

Abstract
Agonists and pseudo-agonists for Toll-like receptor 4 (TLR4) are common in our environment. Thus, human exposure to these agents may result in “priming or sensitization” of TLR4. A body of evidence suggests that LPS-mediated sensitization of TLR4 can increase the magnitude of responses to exogenous agents in multiple tissues. We have previously shown that reactive oxygen and nitrogen species (RONS) stimulate TLR4. There is no evidence that LPS-primed TLR4 can influence the magnitude of responses to oxidants from either endogenous or exogenous sources. In the present study, we directly tested the hypothesis that LPS-primed TLR4 will sensitize primary murine peritoneal macrophages (pM) to oxidant-mediated prostaglandin E2 (PGE2) production. We used potassium peroxychromate (PPC) and potassium peroxynitrite (PPN) as direct in vitro sources of exogenous RONS. Our results showed that a direct treatment with PPC or PPN alone as sources of exogenous oxidants had a limited effect on PGE2 biosynthesis. In contrast, pM sensitized by prior incubation with LPS-EK, a TLR4-specific agonist, followed by oxidant stimulation exhibited increased transcriptional and translational expression of cyclooxygenase-2 (COX-2) with enhanced PGE2 biosynthesis/production only in pM derived from TLR4-WT mice but not in TLR4-KO mice. Thus, we have shown a critical role for LPS-primed TLR4 in oxidant-induced inflammatory phenotypes that have the potential to initiate, propagate and maintain many human diseases.
Keywords: Toll-like receptor 4, Primary murine macrophages, Oxidative stress, Phospholipase A2, Cyclooxygenases
1. INTRODUCTION
A progressive change in receptor sensitivity with prior exposure to an activating ligand is referred to as “priming” or “sensitization” of the receptor [1]. A potential “priming” of TLR4 by pseudo-TLR4 ligands such as common drugs especially opioids [2], tricyclic antidepressants [3,4] and even ethanol [5,6] appears to be commonplace. Furthermore, human exposure to LPS, a native TLR4 ligand, is commonplace via multiple sources. Exposure may be through different sources such as bacterial infection, microbiome translocation of gut microflora, gut injury, dietary alteration and in a variety of occupational and environmental settings [7].
LPS is a complex glycolipid in the outer membrane of Gram-negative and select Gram-positive bacteria and plays a critical role in activating the innate immunity [8,9,10], which includes initiation of macrophage responses that may involve secretion of cytokines, nitric oxide and eicosanoids (prostaglandins). LPS is also a natural ligand for TLR4/MD-2/CD14 receptor complex in many cells especially dendritic, monocytes, macrophages and B-cells [11]. Besides, injured tissue such as dying tumor cells can persistently release endogenous danger signals collectively termed damage associated molecular patterns (DAMPs) such as certain heat shock protein (HSPs), high mobility group box 1(HMGB1) protein that serve as endogenous and exogenous ligands for TLR4 as well [12]. In addition, TLR4 interacts with molecules, e.g. LPS, released by bacteria, which are collectively called pathogen associated molecular patterns (PAMPs). Because TLR4/CD14/MD-2 receptor complex shares a common signaling cascade with IL-1 receptor, we hypothesize that prior exposure to LPS might sensitize prostaglandin-synthesizing enzyme to oxidants in primary murine peritoneal macrophages (pM). LPS can induce the release of prostaglandins in macrophages [13].
Prostaglandin E2 (PGE2), a prostanoid synthesized from arachidonic acid (AA), can increase vascular permeability, induce fever, play a role in muscle regeneration and maintain hyperalgesic responses [14]. Because of multiple important physiological roles of PGE2, many key patents describing inventions for utilization of PGE2 for either diagnostic or therapeutic purposes have been filed [15; also US Pat No. 3,691,216 for PGE2 methyl ester and PGE2 methyl ester diacetate, filed 1972; US Pat No 3,795,697, filed 1974). PGE2 is not stored but is synthesized de novo from membrane-released AA when cells are activated by stimuli such as mechanical trauma, cytokines, and growth factors. AA metabolites play critical roles in initiating and/or terminating inflammatory processes [16,17].
A family member of phospholipase A 2 (PLA2) enzymes initiates PGE2 synthesis. The PLA2 enzyme family catalyzes the hydrolysis of membrane phospholipids at the sn-2 position to liberate AA (a 20-carbon unsaturated fatty acid) to initiate PGE2 synthesis. Both cytosolic PLA2 (cPLA2) and secretory group V PLA2 (sPLA2-V) are involved in regulating AA mobilization in response to macrophage exposure to TLR4 activation [18]. The AA released in the membrane is rapidly oxidized into the unstable metabolite, prostaglandin G2 (PGG2), which is subsequently presented to PGH2 by the cyclooxygenase (COX) enzymes. COX-1, expressed constitutively in most cells, is the dominant source of prostanoids that serve housekeeping functions, whereas COX-2 is the more important source of prostanoids formed in inflammation [19]. The final step in the biosynthesis of PGE2 is catalyzed by prostaglandin E synthases (PGESs), a family of oxido-reductases, which has generated increasing interest as a therapeutic target in the treatment of inflammatory-related diseases. mPGES-1 responds to inflammatory stimuli and is frequently induced concomitantly with COX-2 after stimulation by LPS, TNF-α, or IL-1β [20].
PGE2 then exerts its action locally by binding to one or more of its four cognate receptors, termed EP1-EP4, which are all G-protein-coupled receptors (GPCRs). In the onset of the inflammatory response, PGE2 acts as a vasodilator to facilitate tissue influx of neutrophil of immune cells from the blood stream resulting in swelling and edema at the site of infection or tissue injury [16]. Furthermore, PGE2 can stimulate sensory nerves to increase pain response and act on neurons in the pre-optic area to promote pyrogenic effects [21]. In addition, recent studies underscore that PGE2 exacerbates inflammation by promoting the activation of TH17 cells, a subset of CD4+ helper T cells. PGE2-mediated production of IL-17 can exacerbate the development of multiple inflammatory diseases, such as inflammatory bowel disease (IBD) and collagen-induced arthritis in mice [22,23]. PGE2 plays a key role in inflammation, a common and critical pathologic process with its classical acute symptoms of pain, heat, swelling and loss/gain of function.
An extensive body of evidence suggests that LPS priming of TLR4 can change the magnitude of responses to exogenous agents in the liver, kidney, respiratory tract and lymphoid tissue [7]. Furthermore, reactive oxygen and nitrogen species (RONS) appear to participate in the regulation of TLR4 gene expression [24]. Because prooxidants can regulate TLR4 gene expression [25,26], it is still not understood whether LPS-primed TLR4 can influence the magnitude of responses to oxidants from exogenous sources. Thus, the effect of LPS-primed TLR4 can potentially affect critical events in cells of macrophage lineage. Therefore, we will address two basic questions: i) would prior exposure to LPS sensitize TLR4 to responses to exogenous oxidants? and ii) if so, what mechanism(s) is involved with respect to the transcriptional and/or translational activation of PGE2 biosynthetic enzymes to enhance PGE2 biosynthesis and release? We sensitized pM by prior exposure to LPS-EK (Ultrapure) [a specific TLR4/MD-2/CD-14 receptor complex agonist at 100 ng/ml for 4 h], which we determined empirically as an optimal priming condition in this pM system. In the present study, we directly tested the hypothesis that LPS-mediated priming of TLR4 will sensitize pM to oxidant-induced PGE2 biosynthesis.
2. MATERIALS AND METHODS
2.1 Antibodies
Anti-CD11 b antibody (M1/70), isotype control rat (IgG2b) and anti-mPGES-1 were purchased from Abcam (Cambridge, England CB4 0FL), whereas anti-TLR4 antibody was obtained from Novus Biologicals (Littleton, Colorado, USA). Anti-COX-1 (D2G6) rabbit mAb, anti-COX-2 (D5H5) rabbit mAb and anti-cPLA2 antibody were purchased from Cell Signaling Technology (Cambridge, MA, USA). HRP-conjugated ACTB were purchased from Proteintech Group.
2.2 Oxidants and other chemicals
Potassium peroxychromate (PPC), used in the study as a primary exogenous source of ROS, is not available commercially, but was synthesized in the laboratory according to a previously published protocol [27]. It was characterized by elemental and infrared analyses with a purity of > 98%. PPC has been used as a source of ROS to examine their effects on biochemical and biological functions [28]. PPC decomposes readily in aqueous systems to release several oxygen-centered free radicals including H2O2, hydroxyl radical (•OH), singlet oxygen (1O2) and possibly superoxide anion (O2•−). Potassium peroxynitrite (PPN) (Millipore, Billerica, Mass, USA) was used as a direct donor of peroxynitrite anion (-OONO) under physiological conditions. Linsidomine chloride (SIN-1) was obtained from AdipoGen (San Diego, CA, USA) and produces cell permeable peroxynitrite anions that can react with lipids, DNA and proteins by direct oxidative reaction or by indirect radical-induced mechanisms [29]. The intracellular total antioxidant capacity assay kit and the ELISA kit for PGE2 were purchased from Cayman Chemical (Ann Arbor, MI, USA). The ELISA kit for mouse-specific TNF-α was purchased from BioLegend (San Diego, CA, USA). LPS-EK from E. coli K12 (LPS-EK Ultrapure) was obtained from InvivoGen (San Diego, CA, USA). TRI Reagent for RNA extraction was obtained from Molecular Research Center (Cincinnati, OH, USA). High-capacity cDNA reverse transcription assay kit and Restore Western Blot Stripping Buffer were obtained from ThermoFisher Scientific (Grand Island, NY, USA). Thioglycollate brewer powder was purchased from BD Biosciences (San Jose, CA).
2.3 Isolation and characterization of thioglycollate-elicited peritoneal macrophages (pM)
We isolated primary pM from mouse strain B6.B10ScN-Tlr4 lps-del/JthJ (Jackson Labs) with complete deletion of TLR4 gene (knockout)[TLR4-KO] [30] with a corresponding wild-type (TLR4-WT) control strain C57BL6. These mice were cared for and maintained as approved by UMKC-IACUC in accordance with NIH guidelines. We isolated and characterized primary pM from both strains according to a standard published method [31]. pM were plated in tissue culture plates for 1 h, and media containing floating non-adherent cells were removed and replaced with DMEM/12 medium supplemented with 10 % (v/v) FBS, 50 units/ml penicillin and 50 μg/ml streptomycin for 24 h before they were used in subsequent experiments. Incubation with DMEM/12 medium for 24 h further enriched the purity of pM [32].
