

Abstract
CD11c+ T-bet+ B cells have now been detected and characterized in different experimental and clinical settings, in both mice and humans. Whether such cells are monolithic, or define subsets of B cells with different functions is not yet known. Our studies have identified CD11c+ IgM+ CD19hi splenic IgM memory B cells that appear at approximately three weeks post-ehrlichial infection, and persist indefinitely, during low-level chronic ehrlichial infection. Although the CD11c+T-bet+ B cells we have described are distinct, they appear to share many features with similar cells detected under diverse conditions, including viral infections, aging, and autoimmunity. We propose that CD11c+ T-bet+ B cells as a group share characteristics of memory B cells that are maintained under conditions of inflammation and/or low-level chronic antigen stimulation. In some cases, these cells may be advantageous, by providing immunity to re-infection, but in others may be deleterious, by contributing to aged-associated autoimmune responses.
Introduction1
CD11c+ T-bet-expressing B cells are now recognized as a novel and distinct B cell population that is important for long-term immunity to infections, and for autoimmunity. A diverse set of observations, from different experimental models, as well as clinical studies, have suggested that B cells defined by expression of CD11c, T-bet, and other diagnostic cell surface markers and transcription factors, form a discrete B cell subset. These cells appear to be induced by a common mode of innate activation, and may be maintained for long periods of time by chronic inflammation, or antigen stimulation. Our investigations have been facilitated by the use of a bacterial infection model, which has allowed us to study the genesis, differentiation, and maintenance of CD11c+ T-bet+ B cells, which include novel IgM memory cells, in a unique experimental context. We have observed similar properties of CD11c+ T-bet+ B cells as those described in other studies, but also distinct differences, which may be attributed to different modalities of B cell activation during infection. Our objective in this review is to describe how our studies, in a bacterial infection model, integrate with and inform other studies emerging from this exciting area of B cell biology.
Discovery of CD11c+ B cells
We first identified CD11c+ expressing B cells, fortuitously, in 2008, as part of unrelated studies designed to investigate possible roles for DC subsets during ehrlichial infection [1]. Although a there had been a few prior reports of CD11c transcribed or expressed on B cells, primarily in humans, our finding was unexpected, and indeed, a number of experiments were required to demonstrate that the CD11c+ splenocytes were, indeed B cells. Our studies were performed primarily in an experimental C57BL/6 mouse model of ehrlichiosis, which utilizes a pathogen known as Ehrlichia muris. E. muris is a vector-borne bacterium, transmitted by the Ixodes tick, and, like many related rickettsiae, are obligate intracellular bacteria. E. muris primarily infects monocytes and macrophages [2], although we have also detected the pathogen in DCs [3]. Although E muris was originally described as a mouse pathogen, a closely related, if not identical pathogen, has been shown to infect humans (known as the E. muris-like agent, or EMLA; [4]). As an obligate intracellular pathogen, E. muris cannot replicate outside of host cells, although we have reported that the bacteria can be found extracellularly [5], a property that is presumably necessary to facilitate blood-borne transmission, and which at the same time may predispose the bacteria to clearance by antibodies [6]. E. muris infection, which is not fatal in immunocompetent mice and humans, nonetheless causes a number of clinical manifestations, including anemia, thrombocytopenia, splenomegaly, as well as hematological alterations, including myelopoiesis and perturbation of the hematopoietic stem cell niche [7]. The latter likely develops as a component of host defense, although it is also possible that changes in hematopoietic activity are orchestrated by the bacteria.
Some of the pathologies (i.e., splenomegaly) caused by E. muris infection persist, likely because the bacteria establish a low-level chronic infection [3, 8]. Chronic infection is associated with increased frequencies of CD69+ KLRG-1+ CD4 T cells in the spleen, as well as high expression of IAb on F4/80 splenic macrophages [3], indicating that the spleen may maintain an inflammatory environment that supports long-term B cell responses. Of particular relevance for the current discussion is the additional finding that the ehrlichiae as a group do not encode canonical TLR ligands, including LPS and peptidoglycan [9]. The means whereby the ehrlichiae trigger innate immunity is unknown, and likely involves innate pathways other than TLR signaling.
E. muris infection generates strong cellular and humoral immune responses [10–13]. These responses are capable of protecting infected mice from a second, fatal, challenge infection with a closely related ehrlichia known as Ixodes ovatus ehrlichia (IOE; [14]); in the absence of pre-existing immunity, IOE is fatal within approximately 14 days post-infection. Our studies of immunity using the E. muris model revealed, however, that short-term protection from fatal IOE infection could be achieved independently of CD4 T cells ([15]; later studies showed that CD4-deficient mice eventually succumb within approximately 90 days post-infection). These observations supported our previous studies which demonstrated that antibodies were highly effective in protecting immunodeficient SCID mice from fatal infection with a related ehrlichia, E. chaffeensis. T-independent immunity to E. muris was unexpected, given the well-described role of CD4 T cells and cellular responses for immunity to prototypic intracellular bacteria. Although E. muris infection is associated with robust antigen-specific IgM and IgG responses, we showed that IgM, by itself, was sufficient for long-term immunity to fatal infection [16], observations which supported a major role for T-independent humoral immunity in host defense. The origin of the protective IgM response was unknown, however, prior to our discovery of CD11c+ T-bet+ B cells, which occurred at about the time when we first observed CD4 T cell-independent immunity.
Our first observation relevant to CD11c+ T-bet+ B cells, was the detection of a large population of MHCII+ CD11c+ splenocytes, beginning on about day 7 post-infection [1]. Although the cells were first thought to be dendritic cells, further analysis revealed that they also exhibited surface expression of B220, as well as a number of other classical B cell surface markers, which unambiguously identified the cells as B cells. The population was detected at its highest frequency and number on day 11 post-infection (two days following peak bacterial infection), and persisted until at least day 18 post-infection. The CD11c+ B cells were found in remarkably high frequency and number, as high as 20% of total splenocytes, representing as many as 25 million cells within the spleen. The cells were also found to reside in the splenic red pulp, an observation that was also consistent with their CD4 T cell-independent origin (i.e., lack of a requirement for GCs for their generation).
A key to understanding CD11c+ B cell function was the expression of CD138 (Syndecan-1; a marker of antibody-secreting cells), on a large proportion of the CD11c+ B cells. Indeed, ELISpot analyses of flow cytometrically-purified CD11c+ B cells revealed that nearly all of the antigen-specific ASCs that were detected early during infection in the spleen were found within the CD11c+ B cell population [1]. Moreover, the predominant Ig that was secreted by the B cells was IgM. Later studies from our laboratory showed that most of the IgM produced by these cells recognized an immunodominant outer membrane protein known as OMP-19; however, many of the IgM produced from hybridoma cell lines were polyreactive, that is, these antibodies had the capacity to bind to diverse, structurally-unrelated antigens, including proteins, carbohydrates, and DNA [17]. It is not known whether the capacity of the CD11c+ B cells, which are formally plasmablasts, to generate polyreactive IgM, bears any relationship to their origin as T-bet-expressing B cells. Although the polyreactive antibodies produced bind to DNA and chromatin, in preliminary studies we did not detected any acceleration of autoimmunity in E. muris-infected autoimmune-prone NZB/NZW mice (unpublished data).
Although we did not initially characterize T-bet expression in the CD11c+ B cells we detected during acute ehrlichial infection, the transcription factor is expressed by these cells (Figure 1A). Thus, early CD4 T cell-independent plasmablasts are among the widening examples of B cells that can be distinguished on the basis of both CD11c and T-bet expression. In addition to CD11c and T-bet, the CD11c+ B cells expressed the integrin CD11b, which can be expressed on B-1 cells ([18]; the B cells we characterized were CD5-negative), and exhibited high expression of the VLA-4 subunits CD29 and CD49d [1]. Although CD11b expression is characteristic of B-1 cells, and B-1 cells can be found within the spleen, none of our other studies have supported the conclusion that the CD11c+ T-bet+ B cells we identified are derived from this B cell lineage. The splenic CD11c+ B cells also exhibited low surface expression of CD21, CD23, CD62L, and high expression of CD43, CD44, CD9, CD38, CXCR4, and TACI, relative to canonical follicular CD19+ B cells [1]. In recent studies from our laboratory we have also shown that the CD11c+ T-bet+ plasmablasts, or a common precursor, give rise to long-term CD4 T cell-independent CD11c-negative bone marrow IgM ASCs that we have also detected during ehrlichial infection ([16]; unpublished data). These data revealed that CD11c+ T-bet+ ASCs can be generated independently of CD4 T cells during acute infection, and can contribute to protective immunity during both and acute and chronic infection. Moreover, our work indicates that T-bet expression, per se, does not necessarily dictate B cell effector function, but perhaps is characteristic of a unique lineage or cell population that is elicited in B cells under similar innate inflammatory conditions.
Figure 1. T-bet expression in CD11c+ plasmablasts and IgM memory cells.

A. Infected wild-type mice were analyzed for intracellular T-bet expression on day 10 post-infection in CD11c-negative B220+ B cells (rectangular gate and open histogram) and CD11c-positive B220+ plasmablasts (oval gate and light grey histogram).
B. Similar studies were performed on day 35 post-infection to detect T-bet expression in CD11c-positive B cells (light gray histogram), CD19hi B cells (dark grey histogram), and CD19lo B cells (empty histogram).
C. Representative flow cytometry analysis of CD19+ (open histograms) and CD19hi (light grey histograms) B cells, both CD11c-negative, and CD19+ CD11c-positive B cells (dark grey histograms). The data were acquired on day 25 post-infection, and are representative of data acquired from infected mice that contained IgM memory cells.
