

Abstract
Inosine is an endogenous nucleoside that is produced by metabolic deamination of adenosine. Inosine is metabolically more stable (half-life 15h) than adenosine (half-life <10 s). Inosine exerts anti-inflammatory and immunomodulatory effects similar to those observed with adenosine. These effects are mediated in part through the adenosine A2A receptor (A2AR). Relative to adenosine inosine exhibits a lower affinity towards the A2AR. Therefore, it is generally believed that inosine is incapable of activating the A2AR through direct engagement, but indirectly activates the A2AR upon metabolic conversion to higher affinity adenosine. A handful of studies, however, have provided evidence for direct inosine engagement at the A2AR leading to activation of downstream signaling events and inhibition of cytokine production. Here, we demonstrate that under conditions devoid of adenosine, inosine as well as an analog of inosine 6-S-[(4-Nitrophenyl)methyl]-6-thioinosine selectively and dose-dependently activated A2AR-mediated cAMP production and ERK1/2 phosphorylation in CHO cells stably expressing the human A2AR. Inosine also inhibited LPS-stimulated TNF-α, CCL3 and CCL4 production by splenic monocytes in an A2AR-dependent manner. In addition, we demonstrate that a positive allosteric modulator (PAM) of the A2AR enhanced inosine-mediated cAMP production, ERK1/2 phosphorylation and inhibition of pro-inflammatory cytokine and chemokine production. The cumulative effects of allosteric enhancement of adenosine-mediated and inosine-mediated A2AR activation may be the basis for the sustained anti-inflammatory and immunomodulatory effects observed in vivo and thereby provide insights into potential therapeutic interventions for inflammation- and immune-mediated diseases.
Keywords: Inosine, A2AR, PAM, cAMP, ERK1/2, cytokines
1. Introduction
The endogenous purine nucleoside, inosine is formed through the metabolic conversion of adenosine by the enzyme adenosine deaminase (ADA). It is produced both extracellularly as well as intracellularly during normal cell metabolism. Inosine has a longer half-life (15 h; [1]) relative to adenosine (< 10 s; [2]) and consequently, the basal level of inosine in the interstitial fluid can be 2–7 times higher than that of adenosine. In agreement with this observation, in pathological conditions, there is an increase in tissue inosine levels [3–6].
Resembling adenosine, inosine exerts a wide range of anti-inflammatory and immunomodulatory properties. These include inhibition of proinflammatory cytokine and chemokine production [7–9], induction of anti-inflammatory cytokine production [7], improvement of islet transplant survival [10], reduced multiorgan inflammation and prolonged survival in scurfy mice [11] as well as alleviation of clinical signs of myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis [12], allergic lung inflammation [13], streptozotocin-induced non-obese type 1 diabetes [14], TNBS-induced colitis [15] and glycodeoxycholic acid-induced acute pancreatitis [16].
Inosine exerts anti-inflammatory and immunomodulatory effects through specific membrane bound G protein-coupled receptors (GPCRs) termed P1-purinoceptors, also known as adenosine receptors (AR). There are four AR subtypes termed A1R, A2AR, A2BR and A3R. Among them, A2AR plays a critical nonredundant role in down-regulating inflammation [17]. A2AR is coupled to the stimulatory G protein Gαs [18]. Adenosine engagement at the A2AR leads to an increase in intracellular cAMP levels as well as phosphorylation of signal-regulated kinase-1 and -2 (ERK1/2). Utilizing a combination of label-free, cell-based, and membrane-based functional assays in conjunction with an equilibrium agonist-binding assay we have recently provided in vitro evidence for direct inosine engagement at the A2AR and subsequent induction of downstream cAMP production and ERK1/2 phosphorylation [19].
GPCRs initiate signaling upon binding of cognate ligands at evolutionarily conserved sites termed orthosteric sites. In addition, GPCRs also contain allosteric sites that are topologically distinct from the orthosteric sites. Therefore, structural determinants of ligand binding at the orthosteric and allosteric sites are inherently different. Unlike orthosteric ligands, allosteric ligands have little or no intrinsic ability to activate GPCRs upon engagement at the allosteric site. They only modulate orthosteric ligand-mediated receptor function through conformational changes that manifest as altered affinity and/or efficacy of the receptors towards orthosteric ligands, hence they preserve the endogenous orthosteric ligand-mediated physiological responses. Allosteric ligands that enhance the orthosteric ligand-mediated responses are termed positive allosteric modulators (PAMs).
To examine the potential of allosteric enhancement of the A2AR function to alter inflammatory immune responses in vitro and in vivo, we developed a series of compounds with PAM activity. One of these compounds, AEA061, is a small molecule that meets the stringent criteria of a PAM of the A2AR [20]. AEA061 has no intrinsic activity towards either rat or human A2ARs but enhances the affinity and maximal response of the receptors to adenosine. AEA061 is selective towards A2AR. It does not affect potency or efficacy of A1R or A3R but increases the potency of A2BR by two-fold without altering efficacy (unpublished data). Positive allosteric modulation of the A2AR with AEA061 inhibits inflammatory cytokine and chemokine production in vitro and reduces circulating plasma TNF-α and MCP-1 levels and increases plasma IL-10 in endotoxemic A2AR intact, but not in A2AR deficient, mice [20].
We sought to further establish that inosine directly, and not through its conversion to adenosine via salvage pathways, activates the A2AR. To this end, we examined the ability of inosine as well as the inosine analog 6-S-[(4-Nitrophenyl)methyl]-6-thioinosine (NBMPR) to activate the A2AR in cell-based and cell-free assays in the presence of ADA and AEA061. We demonstrated that inosine as well as NBMPR activates the A2AR in the presence and in the absence of ADA. Moreover, both inosine and NBMPR inhibit pro-inflammatory cytokine and chemokine production by A2AR intact, but not by A2AR deficient mouse splenic monocytes. We next sought to determine if inosine-mediated A2AR activation is amenable to allosteric modulation. We now present data supporting the hypothesis that positive allosteric modulation of the A2AR enhances inosine-mediated A2AR activation as demonstrated by increased cAMP production, ERK1/2 phosphorylation and inhibition of pro-inflammatory cytokine and chemokine production.
2. Materials and Methods
2.1. Mice
Male BALB/cJ mice and A2AR null mice (C;129S-Adora2atm1Jfc/J; Jackson Laboratories) were housed at 68–72 °F with a 12 h light/dark cycle, fed normal rodent chow and water ad libitum and were kept in a pathogen-free environment. A protocol approved by the Animal Care and Use Committee of the Molecular Medicine Research Institute was used in this study.
2.2. Materials
CGS 21680, NBMPR, and ZM 241385 were purchased from Tocris Biosciences. Growth media and adenosine deaminase were obtained from Lonza and Worthington Biochemical Corporation respectively. Rolipram, adenosine, inosine, adenosine 5′-[α,β-methylene] diphosphate and LPS (E. coli O111:B4) and all the reagents (unless otherwise stated) were purchased from Sigma-Aldrich.
