadm.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beck AT, Steer RA, Brown GK. Beck Depression Inventory-II. San Antonio, TX: Pearson; 1996. [Google Scholar]
- 27.Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res. 1982;17(1):37–49. doi: 10.1016/0022-3956(82)90033-4. [DOI] [PubMed] [Google Scholar]
- 28.Spitzer RL, Kroenke K, Williams JB. Validation and utility of a self-report version of PRIME-MD: the PHQ primary care study. Primary Care Evaluation of Mental Disorders. Patient Health Questionnaire. JAMA. 1999;282(18):1737–1744. doi: 10.1001/jama.282.18.1737. [DOI] [PubMed] [Google Scholar]
- 29.Williams DR, Holton JL, Strand C, et al. Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson’s syndrome. Brain. 2007;130(Pt 6):1566–1576. doi: 10.1093/brain/awm104. [DOI] [PubMed] [Google Scholar]
- 30.Murray ME, Przybelski SA, Lesnick TG, et al. Early Alzheimer’s disease neuropathology detected by proton MR spectroscopy. J Neurosci. 2014;34(49):16247–16255. doi: 10.1523/JNEUROSCI.2027-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 32.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 33.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8(1):1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wells CE. Pseudodementia. Am J Psychiatry. 1979;136(7):895–900. doi: 10.1176/ajp.136.7.895. [DOI] [PubMed] [Google Scholar]
- 35.Bonelli RM, Cummings JL. Frontal-subcortical dementias. Neurologist. 2008;14(2):100–107. doi: 10.1097/NRL.0b013e31815b0de2. [DOI] [PubMed] [Google Scholar]
- 36.Esmonde T, Giles E, Gibson M, Hodges JR. Neuropsychological performance, disease severity, and depression in progressive supranuclear palsy. J Neurol. 1996;243(9):638–643. doi: 10.1007/BF00878659. [DOI] [PubMed] [Google Scholar]
- 37.Litvan I, Mega MS, Cummings JL, Fairbanks L. Neuropsychiatric aspects of progressive supranuclear palsy. Neurology. 1996;47(5):1184–1189. doi: 10.1212/wnl.47.5.1184. [DOI] [PubMed] [Google Scholar]
- 38.Menza MA, Cocchiola J, Golbe LI. Psychiatric symptoms in progressive supranuclear palsy. Psychosomatics. 1995;36(6):550–554. doi: 10.1016/S0033-3182(95)71610-3. [DOI] [PubMed] [Google Scholar]
- 39.Fiorenzato E, Weis L, Seppi K, et al. Brain structural profile of multiple system atrophy patients with cognitive impairment. J Neural Transm (Vienna) 2017;124(3):293–302. doi: 10.1007/s00702-016-1636-0. [DOI] [PubMed] [Google Scholar]
- 40.Day GS, Lim TS, Hassenstab J, et al. Differentiating cognitive impairment due to corticobasal degeneration and Alzheimer disease. Neurology. 2017;88(13):1273–1281. doi: 10.1212/WNL.0000000000003770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 42.Schmechel DE, Saunders AM, Strittmatter WJ, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(20):9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strittmatter WJ, Saunders AM, Schmechel D, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(5):1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fiorenzato E, Antonini A, Wenning G, Biundo R. Cognitive impairment in multiple system atrophy. Mov Disord. 2017 doi: 10.1002/mds.27085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: (A) Semi-quantitative score on a four-point scale of PSP-related tau pathology. Representative images of immunohistochemistry for tau on sections of the globus pallidus (neurofibrillary tangles/pretangles and threads), putamen (tufted astrocytes), and cerebellar white matter (coiled bodies). The scores indicate 1 = sparse, 2 = moderate, and 3 = frequent. Bar = 100 μm. (B) Quantitative assessment of tau burden. The upper image shows immunohistochemistry for tau (CP13) in the grey matter of motor cortex and the lower image shows the custom-designed color deconvolution algorithm to highlight tau deposits (shown in red). Pretangles, tufted astrocytes, threads, and coiled bodies are observed and quantified. (C) The Spearman rank correlation test shows strong correlation between semi-quantitative tau burden and quantitative tau burden (Spearman’s rho 0.86, P = 2 × 10−7).
Supplementary Figure 2: Median total tau burden (A), number of senile plaques (B), and number of NFTs (C) in PSP-CI (left bars) and PSP-NC (right bars). * indicates statistical significance after applying a Bonferroni correction for multiple testing in neuropsychological scores (P < 0.0025). Abbreviations: PSP-CI, progressive supranuclear palsy with cognitive impairment; PSP-NC, progressive supranuclear palsy without cognitive impairment.