2.4 Sensitization and treatment of pM
pM were sensitized by incubation with 100 ng/ml LPS-EK for 4 h in culture medium supplemented with 10% heat-inactivated (HI) FBS, rinsed once thoroughly with culture medium supplemented with 1% HI FBS, 50 units/ml penicillin and 50 ug/ml streptomycin. Finally, cells were incubated overnight with fresh medium supplemented with 1% HI FBS without oxidants (control) or with PPC or PPN.
2.5 RNA extraction and reverse transcription
pM were collected in TRI Reagent. Total RNA was extracted from cells using TRI Reagent according to the manufacturer’s instruction. Total RNA was quantified in a Nanodrop spectrophotometer (ThermoFisher Scientific). An aliquot of RNA sample (2 μl) with a ratio of A260/280 nm absorbance of 1.8 to 2.0 was reverse transcribed in a 20 μl reaction volume using a High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific)
2.5 Quantitative polymerase chain reaction (qPCR)
Quantitative-polymerase chain reaction (RT-qPCR) was performed using Step-One TM Real-Time PCR System (Applied Biosystem, Foster City, CA) via standard fluorescent methodology and thermal cycling conditions following the manufacture’s recommendations, including validation of each gene amplification tested by identification of single peaks in the melting curves. The qPCR reaction mixture contained 1 μl cDNA, 10 μl of the Real-time Bullseyes EvaGreen qPCR Mastermix-ROX (MIDSCI), 0.4 μl of primer pairs (10 μM) and 8.6 μl H2O in a complete reaction volume of 20 μl. CT values were normalized to β-Actin or GAPDH as a reference gene. Gene expression was determined as up/down regulation of the gene of interest compared to the control. We followed the guidelines for the minimum information for publication of quantitative real time PCR experiments (MIQE) [33,34], which included uniformity in cell treatment with incubation conditions, RNA extraction protocols, and its essential purity across samples, quantification, storage and uniformity of the # of cycles with linearity of samples.
2.7 Preparation of whole-cell extract
pM cultured in 6-well plates were washed with ice-cold PBS and lysed in whole cell extraction lysis buffer PE LB™ for Mammalian Cells (G-Biosciences, St. Louis, MO) supplemented with appropriate protease inhibitors ProteaseArrest™ (G-Biosciences, St. Louis, MO). Cell lysates were centrifuged at 12,000 × g at 4°C for 20 min to remove cell debris. Protein assay was performed using BCA Protein Assay kit (Thermo Scientific, Waltham, MA) according to the manufacturer’s instructions. Protein samples were diluted at 1:20 with the extraction buffer, and 5 μl of the dilution was added into a 96-well plate, and incubated with 200 μl working solution for 30 min at 37°C. Total protein was quantified by absorbance reading at 562 nm.
2.8 Immunoblot assay
Aliquots of whole cell extracts containing 20-30 μg of total protein were mixed with 5 X SDS loading buffer in a total volume of 20 μl and denatured at 100 °C for 10 min. Equal amount (30 μg) of denatured total protein was loaded per lane, fractionated on a 4-12% Bis-Tris electrophoresis gel, and transferred onto PVDF membranes. After blocking with 5% non-fat milk in TBST (Tris-buffered saline, 0.1% Tween 20), the membranes were probed overnight at 4 °C with primary antibody (Ab). After three washes in TBS-T, the membranes were incubated for 2 h with secondary Ab. Finally, the membranes were developed using Super Signal West Femto Chemiluminescent Substrate kit, and signals were visualized with the Fujifilm LAS-400 imaging system.
To determine the expression of more than one protein on the same membrane, Restore Western Blot Stripping Buffer was used for re-blotting. Fifteen (15) ml of stripping buffer was added to the membrane, and incubated for 15 min with shaking at room temperature. After removal of the stripping buffer, the membrane was washed 3 times for 5 min each in 0.5 % TBS Tween-20, followed by blocking, and incubation with primary Ab and secondary Ab. The effectiveness of initial stripping was tested before blocking.
2.9 Immunocytochemistry
Isolated primary pM were seeded in 8-well chamber slide pretreated with CC2™ (Nunc™ Lab-Tek™ II, Thermo Scientific) to facilitate overnight attachment. Next day, the culture medium was removed and cells were fixed in 10% buffered formalin phosphate for 15 min at room temperature followed by two washes with ice cold PBS. Cells were then incubated with 1% BSA in PBS with 0.1% Tween 20 (PBST) for 30 min at room temperature followed by incubation with primary anti-CD11b Ab or anti-isotype Ab in 1% BSA in PBST for 1 h at room temperature. After three wishes with PBS, cells were incubated with 2nd Ab conjugated with fluorescein isothiocyanate (FITC) in 1% BSA and NucBlue® live cell stain reagent for 1 h at room temperature in dark. After three rinses with PBS, images were acquired using a fluorescence microscope (Axiovert 200M; Zeiss) at excitation and emission wavelengths of ~490/555 nm for FITC and 405/410-550 nm for NucBlue®.
2.10 Flow cytometry
Isolated primary pM were seeded overnight in 6-well tissue culture plates. Next day, cells were harvested by scraping and resuspended approximately at 1.0 × 107 cells/ml in ice cold PBS supplemented with 10% FBS 1 % sodium azide (NaN3). Two tubes of cells suspension (100 μl for each tube) was incubated with primary anti-CD11b Ab and anti-isotype Ab (used as negative control), respectively, in 3% BSA in PBS for 1 h at room temperature in the dark. After three washes with PBS, cells were suspended in ice cold PBS followed by incubation with 2nd Ab conjugated with FITC for 30 min at room temperature in the dark. Cells were then washed three times and resuspended in ice cold PBS supplemented with 3% BSA and 1% of sodium azide.
The acquisition of the flow cytometric data and analyses was conducted with FACSCanto II™flow cytometer (BD Biosciences, San Jose, CA, USA). The fluorescence intensity was determined using FITC filter at excitation and emission wavelengths of 490/555 nm. Cells probed with anti-isotype Ab were used as negative controls for CD11b expression. For each parameter investigated, at least 104 events (cells) were analyzed per sample. The fluorescence intensities as logarithmically amplified data were compared between different treatments.
2.11 Measurement of intracellular total antioxidant capacity (iTAOC)
Cells (5×105/well) were plated in 6-well plates and grown overnight before use. After simulation with oxidants or LPS-EK for 2 h, cells were lysed for the analysis of iTAOC using the antioxidant assay kit as per manufacturer’s instructions. In this assay, we used PPC and SIN-1 to generate ROS and RSN, respectively, instead of PPC and PPN, which we used in all other experiments in the present manuascipt. Our rationale was to maximize the extended RNS-generating capacity of SIN-1 compared with PPN.
Briefly, the assay relies on the ability of antioxidants in the sample to inhibit the formation of oxidized ABTS® +[2,2-Azino-di- (3-ethylbenzthiazoline) sulphonate] by met-myoglobin. The amount of ABTS® +produced was monitored by reading the absorbance at 405 nm with a Bio-Tek microplate reader (Burlington, VT, USA). The capacity of the antioxidants in cell lysate was calculated from Trolox (a water-soluble tocopherol analogue) standard curves.
2.12 Quantification of PGE2 levels
pM cells were plated in 12-well plates and cultured overnight. Cells were primed or not for 4 h as described. The culture media was removed and cells thoroughly rinsed with media, which was discarded. Cells were then treated with or without oxidants, and incubated for 16 h. Aliquots of media were collected in microfuge tubes containing 10-μg/ml indomethacin to prevent oxidation of PGE2 that can occur in vitro. Samples were stored at −80 ° C until analyzed. The levels of PGE2 in the cell culture medium were quantified using an ELISA kit according to manufacturer’s instructions.
2.13 Statistical Analysis
We used the IBM Statistical Package for the Social Sciences (SPSS) version 22 to perform data analysis in all experiments. Data are presented as the mean ± SEM from at least 3 to 6 independent experiments carried out in duplicates or triplicates, where applicable, and analyzed by 1-or 2-way analysis of variance (ANONA) followed by Tukey’s post hoc tests with p ≤ 0.05 considered as significant.
3. RESULTS
3.1 Characterization of primary peritoneal macrophages (pM)
First, we confirmed the genotypes of TLR4-WT and TLR4-KO mice using PCR from genomic DNA extracted from mouse-tail by a standard method (data not shown). We determined the purity of macrophages by measuring the expression of CD11b, generally considered as a cellular surface marker unique to cells of macrophage lineage. To ensure that the observed staining is due to the Ab binding to the desired antigen but not to some general unspecific binding of immunoglobulin to cells, we used anti-isotype Ab as negative control. CD11b expression was visualized using immuno-fluorescent microscopy as shown in Fig. 1A. No fluorescent signal was observed in cells stained with the anti-isotype Ab while almost all of the cells are fluorescent when they were stained with anti-CD11b mAb. CD11b-positive cells were further quantified by flow cytometry. Our results showed that a forward shift in the peak in cells stained with CD11b mAb compared to cells stained with anti-isotype thus confirming the specificity of the mAb. In addition, above 95% of peritoneal exudate cells (PECs) isolated from mice are CD11b positive (Fig. 1B), which is consistent with previous studies [31].
Figure 1. Confirmation of the purity of peritoneal macrophages (pM) isolated from TLR4-WT and TLR4-KO mice.