CD11c expression in B cells
Our initial observation regarding the novel expression of CD11c in B cells in a murine infection model, was preceded by other studies, mostly in humans, that reported CD11c expression in mitogen-activated B cells [19], and in Hairy cell leukemia cells (HCLs). HCLs are typically characterized on the basis of their expression of CD11c, CD25, CD103, and CD123 [20, 21]. Given their characteristic expression of CD11c, it is likely that HCLs are derived from immortalized CD11c+ T-bet+ B cells. Other human B cells, including memory cells, characterized by expression of the Fc Receptor (FcR)-like proteins, FcRL4 and/or FcRL5, have been shown to also express CD11c [22–25]. Although the function and role of these FcR-like proteins is not well-understood [26], FcrRL4 has been reported to enhance TLR signaling [27], and both FcRL4 and FcRL5 both have been reported to bind immunoglobulins [28]. Yet other studies have described a CD21lo B cell subset that bears close resemblance to CD11c+ T-bet+ B cells; although CD11c expression was not analyzed in the studies of CD21lo B cells, CD11c+ B cells have been reported in other studies to exhibit a common feature of low relative expression of CD21 [22, 24, 29–32]. CD21lo human B cells have been proposed by some investigators to be exhausted or anergic B cells [24, 29–33]. Another study demonstrated that murine CCR6hiCD11cint B cells were shown to promote M-cell differentiation in Peyer’s patches [34]. These data together provided an early indication that CD11c+ B cells, or their equivalents, are important in humans. Examples of studies that have reported CD11c expression in B cells in healthy and diseased humans and mice are shown in Table I.
Table I.
Identification of CD11c+ and related B cells in mice and humans
| Condition | References | Comments |
|---|---|---|
| Healthy humans | [19] | CD11c+ detected on activated B cells |
| Ehrlichial infection | [1, 48] | CD11c+ identified on spleen plasmablasts and IgM memory cells |
| Aged humans and SLE patients | [35] [36] | CD11c+ T-bet+ B cells (ABCs); autoreactive Abs detected in aged mice |
| Hairy Cell Leukemia (HCL) and variant | [21, 91, 92] | CD11c expression on IgM+ lymphoma cells; BRAF mutation is associated with cell growth |
| Viral infections | [37] [38] | CD11c+ T-bet+ B cells detected early in response to infections |
| Healthy humans | [22] [23] | CD11c expression detected on FcRL4+ non-germinal center memory B cells in tonsils |
| Healthy humans | [24] | CD11c expression detected on FcRL5 IgM+ CD21low tissue-like memory B cells |
| Healthy humans | [93] | CD11c+ expression on IgM+ memory cells in blood |
| Hepatitis C, B infection | [33] [31], [94] [25] | Accumulation of IgM+ CD11c+ CD21low memory B cells in patients with Mixed Cryoglobuliemia |
| HIV infection | [29] | IgM+ CD21low Tissue-like memory B cells increased in blood of viremic patients |
| Malaria infection | [86] | IgM+ memory B cells detected during infection |
| CVID | [32, 95]; [30] | IgM+ CD21low B cells with low SHM and poly- and auto-reactivity associated with splenomegaly |
| SLE | [89] | IgMlow CD21low B cells a present in the periphery of SLE patients |
The relevance of CD11c+ B cells to human health is supported by other work, published in 2011, that identified CD11c expression in SLE-prone mice, and in elderly autoimmune women [35]. The CD11c+ B cells detected in women were responsible for the production of autoantibodies. Given their detection in and association with aged individuals, the B cells were named Age-related B Cells (ABCs). Among a panel of genes uniquely expressed by the ABCs was T-bet [35]. The authors also reported a major role for TLR7 signaling in driving the development of this population, likely via an antigen-dependent mechanism. The report was the first to identify T-bet expression in CD11c+ B cells in mice. Hao et al., also in 2011, described large populations of ABCs in aged C57BL/6 mice [36]. Although the latter authors did not report CD11c or T-bet expression in the B cells, these were likely the same or similar to the cells described by Rubtsov and colleagues. In the studies by Hao et al., it was reported that the B cells were refractory to stimulation via the B cell receptor. A role for T-bet function in CD11c+ B cells was addressed further by Rubtsova et al. in 2013, where the authors demonstrated that signaling via the BCR, TLR7, and the IFNγ receptor, synergized to activate T-bet, CD11c, and CD11b expression, as well as the production of IgG2c [37]. Additional signaling, via IL-21, in concert with TLR9, was shown to promote T-bet expression in vitro [38, 39]. These studies together have led to the emerging consensus that T-bet expression defines these cells as novel B cell subset. CD11c and T-bet expression are not always co-incident, however [38]; indeed, in our own studies we have also observed T-bet-positive B cells that do not express CD11c (see below).
Although T-bet is now well-known as a factor required for Th1 cell differentiation, T-bet has been known to be expressed by B cells since the discovery of the transcription factor [40]. T-bet expression has been long-recognized to be associated with IgG2a/c production [41] [42, 43], and T-bet is expressed in IgG2a/c memory B cells [44]. Both IFNγ and CpG (via TLR9) have been shown to activate T-bet expression in B cells [45], and it has been demonstrated that IL-27 can play an important role in T-bet expression and class switching to IgG2a [46]. Thus, T-bet expression can be triggered via IFNγ, TLR, and IL-17 receptors on B cells, where it appears to function analogous to its function in T cells, where it drives CD4 T cell polarization [44, 47].
CD11c+ T-bet+ IgM memory cells
Ongoing studies in our experimental model of ehrlichial infection led us to the discovery of a second, perhaps related, CD11c+ T-bet+ B cell population [48]. These CD11c+ B cells were also detected in the spleen, beginning at about 3 weeks post-infection, not long after the CD11c+ plasmablast population described above declines in frequency. Similar to the early plasmablasts described in our earlier studies, these later CD11c+ B cells were present at high frequencies during infection, and were detected primarily in the spleen [48]. Few, if any, CD11c+ B cells were detected in young, uninfected C56BL/6 mice. Spleen CD11c+ B cells analyzed on day 30 post-infection expressed T-bet (Figure1B), IgM, and a number of other cell surface proteins known to be differentially expressed by switched memory B cells elicited by (4-hydroxy-3-nitrophenyl) acetyl (NP) immunization, including CD73, CD80, CD86, CD38, CD95, and PD-L2 [48]. Moreover, the CD11c+ T-bet+ B cells did not exhibit a surface receptor phenotype typically associated with GC B cells (i.e., the were GL7lo and CD38hi); this was a consideration, as GC B cells can be long-lived [49].
We used a number of criteria to establish that the CD11c+ T-bet+ B cells we identified were indeed memory cells, even though, paradoxically, they were detected at much higher frequencies and numbers than was expected for canonical memory B cells. In addition to their expression of cell surface markers characteristic of memory B cells, the CD11c+ B cells were largely quiescent, persisted indefinitely, and were not antibody-secreting cells; approximately half of the population expressed mutated BCRs, indicative of AID expression triggered by antigen [48]. We formally addressed whether the IgM CD11c+T-bet+ B cells in our model infection were memory cells, by depleting the CD11c+ B cells in CD11c-diphtheria toxin (DT)-transgenic mice. The mouse strain was originally generated to assess immune function in mice that lacked CD11c+ DCs [50]. For our studies, we avoided confounding problems associated with DC depletion, by generating bone marrow-chimeric mice, wherein all B cells were DT-sensitive, but most DCs (50–90%) were DT-insensitive (CD11c+ B cell depletion was verified in the treated mice). We administered specific antigen (ehrlichial OMP-19) to ehrlichial-infected mice on day 30 post-infection, and treated the mice with DT on days -1 and 6 post-antigen administration. ELISA performed on day 12 post-antigen challenge revealed a major reduction of the amnestic antigen-specific IgG2c response, formally demonstrating that CD11c+ B cells were required for recall responses, demonstrating that the population contained antigen-specific IgM memory B cells [48]. Similarly-defined memory B cells had been described in the literature [51, 52], but the cells received limited attention in mouse model studies prior to two reports in 2009 and 2011 [49, 53]. Those studies revealed that IgM memory cells were a major reservoir of long-term immunity. Although our studies have focused primarily on IgM memory cells, we have also detected switched T-bet+ memory cells in our model (unpublished data), consistent with other studies that have reported both switched and non-switched CD11c+ T-bet+ B cells [35].
Although the CD11c+ T-bet+ memory cells we identified expressed IgM, as indicated above, we detected antigen-specific IgG2c following challenge [48]. This observation is consistent, however, with other studies of IgM memory cells that demonstrated that, following antigen challenge, IgM memory cells can enter GCs and undergo class switch recombination [49, 53]. Indeed, one possible benefit of IgM memory cells may be their ability to undergo class switching and somatic hypermutation during secondary infection, a fate that differs from switched memory B cells, which are thought to primarily undergo plasma cell differentiation following antigen challenge [54]. The production of IgG2c is also consistent with other reports that demonstrated that this antibody isotype predominates among switched B cells [55–57]. More recent studies from our laboratory have revealed that GC differentiation of CD11c+ IgM memory studies following re-infection is accompanied by down-regulation of CD11c (unpublished data); these findings explain why we had been unable to identify switched CD11c+ memory cells after antigen challenge in our published studies.
We also addressed whether the IgM memory cells in our studies underwent somatic mutation, as has been reported for IgM memory cells in other studies [49, 53, 58, 59]. Our analyses revealed that over 50% of the T-bet+ CD11c+ memory cells we characterized carried B cell receptor heavy chain mutations, albeit at a relatively low frequency (from 1 to 6 mutations per cell). These data revealed that AID was expressed in at least a portion of the IgM memory cells. We confirmed this conclusion in later studies, and demonstrated that AID expression can be used as an additional marker for T-bet+ CD11c+ memory cells elicited during ehrlichial infection (unpublished data). AID mRNA expression was, by comparison with GC B cells, much lower, which may explain why IgM memory cells undergo limited somatic mutation in the absence of class-switch recombination. The apparent low AID activity also suggests that the IgM memory cells develop independent of GCs, although this has not yet been resolved.