2.3. Cell culture
CHO-K1 cells stably expressing human A2AR (CHO-hA2AR; [21]) were grown in DMEM/F-12 (1:1) supplemented with 10% FBS, 2mM glutamine and G418 (0.2 mg/ml). All cells were maintained at 37 °C in a 5% CO2 incubator.
2.4. Cell-based cAMP assay
CHO-hA2AR [21] cells were seeded in 96-well half-area white plates (Greiner bio-one; 5 × 103 cells/well) in the absence of G418 20 h prior to assay. Cells were either pretreated or left untreated (control) prior to stimulation. For the pretreatment, CHO-hA2AR cells were washed twice with Hanks’ balanced salt solution (HBSS) and incubated in HBSS containing adenosine deaminase (ADA; 3 U/ml) and adenosine 5′[α,β-methylene] diphosphate (50 μM) for 15 min at 37°C. Both control and pretreated cells were washed twice with HBSS and incubated with rolipram (50 μM), adenosine 5′-[α,β-methylene] diphosphate (50 μM), adenosine, inosine and NBMPR at indicated concentration(s) in the presence or in the absence of ZM 241385 (100 nM) for 10 min at 37 °C. Pretreated cells were also incubated with the same assay components in the presence of ADA (3 U/ml) for 10 min at 37 °C. Intracellular cAMP levels were quantified using an HTRF assay kit (Cisbio).
2.5. Cell-free membrane-based cAMP assay
HEK293-hA2AR cell membranes (PerkinElmer) were incubated in HBSS, containing adenosine 5′-[α,β-methylene] diphosphate (50 μM) and ADA (3 U/ml) at 37 °C for 20 min. Membranes were washed twice with 33 mM HEPES containing 0.1% Tween 20 and stimulated with the same buffer containing 100 μM ATP, 2 μM GTP, 10 μM GDP, 2 μM MgCl2, 150 mM NaCl, 50 μM adenosine 5′-[α,β-methylene] diphosphate, 50 μM rolipram, ADA (3 U/ml) and NBMPR (0–300 μM) or CGS 21680 (100 nM) in the presence and in the absence of ZM 241385 (100 nM) in half-area white plates (Greiner bio-one; 4.5 μg protein/well) for 30 min at 37 °C. cAMP levels were quantified using an HTRF assay kit (Cisbio).
2.6. ERK1/2 phosphorylation assay
CHO-hA2AR cells [21] were seeded in 96-well plates (Greiner bio-one; 2.5 × 104 cells/well) in the absence of G418 20 h prior to assay. The medium was replaced with medium lacking serum and incubated for an additional 3 h. Cells were either pretreated or left untreated (control) prior to stimulation. For the pretreatment, CHO-hA2AR cells were washed twice with HBSS and incubated in HBSS containing ADA (3 U/ml) and adenosine 5′-[α,β-methylene] diphosphate (50 μM) for 15 min at 37 °C. Control as well as pretreated cells were washed with warm HBSS to remove ADA and incubated with adenosine 5′-[α,β-methylene] diphosphate (50 μM), inosine and AEA061 at indicated concentration(s) in the presence or in the absence of ZM 241385 (100 nM) for 10 min at 37 °C. Pretreated cells were also incubated with the same assay components in the presence of ADA (3 U/ml) for 10 min at 37 °C. The assay was terminated by aspirating the assay buffer and incubating cells with lysis buffer (50 μl/well) at room temperature with shaking for 10 min. Phospho ERK1/2 levels were detected using an Alphascreen Surefire kit (PerkinElmer) according to the manufacturer’s suggested protocol. Briefly, 10μl of the lysate was transferred to a ProxiPlate-384 (PerkinElmer) and incubated with 10 μl of assay detection mixture at room temperature in the dark for 2 h. Fluorescent emissions were quantified using an EnSpire multimode plate reader (PerkinElmer).
2.7. Cytokine assays
Splenic monocytes/macrophages were isolated from 8–10 weeks old male BALB/cJ mice and A2AR null mice by plastic adherence after incubation for 2 h at 37 °C and 5% CO2, followed by washing with warm media to remove nonadherent cells. Monocytes/macrophages were seeded in 96-well plates (2 × 105 cells per well) and stimulated with lipopolysaccharide (LPS; 50 ng/ml) in RPMI 1640 containing 1% heat inactivated FBS for 4 h in the presence and in the absence of ADA (3 U/ml) and indicated concentration(s) of inosine, CGS 21680 and AEA061. Cytokine/chemokine levels in the culture supernatants were quantified using ELISA Max kits (BioLegend) and/or bead-based multiplex immunoassays (Eve Technologies).
2.8. Data analysis
Data were analyzed using GraphPad Prism Software. Dose response curves were generated by non-linear regression with a variable slope. Statistical comparisons of the two groups were compared using a paired t-test. Comparisons of multiple groups were performed using one-way ANOVA followed by Tukey’s multiple comparisons test.
3. Results
3.1. Inosine dose-dependently induces A2AR-mediated cAMP production
Utilizing a combination of label-free, cell-based, and membrane-based functional assays in conjunction with an equilibrium agonist-binding assay we have demonstrated that inosine is a low-affinity agonist at the A2AR [19]. We have also provided evidence to dismiss the notion that exogenous inosine influences extracellular adenosine levels through the equilibrative nucleoside transporter 1 (ENT1) and ENT2 to activate the A2AR in cellular assays [19]. To further establish that inosine directly activates the A2AR, we evaluated inosine-mediated cAMP production by CHO-K1 cells stably transfected with human A2AR (CHO-hA2AR) in the presence of adenosine deaminase (ADA), an enzyme that eliminates adenosine by catalyzing the hydrolytic deamination of adenosine to inosine. To this end, we utilized a previously described approach that isolates and decreases background signaling and thereby enhances the cAMP signal generated by exogenous inosine [19]. In CHO-hA2AR cells, adenosine induced cAMP production (Fig 1A). The A2AR inverse agonist ZM 241385 blocked the adenosine-mediated cAMP production indicating that the adenosine effects are mediated through the A2AR. Addition of ADA at 3 U/ml (Fig 1A) and 6 U/ml (data not shown) both significantly reduced baseline cAMP levels and completely abolished adenosine-mediated cAMP production by CHO-hA2AR cells suggesting that 3 U/ml of ADA is sufficient to rapidly convert high-affinity agonist adenosine to low-affinity agonist inosine. In the presence of the same concentration of ADA, exogenous inosine increased cAMP production by CHO-hA2AR cells that were blocked by ZM 241385 demonstrating that inosine directly engages the A2AR and is a bonafide agonist at this receptor. Moreover, dose response analysis indicated that ADA increased the EC50 without altering Emax for inosine-mediated cAMP production in CHO-hA2AR cells (Fig 1B).
Fig. 1.