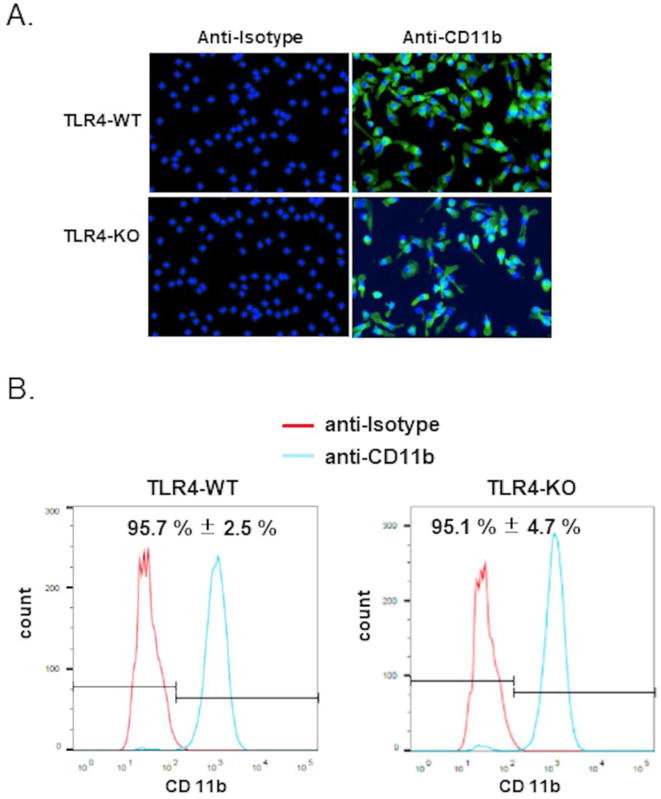
PECs were stained with anti-isotype antibody (Ab) or anti-CD11b monoclonal Ab. (A) Representative immuno-fluorescent image of three independent experiments are shown. Cells were counterstained with DAPI (blue) to identify the cell nuclei. (B) Quantification of CD11b+ cells using flow cytometry. Representative flow cytometry figures of three independent experiments are shown. The data represent mean ± SEM, n = 3.
3.2 Confirmation of TLR4 expression in the pM
We confirmed the expression of TLR4 mRNA and protein in the pM. As Fig. 2A shows, TLR4 cDNA was readily amplified from RNA derived from WT (Lanes 2 and 3) while it was not amplified from RNA derived from TLR4-KO mice (Lanes 4 and 5). β-actin was used as the housekeeping gene (Lanes 6–9). The RT-PCR results confirmed that pM derived from TLR4-KO mice failed to express TLR4 mRNA compared with pM derived from WT mice. The data were consistent with our mouse genotyping results.
Figure 2. Confirmation of the expression and deletion of TLR4 in peritoneal macrophages (pM) isolated from TLR4-WT and TLR4-KO mice, respectively.

(A) TLR4 mRNA expression in pM by RT-PCR analysis. RT-PCR was performed using specific primers for mice TLR4 and β-actin on the total RNA of pM. Lane 1: 100-bp DNA ladder; Lane 2 & 3: TLR4 mRNA from pM derived from TLR4-WT mice with corresponding β-actin mRNA (Lanes 6 & 7) against Lanes 4 & 5: TLR4 mRNA from pM derived from TLR4-KO mice with their corresponding β-actin mRNA (Lanes 8 & 9). (B) TLR4 protein expression by Western blot analysis. (C) TNF-α production by LPS-EK stimulation. pM were treated with LPS-EK (10 ng/ml) for 16 h and levels of TNF-α in culture supernatant was quantified by ELISA. The data represent mean ± SEM from three independent experiments conducted in duplicate, * p ≤ 0.001 compared with untreated control
Then, we assessed TLR4 protein expression in cell lysates of pM derived from TLR4-WT and TLR4-KO mice by Western blot analysis. As shown in Fig. 2B, one robust band corresponding to the size of full length TLR4 95 kDa in pM derived from WT mice was observed whereas this band was almost undetectable in cell lysate of pM derived from TLR4-KO mice. Having confirmed TLR4 expression in pM at both mRNA and protein levels, we next characterized the primary pM with respect to response to TLR4 agonist stimulation.
pM isolated from TLR4-WT and TLR4-KO mice were stimulated with TLR4 specific agonist LPS-EK at 10 ng/ml. Cell culture supernatant was subjected to TNF-α analysis using ELISA. TNF-α was significantly induced upon LPS-EK stimulation in pM derived from TLR4-WT mice but not in those derived from TLR4-KO mice (Fig. 2C). These results confirmed two crucial points: (i) pM derived from TLR4-WT mice responded to the stimulation of TLR4 agonist while pM derived from TLR4-KO mice were completely irresponsive to stimulation of LPS-EK, and (ii) LPS-EK specifically stimulates TLR4 activation.
3.3 Stimulation of TLR4 decreased intracellular total antioxidant capacity (iTAOC)
We confirmed the redox active responses of ROS and RNS from PPC and SIN-1, respectively. The assay choice is based on the rationale that antioxidants would commonly neutralize radicals via a hydrogen atom transfer or single electron transfer mechanism. We treated pM derived from TLR4-WT and TLR4-KO with different concentrations of PPC and SIN-1, and with LPS-EK as positive control. As a burden of ROS or RNS production is largely counteracted by an intricate antioxidant defense system [35], we quantified iTAOC as a primary indicator of response to oxidants stress. Hydrophilic and lipophilic samples are compatible with the assay.
Treatment of pM with PPC (2.5 and 5 μM) for 2 h specifically induced a concentration-dependent decrease in cellular iTAOC in pM expressing TLR4 with decrease of 31 and 44 % compared with control, respectively. Similarly, SIN-1 (0.5 and 1mM) treatment for 2 h decreased iTAOC by 48 and 58 %, respectively, in pM expressing TLR4. However, both PPC and SIN-1 had no effect on iTAOC in comparison with control cells derived from TLR4-KO mice (Fig. 3B). Thus, deletion of TLR4 expression appears to protect cells from oxidant stress. LPS-EK (10 ng/ml) decreased iTAOC by 30% compared with control cells. These results further affirmed that oxidants induced intra-cellular oxidant stress. Consistently, pM derived from TLR4-KO mice are resistant to RONS-mediated increase in iROS levels [36] and decrease in TAOC levels compared with the pM derived from TLR4-WT.
Figure 3. Effects of oxidants on levels of intracellular total antioxidant capacity (iTAOC) following stimulation of pM.
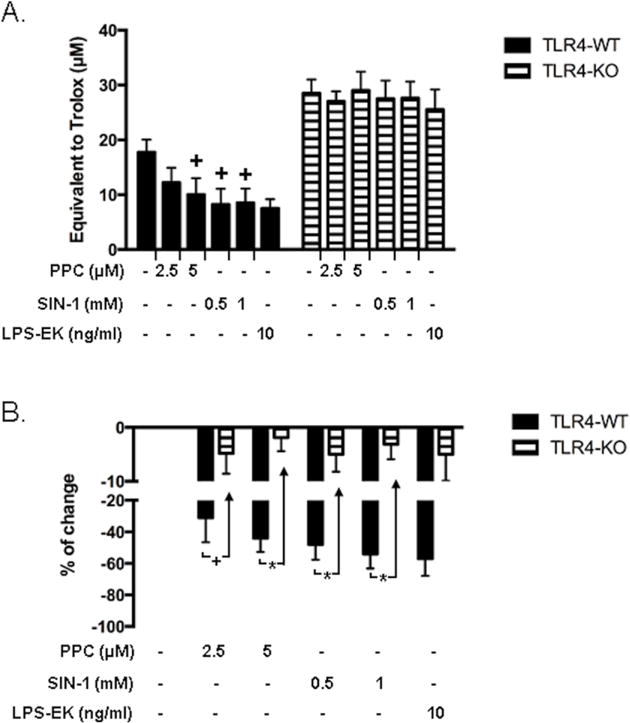
To confirm the redox active properties of ROS from PPC and RNS from SIN-1, respectively, cell lysates were prepared and tested for intracellular total antioxidants capacity. (A) Intracellular total antioxidant capacity (iTAOC) was quantified as μM Trolox equivalents (TE). (B) % Change of control was calculated as [(TEtreatment –TEcontrol) ⁄ TEcontrol ×100%] to represent the effects of oxidants or LPS-EK on iTAOC over control untreated cells. The data represent five (5) independent experiments carried out in triplicate. +p ≤ 0.05 versus untreated pM from TLR4-WT control (0%); *p ≤ 0.001 versus pM from TLR4-KO
3.4 TLR4 priming mediates oxidant-induced PGE2 production
PPC and PPN, which have been proven to release several oxygen-centered free radicals and peroxynitrite, respectively, were used as exogenous sources of reactive oxygen/nitrogen species (RONS). As Fig. 4 shows, PPC (5 μM) or PPN (100 μM) alone induced a limited increase in PGE2 production in pM derived from either TLR4-WT or TLR4-KO mice. pM expressing TLR4 exposed to LPS-EK 100 ng/ml for 4 h and then to culture medium (vehicle) for 16 h exhibited moderately enhanced PGE2 production up to 503.1 ± 86.0 pg/ml compared to un-sensitized pM with PGE2 levels of 126.6 ± 34.2 pg/ml (Fig. 4), which is about a 4-fold increase. In contrast, pM expressing TLR4 primed with LPS-EK (100 ng/ml) for 4 h and then treated with PPC or PPN for 16 h showed a robust PGE2 synthesis. PGE2 was increased up to 897.5 ± 120 pg/ml and 1416.3 ± 140 pg/ml by PPC and PPN, respectively. However, the pM that lacked TLR4 expression was not responsive to LPS-EK sensitization/priming. Treatment with oxidants did not affect PGE2 production after LPS-EK sensitization in pM isolated form TLR4-KO mice. These results demonstrated that TLR4 priming sensitized pM to oxidant-induced PGE2 production.
Figure 4. Effects of oxidants on PGE2 production after prior incubation with LPS-EK.
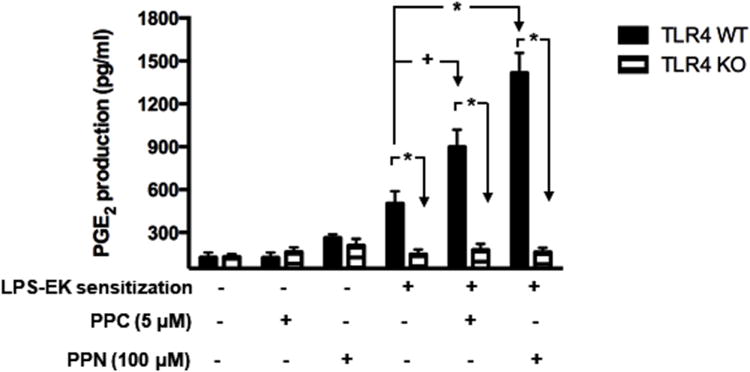
pM derived from TLR4-WT and TLR4-KO mice were sensitized by incubating for 4 h with media containing 100 ng/ml LPS-EK, which was removed and cells rinsed once with fresh medium. The cells were incubated for 16 h with fresh medium or medium containing PPC or PPN. We used ELISA kit to quantify PGE2 present in aliquots of the culture media. * p ≤ 0.01, + p ≤ 0.05, n = 3-6.