Generation of the CD11c+ B cells in our infection model was distinct from the early day 10 T cell-independent CD11c+ plasmablasts described above, in that the memory cells required both CD4 T cells and IL-21, for their generation [48]. The CD4 and IL-21 dependency suggested a role for T follicular helper (Tfh) T cells; a requirement for IL-21 for the development of T-bet+ B cells has been demonstrated in recent studies by Cancro and colleagues [38]. Analysis of IgM memory cells has also revealed that a proportion of cells present in our experimental infection model do not express CD11c. These cells are likely closely related, because they express most of the other cell surface markers that we used to characterize the IgM memory cells, including PD-L2, CD73, CD95, CD80, and TACI; however, the CD11c-negative cells exhibited lower expression of CD11b (Figure 1C). This difference, and likely others, suggests that T-bet+ CD11c+ and CD11c-negative T-bet+ IgM memory cells may perform different functions, have different fates, or simply reflect developmental changes in CD11c expression. With regard to the latter interpretation, our observation that DT treatment in CD11c-promoter DT transgenic mice largely ablated secondary IgG2c responses to antigen [48] suggests that the CD11c-negative cells transition to CD11c-positive cells, although this remains to be confirmed.
TLRs and innate signals
One likely important difference between our studies of CD11c+ T-bet+ plasmablasts and IgM memory cells, and other reports, is the apparent lack of a requirement for TLR signaling for the generation and/or maintenance of the cells during ehrlichial infection. As indicated in the discussion above, the ehrlichiae do not encode canonical TLR ligands, such as LPS or peptidoglycan [9]. Moreover, our studies of MyD88 mice failed to reveal any change in the generation of CD11c+ T-bet+ plasmablasts in the absence of MyD88-dependent TLR signaling (unpublished data). We performed additional studies to address whether TLR signaling played any role in the generation of CD11c+ T-bet+ IgM memory cell, by utilizing Unc93b-deficient mice, which are deficient in TLR3, 7, 8, and 9 signaling [60, 61]. Generation of the CD11c+ T-bet+ IgM memory B cells was unaffected by the absence of Unc93b (Figure 2). These data indicated that TLR signals are not necessary drive the generation, and likely function, of CD11c+ T-bet+ B cells. This finding was unexpected, given that TLR signaling has been shown to be very important in several studies of CD11c+ T-bet+ B cells [35, 38, 62].
Figure 2. CD11c+ IgM memory cells were generated at normal frequencies in Unc93b-deficient mice.
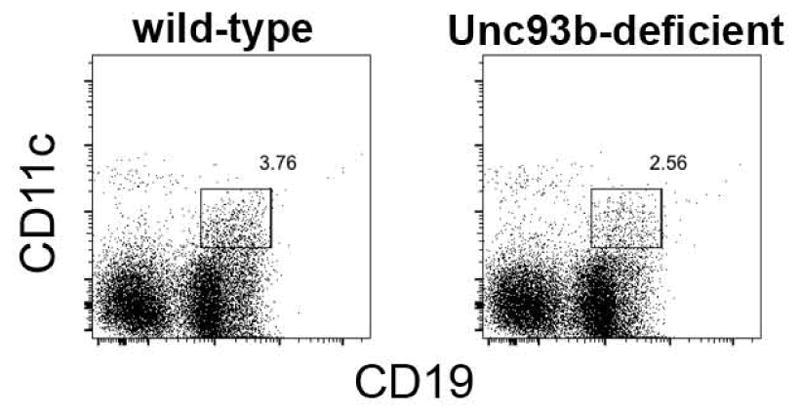
CD11c+ IgM memory cells were monitored in infected wild-type, and Unc93b-deficient mice, as in Figure 1. The data are representative of several studies, where no statistical differences in cell frequency between strains were noted (P=0.1).
One possible explanation for this difference is that T-bet expression in our studies is driven solely by IFNγ, perhaps produced by CD4 T cells, or innate cells [3]. Alternatively, CD11c+ T-bet+ memory B cells may be generated or activated via distinctly different innate signals. The signals that drive innate immunity and B cell responses in our experimental model remain unknown, but these data reveal that TLR signaling is not always required to generate CD11c+ T-bet+ B cells. We have not yet demonstrated formally that TLR signaling is required for activating CD11c+ T-bet+ B cell functions during ehrlichial infection, although CD11c+ T-bet+ B cells can undergo secondary responses in the apparent absence of TLRs [48]. The identification of the innate signals that support the development of CD11c+ T-bet+ B cells during bacterial infection is an active area of investigation.
Role of Chronic infection and/or inflammation
Another important feature of our experimental model is that E. muris infection is chronic. Although the bacteria are difficult to detect in spleen and liver, using quantitative PCR, after about day 30 post-infection, transfer of unseparated spleen cells from infected to uninfected mice results in infection, which is fatal in immunocompromised SCID or RAG-deficient mice. Although infection is chronic in C57BL/6 mice, there are few indications of chronic pathology; however, we have observed higher frequencies of splenic CD4 and CD8 T cells during chronic infection [3]. Although bacteria are present in the spleen, likely in low numbers, it does not appear that the IgM memory cells are in continual contact with antigen, because the bacteria are largely residing inside host cells; moreover, the cell surface characteristics of the IgM memory cells suggest that they are not chronically activated, nor are they proliferating [48]. Moreover, the IgM memory cells in our model are maintained following antibiotic treatment, albeit at a lower frequency [48]. Several studies have reported antigen-independent activation of T-bet+ B cells, but it is difficult to address whether this occurs in vivo. Although the role of antigen in the maintenance of CD11c+T-bet+ B cells, in our model, and likely others, is unresolved, we propose, that the B cells in our model are maintained by the presence of low level chronic antigen, and that this may account for the relatively high frequency of these IgM memory cells. Alternatively, the B cells may be maintained by chronic inflammation, perhaps via TLRs or other inflammatory signaling, chronic inflammation associated with autoimmunity, or aged-related inflammation. Our work is inconsistent with the hypothesis that CD11c+T-bet+ B cells are exhausted or anergic, as has been suggested in other studies. Indeed, our published and ongoing studies of CD11c+ IgM memory cells have indicated that at least a portion of the B cells undergo expansion and GC differentiation following secondary challenge with antigen, or secondary infection [48]; unpublished data), that is, they formally act as functional memory cells.
Transcriptome analyses
As part of our efforts to identify novel functions of CD11c+ T-bet+ B cells, we have performed analyses of the transcriptomes of these cells. For these studies, we compared flow cytometrically-purified CD11c+ and CD11c-negative B cells from infected mice, isolated after day 30 post-infection. The CD11c+ population consisted of CD11c+ T-bet+ IgM memory cells; the CD11c-negative cells contained primarily canonical naive B cells, as these cells were only distinguished only by expression of CD19. The populations were analyzed using RNAseq, which clearly distinguished the CD11c+ and -negative cells on the basis of the differential expression of more than 900 genes (Figure 3).
Figure 3. Transcriptome analysis of CD11c+ T-bet+ IgM memory cells.
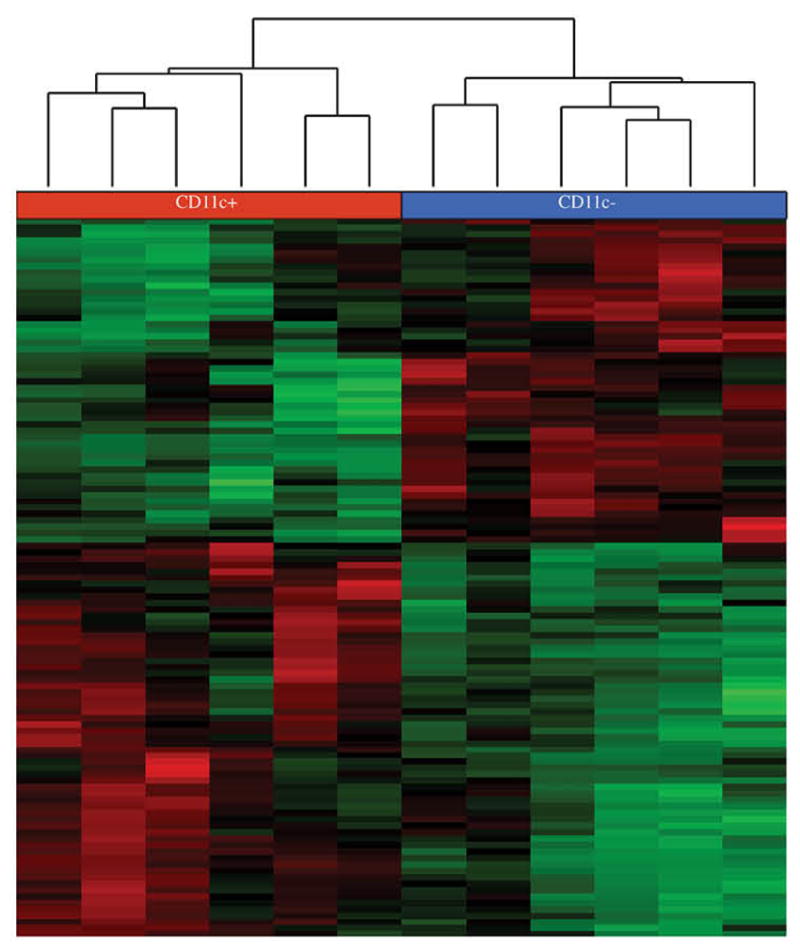
CD19+ CD11c-positive and CD19+ CD11c-negative B cells from C57BL/6J mice 30 days post infection, were isolated by flow cytometric cell sorting. The cells, which were deposited directly into lysis buffer, were collected from 4 different experiments, each containing 1 to 3 mice. Sample libraries were generated using an Illumina Truseq Stranded mRNA Prep kit. Samples were analyzed using either paired-end reagents, using an Illumina MiSeq instrument, or using single-end reagents, using an Illumina Nextseq instrument. The data were analyzed using Partek Flow software as follows: Reads were aligned using TopHat2-2.1.0 with the mm10 assembly, the data were quantified using Partek E/M, and were normalized using RPKM, followed by Quantile normalization. The Partek Gene-Specific Analysis algorithm was used to identify genes between CD11c+ T-bet+ IgM memory cells and CD19+ CD11c-negative B cells. Genes were filtered to limit the analysis to those that exhibited statistically-significant differences (p-value less than or equal to 0.05). Red and green indicate positive or negative differential expression, respectively. The color intensity indicates the standard deviation from the mean, for each gene. The dendrogram was created using average linkage clustering, and a Euclidean point distance metric.