Inosine directly and dose-dependently stimulates hA2AR-mediated cAMP production. CHO-hA2AR cells were incubated with adenosine (control) or inosine with and without ADA (3 U/ml) and in the presence/absence of the hA2AR-selective inverse agonist ZM 241385 for 10 min (A). Mean intracellular cAMP levels ± SEM of a representative experiment are shown (n=3; ***, < 0.001 vs DMSO; θθθ, p < 0.001 vs without ADA; ###, p < 0.001 vs DMSO with ADA; τττ, < 0.001 vs without ADA and ZM 241385; ^^, p < 0.001 vs with ADA but without ZM 241385). Dose response of hA2AR-mediated cAMP production to inosine (B). Mean intracellular cAMP levels ± SEM of a representative experiment are shown (n=3; ***, < 0.001 vs ADA).
3.2. Inosine analog 6-S-[(4-Nitrophenyl)methyl]-6-thioinosine induces A2AR-mediated cAMP production
The inosine analog 6-S-[(4-Nitrophenyl)methyl]-6-thioinosine (NBMPR) is a high- and low-affinity inhibitor of ENT1 and ENT2 respectively [22]. ENT1 and ENT2 are major transporters of adenosine and inosine across cell membranes [23]. Therefore, NBMPR has been used in cell-based AR activation assays to reduce the potential impact of cellular uptake, intracellular production and transport of adenosine and inosine. As NBMPR structurally resembles inosine, we examined whether NBMPR induces cAMP production by CHO-hA2AR cells. As shown in Fig 2A, both adenosine as well as NBMPR activated A2AR-mediated cAMP production by CHO-hA2AR cells. To rule out the possibility that NBMPR-dependent activation of the A2AR is not due to an increase in extracellular adenosine upon NBMPR addition via some hitherto unknown mechanism(s), we included ADA in the assay. The addition of ADA at 3 U/ml reduced basal as well as adenosine-induced cAMP production by CHO-hA2AR cells (Fig 2A). In addition, both adenosine- and NBMPR-mediated cAMP production were inhibited by the A2AR inverse agonist ZM 241385 in the presence and in the absence of ADA indicating selectivity of the two agonists. These results suggest that the inosine analog NBMPR stimulates the A2AR resembling the activation seen with inosine.
Fig. 2.


Inosine analog NBMPR dose-dependently induces hA2AR-mediated cAMP production in cellular and cell-free membrane assays.
CHO-hA2AR cells were incubated with adenosine (control) or NBMPR with and without ADA (3 U/ml) and in the presence/absence of the hA2AR-selective inverse agonist ZM 241385 for 10 min (A). Mean intracellular cAMP levels ± SEM of a representative experiment are shown (n=3; **, < 0.01 vs DMSO; ***, < 0.001 vs DMSO; θθθ, P < 0.001 vs without ADA; ###, p < 0.001 vs DMSO with ADA; τττ, < 0.001 vs without ADA and ZM 241385; ^^^, p < 0.001 vs with ADA but without ZM 241385). CHO-hA2AR cell membranes were incubated with indicated concentrations of NBMPR and CGS 21680 (CGS) in the presence and in the absence of the A2AR-selective inverse agonist ZM 241385 for 30 min (B). Mean cAMP production ± SEM of representative experiments are shown (n=6; ** and ***, p < 0.05 and p < 0.001 vs DMSO respectively; ^^ and ^^^, p < 0.05 and p < 0.001 vs without ZM 241385 respectively).
We demonstrated previously that inosine activates cAMP production by HEK293-hA2AR cytosol-free membrane preparations indicating that inosine directly activates the A2AR [19]. This cell-free assay is completely devoid of potential confounding variables such as influx, intracellular production and efflux of adenosine and inosine. To further establish inosine agonism at the A2AR we evaluated the efficacy of the inosine analog NBMPR on A2AR activation in HEK293-hA2AR membrane preparations. As shown in Fig 2B, NBMPR dose-dependently increased cAMP production and the A2AR inverse agonist ZM 241385 blocked NBMPR-induced cAMP production by HEK293-hA2AR cytosol-free membrane preparations. These results indicate that the inosine analog NBMPR specifically and directly activates the A2AR.
3.3. Positive allosteric modulation enhances inosine-mediated hA2AR activation
AEA061, a small molecule that meets the stringent criteria of a positive allosteric modulator (PAM) of the A2AR, enhances adenosine-mediated A2AR activation and exerts pharmacological activity in a mouse model of endotoxemia [20]. We sought to examine whether inosine-mediated A2AR activation is also amenable to functional enhancement by AEA061. To this end, we evaluated the effects of AEA061 on inosine-mediated cAMP production by CHO-hA2AR cells, doing so in the presence of ADA to eliminate adenosine. As shown in Figure 3A, inosine induced cAMP production (p < 0.001) and the A2AR inverse agonist ZM 241385 blocked this induction (p < 0.001). AEA061 by itself did not induce cAMP production. However, it enhanced inosine-mediated cAMP production (p < 0.05) that was again blocked by the A2AR inverse agonist ZM 241385 (p< 0.01). These results demonstrate that AEA061 has no intrinsic activity towards hA2AR-mediated cAMP production, but can allosterically augment inosine-driven hA2AR-mediated cAMP production.
Fig. 3.



PAM of the A2AR AEA061 enhances inosine-inducible, A2AR-mediated cAMP production. CHO-hA2aR cells were incubated with inosine (Ino; 1000 μM) and AEA061 (10 μM) in the presence and in the absence of ZM 241385 (100 nM) for 10 min (A). Mean cAMP levels ± SEM of a representative experiment is shown (n=3; ***, p < 0.001 vs DMSO; ###, < p < 0.001 vs inosine; &&&, p < 0.001 vs without ZM 241385). AEA061 increases Emax (B) and alters EC50 (C) of inosine-mediated A2AR activation. CHO-A2AR cells were incubated with AEA061 and varying concentrations of inosine for 10 min. Mean Emax (B) and EC50 (C) values of four experiments with SEM are shown (**, < 0.01 vs DMSO; # and ##, p < 0.05 and 0.01 vs without ADA pretreatment; ^^, p < 0.01 vs ADA pretreatment without subsequent ADA addition respectively).
ADA is a multifunctional protein with intracellular and extracellular localization. In addition to converting adenosine to inosine, ADA physically interacts with the A2AR and allosterically modulates adenosine binding to the A2AR [24, 25]. To fully understand the effect of AEA061 on inosine-mediated A2AR activation in the context of the natural cellular milieu, we examined the dose response of inosine at the hA2AR in CHO-hA2AR cells under several assay conditions. CHO-hA2AR cells were pretreated with ADA and adenosine 5′-[α,β-methylene] diphosphate, an inhibitor of ecto-5′-nucleotidase to prevent exogenous production of adenosine, then washed and stimulated in the presence or in the absence of ADA with inosine. Regardless of the treatment condition, AEA061 increased the Emax (maximal response) for inosine-induced hA2AR-mediated cAMP production (Fig 3B). Relative to the control (without the pretreatment), both ADA pretreatment and ADA pretreatment plus subsequent ADA addition increased the EC50 of inosine-mediated cAMP production (decreased affinity of the hA2AR; Fig 3C). AEA061 reduced the EC50 of inosine (increased hA2AR affinity to inosine) in ADA pre-treated CHO-hA2AR cells (p < 0.01) but not in cells without the ADA pretreatment alone or with ADA pretreatment and subsequent ADA addition. These results suggest that the positive allosteric modulator AEA061 exhibits differential effects on the affinity and efficacy of the A2AR to inosine. AEA061 enhances efficacy of the A2AR towards inosine both in the presence and absence of membrane-bound adenosine and ADA. However, AEA061 enhances A2AR affinity to inosine only in the absence of membrane-bound adenosine and ADA.