3.5 TLR4 priming mediates oxidant-induced PLA2 mRNA expression
We examined the potential mechanism(s) by which TLR4 priming with its native ligand might sensitize pM to oxidant-induced PGE2 biosynthesis. We examined the transcriptional and translational effects of treatments on the enzymes involved in PGE2 biosynthesis. Liberation of AA from membrane glycerophospholipids by PLA2 is an initial step in PGE2 production. Expression of PLA2 is essential for PGE2 production. It has been reported that sPLA2 type II gene is naturally defective in C57BL/6 mice [37]. In addition, cPLA2 and sPLA2 type V have been reported to be up-regulated following TLR4 activation in pM [38]. Therefore, we examined the gene expression of cPLA2 and sPLA2 following treatment with oxidants in LPS-EK-sensitized pM.
Consistent with PGE2 production, Fig. 5A showed that: (i) PPC or PPN treatment alone had limited effects on the level of cPLA2 mRNA in pM derived from either TLR4-WT or TLR4-KO mice; (ii) cells sensitized with LPS-EK following medium incubation showed a 2.7-fold increase in cPLA2 mRNA levels compared with un-sensitized pM expressing TLR4; (iii) cells sensitized with LPS-EK followed by 16 h incubation with PPC or PPN did not show further increase in cPLA2 mRNA levels in comparison with those from primed pM following incubation in the medium alone; and finally, (iv) the induction of cPLA2 was only observed in pM that express TLR4, unlike pM with a complete deletion of TLR4, which showed no demonstrable response to LPS-EK sensitization following priming.
Figure 5. Effects of oxidants on mRNA expression for cPLA2, sPLA2 (Type V), COX-2, COX-1, mPGES-1 and mPGES-2 following priming with LPS-EK.
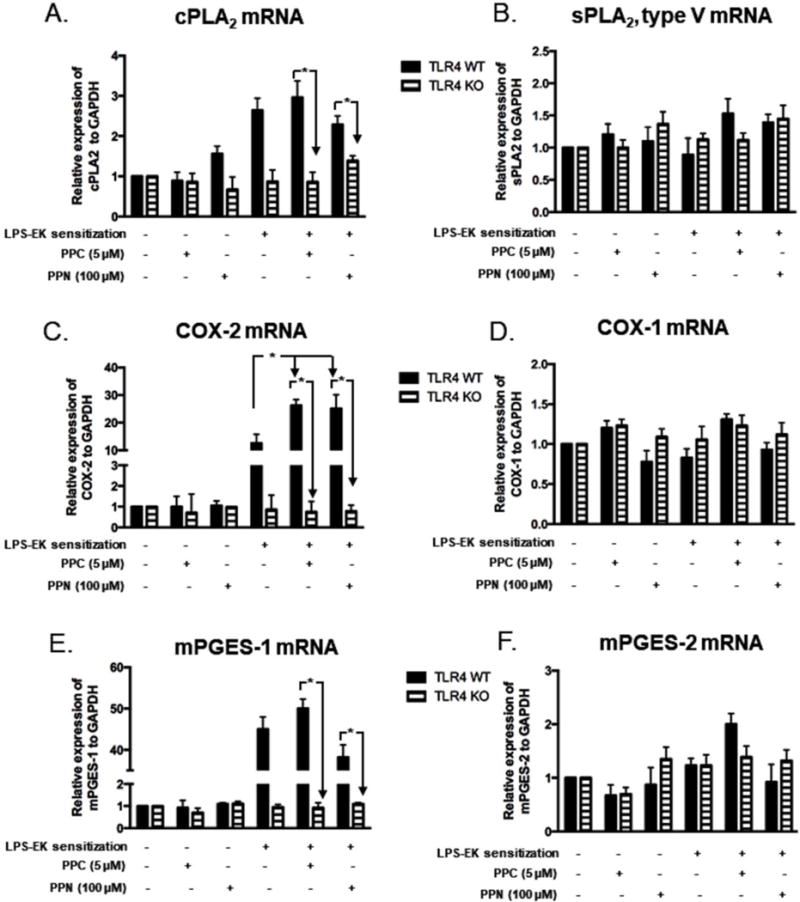
pM were sensitized by incubating for 4 h in media containing 100 ng/ml LPS-EK, cells were rinsed once in fresh media, and then incubated for 6 h with fresh medium or media containing PPC or PPN. Cellular mRNA levels were analyzed and quantified by RT-quantitative PCR analyses. * p ≤ 0.01, + p ≤ 0.05, n = 3-5.
Compared with the mRNA of cPLA2, the mRNA of sPLA2 type V is not inducible following LPS-EK sensitization. As Fig. 5B shows, PPC or PPN treatment alone had limited effects on the level of sPLA2 type V mRNA in pM derived from either TLR4-WT or TLR4-KO mice. Furthermore, cells sensitized with LPS-EK followed by incubation in the medium alone or oxidant (PPC or PPN) treatment showed no significantly demonstrable effects on the levels of sPLA2 type V mRNA in pM derived from either TLR4-WT or TLR4-KO mice.
3.6 TLR4 priming enhances oxidant-induced COX gene expression
Fig. 5C shows that: (i) PPC or PPN treatment alone had limited effects on the level of COX-2 mRNA in pM derived from either TLR4-WT or TLR4-KO mice; (ii) cells sensitized with LPS-EK following medium incubation showed a 13.7-fold increase of COX-2 mRNA levels compared with un-sensitized pM expressing TLR4; (iii) cells sensitized with LPS-EK followed by stimulation with PPC or PPN for 16 h exhibited a 28.5- and 28.3-fold increases in COX-2 mRNA expression, respectively, which was higher than those in sensitized pM following medium incubation; and (iv) induction of COX-2 was only observed in pM that express TLR4, but not in pM with complete deletion of TLR4 gene. These results again suggest that oxidants stimulate PGE2 production in LPS-EK-sensitized pM through upregulation of COX-2 gene expression.
Compared with COX-2, the mRNA for COX-1 did not respond at all to LPS-EK sensitization. PPC or PPN treatment alone had no effects on the level of COX-1 mRNA in pM derived from either TLR4-WT or TLR4-KO mice (Fig. 5D). Furthermore, the pM sensitized with LPS-EK followed by incubation with medium alone or medium with oxidant (PPC or PPN) treatment showed no effects on the levels of COX-1 mRNA in pM derived from either TLR4-WT or TLR4-KO mice.
3.7 Role of TLR4 priming in mediating oxidant-induced mPGES mRNA expression
We also examined the effect of TLR4 priming on mPGES-1 and mPGES-2 gene expression. mPGES-1 has been shown to respond to inflammatory stimuli and is frequently induced concomitantly with COX-2 after stimulation by LPS, TNF-α, or IL-1β [20]. In contrast, mPGES-2 is constitutively expressed in a most tissues and equally expressed in both the normal and pathological samples.
As shown in Fig. 5E, (i) PPC or PPN treatment alone had limited effects on the level of mPGES-1 mRNA in pM derived from either TLR4-WT or TLR4-KO mice; (ii) cells sensitized with LPS-EK followed by incubation in the media alone showed a 43.7-fold increase in mPGES-1 mRNA expression levels compared with un-sensitized pM from TLR4-WT mice; (iii) cells sensitized with LPS-EK followed by PPC or PPN stimulation for 16 h exhibited an increase in mPGES-1 mRNA expression of 50.5- and 40.1-fold, respectively; (iv) cells sensitized with LPS-EK followed by PPN stimulation for 16 h did not show a further increase in mPGES-1 mRNA expression compared with those in LPS-EK-sensitized pM following incubation in the medium alone; and (v) induction of mPGES-1 gene expression was only observed in pM expressing TLR4, whereas pM with deletion of TLR4 gene showed no response to LPS-EK sensitization.
Compared with mPGES-1, the mRNA for mPGES-2 was not responsive to LPS-EK sensitization. Treatment with oxidants (PPC or PPN) alone had no discernible effects on the level of mPGES-2 mRNA in pM derived from both TLR4-WT and TLR4-KO mice (Fig. 5F). Furthermore, treatment with LPS-EK followed by incubation in the media alone had no effects on the levels of mPGES-2 mRNA in pM derived from either TLR4-WT or TLR4-KO mice. Our results suggest that in spite of increased mPGES-1mRNA expression, the upregulation of mPGES-2 protein expression may not play a role in oxidant-mediated PGE2 production in LPS-sensitized pM.
3.8 LPS-mediated TLR4 priming facilitates oxidant-induced protein expression
We examined the protein expressions of the key enzymes responsible for PGE2 biosynthesis, including cPLA2, COX-2, COX-1, and mPGES-1. Consistent with mRNA expression profile in pM derived from TLR4-WT mice, Fig. 6A and 6B show that: (i) PPC or PPN treatment alone had limited effects on the level of cPLA2 protein in pM; (ii) pM sensitized with LPS-EK followed by incubation in fresh medium alone showed a negligible (1.4-fold) increase in cPLA2 protein expression compared with the un-sensitized pM; and (iii) cells sensitized with LPS-EK followed by incubation with PPC or PPN did not exhibit significantly further increase of cPLA2 protein levels compared with those in sensitized pM following medium incubation. These results suggest that cPLA2 may not contribute to the oxidant-induced PGE2 production in LPS-EK-sensitized pM, although cPLA2 mRNA in this treatment paradigm was significantly increased. This suggests apparent instability of cPLA2 mRNA that was not translated into functional protein.
Figure 6. Western (Immuno) blot analyses for cPLA2, COX-1, COX-2 and mPGES-1 with relative expression ratios to β-actin following prior priming with LPS-EK.