We identified a number of differentially-expressed genes (i.e., those expressed at levels 2-fold higher or lower, and that exhibited statistically-significant differences) that may play an important role in the function of CD11c+ T-bet+ B cells, including IgM memory cells (Table II). The table only shows a subset of genes with known or potentially important B cell functions, and is only intended to be a point of departure, because RNA expression in the cells has not yet been validated using other methods, and because changes in RNA expression does not always correlate with changes in protein expression [63]. The genes were arbitrarily classified into groups based on protein functions, and included integrins, cell surface receptors, innate receptors, Fc Receptors, and transcription factors. Among the factors encoded by genes that exhibited striking differences in expression between IgM memory cells and canonical CD19-positive B cells, were CD11c and T-bet, validating the integrity of the cell separations and analysis. Other differentially expressed integrins, included CD29 (encoded by Itgb1), which, with the alpha 4 integrin CD49d, forms VLA-4, and CD18 (Itgb2), which with CD11c forms the Complement Receptor 4 (CR4). In contrast, another integrin, Itgb7, which encodes a gut homing receptor, was down-regulated on CD11c+ IgM memory cells, consistent with the spleen tropism of the memory cells. Although not detected in the transcriptome analyses, CD11b is also characteristically expressed on CD11c+ IgM memory cells (see Figure 1c). These data together demonstrate that integrin expression is characteristic of these B cells; the role of integrin expression on B cells is not known, although some studies have indicated that integrins can regulate B cell migration within the spleen [64, 65]. CD11c also binds iC3b, fibrinogen, and possibly other ligands [66], but the significance of these possible interactions for CD11c+ IgM memory cell function is presently unclear.
Table II.
Differential Expression of Select Immune-Related Genes in CD11c+ T-bet+ IgM Memory Cells
| Group | Gene symbol | Gene Name | Entrez ID | Fold change | Comments/Possible functions in B cells | References |
|---|---|---|---|---|---|---|
| Integrins | Itgax | CD11c | 3687 | 47.2 | Binds fibrinogen; component of CR4; implicated in phagocytosis | [96, 97] |
| Itgb1 | CD29 | 3688 | 5.1 | Adhesion of B cells to germinal centers; inhibition of apoptosis | [98, 99] | |
| Itgb2 | CD18 | 3689 | 3.2 | Integrin subunit; Component of LFA-1, CR3, CR4 | ||
| Itgb7 | LPAM-1 (β subunit) | 3695 | −2.5 | Lymphocyte homing to Peyer’s patch | [100] | |
|
| ||||||
| Cell Surface Receptors | S1pr5 | Sphingosine-1-Phosphate Receptor 5 | 53637 | 15.3 | Limits lymphocyte egress from lymphoid tissues; T-bet inducible in NK cells | [67, 68] |
| Adora2a | adenosine A2a receptor | 135 | 9.8 | Differentially expressed in memory B cells; Germinal center T cell help | [71, 72] | |
| Il2rb | β subunit of IL-2 receptor | 3560 | 6.1 | Proliferation and differentiation; can synergize with IL-10 | [101, 102] [35] | |
| Cxcr3 | Chemokine Receptor 3 | 2833 | 4.1 | Chemotaxis; Highly expressed in CD27+ B cells during chronic HCV | [103] [104] |
|
| Tnfrsf1b | TNF Receptor 2 | 7133 | 2.5 | May affect B cell migration | [105] | |
| Il10ra | IL10 receptor | 3587 | 2.2 | Il10 may Induce apoptosis or differentiation | [106, 107] | |
| CD72 | CD antigen | 971 | 2.1 | Negatively regulates BCR signaling; Prevent B cell differentiation | [108] [109] | |
| Siglecg | Sialic Acid Binding Ig-Like Lectin | 243958 | −3.4 | Binds sialic acid; involvement in B cell tolerance and TLR response | [79] [110] |
|
|
| ||||||
| Innate Immune Receptors | C1qa, b, c | A, B and C chains of the C1q complex | 712, 713, 714 | 8.6–7.7 | Promotes antibody responses | [111] |
| Cfb | complement factor B | 629 | 5.8 | Regulated by TLR signaling | [112] | |
| Tlr9 | toll-like receptor 9 | 54106 | 3.9 | Innate B cell response to LPS | [113] | |
| Nod1 | nucleotide-binding oligomerization domain containing-1 | 10392 | 3.5 | Intracellular pattern recognition receptor B cell activation | [114] | |
| Cfp | Properdin | 5199 | 2.1 | Positive regulator of complement activation | [115] | |
| Cr2 | CD21 | 1380 | −2.6 | Binds to C3d. Acts with CD19 to enhance | [116] | |
|
| ||||||
| Fc Receptors and related | Fcgr2b | Fc RIIb | 2213 | 3.7 | Inhibitory FcR expressed on B cells l | [117] |
| Fcgr4 | Fc RIV | 246256 | 3.6 | Low affinity IgG receptor | [118] | |
| Fcer1g | FceRI | 2207 | 2.8 | FcR common gamma chain; required signaling component of FcRs | ||
| Fcmr/Faim3 | IgM Fc Receptor | 9214 | −3.9 | Reported IgM receptor; Inhibitor of apoptosis | [69] [119] | |
| Fcer2a | CD23 | 2208 | −10.2 | Negative regulator of BCR signaling | [120] | |
|
| ||||||
| Transcription Factors | Tbx21 | T-Bet | 30009 | 7.8 | Responsible for IgG2a synthesis in response to IFNγ | [45] |
| Bcl6 | B cell lymphoma-6 | 604 | −2.6 | Required in germinal center B cells | [80, 121] | |
We also observed high relative expression of Fc Receptors on CD11c+ IgM memory cells, including FcγRIIb, FcγR4, as well as the FcR common gamma chain, FcγR1g (Table II). FcγRIIb, an inhibitory FcR, and the only FcR reported to be expressed by B cells, exhibited 4-fold higher surface expression on CD11c+ T-bet+ IgM memory cells, relative to canonical CD19+ B cells. In the absence of this inhibitory FcR, IgM memory cells undergo a major expansion, relative to wild-type mice (unpublished data). This observation may be important, because it suggests that FcγRIIb binds immune complexes during ehrlichial infection, and that these complexes negatively regulate IgM memory cells, as has been suggested in other studies [53]. FcγR4 has not been shown to be expressed on canonial B cells, although expression of this receptor has not yet been monitored on IgM memory cells.
Other differences between CD11c IgM memory cells and canonical CD19+ B cells include relative high gene expression of a member of the sphingosine-phosphate receptor (S1PR) superfamily, S1PR5. S1PRs act to regulate egress of lymphocytes from lymphoid tissues, but these receptors also play a role in intra-lymphoid tissue migration of B cells [67, 68]. Expression Faim3, which encodes a putative IgM Fc receptor [69], was negatively-regulated on IgM memory cells (Table II), which would suggest that secreted IgM does not regulate IgM memory cell function.
Another potentially important cellular pathway identified in CD11c+ T-bet+ IgM memory cells is adenosine metabolism; the IgM memory cells express on their surface relatively high amounts of both CD73 [48] and CD39 (unpublished data). CD73 catalyzes the extracellular hydrolysis of AMP to adenosine, and CD39 acts upstream, by catalyzing the conversion of ATP to ADP and AMP [70]. These observations are consistent with other studies that demonstrated that NP-specific switched memory cells characteristically expressed CD73 [71]. CD11c+ T-bet+ IgM memory cells also exhibit relatively high expression of Adora2a, which encodes a G protein-coupled adenosine receptor of the A2A subtype [72, 73]. Adenosine has been reported to exhibit anti-inflammatory activity [74]. It seems likely, then, that IgM memory cells are regulated via adenosine receptors, which likely function by regulating inflammation, as has been reported for CD73. However, our initial studies of CD73-deficient mice have failed to reveal a requirement for this receptor for the generation or function of IgM memory cells (unpublished data), findings which are consistent with other published studies that have reported relatively modest effects of CD73-deficiency in B cells [75]. Nevertheless, further investigation into a role for adenosine and related receptors on memory B cells will likely be informative. Yet other potentially important cell surface receptors positively regulated on IgM memory cells include CXCR3 (which is regulated by T-bet; [76] [77]), TNFR2, the IL-9 Receptor, IL-10R, and CD72. CD72 has been implicated in B cell regulation; in its absence, anergic B cells proliferated and survived in response to self-antigen [78]. mRNA encoding both sialic acid binding Ig-like lectin-G (SIGLEC-G) and CD24 were also down-regulated in CD11c+ T-bet+ IgM memory cells; the former has been reported to be involved in B cell tolerance to self-antigens [79]. Genes encoding innate factors and receptors that were differentially regulated in CD11c+ T-bet+ IgM memory cells also included components of the C1q complex, complement factor B, and properdin, all of which suggest a possible role for complement in B cell regulation, even though complement receptor CR2 mRNA was down-regulated (Table II). RNA encoding TLR9 and NOD1 were also upregulated in CD11c+ T-bet+ IgM memory cells, although a role for the TLR or NOD innate pathways has not been described in innate immunity to ehrlichial infection. The NOD pathway, as well as other innate intracellular signaling pathways, may provide an alternative means of innate activation of CD11c+ T-bet+ cells during ehrlichial infection, but the bacteria are not known to access the cytosolic compartment. Finally, Bcl6 mRNA was downregulated in IgM memory cells. Because BCL-6 regulates GC B cell function [80], this finding is consistent with our data that has shown, on the basis of cell surface expression of CD38 and GL-7, that IgM memory cells are not long-term GC cells. Together, our transcriptome analyses suggest an important role for interactions of CD11c+ T-bet+ IgM memory cells with various components, including FcRs, the extracellular matrix, and innate factors, including complement. These interactions may provide a key to how these cells are maintained during low-level chronic infection, and in autoimmune or aged individuals.