3.4. Positive allosteric modulation enhances inosine-mediated hA2AR-dependant ERK1/2 activation
We demonstrated previously that A2AR activation by both adenosine and inosine leads to ERK1/2 phosphorylation [19]. To determine whether the PAM of the A2AR, AEA061, potentiates inosine-inducible, A2AR-mediated ERK1/2 activation, we evaluated the effects of AEA061 on ERK1/2 phosphorylation in CHO-hA2AR cells. Consistent with our previous observations, inosine increased ERK1/2 phosphorylation (Fig 4A) indicating that inosine effects are mediated through the A2AR. Moreover, AEA061 increased both basal and inosine-induced ERK1/2 phosphorylation (Fig 4A). The A2AR inverse agonist ZM 241385 reversed inosine-, AEA061- and inosine plus AEA061-mediated ERK1/2 phosphorylation. Dose response analyses indicated that AEA061 alone enhanced the Emax (maximal response; Fig 4B) regardless of the assay conditions. AEA061 reduced EC50 (increased affinity) to inosine with and without the ADA pre-treatment (Fig 4C). Although not statistically significant, AEA061 lowered the EC50 to inosine in CHO-hA2AR cells with ADA pre-treatment and subsequent ADA addition. These results collectively suggest that both AEA061- and inosine-induced ERK1/2 phosphorylation are mediated through the A2AR and that AEA061 functions both as an agonist as well as a PAM with respect to ERK1/2 activation, a sharp contrast to its effect on cAMP activation where AEA061 functions solely as a PAM.
Fig. 4.



AEA061 potentiates inosine-inducible, A2AR-mediated ERK1/2 phosphorylation. CHO-hA2AR cells were incubated with inosine (Ino; 1000 μM) and AEA061 (10 μM) in the presence and in the absence of ZM 241385 for 10 min (A). Phospho ERK1/2 levels were quantified using a FRET-based detection kit. Mean FRET ratios ± SEM of a representative experiment are shown (n=3; ***, p < 0.001 vs DMSO; ##, < 0.01 vs AEA061; && and &&&, p < 0.01 and 0.001 vs without ZM 241385 respectively). AEA061 enhances Emax (B) and reduces EC50 (C) of inosine-mediated ERK1/2 phosphorylation. CHO-A2aR cells were pre-treated and incubated with AEA061 and varying concentrations of inosine for 10 min. Mean Emax (B) and EC50 (C) values of three independent experiments with SEM are shown (* and ***, < 0.05 and 0.001 vs DMSO respectively).
3.5. Inosine suppresses the production of pro-inflammatory cytokines through the A2AR
Inosine exerts anti-inflammatory effects through inhibition of the production of pro-inflammatory cytokines and chemokines [7, 26–28]. Using pharmacological tools, Haskó et al. [7] demonstrated that inosine-mediated suppression of TNF-α production is mediated through both A1R and A2AR in vitro. To further assess the role of A2AR in inosine-mediated suppression of cytokine production, we investigated inosine’s effects on cytokine production by monocytes isolated from A2AR intact and deficient mice. To distinguish adenosine effects from those of inosine, we utilized ADA which converts adenosine to inosine. Consistent with the role of the A2AR, activation of the receptor with the selective agonist CGS 21680 in the absence and in the presence of ADA inhibited TNF-α production by A2AR-intact but not A2AR-deficient monocytes (Fig 5A and 5B). In the absence of ADA, inosine at 30 μM and 100 μM inhibited LPS-stimulated TNF-α production by 18.6 ± 3.7% (p < 0.01) and 30.1 ± 1.3% (p < 0.001) by A2AR intact monocytes respectively (Fig 5A). The same concentrations of inosine inhibited LPS-stimulated TNF-α production by 5.2 ± 1.3% and 16.3 ± 0.7% (p < 0.001) in A2AR deficient monocytes. These results indicate that inosine-mediated inhibition of TNF-α production is reduced by 46% (p < 0.01) in A2AR-deficient monocytes compared with receptor-intact monocytes suggesting that inosine-mediated inhibition of TNF-α at least in part mediated through the A2AR. The inclusion of ADA in the assay prevents A2AR activation by adenosine as it converts extracellular adenosine produced by monocytes to inosine. When the confounding effects of adenosine were eliminated, inosine-mediated inhibition of LPS-stimulated TNF-α production continued to be reduced in both A2AR intact and A2AR deficient monocytes (Fig 5B). In the presence of ADA, inosine at 30 μM and 100 μM inhibited LPS-stimulated TNF-α production by 13.2 ± 1.9% (p < 0.01) and 16.8 ± 1.6% (p < 0.01) in A2AR intact monocytes respectively (Fig 5B). The same concentrations of inosine inhibited LPS-stimulated TNF-α production by 5.2 ± 2.2% and 12.4 ± 1.2% (P < 0.01) in A2AR deficient monocytes, indicating a 2.5- and a 1.4-fold reduction in the inhibition of TNF-α production in A2AR deficient monocytes in comparison with A2AR-intact monocytes at 30 μM and 100 μM inosine respectively. These results indicate that inosine-mediated A2AR activation leads to inhibition of TNF-α production as is the case for adenosine-mediated A2AR activation. Inhibition of TNF-α production in A2AR deficient monocytes by inosine and not by A2AR selective agonist CGS 21680 suggests that inhibitory effects of inosine are also mediated through other adenosine receptor subtype(s).
Fig. 5.