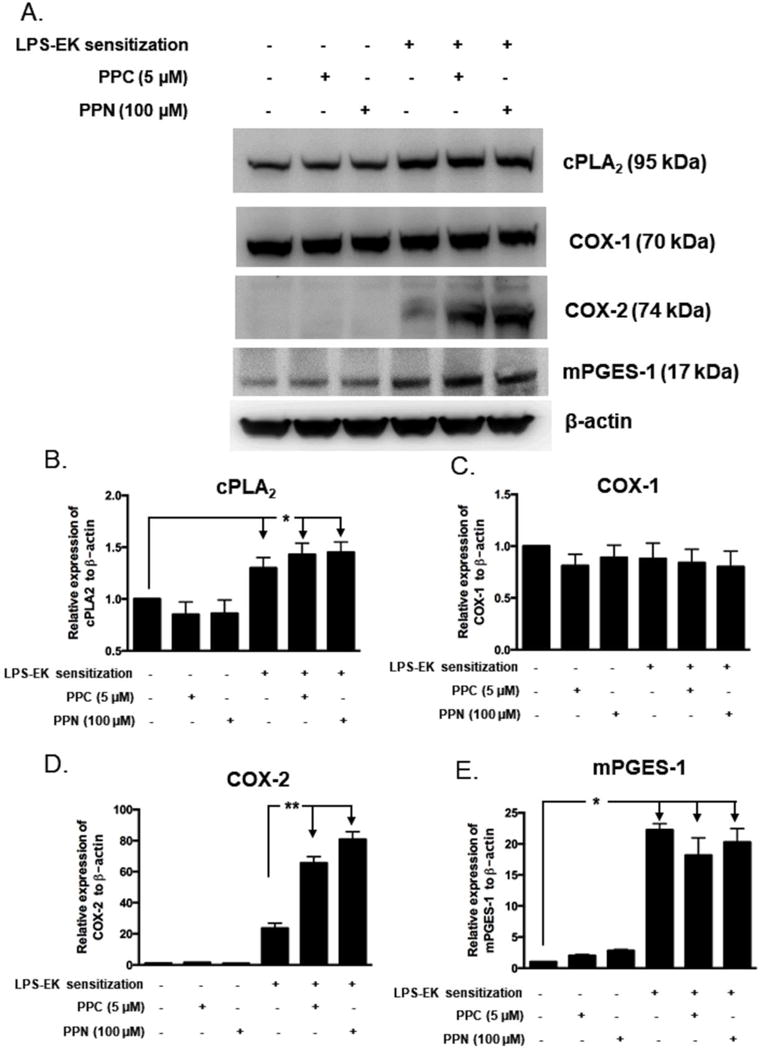
pM derived from TLR4-WT mice were sensitized for 4 h with 100 ng/ml LPS-EK. Initial incubation media containing LPS-EK was removed. Cells were thoroughly rinsed once with fresh media and then incubated for 16 h with either fresh medium alone or media containing either PPC or PPN. Total cellular lysates were prepared and subjected to Western blot analysis. (A) Representative immunoblot results are shown. The histograms (B – E) represent the OD ratio of target immunoblot signal from (A) after normalization to the housekeeping protein β-actin. The experimental data are presented as the mean ± SEM from three independent experiments. * p ≤ 0.05, ** p ≤ 0.01, n = 3.
Fig. 6A and 6D shows that: (i) incubation with PPC or PPN alone did not induce COX-2 protein expression in pM derived from TLR4-WT; (ii) pM cells derived from TLR4-WT mice sensitized with LPS-EK follow by medium incubation for 16 h showed a ~24-fold increase in COX-2 protein expression compared with un-sensitized pM; (iii) Furthermore, pM derived from TLR4-WT sensitized with LPS-EK followed by PPC and PPN stimulation for 16 h exhibited an increased expression of COX-2 protein of ~66- and ~80-fold, respectively, which was remarkably higher than those in sensitized pM from TLR4-WT followed by further medium incubation alone in the absence of oxidants. Our results demonstrated that oxidants stimulate PGE2 production in LPS-EK-sensitized pM from TLR4-WT by a very robust upregulation of COX-2 gene expression.
Compared with COX-2, the expression of COX-1 did not respond to LPS-EK sensitization. As Fig. 6A and 6C shows, PPC or PPN treatment alone had no effects on the levels of COX-1 protein in pM derived from TLR4-WT mice. Additionally, pM sensitized with LPS-EK followed by medium incubation or oxidant (PPC or PPN) treatment had no effects on the levels of COX-1 protein in pM. These results suggest that COX-1 may not contribute to oxidant-induced PGE2 production in LPS-EK sensitized pM.
We finally examined the protein expression of mPGES-1. As shown in Fig. 6A and 6E, (i) PPC or PPN treatment alone failed to induce mPGES-1 protein expression in pM derived from TLR4-WT; (ii) pM sensitized with LPS-EK followed by medium incubation showed a 22.7-fold increase in mPGES-1 protein expression levels compared with un-sensitized pM from TLR4-WT mice; and (iii) pM sensitized with LPS-EK followed by PPC and PPN stimulation for 16 h did not exhibit further increase of mPGES-1 protein expression different from LPS-EK stimulation suggesting that mPGES-1 may not be a major contributor to oxidant-induced PGE2 production through TLR4-stimulation.
Incidentally, pM derived from TLR4-KO mice showed no response whatsoever to either oxidant treatment alone or LPS-EK sensitization with respect to protein expression of cPLA2, COX-1, COX-2, or mPGES-1 (Fig. 7A, 7B & 7C)).
Figure 7. Western (Immuno) blot analyses for cPLA2 and COX-1 with relative expression ratios to β-actin following prior priming with LPS-EK.
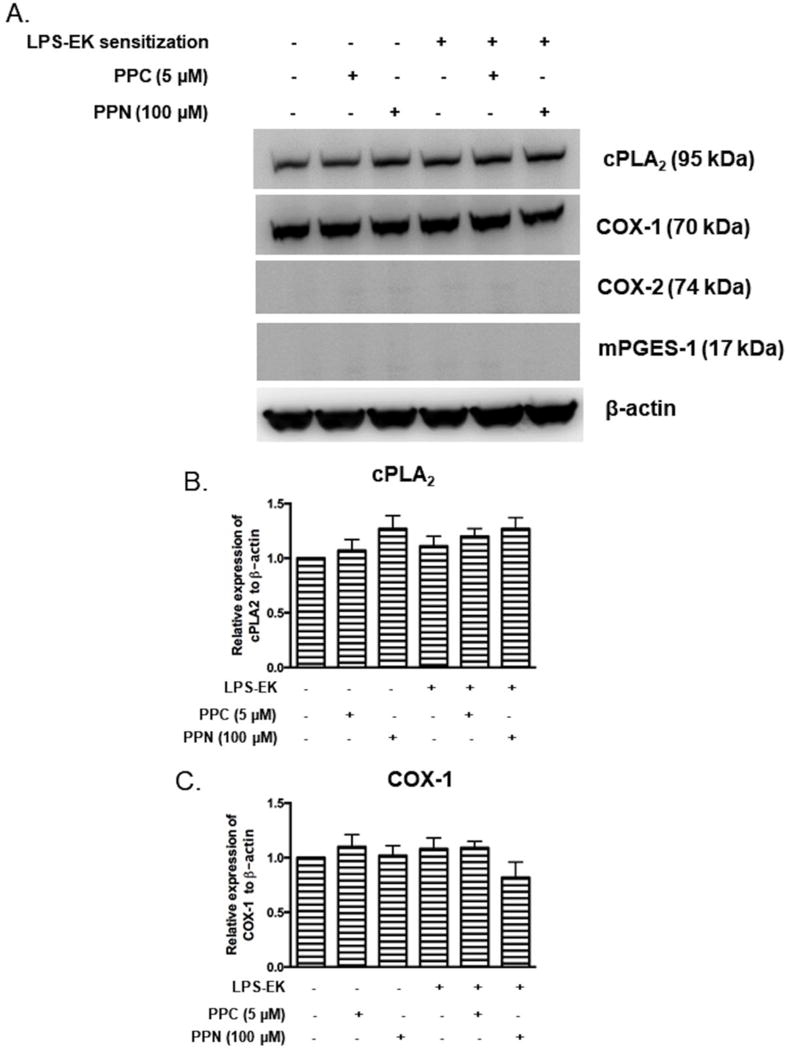
pM derived from TLR4-KO mice were sensitized for 4 h with 100 ng/ml LPS-EK, then incubated for 16 h in fresh medium or media containing PPC or PPN. Total cellular lysates were subjected to Western blot analysis. (A) Representative immunoblot results are shown. (B and C) The histograms represent the OD ratio of target immunoblot signal from (A) after normalization to housekeeping protein β-actin. The experimental data are presented as the mean ± SEM from three independent experiments.
4. DISCUSSION
We have shown that priming of TLR4 by pre-incubation with its native ligand LPS-EK would affect the magnitude of responses to oxidants in primary macrophages. For the first time we have shown that: (i) PPC or PPN treatment alone as RONS generators had limited effects on PGE2 production and gene expression of enzymes responsible for PGE2 production which includes PLA2, COX, and mPGES in pM derived from both TLR4-WT and TLR4-KO mice; (ii) pM sensitized with LPS-EK, followed by rinsing with fresh media and re-incubation in fresh media showed a moderate increase in PGE2 production and gene expression for cPLA2, COX-2 and mPGES-1 in pM derived from TLR4-WT, but not pM from TLR4-KO mice; (iii) pM sensitized with LPS-EK followed by oxidant stimulation exhibited a significantly higher increase in PGE2 production and expression of COX-2 protein only in pM derived from TLR4-WT mice.
Activation of the innate immune system might potentially predispose a host to toxicant-induced tissue injury. The in vitro results presented here have demonstrated that oxidants can activate the gene expression of COX-2 resulting in enhanced production of PGE2 in pM derived from TLR4-WT mice sensitized with LPS. Our findings confirmed in vivo studies, which revealed that LPS pre-treatment sensitized animal to xenobiotic chemical-induced cytokines storm [39]. Our results indicate that episodic exposure to LPS and other TLR4 agonists might induce a state of enhanced sensitivity to oxidants in a host, ultimately leading to proinflammatory responses or heightened proinflammatory phenotype that is primed for disease initiation, propagation and maintenance.