Key Questions
Given both the similarities observed between the CD11c+ T-bet+ B cells in our model, and those described in the field, we propose that CD11c+ and/or T-bet+ expression articulate a novel polarized B cell subset. Such polarization is likely analogous to that encountered by T helper cells, which are polarized based on the conditions that the T cells encounter during activation, as well as by other factors, such as receptor affinity [47][40], or B cell origin (i.e., as Follicular, B-1, or MZ cells). Indeed, T-bet is well-known to be triggered by IFNγ expression, and in an analogous fashion, IFNγ polarizes Th1 cells. In both ours and other studies, CD11c+ T-bet+ B cells are elicited during acute bacterial and viral infections [25, 37, 48], suggesting that, as for T cells, the early milieu encountered by the developing B cells plays an important roles in B cell polarization during infection.
Although CD11c+ T-bet+ B cells were described as ABCs in other studies [35, 36], this moniker may simply reflect that these B cells persist, in aged or autoimmune individuals, under conditions of chronic antigen persistence and/or chronic inflammation. The identification of these B cells during long-term bacterial and viral infections is consistent with this interpretation. It is even possible that CD11c+ T-bet+ B cells that have been identified under chronic conditions may be a novel type of memory B cell. Therefore, we propose that CD11c+ and/or T-bet+ cells, as a group, reflect a similar or identical subset of B cells that is polarized early during early infections by IFNγ, and other pro-inflammatory cytokines, and is maintained long-term, perhaps as memory cells, by chronic antigen, or by chronic low-level innate receptor stimulation, via TLRs, other innate receptors on B cells. Included in these conditions is inflammation associated with aging, also known as “Inflammaging” [81], which may also explain why CD11c+ T-bet+ B cells were detected in healthy aged mice [36].
Our studies have also suggested that the CD11c+ T-bet+ IgM memory cells may be generated independently of GCs, based in part, on our observation that GCs are inhibited during ehrlichial infection [82, 83], and because the B cells do not undergo class switching, which typically occurs in GCs. GC-independent B cell memory is now considered to represent a distinct pathway for generating memory B cells [84], so it is possible that all chronic CD11c+ T-bet+ B cells represent GC-independent memory cells. In this regard, there have been to date only limited studies of BCR repertoire and/or somatic mutation among CD11c+T-bet+ B cells under chronic conditions (Knode et al., 2017). Such information will provide additional clues regarding the origin and maintenance of these novel B cells.
Although the concept that CD11c+ T-bet+ memory B cells may be maintained under conditions of chronic inflammation or antigen may seem oxymoronic, because memory lymphocytes have usually been defined as cells that are maintained in the complete absence of antigen [85]. Such a definition may be far too restrictive in this case, however, for several reasons. First, some chronic viral and bacterial infections generate and maintain memory B cells, including IgM memory cells [25, 86]. Second, antigens have been shown to persist for long periods, on follicular dendritic cells, and antigens so displayed may be important for maintenance of B cell memory [87]. Whether memory B cells require antigen for maintenance has been a topic of debate [88]. In our experimental model, where infection is chronic, the CD11c+ T-bet+ B cells likely have limited contact with antigen, because the bacteria are rare, and are likely sequestered, as obligate intracellular bacteria, inside host cells. Nevertheless, we propose that limited contact with antigen may, in fact, be key for maintaining these cells for long periods. In a similar fashion, autoantigens may be sequestered or present in low quantities that are nevertheless sufficient to maintain long-term CD11c+ T-bet+ memory B cells. Finally, even aged healthy humans appear to support CD11c+ T-bet+ B cells, i.e., ABCs, for long periods, perhaps via long-term antigen persistence [62].
Clinical implications
A final consideration is the relevance of CD11c+ T-bet+ B cells in human health. The studies from Kappler, Marrack, and colleagues suggested an important role in autoimmunity [62], and ours and other studies suggest an important role in long-term host defense. As for other lymphocyte subsets, CD11c+ T-bet+ B cells may play opposing roles, depending on immunological context. Approaches to limit the activity of CD11c+ T-bet+ B cells may therefore be helpful for ameliorating autoimmune diseases such as SLE, where B cells play major roles [35, 89, 90]. Alternatively, it may be advantageous to target CD11c+ T-bet+ B cells with vaccines; knowledge of the factors that drive the development and differentiation of these cells will be important in this context. IgM memory cells, perhaps similar to the CD11c+ T-bet+ B cells we have been described, have now been identified in several infection models and in humans, and likely provide a component of protection, especially for chronic infections. Ongoing and future studies from the field will help to clarify the role of CD11c+ T-bet+ B cells in many different disease contexts.
Highlights.
CD11c+ T-bet+ IgM memory cells are generated during bacterial infection
The B cells are derived independently of TLR signaling
CD11c+ T-bet+ IgM memory B cells exhibit a distinct transcriptome
Are important for long-term immunity during chronic infections and diseases
Acknowledgments
We gratefully acknowledge Drs. Elizabeth Leadbetter (U. of Texas, San Antonio), and Mary Tomayko (Yale University) for helpful comments regarding the manuscript, Karen Gentile and Dr. Frank Middleton (Upstate Medical University), for assistance with the transcriptome analyses, and Drs. Anne Marshak-Rothstein (UMASS Medical School) and Bruce Beutler (UT Southwestern), for providing the Unc93b-deficient mice. This work was supported by U.S. Department of Health and Human Services grant R01AI064678 to G.M.W.
Footnotes
Abbreviations: ABCs, Age-related B cells; AID, activation-induced cytidine deaminase; BCR, B cell receptor; DT, diphtheria toxin; EMLA, E. muris-like agent; FcR, Fc receptor; GC, germinal center; HCL, hairy cell leukemia; IOE, Ixodes ovatus ehrlichia; LPS, lipopolysaccharide; MZ, marginal zone; NP, (4-hydroxy-3-nitrophenyl) acetyl; OMP-19, outer membrane protein 19; SCID, severe combined immunodeficiency; SLE, Systemic lupus erythematosus; Tfh, T follicular helper cell; TLR, toll-like receptor
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Racine R, Chatterjee M, Winslow GM. CD11c expression identifies a population of extrafollicular antigen-specific splenic plasmablasts responsible for CD4 T-independent antibody responses during intracellular bacterial infection. J Immunol. 2008;181:1375–1385. doi: 10.4049/jimmunol.181.2.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker DH, Ismail N, Olano JP, McBride JW, Yu XJ, Feng HM. Ehrlichia chaffeensis: a prevalent, life-threatening, emerging pathogen. Trans Am Clin Climatol Assoc. 2004;115:375–384. [PMC free article] [PubMed] [Google Scholar]
- 3.Nandi B, Chatterjee M, Hogle K, McLaughlin M, MacNamara K, Racine R, Winslow GM. Antigen display, T-cell activation, and immune evasion during acute and chronic ehrlichiosis. Infect Immun. 2009;77:4643–4653. doi: 10.1128/IAI.01433-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pritt BS, Sloan LM, Johnson DK, Munderloh UG, Paskewitz SM, McElroy KM, McFadden JD, Binnicker MJ, Neitzel DF, Liu G, Nicholson WL, Nelson CM, Franson JJ, Martin SA, Cunningham SA, Steward CR, Bogumill K, Bjorgaard ME, Davis JP, McQuiston JH, Warshauer DM, Wilhelm MP, Patel R, Trivedi VA, Eremeeva ME. Emergence of a new pathogenic ehrlichia species, Wisconsin and Minnesota, 2009. The New England journal of medicine. 2011;365:422–429. doi: 10.1056/NEJMoa1010493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li JS, Winslow G. Survival, replication, and antibody susceptibility of Ehrlichia chaffeensis outside of host cells. Infection and Immunity. 2003;71:4229–4237. doi: 10.1128/IAI.71.8.4229-4237.2003. see Commentary Inf. Immun. 4271:4225–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li JS, Yager E, Reilly M, Freeman C, Reddy GR, Chu FK, Winslow G. Outer membrane protein specific monoclonal antibodies protect SCID mice from fatal infection by the obligate intracellular bacterial pathogen Ehrlichia chaffeensis. J Immunol. 2001;166:1855–1862. doi: 10.4049/jimmunol.166.3.1855. [DOI] [PubMed] [Google Scholar]
- 7.MacNamara KC, Oduro K, Martin O, Jones DD, McLaughlin M, Choi K, Borjesson DL, Winslow GM. Infection-induced myelopoiesis during intracellular bacterial infection is critically dependent upon IFN-gamma signaling. J Immunol. 2011;186:1032–1043. doi: 10.4049/jimmunol.1001893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawahara M, Suto C, Shibata S, Futohashi M, Rikihisa Y. Impaired antigen specific responses and enhanced polyclonal stimulation in mice infected with Ehrlichia muris. Microbiol Immunol. 1996;40:575–581. doi: 10.1111/j.1348-0421.1996.tb01111.x. [DOI] [PubMed] [Google Scholar]