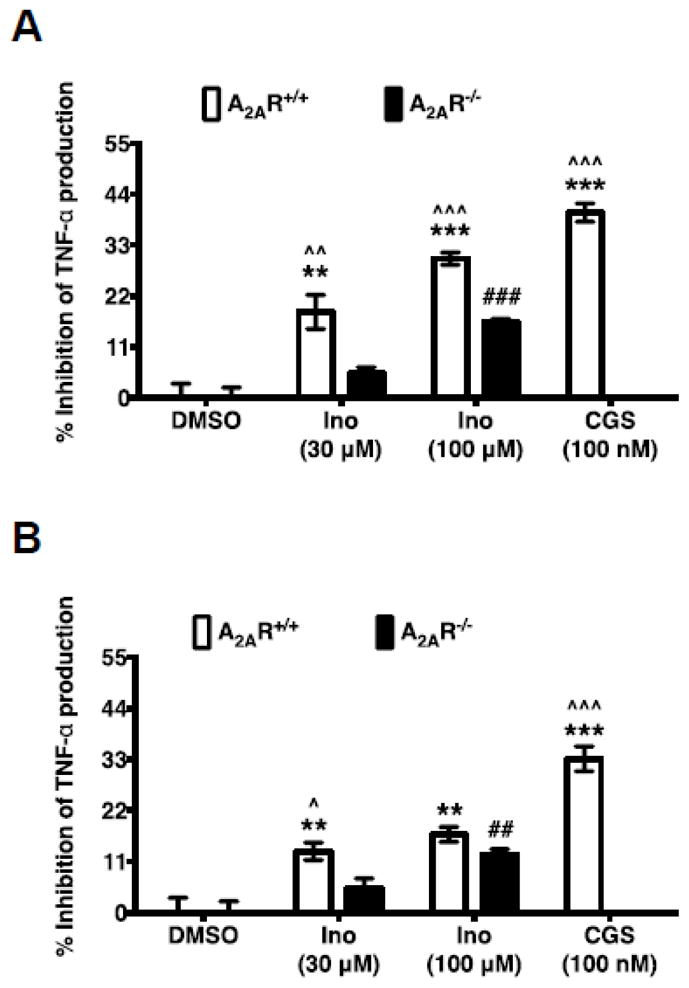
Inosine-mediated A2AR activation inhibits TNF-α production by mouse splenic monocytes. A2AR intact and deficient mouse splenic monocytes were stimulated with LPS (50 ng/ml), inosine (Ino) and CGS 21680 (CGS) in the absence (A) and in presence (B) of ADA (3 U/ml) for 4 h. Mean % inhibition of TNF-α production relative to DMSO (control) of a representative experiment performed in triplicate with SEM are shown (n=3; ** and ***, p < 0.01 and 0.001 vs A2AR+/+ DMSO; ## and ###, p < 0.01 and 0.001 vs A2AR−/− DMSO; ^, ^^ and ^^^, p < 0.05, 0.01 and 0.001 vs A2AR−/− respectively).
3.6. PAM of the A2AR enhances inosine-mediated suppression of pro-inflammatory cytokines production
To examine whether allosteric enhancement of inosine-mediated A2AR stimulation leads to increased suppression of cytokine production, we evaluated the effects of inosine in the presence and in the absence of AEA061 on TNF-α production by LPS-stimulated monocytes. To assess the potential for A2AR activation by adenosine produced via the salvage pathway, we performed the assays with and without ADA. Consistent with the anti-inflammatory role of the A2AR, selective activation of this receptor with CGS 21680 in the absence of ADA inhibited TNF-α production by LPS-stimulated A2AR-intact monocytes by 39% and the PAM of the A2AR enhanced this CGS 21680-mediated inhibition to 49% (Fig 6A; p < 0.001). Under the same conditions, CGS 21680 alone and in combination with AEA061 reduced LPS-stimulated production in A2AR deficient monocytes by only 6.7% and 14% respectively (p < 0.001; Fig 6A) demonstrating the inhibitory effects of these agents are mediated through the A2AR. Similarly, both inosine and AEA061 as single agents dose-dependently inhibited TNF-α production by LPS-stimulated A2AR intact monocytes in the absence of ADA. In combination, they produced even greater inhibition than either agent alone indicating allosteric enhancement (Fig 6A). Although the higher concentration of AEA061 (0.3 μM) both alone and in combination produced a greater percent inhibition of TNF-α production by A2AR intact monocytes than AEA061 at 0.1 μM, the difference in percent inhibition of TNF-α production between A2AR intact and deficient monocytes had narrowed. This suggests loss of selectivity of AEA061 towards the A2AR at higher concentrations. In the presence of ADA (Fig 6B), inosine and AEA061 produced the same general pattern of inhibition of TNF-α production by A2AR intact and deficient monocytes. However, the overall percent inhibition of TNF-α production was somewhat lower in the presence of ADA relative to the absence of ADA. Inhibition of production of the pro-inflammatory chemokine CCL3 by inosine and AEA061 in the absence of ADA (Fig 6C) closely resembles that of TNF-α (Fig 6A) in A2AR intact and deficient monocytes under the same conditions. However, in the presence of ADA, the difference in CCL3 inhibition (Fig 6D) between A2AR intact and deficient monocytes was more prominent. Inosine and AEA061 also individually inhibited CCL4 production by monocytes (Fig 6E) and the combination of these two agents produced greater inhibition than each agent alone suggesting allosteric potentiation of the A2AR. The addition of ADA reduced the inhibition of CCL4 production under the same conditions, but increased the difference in inhibition between A2AR intact and deficient monocytes (Fig 6F). Collectively, these results indicate that positive allosteric potentiation of inosine-mediated A2AR activation inhibits pro-inflammatory cytokine/chemokine production by monocytes.
Fig. 6.



PAM of the A2AR potentiates inosine-mediated inhibition of TNF-α in an A2AR dependent manner. Splenic monocytes isolated from A2AR intact and deficient mice were stimulated with LPS (50 ng/ml), inosine (Ino), CGS 21680 (CGS) in the absence (A, C, E) and in presence (B, D, F) of ADA (3 U/ml) for 4 h. Mean percent inhibition of TNF-α (A & B), CCL3 (C & D) and CCL4 (E & F) production relative to DMSO (control) of a representative experiment performed in replicates of four with SEM are shown (n=4; *, ** and ***, p < 0.05, 0.01 and 0.001 vs DMSO; ## and ###, p < 0.01 and 0.001 vs A2AR+/+ AEA061; ^^^, p < 0.001 vs CGS 21680; φ, φφ and φφφ, < 0.05, 0.01 and 0.001 vs A2AR+/+ respectively).
Discussion
Inosine, a metabolite of adenosine, exerts anti-inflammatory and immunomodulatory effects in vivo [28]. These effects are at least in part mediated through the A2AR, a member of a class of purinergic G protein-coupled receptors. Inosine is less potent than adenosine in activating the A2AR [29]. Hence, the prevailing notion is that the inosine effects observed in vivo are not generated by direct inosine engagement at the A2AR but rather mediated indirectly through receptor activation by adenosine produced from the metabolic conversion of inosine. However, a handful of studies have provided initial circumstantial evidence for direct activation of A2AR by inosine [7, 13, 27]. Recently, utilizing a combination of label-free, cell-based, and membrane-based functional assays in conjunction with an equilibrium agonist-binding assay we demonstrated that inosine directly engages the A2AR and activates signaling events downstream of the receptor leading to cAMP production and ERK1/2 phosphorylation [19]. In the present study, we sought to provide additional proof for A2AR activation through direct engagement of inosine at the receptor as well as determine if inosine-mediated engagement of the receptor was amenable to allosteric modulation.
We employed several approaches to rule out indirect and confounding effects of adenosine on A2AR activation. We pretreated cells with adenosine deaminase (ADA) to rid the cells of endogenously produced adenosine and adenosine 5′-[α,β-methylene] diphosphate, an inhibitor of ecto-5′-nucleotidase, to halt endogenous/exogenous production of adenosine and inosine. Moreover, we utilized ADA during the treatment phase of the assay to eliminate adenosine produced during the short duration of the A2AR activation assay. Under these conditions, exogenous inosine as well as the inosine analog NBMPR but not adenosine stimulated activation of the A2AR. This inosine- and NBMPR-mediated A2AR activation was dose-dependent and was inhibitable by the A2AR inverse agonist ZM 241385 providing strong evidence for inosine agonism at this receptor.