We have shown that macrophages sensitized by pretreatment with LPS-EK exhibited enhanced responsiveness to oxidants with respect to PGE2 production (Fig. 4). Our reports are in accord with other reports. The oxidant hydrogen peroxide (H2O2) facilitated the production of COX-2 and PGE2 only in LPS-stimulated human primary monocytes, whereas H2O2 alone did not activate monocytes [40]. These findings demonstrate that the state of cellular activation may be critical in determining the cellular responses to oxidants, and thus initiation and propagation of inflammatory phenotype.
We further examined the potential mechanism(s) by which oxidant-induced PGE2 production in the pM sensitized by prior exposure to LPS-EK. The three key enzymes that mediate PGE2 production are PLA2, COX-2 and mPGES [14]. Our results indicate that the gene expression of cPLA2, COX-2 and mPGES-1 were significantly induced following LPS-EK treatment, thereby confirming that the genes for these enzymes are inducible by TLR4 activation. In contrast, the gene expression for sPLA2, COX-1 and mPGES-2 were not affected in the presence of TLR4 gene upon LPS-EK treatment. This confirmed that the genes for these PGE2-producing enzymes are less stable due to inflammatory stimulus alone. The upregulation of mPGES-2 mRNA induced by PPC treatment may be due to the instability of its mRNA caused by increased intracellular ROS. Under stress conditions including oxidative stress, mRNA stability changes precede changes in steady-state mRNA amounts [41].
It has been documented that increased expression of cPLA2 is associated with a robust increase in cPLA2 activities in the cytosolic fraction [42]. Epithelial cells with higher expression of cPLA2 than sPLA2 are more susceptible to oxidative stress, which suggests that cPLA2 is an important endogenous mediator of oxidant-induced cell injury [42]. Our studies showed that cPLA2 expression was significantly upregulated by LPS-EK sensitization. This would theoretically result in increased availability of cytosolic AA released from the membrane phospholipids for PGE2 production. This also means that increased cPLA2 activity by LPS-EK sensitization might provide more substrates for subsequent enzymes such as COX-2, which is significantly increased by oxidants (Figs. 5 and 6). In the present study, we did not detect a further significant increase in cPLA2 expression by oxidants in sensitized pM that may result from increased intracellular Ca2+ due to oxidant-mediated mitochondria dysfunction. This may be necessary, but not sufficient for inducing of cPLA2 [43]. Thus, more empirical data will be necessary to confirm whether oxidants can change cPLA2 enzymatic activity.
RONS from PPC and PPN markedly up-regulated COX-2 expression at both the mRNA and protein levels in pM sensitized with LPS-EK, but had no effects on COX-1 gene expression (Figs. 2 and 3). These results demonstrated that the induction of COX-2 in pM may be responsible for oxidative stress-induced PGE2 production as it only occurred in pM that express TLR4. The dramatic increase in COX-2 expression upon stimulation of inflammatory cells and inflamed tissues, and the assumption that the side effects of COX-1 inhibition provided the rationale for developing a selective COX-2 inhibitor for treating arthritis and other chronic inflammatory diseases [44]. Indeed, induction of COX enzymes by oxidants appears to be cell- or tissue-dependent or specific. For example, murine placenta, which experience oxidative stress during gestation, exhibited higher expression of COX-1 and COX-2 [45].
mPGES-1 is a perinuclear protein that is markedly induced by cytokines and growth factors as in the case of COX-2 [43]. In addition, mPGES-1 is functionally coupled to COX-2 in marked preference to COX-1 resulting in PGE2 production [46]. Induction of COX-2 and mPGES-1 by pro-inflammatory factors and their cooperation in converting AA to PGE2 in vitro suggests that both enzymes are important for PGE2 biosynthesis and that inhibition of either is sufficient to inhibit PGE2 production [47]. mPGES-1 and oxidative stress are associated in inflammatory disorders. Reduced atherogenesis was concomitant with a reduction in oxidative stress in mice lacking mPGES-1 conditionally in myeloid cells [48]. Additionally, mPGES-1 deletion suppressed oxidative stress and formation of angiotensin II-induced abdominal aortic aneurysm, which is an inflammatory disorder characterized by localized connective tissue degradation and smooth muscle cell apoptosis [49]. These studies present mPGES-1 as a potential drug target for treating multiple inflammatory conditions. In accord with studies [48,49], our data show that mPGES-1 is robustly induced upon TLR4-activation. However, studies on the effects of oxidants on both mPGES-1 and mPGES-2 remain limited or largely nonexistent.
In the present study, however, treatment with oxidants failed to further increase the mRNA and protein of mPGES-1 at 6 h and 16 h, respectively, in sensitized pM derived from both TLR4-WT and TLR4-KO mice. mPGES-1 protein expression is induced by LPS as early as 6 h and persisted to 48 h in microglia [50], and syntheses of mRNA and protein are dynamic processes. To further confirm the effects of oxidant on mPGES-1, expression (at the mRNA and protein levels) and enzymatic activities of mPGES-1 would need be examined at different time points in future experiments. Compared with mPGES-1, mPGES-2 is constitutively expressed in various cells and tissues. It is functionally coupled to both COX-1 and COX-2 [51]. Mice with mPGES-2 deficiency show no specific phenotype and no alteration in PGE2 levels in several tissues or in LPS-stimulated macrophages [52]. Our results show that LPS-EK did not stimulate mPGES-2 expression in pM, which is consistent with these observations.
Among the three inducible enzymes (cPLA2, COX-2 and mPGES-1), COX-2 is the only enzyme whose expression was further increased by oxidants in pM of TLR4-WT mice. This further confirmed that COX-2 remains a rate-limiting enzyme in PGE2 production [14] in spite of oxidants. Our data suggest that oxidant-mediated upregulation of COX-2 is sufficient to result in PGE2 production in sensitized pM.
The underlying mechanism (s) by which TLR4 agonists sensitize macrophages to oxidant are still not well understood. It has been proposed that TLR4 activation serve as an initial danger signal, thereby reprogramming the macrophage to a phenotype that is exquisitely sensitive to secondary danger signals generated by subsequent exposure to xenobiotics or oxidants [7]. Our in vitro findings provide strong evidence for this hypothesis. There is a growing evidence for functional plasticity of macrophages and their ability to adapt to changing microenvironments [53]. Macrophages can be selectively reprogrammed to a specific phenotype of immune response following relatively short-term exposure to microbial ligands [54]. LPS priming through up-regulation of TLR4, prolonged and enhanced MAPK and NF-κB activation, and chromatin remodeling can indeed result in macrophage reprogramming [54].
Our results showed that oxidants stimulated the expression of the mRNAs of three inducible genes encoding enzymes in pM sensitized with LPS-EK. COX-2 is induced by TLR4 activation [18,40]. Increased expression of TLR4 and its co-receptor by LPS would partially explain the increased responsiveness. First, LPS would up-regulate the expression of TLR4 and its co-receptors, which would increase the sensitivity of macrophages to exogenous oxidants. It has been reportedly shown for some time that LPS can directly increase mRNA levels in human monocytes and neutrophils [55]. LPS in combination with anti-CD40 mAb also increased TLR4-MD-2 complex surface expression [56]. Additionally, LPS resulted in an initial but transient increase in CD14 expression (that serves as co-receptor for TLR4 activation), which was associated with distribution of TLR4 in enterocytes [57]. Simultaneously, pro-inflammatory cytokines induced by LPS sensitization modulated TLR4 expression. IL-6 incubation upregulated TLR4 cell surface protein leading to increased responsiveness to TLR4 activation in human monocytes [58].
Second, COX-2 appears to be induced by the dominant transcriptional factor NF-κB activation [59]. It has been proposed that oxidant modulation of NF-κB activity is largely dependent on the degradation of IκBα and subsequent NF-κB activation, a process that requires an activating stimulus, such as LPS. Here, LPS priming might lead to prolonged and enhanced NF-κB activation, which in turn facilitates oxidant-mediated NF-κB activation. Our findings are in accord with other studies because H2O2 increased the degradation of IκBα, the nuclear localization of p50, and the activation of NF-κB only in LPS-activated human primary monocytes [40]. Interestingly, there was no induction of COX-2 and mPGES-1genes by prior incubation with LPS-EK in pM derived from TLR4-KO. Furthermore, there was no concurrent induction of COX-2 and mPGES-1genes by prior incubation with LPS-EK in pM derived from TLR4-WT [60]. Thus, this does not appear to support the hypothesis that a primary trigger for a concurrent activation on COX-2 and mPGES-1 is functional TLR4 stimulation. Nonetheless, others have shown that NF-κB can regulate the expression of both COX-2 and mPGES-1 in macrophages induced by LPS, a native ligand for TLR4 [61].
Taken together, LPS-mediated TLR4 priming sensitized pM to oxidant-induced expression of COX-2 and PGE2 biosynthesis. Our data provide a potential mechanism(s) by which exogenous oxidants may facilitate human disease states (inflammatory processes in the presence of bacterial LPS or other TLR4 primers/activators such as DAMPs or PAMPs and including the ubiquitous air pollutants). Our results provide further empirical evidence for a potential role of TLR4 ligands/pseudo ligands in initiating, propagating and maintaining many disease states.
Table 1.