- 9.Lin M, Rikihisa Y. Ehrlichia chaffeensis and Anaplasma phagocytophilum lack genes for lipid A biosynthesis and incorporate cholesterol for their survival. Infect Immun. 2003;71:5324–5331. doi: 10.1128/IAI.71.9.5324-5331.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ismail N, Soong L, McBride JW, Valbuena G, Olano JP, Feng HM, Walker DH. Overproduction of TNF-alpha by CD8+ type 1 cells and down-regulation of IFN-gamma production by CD4+ Th1 cells contribute to toxic shock-like syndrome in an animal model of fatal monocytotropic ehrlichiosis. J Immunol. 2004;172:1786–1800. doi: 10.4049/jimmunol.172.3.1786. [DOI] [PubMed] [Google Scholar]
- 11.Mattner J, Debord KL, Ismail N, Goff RD, Cantu C, 3rd, Zhou D, Saint-Mezard P, Wang V, Gao Y, Yin N, Hoebe K, Schneewind O, Walker D, Beutler B, Teyton L, Savage PB, Bendelac A. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. 2005;434:525–529. doi: 10.1038/nature03408. [DOI] [PubMed] [Google Scholar]
- 12.Bitsaktsis C, Huntington J, Winslow G. Production of IFN-gamma by CD4 T cells is essential for resolving ehrlichia infection. J Immunol. 2004;172:6894–6901. doi: 10.4049/jimmunol.172.11.6894. [DOI] [PubMed] [Google Scholar]
- 13.Bitsaktsis C, Winslow G. Fatal recall responses mediated by CD8 T cells during intracellular bacterial challenge infection. J Immunol. 2006;177:4644–4651. doi: 10.4049/jimmunol.177.7.4644. [DOI] [PubMed] [Google Scholar]
- 14.Shibata S, Kawahara M, Rikihisa Y, Fujita H, Watanabe Y, Suto C, Ito T. New Ehrlichia species closely related to Ehrlichia chaffeensis isolated from Ixodes ovatus ticks in Japan. J Clin Microbiol. 2000;38:1331–1338. doi: 10.1128/jcm.38.4.1331-1338.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bitsaktsis C, Nandi B, Racine R, MacNamara KC, Winslow G. T-Cell-independent humoral immunity is sufficient for protection against fatal intracellular ehrlichia infection. Infect Immun. 2007;75:4933–4941. doi: 10.1128/IAI.00705-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Racine R, McLaughlin M, Jones DD, Wittmer ST, MacNamara KC, Woodland DL, Winslow GM. IgM production by bone marrow plasmablasts contributes to long-term protection against intracellular bacterial infection. J Immunol. 2011;186:1011–1021. doi: 10.4049/jimmunol.1002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones DD, DeIulio GA, Winslow GM. Antigen-driven induction of polyreactive IgM during intracellular bacterial infection. J Immunol. 2012;189:1440–1447. doi: 10.4049/jimmunol.1200878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y, Tung JW, Ghosn EE, Herzenberg LA, Herzenberg LA. Division and differentiation of natural antibody-producing cells in mouse spleen. Proc Natl Acad Sci U S A. 2007;104:4542–4546. doi: 10.1073/pnas.0700001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Postigo AA, Corbi AL, Sanchez-Madrid F, de Landazuri MO. Regulated expression and function of CD11c/CD18 integrin on human B lymphocytes. Relation between attachment to fibrinogen and triggering of proliferation through CD11c/CD18. J Exp Med. 1991;174:1313–1322. doi: 10.1084/jem.174.6.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta AK, Sachdeva MU, Ahluwalia J, Das R, Naseem S, Sharma P, Kumar N, Malhotra P, Varma N, Varma S. Haematological profile of 21 patients with hairy cell leukaemia in a tertiary care centre of north India. Indian J Med Res. 2015;142:426–429. doi: 10.4103/0971-5916.169204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matutes E, Martinez-Trillos A, Campo E. Hairy cell leukaemia-variant: Disease features and treatment. Best Pract Res Clin Haematol. 2015;28:253–263. doi: 10.1016/j.beha.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Ehrhardt GR, Hijikata A, Kitamura H, Ohara O, Wang JY, Cooper MD. Discriminating gene expression profiles of memory B cell subpopulations. J Exp Med. 2008;205:1807–1817. doi: 10.1084/jem.20072682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ehrhardt GR, Davis RS, Hsu JT, Leu CM, Ehrhardt A, Cooper MD. The inhibitory potential of Fc receptor homolog 4 on memory B cells. Proc Natl Acad Sci U S A. 2003;100:13489–13494. doi: 10.1073/pnas.1935944100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Borrego F, Nagata S, Tolnay M. Fc Receptor-like 5 Expression Distinguishes Two Distinct Subsets of Human Circulating Tissue-like Memory B Cells. J Immunol. 2016;196:4064–4074. doi: 10.4049/jimmunol.1501027. [DOI] [PubMed] [Google Scholar]
- 25.Chang LY, Li Y, Kaplan DE. Hepatitis C viraemia reversibly maintains subset of antigen-specific T-bet+ tissue-like memory B cells. J Viral Hepat. 2016 doi: 10.1111/jvh.12659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis RS. Fc receptor-like molecules. Annu Rev Immunol. 2007;25:525–560. doi: 10.1146/annurev.immunol.25.022106.141541. [DOI] [PubMed] [Google Scholar]
- 27.Sohn HW, Krueger PD, Davis RS, Pierce SK. FcRL4 acts as an adaptive to innate molecular switch dampening BCR signaling and enhancing TLR signaling. Blood. 2011;118:6332–6341. doi: 10.1182/blood-2011-05-353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson TJ, Fuchs A, Colonna M. Cutting edge: human FcRL4 and FcRL5 are receptors for IgA and IgG. J Immunol. 2012;188:4741–4745. doi: 10.4049/jimmunol.1102651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moir S, Ho J, Malaspina A, Wang W, DiPoto AC, O'Shea MA, Roby G, Kottilil S, Arthos J, Proschan MA, Chun TW, Fauci AS. Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J Exp Med. 2008;205:1797–1805. doi: 10.1084/jem.20072683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rakhmanov M, Keller B, Gutenberger S, Foerster C, Hoenig M, Driessen G, van der Burg M, van Dongen JJ, Wiech E, Visentini M, Quinti I, Prasse A, Voelxen N, Salzer U, Goldacker S, Fisch P, Eibel H, Schwarz K, Peter HH, Warnatz K. Circulating CD21low B cells in common variable immunodeficiency resemble tissue homing, innate-like B cells. Proc Natl Acad Sci U S A. 2009;106:13451–13456. doi: 10.1073/pnas.0901984106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Visentini M, Conti V, Cagliuso M, Siciliano G, Scagnolari C, Casato M, Fiorilli M. Persistence of a large population of exhausted monoclonal B cells in mixed cryoglobuliemia after the eradication of hepatitis C virus infection. J Clin Immunol. 2012;32:729–735. doi: 10.1007/s10875-012-9677-0. [DOI] [PubMed] [Google Scholar]
- 32.Isnardi I, Ng YS, Menard L, Meyers G, Saadoun D, Srdanovic I, Samuels J, Berman J, Buckner JH, Cunningham-Rundles C, Meffre E. Complement receptor 2/CD21- human naive B cells contain mostly autoreactive unresponsive clones. Blood. 2010;115:5026–5036. doi: 10.1182/blood-2009-09-243071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Charles ED, Brunetti C, Marukian S, Ritola KD, Talal AH, Marks K, Jacobson IM, Rice CM, Dustin LB. Clonal B cells in patients with hepatitis C virus-associated mixed cryoglobulinemia contain an expanded anergic CD21low B-cell subset. Blood. 2011;117:5425–5437. doi: 10.1182/blood-2010-10-312942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ebisawa M, Hase K, Takahashi D, Kitamura H, Knoop KA, Williams IR, Ohno H. CCR6hiCD11c(int) B cells promote M-cell differentiation in Peyer's patch. Int Immunol. 2011;23:261–269. doi: 10.1093/intimm/dxq478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, Marrack P. TLR7-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood. 2011;118:1305–1311. doi: 10.1182/blood-2011-01-331462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hao Y, O'Neill P, Naradikian MS, Scholz JL, Cancro MP. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood. 2011;118:1294–1304. doi: 10.1182/blood-2011-01-330530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rubtsova K, Rubtsov AV, van Dyk LF, Kappler JW, Marrack P. T-box transcription factor T-bet, a key player in a unique type of B-cell activation essential for effective viral clearance. Proc Natl Acad Sci U S A. 2013;110:E3216–3224. doi: 10.1073/pnas.1312348110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naradikian MS, Myles A, Beiting DP, Roberts KJ, Dawson L, Herati RS, Bengsch B, Linderman SL, Stelekati E, Spolski R, Wherry EJ, Hunter C, Hensley SE, Leonard WJ, Cancro MP. Cutting Edge: IL-4, IL-21, and IFN-gamma interact to govern T-bet and CD11c expression in TLR-activated B cells. J Immunol. 2016;197:1023–1028. doi: 10.4049/jimmunol.1600522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kovesdi D, Angyal A, Huber K, Szili D, Sarmay G. T-bet is a new synergistic meeting point for the BCR and TLR9 signaling cascades. Eur J Immunol. 2014;44:887–893. doi: 10.1002/eji.201343841. [DOI] [PubMed] [Google Scholar]
- 40.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 41.Gerth AJ, Lin L, Peng SL. T-bet regulates T-independent IgG2a class switching. Int Immunol. 2003;15:937–944. doi: 10.1093/intimm/dxg093. [DOI] [PubMed] [Google Scholar]
- 42.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 43.Barnett BE, Staupe RP, Odorizzi PM, Palko O, Tomov VT, Mahan AE, Gunn B, Chen D, Paley MA, Alter G, Reiner SL, Lauer GM, Teijaro JR, Wherry EJ. Cutting Edge: B Cell-Intrinsic T-bet Expression Is Required To Control Chronic Viral Infection. J Immunol. 2016;197:1017–1022. doi: 10.4049/jimmunol.1500368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang NS, McHeyzer-Williams LJ, Okitsu SL, Burris TP, Reiner SL, McHeyzer-Williams MG. Divergent transcriptional programming of class-specific B cell memory by T-bet and RORalpha. Nat Immunol. 2012;13:604–611. doi: 10.1038/ni.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harris DP, Goodrich S, Gerth AJ, Peng SL, Lund FE. Regulation of IFN-gamma production by B effector 1 cells: essential roles for T-bet and the IFN-gamma receptor. J Immunol. 2005;174:6781–6790. doi: 10.4049/jimmunol.174.11.6781. [DOI] [PubMed] [Google Scholar]