Our data indicate that inosine inhibits pro-inflammatory cytokine and chemokine production in an A2AR-dependent manner in vitro. The level of inhibition of pro-inflammatory cytokine production achieved with inosine in mouse monocytes is consistent with that published by Haskó et al. [7] and is also in agreement with the EC50 values for inosine-mediated A2AR activation in CHO-hA2AR cells reported here. These results strongly support the contention that the activation of the A2AR by inosine leads to a reduction in the production of pro-inflammatory cytokines and chemokines. Our results also indicate that ADA in general reduced the inhibition of pro-inflammatory cytokine and chemokine production in both A2AR intact and deficient monocytes. This is consistent with the fact that extracellular adenosine produced by monocytes/macrophages during the assay is rapidly deaminated by exogenous ADA to inosine that has lower affinity than adenosine at the A2AR, hence lower inhibition of TNF-α production by A2AR intact monocytes. The reduced inhibition of TNF-α production in A2AR-deficient monocytes in the presence of ADA suggests that inosine is also less potent than adenosine in activating A2AR-independent pathways [30, 31] leading to inhibition of TNF-α production.
On the basis of crystal structure predictions and site-directed mutagenesis studies, the critical residues that form the adenosine binding pocket in the A2AR have been identified [32, 33]. The nature of the interactions of inosine with these residues may explain the observed low potency of the receptor to inosine [19, 34]. To better understand the molecular mechanism of A2AR recognition by adenosine and inosine, Deganutti et al. [35] utilized supervised molecular dynamics simulation, a computational method that allows identification and characterization of multiple stable receptor conformations such as orthosteric, allosteric and meta-binding states. They reported an overlap of meta-stable states predicted for binding of inosine and adenosine to the A2AR suggesting that both agonists share a common molecular mechanism for receptor activation [35]. Their findings are in agreement with our previous [19] as well as present experimental evidence for functional agonism of inosine at the A2AR and explain the molecular mechanism of inosine-mediated receptor activation.
Inosine and the inverse agonist ZM 241385 both bind to the same site at the A2AR with different affinities (inosine has a lower affinity). Consistent with these observations, ZM 241385 at 100 nM completely reversed the effects of 100 μM inosine in CHO-hA2AR cells. However, the same concentration of ZM 241385 (100 nM) was not able to fully reverse the effects mediated by inosine at a higher concentration (1000 μM). This incomplete inhibition is most likely due to mass action as the parental CHO-K1 cells do not express any adenosine receptor subytpes and in addition, the ZM 241385 concentration used was 10,000–fold less than that of inosine. Partial inhibition of NBMPR-mediated A2AR activation by ZM 241385 may be explained also by the law of mass action.
The molecular mechanism of A2AR activation is consistent with conformational selection where binding of agonists and inverse agonists/antagonists stabilizes the receptor in active and inactive conformations respectively [36]. The active conformation(s) engage Gs and adenylate cyclase to produce cAMP whereas the inactive conformation(s) do not. Binding of a PAM such as AEA061 to the A2AR facilitates this process as indicated by enhanced efficacy and potency of the agonist at the receptor [20]. Since AEA061 does not activate A2AR-mediated cAMP production in the absence of an agonist [20], AEA061 by itself does not stabilize A2AR conformation(s) that engage Gs. Depending on the cell type, agonist engagement at the A2AR leads to Gs-mediated, cAMP-dependent and small G protein p21ras-mediated, cAMP-independent ERK1/2 phosphorylation [37, 38]. Therefore, it is conceivable that AEA061 by itself, in the absence of an agonist, stabilizes an A2AR conformation(s) that is capable of engaging p21ras to activate ERK1/2, thereby exhibiting an agonist-like behavior.
Previously we demonstrated that the synthetic small molecule AEA061 does not activate the A2AR by itself but augments adenosine-mediated A2AR activation, and thus bears the hallmarks of positive allosteric modulation [20]. Our data indicate that AEA061 also enhances inosine-mediated A2AR responsiveness in the base-line state as well as under conditions that isolate and eliminate the confounding effects of adenosine. Although the PAM of the A2AR, AEA061, enhances the maximal response of inosine-mediated cAMP production and ERK1/2 phosphorylation under all three assay conditions, it exhibits differential effects with respect to EC50 for cAMP production and ERK1/2 activation. AEA061 reduces the EC50 for inosine-mediated ERK1/2 phosphorylation regardless of the assay condition. In contrast, AEA061 reduces the EC50 for inosine-mediated cAMP production with ADA pretreatment and subsequent ADA addition. It is possible that ADA, in itself an allosteric modulator of the A2AR [24, 25], differentially influences AEA061 effects on cAMP and ERK1/2 pathways.
Our data indicate that positive allosteric modulation of the A2AR by AEA061 enhances inosine-mediated inhibition of pro-inflammatory cytokine and chemokine production by splenic monocytes/macrophages. Inhibition of cytokine and chemokine production by AEA061 alone in the absence of an exogenously added agonist is consistent with the fact that cells produce extracellular adenosine and inosine during normal cell metabolism in amounts sufficient to engage the A2AR allowing allosteric enhancement with AEA061. As AEA061 exhibits no allosteric effects towards A1R and A3R and weak activity towards A2BR (unpublished data), the A2AR-independent inhibition of cytokine production by AEA061 at higher concentrations may be mediated through the low-affinity A2BR. Although inosine does not activate A2BR in stable cell lines overexpressing the receptor in vitro [19, 29], A2BR is the principal mediator of inosine activity in the bladder [39]. Therefore, it is conceivable that the inhibition of cytokine and chemokine production observed in A2AR deficient monocytes with AEA061 in the presence of exogenously added ADA is due to enhancement of extracellularly produced inosine mediating A2BR activation. Further investigation will be necessary to rigorously examine this possibility.
In summary, the major outcomes of the present investigation are the demonstration of inosine agonism at the A2AR, the amenability of inosine-mediated A2AR activation to allosteric modulation and the effect of inosine- and PAM- mediated A2AR activation on pro-inflammatory cytokine and chemokine production. Our data indicate that, as is the case for adenosine, positive allosteric modulation of the A2AR with AEA061 potentiates inosine-mediated cAMP production, ERK1/2 phosphorylation and inhibition of pro-inflammatory cytokine and chemokine production. We previously proposed that both adenosine and inosine are natural agonists of the A2AR and are important to mount a robust and effective immunomodulatory response. Adenosine with a shorter half-life (<10 sec) initiates A2AR activation and upon conversion to metabolically more stable inosine (half-life 15 h) sustains A2AR signaling to prolong anti-inflammatory and immunomodulatory responses. The PAM of the A2AR AEA061 potentiates and preserves the natural temporal and physical pattern of both adenosine-mediated and inosine-mediated A2AR activation to harness the full potential of the endogenous A2AR-dependent mechanisms tasked to effectively downmodulate inflammation. Given the intricacies of GPCR signaling, it is conceivable that potentiation of adenosine- and inosine-mediated A2AR activation through a PAM may have qualitatively and quantitatively different immunomodulatory responses as opposed to A2AR activation through a high-affinity agonist. If proven, positive allosteric modulation of the A2AR may provide new insights into potential therapeutic interventions for immune- and inflammation-mediated diseases.