Primers used in the polymerase chain reaction (PCR) experiments.
| TLR4 | Forward 5′-3′ | AACCAGCTGTATTCCCTCAGCACT | 175 bp |
| Reverse 5′-3′ | ACTGCTTCTGTTCCTTGACCCACT | ||
| GAPDH | Forward 5′-3′ | TGTGATGGGTGTGAACCACGAGAA | 154 bp |
| Reverse 5′-3′ | GAGCCCTTCCACAATGCCAAAGTT | ||
| β-actin | Forward 5′-3′ | GTTGGAGCAAACATCCCCCA | 187 bp |
| Reverse 5′-3′ | ACGCGACCATCCTCCTCTTA | ||
| COX-1 | Forward 5′-3′ | AAGATGGGTCCTGGCTTTAC | 88 bp |
| Reverse 5′-3′ | GGTGATACTGTCGTTCCAGATT | ||
| COX-2 | Forward 5′-3′ | CGGACTGGATTCTATGGTGAAA | 111 bp |
| Reverse 5′-3′ | CTTGAAGTGGGTCAGGATGTAG | ||
| sPLA2 type V | Forward 5′-3′ | CAGGGGGCTTGCTAGAACTCAA | 326 bp |
| Reverse 5′-3′ | AAGAGGGTTGTAAGTCCAGAGG | ||
| cPLA2 | Forward 5′-3′ | GCCGAGGAAGAGGAAAGGATAG | 63 bp |
| Reverse 5′-3′ | TTCGCCCACTTCTCTGCAA | ||
| mPGES-1 | Forward 5′-3′ | ATGAGGCTGCGGAAGAAGG | 149 bp |
| Reverse 5′-3′ | GCCGAGGAAGAGGAAAGGATAG | ||
| mPGES-2 | Forward 5′-3′ | GCTGGGGCTGTACCACA | 193 bp |
| Reverse 5′-3′ | GATTCACCTCCACCACCTGA |
HIGHLIGHTS.
Reactive oxygen and nitrogen species (RONS) activate TLR4.
We treated primary macrophages (pM) with PPC and PPN as in vitro sources of RONS.
Treatment of pM with RONS alone had a minimal effect on PGE2 biosynthesis.
RONS significantly increased COX-2 expression in pM with prior LPS-EK treatment.
RONS increased PGE2 only in pM derived from TLR4-WT mice, but not in TLR4-KO pM.
Acknowledgments
The authors will like to thank Dr. Michael Wacker for use of his immunofluorescence microscope and Dr. Mingui Fu for use of his RT-PCR equipment. The project was supported by NIH-NIDCR 021888 (OJI). This work is part of the doctoral thesis for YZ.
Abbreviations
- ONS
oxidative/nitrosative stress
- TLR
toll-like receptor
- LPS-EK (Ultrapure)
lipopolysaccharide from E. coli K12
- TLR4-WT macrophages
macrophages derived from wild-type mice
- TLR4-KO macrophages
macrophages derived from complete TLR4 knock-out mice
- TAOC
total antioxidant capacity
- pM
primary peritoneal macrophages
- PPC
potassium peroxychromate
- PPN
potassium peroxynitrite
- cPLA2
cytosolic phospholipase A 2
- sPLA2
secretory phospholipase A 2
- COX-1
cyclooxygenase 1
- COX-2
cyclooxygenase 2
- mPGES-1
microsomal prostaglandin E synthase-1
- mPGES-2
microsomal prostaglandin E synthase-2
- FBS
fetal bovine serum
- DMEM
Dulbecco’s modified Eagle’s medium
- ANOVA
analysis of variance
- iROS
intracellular reactive oxygen species
- ELISA
enzyme-linked immuno-sorbent assay
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The project described was supported by Award Number DE021888 from the National Institute of Dental & Craniofacial Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Dental & Craniofacial Research or the National Institutes of Health.
Conflicts of interest:
The authors declare no conflicts of interest.
References
- 1.Criswell H, Mueller RA, Breese GR. Priming of D1-dopamine receptor responses: long-lasting behavioral supersensitivity to a D1-dopamine agonist following repeated administration to neonatal 6-OHDA-lesioned rats. J Neurosci. 1989;9(1):125–133. doi: 10.1523/JNEUROSCI.09-01-00125.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, Slivka PF, Coats BD, Rezvani N, Wieseler J, Hughes TS, Landgraf KE, Chan S, Fong S, Phipps S, Falke JJ, Leinwand LA, Maier SF, Yin H, Rice KC, Watkins LR. Evidence that opioids may have toll-like 4 and MD-2 effects. Brain Behav Immun. 2010;24(1):83–95. doi: 10.1016/j.bbi.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hutchinson MR, Loram LC, Zhang Y, Shridhar M, Rezvani N, Berkelhammer D, Phipps S, Foster PS, Landgraf K, Falke JJ, Rice KC, Maier SF, Yin H, Watkins LR. Evidence that tricyclic small molecules may possess toll-like receptor and myeloid differentiation protein 2 activity. Neuroscience. 2010;168(2):551–563. doi: 10.1016/j.neuroscience.2010.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manček-Keber M, Jerala R. Postulates for validating TLR4 agonists. Eur J Immunol. 2015;45(2):356–370. doi: 10.1002/eji.201444462. [DOI] [PubMed] [Google Scholar]
- 5.Pascual M, Baliño P, Alfonso-Loeches S, Aragón CM, Guerri C. Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage. Brain Behav Immun. 2011;25(Suppl 1):S80–S91. doi: 10.1016/j.bbi.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 6.Kelley KW, Dantzer R. Alcoholism and inflammation: neuroimmunology of behavioral and mood disorders. Brain Behav Immun. 2011;25(Suppl 1):S13–S20. doi: 10.1016/j.bbi.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pestka J, Zhou HR. Toll-like receptor priming sensitizes macrophages to proinflammatory cytokine gene induction by deoxynivalenol and other toxicants. Toxicol Sci. 2006;92:445–455. doi: 10.1093/toxsci/kfl012. [DOI] [PubMed] [Google Scholar]
- 8.Chandler CE, Ernst RK. Bacterial lipids: powerful modifiers of the innate immune response. F1000Res. 2017:6. doi: 10.12688/f1000research.11388.1. pii: F1000 Faculty Rev-1334. doi:10.12688/f1000research.11388.1. eCollection 2017. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Draing C, Sigel S, Deininger S, Traub S, Munke R, Mayer C, Hareng L, Hartung T, von Aulock S, Hermann C. Cytokine induction by Gram-positive bacteria. Immunobiology. 2008;213(3–4):285–296. doi: 10.1016/j.imbio.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Declue AE, Johnson PJ, Day JL, Amorim JR, Honaker AR. Pathogen associated molecular pattern motifs from Gram-positive and Gram-negative bacteria induce different inflammatory mediator profiles in equine blood. Vet J. 2012;192(3):455–460. doi: 10.1016/j.tvjl.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Mockenhaupt FP, Cramer JP, Hamann L, Stegemann MS, Eckert J, Oh NR, Otchwemah RN, Dietz E, Ehrhardt S, Schröder NW, Bienzle U, Schumann RR. Toll-like receptor (TLR) polymorphisms in African children: Common TLR-4 variants predispose to severe malaria. Proc Natl Acad Sci U S A. 2006;103(1):177–182. doi: 10.1073/pnas.0506803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prakash H, Nadella V, Singh S, Schmitz-Winnenthal H. CD14/TLR4 priming potentially recalibrates and exerts anti-tumor efficacy in tumor associated macrophages in a mouse model of pancreatic carcinoma. Sci Rep. 2016;6:31490. doi: 10.1038/srep31490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raddassi K, Petit JF, Lemaire G. LPS-induced activation of primed murine peritoneal macrophages is modulated by prostaglandins and cyclic nucleotides. Cell Immunol. 1993;149:50–64. doi: 10.1006/cimm.1993.1135. [DOI] [PubMed] [Google Scholar]
- 14.Chizzolini C, Brembilla NC. Prostaglandin E2: igniting the fire. Immunol Cell Biol. 2009;87:510–511. doi: 10.1038/icb.2009.56. [DOI] [PubMed] [Google Scholar]
- 15.Ganesh T. Evaluation of WO 2012/177618 A1 and US-2014/0179750 A1: novel small molecule antagonists of prostaglandin-E2 receptor EP2. Expert Opin Ther Pat. 2015;25(7):837–844. doi: 10.1517/13543776.2015.1025752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aghazadeh-Habashi A, Asghar W, Jamali F. Drug-Disease Interaction: Effect of inflammation and Nonsteroidal Anti-Inflammatory Drugs on Cytochrome P450 Metabolites of Arachidonic Acid. J Pharm Sci. 2017:5. doi: 10.1016/j.xphs.2017.09.020. pii: S0022-3549(17)30677-30679. [DOI] [PubMed] [Google Scholar]
- 17.Sacerdoti D, Gatta A, McGiff JC. Role of cytochrome P450-dependent arachidonic acid metabolites in liver physiology and pathophysiology. Prostaglandins Other Lipid Mediat. 2003;72(1–2):51–71. doi: 10.1016/s1098-8823(03)00077-7. Review. [DOI] [PubMed] [Google Scholar]
- 18.Ruiperez V, Astudillo AM, Balboa MA, Balsinde J. Coordinate regulation of TLR-mediated arachidonic acid mobilization in macrophages by group IVA and group V phospholipase A2s. J Immunol. 2009;182:3877–3883. doi: 10.4049/jimmunol.0804003. [DOI] [PubMed] [Google Scholar]
- 19.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59:207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 21.Nakanishi M, Rosenberg DW. Multifaceted roles of PGE2 in inflammation and cancer. Semin Immunopathol. 2013;35:123–137. doi: 10.1007/s00281-012-0342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheibanie AF, Yen JH, Khayrullina T, Emig F, Zhang M, Tuma R, Ganea D. The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23–>IL-17 axis. J Immunol. 2007;178:8138–8147. doi: 10.4049/jimmunol.178.12.8138. [DOI] [PubMed] [Google Scholar]
- 23.Sheibanie AF, Khayrullina T, Safadi FF, Ganea D. Prostaglandin E2 exacerbates collagen-induced arthritis in mice through the inflammatory interleukin-23/interleukin-17 axis. Arthritis Rheum. 2007;56:2608–2619. doi: 10.1002/art.22794. [DOI] [PubMed] [Google Scholar]
- 24.Jin X, Wang L, Wu HS, Zhang L, Wang CY, Tian Y, Zhang JH. N-Acetylcysteine inhibits activation of toll-like receptor 2 and 4 gene expression in the liver and lung after partial hepatic ischemia-reperfusion injury in mice. Hepatobiliary Pancreat Dis Int. 2007;6(3):284–289. [PubMed] [Google Scholar]