- 46.Yoshimoto T, Okada K, Morishima N, Kamiya S, Owaki T, Asakawa M, Iwakura Y, Fukai F, Mizuguchi J. Induction of IgG2a class switching in B cells by IL-27. J Immunol. 2004;173:2479–2485. doi: 10.4049/jimmunol.173.4.2479. [DOI] [PubMed] [Google Scholar]
- 47.Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol. 2013;13:777–789. doi: 10.1038/nri3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yates JL, Racine R, McBride KM, Winslow GM. T cell-dependent IgM memory B cells generated during bacterial infection are required for IgG responses to antigen challenge. J Immunol. 2013;191:1240–1249. doi: 10.4049/jimmunol.1300062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dogan I, Bertocci B, Vilmont V, Delbos F, Megret J, Storck S, Reynaud CA, Weill JC. Multiple layers of B cell memory with different effector functions. Nat Immunol. 2009;10:1292–1299. doi: 10.1038/ni.1814. [DOI] [PubMed] [Google Scholar]
- 50.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yefenof E, Sanders VM, Snow EC, Noelle RJ, Oliver KG, Uhr JW, Vitetta ES. Preparation and analysis of antigen-specific memory B cells. J Immunol. 1985;135:3777–3784. [PubMed] [Google Scholar]
- 52.Zan-Bar I, Strober S, Vitetta ES. The relationship between surface immunoglobulin isotype and immune function of murine B lymphocytes. IV. Role of IgD-bearing cells in the propagation of immunologic memory. J Immunol. 1979;123:925–930. [PubMed] [Google Scholar]
- 53.Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science. 2011;331:1203–1207. doi: 10.1126/science.1201730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seifert M, Kuppers R. Molecular footprints of a germinal center derivation of human IgM+(IgD+)CD27+ B cells and the dynamics of memory B cell generation. J Exp Med. 2009;206:2659–2669. doi: 10.1084/jem.20091087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nguyen HV, Mouly E, Chemin K, Luinaud R, Despres R, Fermand JP, Arnulf B, Bories JC. The Ets-1 transcription factor is required for Stat1-mediated T-bet expression and IgG2a class switching in mouse B cells. Blood. 2012;119:4174–4181. doi: 10.1182/blood-2011-09-378182. [DOI] [PubMed] [Google Scholar]
- 56.Peng SL, Szabo SJ, Glimcher LH. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci U S A. 2002;99:5545–5550. doi: 10.1073/pnas.082114899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu N, Ohnishi N, Ni L, Akira S, Bacon KB. CpG directly induces T-bet expression and inhibits IgG1 and IgE switching in B cells. Nat Immunol. 2003;4:687–693. doi: 10.1038/ni941. [DOI] [PubMed] [Google Scholar]
- 58.Mamani-Matsuda M, Cosma A, Weller S, Faili A, Staib C, Garcon L, Hermine O, Beyne-Rauzy O, Fieschi C, Pers JO, Arakelyan N, Varet B, Sauvanet A, Berger A, Paye F, Andrieu JM, Michel M, Godeau B, Buffet P, Reynaud CA, Weill JC. The human spleen is a major reservoir for long-lived vaccinia virus-specific memory B cells. Blood. 2008;111:4653–4659. doi: 10.1182/blood-2007-11-123844. [DOI] [PubMed] [Google Scholar]
- 59.Della Valle L, Dohmen SE, Verhagen OJ, Berkowska MA, Vidarsson G, van der Schoot Ellen C. The majority of human memory B cells recognizing RhD and tetanus resides in IgM+ B cells. J Immunol. 2014;193:1071–1079. doi: 10.4049/jimmunol.1400706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim YM. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. The Journal of Cell Biology. 2007;177:265–275. doi: 10.1083/jcb.200612056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pelka K, Phulphagar K, Zimmermann J, Stahl R, Schmid-Burgk JL, Schmidt T, Spille JH, Labzin LI, Agrawal S, Kandimalla ER, Casanova JL, Hornung V, Marshak-Rothstein A, Honing S, Latz E. Cutting edge: the UNC93B1 tyrosine-based motif regulates trafficking and TLR responses via separate mechanisms. J Immunol. 2014;193:3257–3261. doi: 10.4049/jimmunol.1301886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rubtsov AV, Rubtsova K, Kappler JW, Marrack P. TLR7 drives accumulation of ABCs and autoantibody production in autoimmune-prone mice. Immunologic research. 2013;55:210–216. doi: 10.1007/s12026-012-8365-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guo Y, Xiao P, Lei S, Deng F, Xiao GG, Liu Y, Chen X, Li L, Wu S, Chen Y, Jiang H, Tan L, Xie J, Zhu X, Liang S, Deng H. How is mRNA expression predictive for protein expression? A correlation study on human circulating monocytes. Acta Biochim Biophys Sin (Shanghai) 2008;40:426–436. doi: 10.1111/j.1745-7270.2008.00418.x. [DOI] [PubMed] [Google Scholar]
- 64.Lu TT, Cyster JG. Integrin-mediated long-term B cell retention in the splenic marginal zone. Science. 2002;297:409–412. doi: 10.1126/science.1071632. [DOI] [PubMed] [Google Scholar]
- 65.Lo CG, Lu TT, Cyster JG. Integrin-dependence of lymphocyte entry into the splenic white pulp. J Exp Med. 2003;197:353–361. doi: 10.1084/jem.20021569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sadhu C, Ting HJ, Lipsky B, Hensley K, Garcia-Martinez LF, Simon SI, Staunton DE. CD11c/CD18: novel ligands and a role in delayed-type hypersensitivity. J Leukoc Biol. 2007;81:1395–1403. doi: 10.1189/jlb.1106680. [DOI] [PubMed] [Google Scholar]
- 67.Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94. doi: 10.1146/annurev-immunol-020711-075011. [DOI] [PubMed] [Google Scholar]
- 68.Jenne CN, Enders A, Rivera R, Watson SR, Bankovich AJ, Pereira JP, Xu Y, Roots CM, Beilke JN, Banerjee A, Reiner SL, Miller SA, Weinmann AS, Goodnow CC, Lanier LL, Cyster JG, Chun J. T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J Exp Med. 2009;206:2469–2481. doi: 10.1084/jem.20090525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choi SC, Wang H, Tian L, Murakami Y, Shin DM, Borrego F, Morse HC, 3rd, Coligan JE. Mouse IgM Fc receptor, FCMR, promotes B cell development and modulates antigen-driven immune responses. J Immunol. 2013;190:987–996. doi: 10.4049/jimmunol.1202227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Antonioli L, Pacher P, Vizi ES, Hasko G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19:355–367. doi: 10.1016/j.molmed.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tomayko MM, Anderson SM, Brayton CE, Sadanand S, Steinel NC, Behrens TW, Shlomchik MJ. Systematic comparison of gene expression between murine memory and naive B cells demonstrates that memory B cells have unique signaling capabilities. J Immunol. 2008;181:27–38. doi: 10.4049/jimmunol.181.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Abbott RK, Silva M, Labuda J, Thayer M, Cain DW, Philbrook P, Sethumadhavan S, Hatfield S, Ohta A, Sitkovsky M. The GS protein-coupled A2a adenosine receptor controls T cell help in the germinal center. J Biol Chem. 2017;292:1211–1217. doi: 10.1074/jbc.C116.764043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lukashev D, Ohta A, Apasov S, Chen JF, Sitkovsky M. Cutting edge: Physiologic attenuation of proinflammatory transcription by the Gs protein-coupled A2A adenosine receptor in vivo. J Immunol. 2004;173:21–24. doi: 10.4049/jimmunol.173.1.21. [DOI] [PubMed] [Google Scholar]
- 74.Kaku H, Cheng KF, Al-Abed Y, Rothstein TL. A novel mechanism of B cell-mediated immune suppression through CD73 expression and adenosine production. J Immunol. 2014;193:5904–5913. doi: 10.4049/jimmunol.1400336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Conter LJ, Song E, Shlomchik MJ, Tomayko MM. CD73 expression is dynamically regulated in the germinal center and bone marrow plasma cells are diminished in its absence. PLoS One. 2014;9:e92009. doi: 10.1371/journal.pone.0092009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Serre K, Cunningham AF, Coughlan RE, Lino AC, Rot A, Hub E, Moser K, Manz R, Ferraro A, Bird R, Toellner KM, Demengeot J, MacLennan IC, Mohr E. CD8 T cells induce T-bet-dependent migration toward CXCR3 ligands by differentiated B cells produced during responses to alum-protein vaccines. Blood. 2012;120:4552–4559. doi: 10.1182/blood-2012-03-417733. [DOI] [PubMed] [Google Scholar]
- 78.Li DH, Winslow MM, Cao TM, Chen AH, Davis CR, Mellins ED, Utz PJ, Crabtree GR, Parnes JR. Modulation of peripheral B cell tolerance by CD72 in a murine model. Arthritis Rheum. 2008;58:3192–3204. doi: 10.1002/art.23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Poe JC, Tedder TF. CD22 and Siglec-G in B cell function and tolerance. Trends Immunol. 2012;33:413–420. doi: 10.1016/j.it.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, Verma NK, Smyth MJ, Rigby RJ, Vinuesa CG. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med. 2010;207:353–363. doi: 10.1084/jem.20091738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nikolich-Zugich J, Li G, Uhrlaub JL, Renkema KR, Smithey MJ. Age-related changes in CD8 T cell homeostasis and immunity to infection. Semin Immunol. 2012;24:356–364. doi: 10.1016/j.smim.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Racine R, Jones DD, Chatterjee M, McLaughlin M, Macnamara KC, Winslow GM. Impaired germinal center responses and suppression of local IgG production during intracellular bacterial infection. J Immunol. 2010;184:5085–5093. doi: 10.4049/jimmunol.0902710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.MacNamara KC, Racine R, Chatterjee M, Borjesson D, Winslow GM. Diminished hematopoietic activity associated with alterations in innate and adaptive immunity in a mouse model of human monocytic ehrlichiosis. Infect Immun. 2009;77:4061–4069. doi: 10.1128/IAI.01550-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol. 2015;15:149–159. doi: 10.1038/nri3802. [DOI] [PubMed] [Google Scholar]