Highlights.
Under conditions devoid of adenosine, inosine dose-dependently activates the A2AR.
Inosine-mediated A2AR activation increases cAMP and phospho-ERK1/2 levels.
Inosine inhibits TNF-α, CCL3 and CCL4 production by monocytes via A2AR activation.
PAM of the A2AR potentiates inosine-mediated receptor signaling.
PAM of the A2AR inhibits TNF-α, CCL3 and CCL4 production.
Acknowledgments
This work was supported in part by NIH grant 5R21AI105518 from the National Institute of Allergy and Infectious Diseases.
Abbreviations
- ADA
adenosine deaminase
- Ado
adenosine
- AR
adenosine receptor
- A1R
adenosine A1R
- A2AR
adenosine A2A receptor
- A2BR
adenosine A2B receptor
- A3R
adenosine A3 receptor
- cAMP
cyclic adenosine monophosphate
- CGS 21680
4-[2-[[6-Amino-9-(N-ethyl-β-D-ribofuranuronamidosyl)-9H-purin-2-yl]amino]ethyl] benzenepropanoic acid hydrochloride
- ENT1
equilibrative nucleoside transporter 1
- ENT2
equilibrative nucleoside transporter 2
- ERK1/2
extracellular signal-regulated kinase-1 and -2
- GPCR
G protein-coupled receptor
- HBSS
Hanks’ balanced salt solution
- HEPES
4-(2-Hydroxyethyl) piperazine-1-ethanesulfonic acid, N-(2-Hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid
- HTRF
homogeneous time resolved fluorescence
- Ino
inosine
- LPS
lipopolysaccharide
- NBMPR
6-S-[(4-Nitrophenyl)methyl]-6-thioinosine
- PAM
positive allosteric modulator
- PBS
phosphate buffered saline
- SD
standard deviation
- SEM
standard error of the mean
- TNBS
2,4,6-trinitrobenzenesulfonic acid
- ZM 241385
4-(2-[7-Amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest. Research reported in this publication was supported in part by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R21AI105518. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Viegas TX, Omura GA, Stoltz RR, Kisicki J. Pharmacokinetics and pharmacodynamics of peldesine (BCX-34), a purine nucleoside phosphorylase inhibitor, following single and multiple oral doses in healthy volunteers. J Clin Pharmacol. 2000;40:410–420. doi: 10.1177/00912700022008991. [DOI] [PubMed] [Google Scholar]
- 2.Möser GH, Schrader J, Deussen A. Turnover of adenosine in plasma of human and dog blood. Am J Physiol. 1989;256:C799–806. doi: 10.1152/ajpcell.1989.256.4.C799. [DOI] [PubMed] [Google Scholar]
- 3.Phillis JW, Walter GA, O’Regan MH, Stair RE. Increases in cerebral cortical perfusate adenosine and inosine concentrations during hypoxia and ischemia. J Cereb Blood Flow Metab. 1987;7:679–686. doi: 10.1038/jcbfm.1987.122. [DOI] [PubMed] [Google Scholar]
- 4.Phillis JW, O’Regan MH, Walter GA. Effects of nifedipine and felodipine on adenosine and inosine release from the hypoxemic rat cerebral cortex. J Cereb Blood Flow Metab. 1988;8:179–185. doi: 10.1038/jcbfm.1988.47. [DOI] [PubMed] [Google Scholar]
- 5.Phillis JW, O’Regan MH, Walter GA. Effects of deoxycoformycin on adenosine, inosine, hypoxanthine, xanthine, and uric acid release from the hypoxemic rat cerebral cortex. J Cereb Blood Flow Metab. 1988;8:733–741. doi: 10.1038/jcbfm.1988.121. [DOI] [PubMed] [Google Scholar]
- 6.Bell MJ, Kochanek PM, Carcillo JA, Mi Z, Schiding JK, Wisniewski SR, Clark RS, Dixon CE, Marion DW, Jackson E. Interstitial adenosine, inosine, and hypoxanthine are increased after experimental traumatic brain injury in the rat. J Neurotrauma. 1998;15:163–170. doi: 10.1089/neu.1998.15.163. [DOI] [PubMed] [Google Scholar]
- 7.Haskó G, Kuhel DG, Németh ZH, Mabley JG, Stachlewitz RF, Virág L, Lohinai Z, Southan GJ, Salzman AL, Szabó C. Inosine inhibits inflammatory cytokine production by a posttranscriptional mechanism and protects against endotoxin-induced shock. J Immunol. 2000;164:1013–1019. doi: 10.4049/jimmunol.164.2.1013. [DOI] [PubMed] [Google Scholar]
- 8.Liaudet L, Mabley JG, Pacher P, Virág L, Soriano FG, Marton A, Haskó G, Deitch EA, Szabó C. Inosine exerts a broad range of antiinflammatory effects in a murine model of acute lung injury. Ann Surg. 2002;235:568–578. doi: 10.1097/00000658-200204000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mabley JG, Pacher P, Liaudet L, Soriano FG, Haskó G, Marton A, Szabo C, Salzman AL. Inosine reduces inflammation and improves survival in a murine model of colitis. Am J Physiol Gastrointest Liver Physiol. 2003;284:G138–144. doi: 10.1152/ajpgi.00060.2002. [DOI] [PubMed] [Google Scholar]
- 10.Schneider S, Klein HH. Inosine improves islet xenograft survival in immunocompetent diabetic mice. Eur J Med Res. 2005;10:283–286. [PubMed] [Google Scholar]
- 11.He B, Hoang TK, Wang T, Ferris M, Taylor CM, Tian X, Luo M, Tran DQ, Zhou J, Tatevian N, Luo F, Molina JG, Blackburn MR, Gomez TH, Roos S, Rhoads JM, Liu Y. Resetting microbiota by Lactobacillus reuteri inhibits T reg deficiency-induced autoimmunity via adenosine A2A receptors. J Exp Med. 2017;214:107–123. doi: 10.1084/jem.20160961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Junqueira SC, dos Santos Coelho I, Lieberknecht V, Cunha MP, Calixto JB, Rodrigues ALS, Santos ARS, Dutra RC. Inosine, an Endogenous Purine Nucleoside, Suppresses Immune Responses and Protects Mice from Experimental Autoimmune Encephalomyelitis: a Role for A2A Adenosine Receptor. Molecular Neurobiology. 2016;54:3271–3285. doi: 10.1007/s12035-016-9893-3. [DOI] [PubMed] [Google Scholar]
- 13.da Rocha Lapa F, de Oliveira AP, Accetturi BG, de Oliveira Martins I, Domingos HV, de Almeida Cabrini D, de Lima WT, Santos AR. Anti-inflammatory effects of inosine in allergic lung inflammation in mice: evidence for the participation of adenosine A2A and A3 receptors. Purinergic Signal. 2013;9:325–336. doi: 10.1007/s11302-013-9351-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mabley JG, Rabinovitch A, Suarez-Pinzon W, Haskó G, Pacher P, Power R, Southan G, Salzman A, Szabó C. Inosine protects against the development of diabetes in multiple-low-dose streptozotocin and nonobese diabetic mouse models of type 1 diabetes. Mol Med. 2003;9:96–104. doi: 10.2119/2003-00016.mabley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rahimian R, Fakhfouri G, Daneshmand A, Mohammadi H, Bahremand A, Rasouli MR, Mousavizadeh K, Dehpour AR. Adenosine A(2A) receptors and uric acid mediate protective effects of inosine against TNBS-induced colitis in rats. Eur J Pharmacol. 2010;649:376–381. doi: 10.1016/j.ejphar.2010.09.044. [DOI] [PubMed] [Google Scholar]