- 25.Pushpakumar S, Ren L, Kundu S, Gamon A, Tyagi SC, Sen U. Toll-like receptor 4 deficiency reduces oxidative stress and macrophage mediated inflammation in hypertensive kidney. Sci Rep. 2017;7(1):6349. doi: 10.1038/s41598-017-06484-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asehnoune K, Strassheim D, Mitra S, Kim JY, Abraham E. Involvement of reactive oxygen species in Toll-like receptor 4-dependent activation of NF-kappa B. J Immunol. 2004;172(4):2522–2529. doi: 10.4049/jimmunol.172.4.2522. [DOI] [PubMed] [Google Scholar]
- 27.Miesel R, Kroger H, Kurpisz M, Weser U. Induction of arthritis in mice and rats by potassium peroxochromate and assessment of disease activity by whole blood chemiluminescence and 99mpertechnetate-imaging. Free Radic Res. 1995;23:213–227. doi: 10.3109/10715769509064035. [DOI] [PubMed] [Google Scholar]
- 28.Edwards JC, Quinn PJ. Decomposing potassium peroxychromate produces hydroxyl radical (.OH) that can peroxidize the unsaturated fatty acids of phospholipid dispersions. J Lipid Res. 1982;23:994–1000. [PubMed] [Google Scholar]
- 29.Trackey JL, Uliasz TF, Hewett SJ. SIN-1-induced cytotoxicity in mixed cortical cell culture: peroxynitrite-dependent and -independent induction of excitotoxic cell death. J Neurochem. 2001;79:445–455. doi: 10.1046/j.1471-4159.2001.00584.x. [DOI] [PubMed] [Google Scholar]
- 30.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008 doi: 10.1002/0471142735.im1401s83. Chapter 14, Unit 14 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Misharin AV, Saber R, Perlman H. Eosinophil contamination of thioglycollate-elicited peritoneal macrophage cultures skews the functional readouts of in vitro assays. J Leukoc Biol. 2012;92:325–331. doi: 10.1189/jlb.1111560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 34.Johnson G, Nour AA, Nolan T, Huggett J, Bustin S. Minimum information necessary for quantitative real-time PCR experiments. Methods Mol Biol. 2014;160:5–17. doi: 10.1007/978-1-4939-0733-5_2. [DOI] [PubMed] [Google Scholar]
- 35.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Karki R, Igwe OJ. Toll-like receptor 4 signaling: A common pathway for interactions between prooxidants and extracellular disulfide high mobility group box 1 (HMGB1) protein-coupled activation. Biochem Pharmacol. 2015;98(1):132–143. doi: 10.1016/j.bcp.2015.08.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kennedy BP, Payette P, Mudgett J, Vadas P, Pruzanski W, Kwan M, Tang C, Rancourt DE, Cromlish WA. A natural disruption of the secretory group II phospholipase A2 gene in inbred mouse strains. J Biol Chem. 1995;270:22378–22385. doi: 10.1074/jbc.270.38.22378. [DOI] [PubMed] [Google Scholar]
- 38.Kuroda E, Yamashita U. Mechanisms of enhanced macrophage-mediated prostaglandin E2 production and its suppressive role in Th1 activation in Th2-dominant BALB/c mice. J Immunol. 2003;170:757–764. doi: 10.4049/jimmunol.170.2.757. [DOI] [PubMed] [Google Scholar]
- 39.Ganey PE, Roth RA. Concurrent inflammation as a determinant of susceptibility to toxicity from xenobiotic agents. Toxicology. 2001;169:195–208. doi: 10.1016/s0300-483x(01)00523-6. [DOI] [PubMed] [Google Scholar]
- 40.Lu Y, Wahl LM. Oxidative stress augments the production of matrix metalloproteinase-1, cyclooxygenase-2, and prostaglandin E2 through enhancement of NF-kappa B activity in lipopolysaccharide-activated human primary monocytes. J Immunol. 2005;175:5423–5429. doi: 10.4049/jimmunol.175.8.5423. [DOI] [PubMed] [Google Scholar]
- 41.Molin C, Jauhiainen A, Warringer J, Nerman O, Sunnerhagen P. mRNA stability changes precede changes in steady-state mRNA amounts during hyperosmotic stress. RNA. 2009;15:600–614. doi: 10.1261/rna.1403509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sapirstein A, Spech RA, Witzgall R, Bonventre JV. Cytosolic phospholipase A2 (PLA2), but not secretory PLA2, potentiates hydrogen peroxide cytotoxicity in kidney epithelial cells. J Biol Chem. 1996;271:21505–21513. doi: 10.1074/jbc.271.35.21505. [DOI] [PubMed] [Google Scholar]
- 43.Robb SJ, Gaspers LD, Wright KJ, Thomas AP, Connor JR. Influence of nitric oxide on cellular and mitochondrial integrity in oxidatively stressed astrocytes. J Neurosci Res. 1999;56:166–176. doi: 10.1002/(sici)1097-4547(19990415)56:2<166::aid-jnr6>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 44.Mardini IA, FitzGerald GA. Selective inhibitors of cyclooxygenase-2: a growing class of anti-inflammatory drugs. Mol Interv. 2001;1:30–38. [PubMed] [Google Scholar]
- 45.Burdon C, Mann C, Cindrova-Davies T, Ferguson-Smith AC, Burton GJ. Oxidative stress and the induction of cyclooxygenase enzymes and apoptosis in the murine placenta. Placenta. 2007;28:724–733. doi: 10.1016/j.placenta.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Ikeda T, Fueki M, Ueno A, Oh S, Kudo I. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 47.Uematsu S, Matsumoto M, Takeda K, Akira S. Lipopolysaccharide-dependent prostaglandin E(2) production is regulated by the glutathione-dependent prostaglandin E (2) synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J Immunol. 2002;168(11):5811–5816. doi: 10.4049/jimmunol.168.11.5811. [DOI] [PubMed] [Google Scholar]
- 48.Chen L, Yang G, Monslow J, Todd L, Cormode DP, Tang J, Grant GR, DeLong JH, Tang SY, Lawson JA, Pure E, Fitzgerald GA. Myeloid cell microsomal prostaglandin E synthase-1 fosters atherogenesis in mice. Proc Natl Acad Sci U S A. 2014;111:6828–6833. doi: 10.1073/pnas.1401797111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang M, Lee E, Song W, Ricciotti E, Rader DJ, Lawson JA, Pure E, FitzGerald GA. Microsomal prostaglandin E synthase-1 deletion suppresses oxidative stress and angiotensin II-induced abdominal aortic aneurysm formation. Circulation. 2008;117:1302–1309. doi: 10.1161/CIRCULATIONAHA.107.731398. [DOI] [PubMed] [Google Scholar]
- 50.Ikeda-Matsuo Y, Ikegaya Y, Matsuki N, Uematsu S, Akira S, Sasaki Y. Microglia-specific expression of microsomal prostaglandin E2 synthase-1 contributes to lipopolysaccharide-induced prostaglandin E2 production. J Neurochem. 2005;94:1546–1558. doi: 10.1111/j.1471-4159.2005.03302.x. [DOI] [PubMed] [Google Scholar]
- 51.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 52.Jania LA, Chandrasekharan S, Backlund MG, Foley NA, Snouwaert J, Wang IM, Clark P, Audoly LP, Koller BH. Microsomal prostaglandin E synthase-2 is not essential for in vivo prostaglandin E2 biosynthesis. Prostaglandins Other Lipid Mediat. 2009;88:73–81. doi: 10.1016/j.prostaglandins.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stout RD, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76:509–513. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Malyshev IY, Shnyra A. Controlled modulation of inflammatory, stress and apoptotic responses in macrophages. Curr Drug Targets Immune Endocr Metabol Disord. 2003;3:1–22. doi: 10.2174/1568008033340342. [DOI] [PubMed] [Google Scholar]
- 55.Bosisio D, Polentarutti N, Sironi M, Bernasconi S, Miyake K, Webb GR, Martin MU, Mantovani A, Muzio M. Stimulation of toll-like receptor 4 expression in human mononuclear phagocytes by interferon-gamma: a molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood. 2002;99:3427–3431. doi: 10.1182/blood.v99.9.3427. [DOI] [PubMed] [Google Scholar]
- 56.Frleta D, Noelle RJ, Wade WF. CD40-mediated up-regulation of Toll-like receptor 4-MD2 complex on the surface of murine dendritic cells. J Leukoc Biol. 2003;74:1064–1073. doi: 10.1189/jlb.0203062. [DOI] [PubMed] [Google Scholar]
- 57.Hornef MW, Frisan T, Vandewalle A, Normark S, Richter-Dahlfors A. Toll-like receptor 4 resides in the Golgi apparatus and colocalizes with internalized lipopolysaccharide in intestinal epithelial cells. J Exp Med. 2002;195:559–570. doi: 10.1084/jem.20011788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tamandl D, Bahrami M, Wessner B, Weigel G, Ploder M, Furst W, Roth E, Boltz-Nitulescu G, Spittler A. Modulation of toll-like receptor 4 expression on human monocytes by tumor necrosis factor and interleukin-6: tumor necrosis factor evokes lipopolysaccharide hyporesponsiveness, whereas interleukin-6 enhances lipopolysaccharide activity. Shock. 2003;20:224–229. doi: 10.1097/00024382-200309000-00005. [DOI] [PubMed] [Google Scholar]
- 59.Jaulmes A, Thierry S, Janvier B, Raymondjean M, Maréchal V. Activation of sPLA2-IIA and PGE2 production by high mobility group protein B1 in vascular smooth muscle cells sensitized by IL-1β. The FASEB Journal. 2006;20:1727–1729. doi: 10.1096/fj.05-5514fje. [DOI] [PubMed] [Google Scholar]
- 60.de Oliveira AC, Candelario-Jalil E, Bhatia HS, Lieb K, Hüll M, Fiebich BL. Regulation of prostaglandin E2 synthase expression in activated primary rat microglia: evidence for uncoupled regulation of mPGES-1 and COX-2. Glia. 2008;56(8):844–855. doi: 10.1002/glia.20658. [DOI] [PubMed] [Google Scholar]
- 61.D’Acquisto F, Iuvone T, Rombolà L, Sautebin L, Di Rosa M, Carnuccio R. Involvement of NF-kappaB in the regulation of cyclooxygenase-2 protein expression in LPS-stimulated J774 macrophages. FEBS Lett. 1997;418(1–2):175–178. doi: 10.1016/s0014-5793(97)01377-x. [DOI] [PubMed] [Google Scholar]