- 85.Maruyama M, Lam KP, Rajewsky K. Memory B-cell persistence is independent of persisting immunizing antigen. Nature. 2000;407:636–642. doi: 10.1038/35036600. [DOI] [PubMed] [Google Scholar]
- 86.Krishnamurty AT, Thouvenel CD, Portugal S, Keitany GJ, Kim KS, Holder A, Crompton PD, Rawlings DJ, Pepper M. Somatically hypermutated Plasmodium-specific IgM(+) memory B cells are rapid, plastic, early responders upon malaria rechallenge. Immunity. 2016;45:402–414. doi: 10.1016/j.immuni.2016.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bachmann MF, Odermatt B, Hengartner H, Zinkernagel RM. Induction of long-lived germinal centers associated with persisting antigen after viral infection. J Exp Med. 1996;183:2259–2269. doi: 10.1084/jem.183.5.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gray D. A role for antigen in the maintenance of immunological memory. Nature Rev Immunol. 2002;2:60–65. doi: 10.1038/nri706. [DOI] [PubMed] [Google Scholar]
- 89.Wehr C, Eibel H, Masilamani M, Illges H, Schlesier M, Peter HH, Warnatz K. A new CD21low B cell population in the peripheral blood of patients with SLE. Clin Immunol. 2004;113:161–171. doi: 10.1016/j.clim.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 90.Kosalka J, Jakiela B, Musial J. Changes of memory B- and T-cell subsets in lupus nephritis patients. Folia Histochem Cytobiol. 2016;54:32–41. doi: 10.5603/FHC.a2016.0005. [DOI] [PubMed] [Google Scholar]
- 91.Wang X, Spielberger R, Huang Q. Hairy cell leukemia variant, a new entity of the WHO 2008. J Clin Oncol. 2011;29:e864–866. doi: 10.1200/JCO.2011.37.8497. [DOI] [PubMed] [Google Scholar]
- 92.Arons E, Suntum T, Stetler-Stevenson M, Kreitman RJ. VH4–34+ hairy cell leukemia, a new variant with poor prognosis despite standard therapy. Blood. 2009;114:4687–4695. doi: 10.1182/blood-2009-01-201731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vasquez C, Franco MA, Angel J. Rapid proliferation and differentiation of a subset of circulating IgM memory B cells to a CpG/cytokine stimulus in vitro. PLoS One. 2015;10:e0139718. doi: 10.1371/journal.pone.0139718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Visentini M, Pascolini S, Mitrevski M, Marrapodi R, Del Padre M, Todi L, Camponeschi A, Axiotis E, Carlesimo M, De Santis A, Fiorilli M, Casato M. Hepatitis B virus causes mixed cryoglobulinaemia by driving clonal expansion of innate B-cells producing a VH1–69-encoded antibody. Clin Exp Rheumatol. 2016;34:S28–32. [PubMed] [Google Scholar]
- 95.Warnatz K, Wehr C, Drager R, Schmidt S, Eibel H, Schlesier M, Peter HH. Expansion of CD19(hi)CD21(lo/neg) B cells in common variable immunodeficiency (CVID) patients with autoimmune cytopenia. Immunobiology. 2002;206:502–513. doi: 10.1078/0171-2985-00198. [DOI] [PubMed] [Google Scholar]
- 96.Sadhu C, Ting HJ, Lipsky B, Hensley K, Garcia-Martinez LF, Simon SI, Staunton DE. CD11c/CD18: novel ligands and a role in delayed-type hypersensitivity. Journal of leukocyte biology. 2007;81:1395–1403. doi: 10.1189/jlb.1106680. [DOI] [PubMed] [Google Scholar]
- 97.Taborda CP, Casadevall A. CR3 (CD11b/CD18) and CR4 (CD11c/CD18) are involved in complement-independent antibody-mediated phagocytosis of Cryptococcus neoformans. Immunity. 2002;16:791–802. doi: 10.1016/s1074-7613(02)00328-x. [DOI] [PubMed] [Google Scholar]
- 98.Freedman AS, Munro JM, Rice GE, Bevilacqua MP, Morimoto C, McIntyre BW, Rhynhart K, Pober JS, Nadler LM. Adhesion of human B cells to germinal centers in vitro involves VLA-4 and INCAM-110. Science. 1990;249:1030–1033. doi: 10.1126/science.1697696. [DOI] [PubMed] [Google Scholar]
- 99.Hayashida K, Shimaoka Y, Ochi T, Lipsky PE. Rheumatoid arthritis synovial stromal cells inhibit apoptosis and up-regulate Bcl-xL expression by B cells in a CD49/CD29-CD106-dependent mechanism. J Immunol. 2000;164:1110–1116. doi: 10.4049/jimmunol.164.2.1110. [DOI] [PubMed] [Google Scholar]
- 100.Holzmann B, McIntyre BW, Weissman IL. Identification of a murine Peyer's patch--specific lymphocyte homing receptor as an integrin molecule with an alpha chain homologous to human VLA-4 alpha. Cell. 1989;56:37–46. doi: 10.1016/0092-8674(89)90981-1. [DOI] [PubMed] [Google Scholar]
- 101.Fluckiger AC, Garrone P, Durand I, Galizzi JP, Banchereau J. Interleukin 10 (IL-10) upregulates functional high affinity IL-2 receptors on normal and leukemic B lymphocytes. J Exp Med. 1993;178:1473–1481. doi: 10.1084/jem.178.5.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Muraguchi A, Kehrl JH, Longo DL, Volkman DJ, Smith KA, Fauci AS. Interleukin 2 receptors on human B cells. Implications for the role of interleukin 2 in human B cell function. J Exp Med. 1985;161:181–197. doi: 10.1084/jem.161.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hauser AE, Debes GF, Arce S, Cassese G, Hamann A, Radbruch A, Manz RA. Chemotactic responsiveness toward ligands for CXCR3 and CXCR4 is regulated on plasma blasts during the time course of a memory immune response. J Immunol. 2002;169:1277–1282. doi: 10.4049/jimmunol.169.3.1277. [DOI] [PubMed] [Google Scholar]
- 104.Mizuochi T, Ito M, Saito K, Kasai M, Kunimura T, Morohoshi T, Momose H, Hamaguchi I, Takai K, Iino S, Suzuki M, Mochida S, Ikebuchi K, Yamaguchi K. Possible recruitment of peripheral blood CXCR3+ CD27+ CD19+ B cells to the liver of chronic hepatitis C patients. J Interferon Cytokine Res. 2010;30:243–252. doi: 10.1089/jir.2009.0047. [DOI] [PubMed] [Google Scholar]
- 105.Corcione A, Ottonello L, Tortolina G, Tasso P, Ghiotto F, Airoldi I, Taborelli G, Malavasi F, Dallegri F, Pistoia V. Recombinant tumor necrosis factor enhances the locomotion of memory and naive B lymphocytes from human tonsils through the selective engagement of the type II receptor. Blood. 1997;90:4493–4501. [PubMed] [Google Scholar]
- 106.Rousset F, Garcia E, Defrance T, Peronne C, Vezzio N, Hsu DH, Kastelein R, Moore KW, Banchereau J. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc Natl Acad Sci U S A. 1992;89:1890–1893. doi: 10.1073/pnas.89.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Itoh K, Hirohata S. The role of IL-10 in human B cell activation, proliferation, and differentiation. J Immunol. 1995;154:4341–4350. [PubMed] [Google Scholar]
- 108.Adachi T, Wakabayashi C, Nakayama T, Yakura H, Tsubata T. CD72 negatively regulates signaling through the antigen receptor of B cells. J Immunol. 2000;164:1223–1229. doi: 10.4049/jimmunol.164.3.1223. [DOI] [PubMed] [Google Scholar]
- 109.Yamazaki T, Nagumo H, Hayashi T, Sugane K, Agematsu K. CD72-mediated suppression of human naive B cell differentiation by down-regulating X-box binding protein 1. Eur J Immunol. 2005;35:2325–2334. doi: 10.1002/eji.200425639. [DOI] [PubMed] [Google Scholar]
- 110.Jellusova J, Nitschke L. Regulation of B cell functions by the sialic acid-binding receptors siglec-G and CD22. Front Immunol. 2011;2:96. doi: 10.3389/fimmu.2011.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sorman A, Zhang L, Ding Z, Heyman B. How antibodies use complement to regulate antibody responses. Mol Immunol. 2014;61:79–88. doi: 10.1016/j.molimm.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 112.Zou L, Feng Y, Li Y, Zhang M, Chen C, Cai J, Gong Y, Wang L, Thurman JM, Wu X, Atkinson JP, Chao W. Complement factor B is the downstream effector of TLRs and plays an important role in a mouse model of severe sepsis. J Immunol. 2013;191:5625–5635. doi: 10.4049/jimmunol.1301903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Avalos AM, Busconi L, Marshak-Rothstein A. Regulation of autoreactive B cell responses to endogenous TLR ligands. Autoimmunity. 2010;43:76–83. doi: 10.3109/08916930903374618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Petterson T, Jendholm J, Mansson A, Bjartell A, Riesbeck K, Cardell LO. Effects of NOD-like receptors in human B lymphocytes and crosstalk between NOD1/NOD2 and Toll-like receptors. J Leukoc Biol. 2011;89:177–187. doi: 10.1189/jlb.0210061. [DOI] [PubMed] [Google Scholar]
- 115.Hourcade DE. The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem. 2006;281:2128–2132. doi: 10.1074/jbc.M508928200. [DOI] [PubMed] [Google Scholar]
- 116.Poe JC, Hasegawa M, Tedder TF. CD19, CD21, and CD22: multifaceted response regulators of B lymphocyte signal transduction. Int Rev Immunol. 2001;20:739–762. doi: 10.3109/08830180109045588. [DOI] [PubMed] [Google Scholar]
- 117.Espeli M, Smith KG, Clatworthy MR. FcgammaRIIB and autoimmunity. Immunol Rev. 2016;269:194–211. doi: 10.1111/imr.12368. [DOI] [PubMed] [Google Scholar]
- 118.Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcgammaRIV: a novel FcR with distinct IgG subclass specificity. Immunity. 2005;23:41–51. doi: 10.1016/j.immuni.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 119.Kubagawa H, Oka S, Kubagawa Y, Torii I, Takayama E, Kang DW, Gartland GL, Bertoli LF, Mori H, Takatsu H, Kitamura T, Ohno H, Wang JY. Identity of the elusive IgM Fc receptor (FcmuR) in humans. J Exp Med. 2009;206:2779–2793. doi: 10.1084/jem.20091107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu C, Richard K, Wiggins M, Zhu X, Conrad DH, Song W. CD23 can negatively regulate B-cell receptor signaling. Sci Rep. 2016;6:25629. doi: 10.1038/srep25629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cattoretti G, Chang CC, Cechova K, Zhang J, Ye BH, Falini B, Louie DC, Offit K, Chaganti RS, Dalla-Favera R. BCL-6 protein is expressed in germinal-center B cells. Blood. 1995;86:45–53. [PubMed] [Google Scholar]