- 16.Schneider L, Pietschmann M, Hartwig W, Marcos SS, Hackert T, Gebhard MM, Uhl W, Büchler MW, Werner J. Inosine reduces microcirculatory disturbance and inflammatory organ damage in experimental acute pancreatitis in rats. Am J Surg. 2006;191:510–514. doi: 10.1016/j.amjsurg.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 17.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 18.Olah ME. Identification of A2A adenosine receptor domains involved in selective coupling to Gs. Analysis of chimeric A1/A2A adenosine receptors. J Biol Chem. 1997;272:337–344. doi: 10.1074/jbc.272.1.337. [DOI] [PubMed] [Google Scholar]
- 19.Welihinda AA, Kaur M, Greene K, Zhai Y, Amento EP. The adenosine metabolite inosine is a functional agonist of the adenosine A2A receptor with a unique signaling bias. Cellular Signalling. 2016;28:552–560. doi: 10.1016/j.cellsig.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Welihinda AA, Amento EP. Positive allosteric modulation of the adenosine A2A receptor attenuates inflammation. Journal of Inflammation. 2014;11 doi: 10.1186/s12950-014-0037-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ. Comparative pharmacology of human adenosine receptor subtypes-characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1997;357:1–9. doi: 10.1007/pl00005131. [DOI] [PubMed] [Google Scholar]
- 22.Ward JL, Sherali A, Mo ZP, Tse CM. Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells. Ent2 exhibits a low affinity for guanosine and cytidine but a high affinity for inosine. J Biol Chem. 2000;275:8375–8381. doi: 10.1074/jbc.275.12.8375. [DOI] [PubMed] [Google Scholar]
- 23.Baldwin SA, Beal PR, Yao SY, King AE, Cass CE, Young JD. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch. 2004;447:735–743. doi: 10.1007/s00424-003-1103-2. [DOI] [PubMed] [Google Scholar]
- 24.Gracia E, Pérez-Capote K, Moreno E, Barkešová J, Mallol J, Lluís C, Franco R, Cortés A, Casadó V, Canela EI. A2A adenosine receptor ligand binding and signalling is allosterically modulated by adenosine deaminase. Biochem J. 2011;435:701–709. doi: 10.1042/BJ20101749. [DOI] [PubMed] [Google Scholar]
- 25.Gracia E, Farre D, Cortes A, Ferrer-Costa C, Orozco M, Mallol J, Lluis C, Canela EI, McCormick PJ, Franco R, Fanelli F, Casado V. The catalytic site structural gate of adenosine deaminase allosterically modulates ligand binding to adenosine receptors. The FASEB Journal. 2012;27:1048–1061. doi: 10.1096/fj.12-212621. [DOI] [PubMed] [Google Scholar]
- 26.Liaudet L, Mabley JG, Soriano FG, Pacher P, Marton A, Haskó G, Szabó C. Inosine reduces systemic inflammation and improves survival in septic shock induced by cecal ligation and puncture. Am J Respir Crit Care Med. 2001;164:1213–1220. doi: 10.1164/ajrccm.164.7.2101013. [DOI] [PubMed] [Google Scholar]
- 27.Gomez G, Sitkovsky MV. Differential requirement for A2A and A3 adenosine receptors for the protective effect of inosine in vivo. Blood. 2003;102:4472–4478. doi: 10.1182/blood-2002-11-3624. [DOI] [PubMed] [Google Scholar]
- 28.Haskó G, Sitkovsky MV, Szabó C. Immunomodulatory and neuroprotective effects of inosine. Trends Pharmacol Sci. 2004;25:152–157. doi: 10.1016/j.tips.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Fredholm BB, Irenius E, Kull B, Schulte G. Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem Pharmacol. 2001;61:443–448. doi: 10.1016/s0006-2952(00)00570-0. [DOI] [PubMed] [Google Scholar]
- 30.Haskó G, Szabó C, Németh ZH, Kvetan V, Pastores SM, Vizi ES. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J Immunol. 1996;157:4634–4640. [PubMed] [Google Scholar]
- 31.Haskó G, Kuhel DG, Chen JF, Schwarzschild MA, Deitch EA, Mabley JG, Marton A, Szabó C. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. FASEB J. 2000;14:2065–2074. doi: 10.1096/fj.99-0508com. [DOI] [PubMed] [Google Scholar]
- 32.Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun B, Bachhawat P, Chu ML, Wood M, Ceska T, Sands ZA, Mercier J, Lebon F, Kobilka TS, Kobilka BK. Crystal structure of the adenosine A2A receptor bound to an antagonist reveals a potential allosteric pocket. Proceedings of the National Academy of Sciences. 2017;114:2066–2071. doi: 10.1073/pnas.1621423114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deganutti G, Welihinda A, Moro S. Comparison of the Human A2A Adenosine Receptor Recognition by Adenosine and Inosine: New Insight from Supervised Molecular Dynamics Simulations. ChemMedChem. 2017;12:1319–1326. doi: 10.1002/cmdc.201700200. [DOI] [PubMed] [Google Scholar]
- 36.Ye L, Van Eps N, Zimmer M, Ernst OP, Prosser RS. Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature. 2016;533:265–268. doi: 10.1038/nature17668. [DOI] [PubMed] [Google Scholar]
- 37.Sexl V, Mancusi G, Höller C, Gloria-Maercker E, Schütz W, Freissmuth M. Stimulation of the mitogen-activated protein kinase via the A2A-adenosine receptor in primary human endothelial cells. J Biol Chem. 1997;272:5792–5799. doi: 10.1074/jbc.272.9.5792. [DOI] [PubMed] [Google Scholar]
- 38.Seidel MG. Activation of Mitogen-activated Protein Kinase by the A2A-adenosine Receptor via a rap1-dependent and via a p21ras-dependent Pathway. Journal of Biological Chemistry. 1999;274:25833–25841. doi: 10.1074/jbc.274.36.25833. [DOI] [PubMed] [Google Scholar]
- 39.Doyle C, Cristofaro V, Sack BS, Lukianov SN, Schäfer M, Chung YG, Sullivan MP, Adam RM. Inosine attenuates spontaneous activity in the rat neurogenic bladder through an A2B pathway. Scientific Reports. 2017;7:44416. doi: 10.1038/srep44416. [DOI] [PMC free article] [PubMed] [Google Scholar]