

Abstract
Long bone formation is a complex process that requires precise transcriptional control of gene expression programs in mesenchymal progenitor cells. Histone deacetylases (Hdacs) coordinate chromatin structure and gene expression by enzymatically removing acetyl groups from histones and other proteins. Hdac inhibitors are used clinically to manage mood disorders, cancers and other conditions, but are teratogenic to the developing skeleton and increase fracture risk in adults. In this study, the functions of Hdac3, one of the enzymes blocked by current Hdac inhibitor therapies, in skeletal mesenchymal progenitor cells were determined. Homozygous deletion of Hdac3 in Prrx1-expressing cells prevented limb lengthening, altered pathways associated with endochondral and intramembranous bone development, caused perinatal lethality, and slowed chondrocyte and osteoblast differentiation in vitro. Transcriptomic analysis revealed that Hdac3 regulates vastly different pathways in mesenchymal cells expressing the Prrx1-Cre driver than those expressing the Col2-CreERT driver. Notably, Fgf21 was elevated in Hdac3-CKOPrrx1 limbs as well as in chondrogenic cells exposed to Hdac3 inhibitors. Elevated expression of Mmp3 and Mmp10 transcripts was also observed. In conclusion, Hdac3 regulates distinct pathways in mesenchymal cell populations and is required for mesenchymal progenitor cell differentiation and long bone development.
Keywords: Hdac3, Fgf21, limb development, chondrogenesis, Mmp3, Mmp10, vorinostat, RGFP966
Introduction
Long bones are formed through the process of endochondral ossification in which mesenchymal progenitor cells condense and differentiate into chondrocytes that create a temporary cartilaginous scaffold. Longitudinal bone growth is dependent on the proliferation and terminal hypertrophic differentiation of chondrocytes at growth plates. The chondrogenic scaffold is eventually degraded and osteoblasts are recruited and produce a mineralized matrix. A variety of extracellular factors, including fibroblast growth factors (Fgf), Indian hedgehog (Ihh), parathyroid hormone related protein (PTHrP), transforming growth factors (Tgf), and Wnts coordinate growth plate chondrocyte differentiation and thereby control limb length. Changes in signaling pathways that delay endochondral bone formation cause skeletal deformities (1–4).
Extracellular stimuli control cell fate and function through signal transduction pathways that change gene expression programs. The accessibility of gene regulatory regions to transcription factors, cofactors, and polymerases depends on histone organization and structure. Gene expression is temporally and spatially controlled by spacing and post-translational modifications of histones, which can facilitate or hamper the recruitment of transcription factors and RNA polymerase complexes. Histone acetylation is a post-translational modification that relaxes chromatin structure and recruits acetyl binding factors (bromodomain- and YEATs domain- containing proteins) and associated transcriptional co-activators. Histone deacetylases (Hdacs) control the timing and extent of transcriptional activity by removing acetyl groups from proteins and promoting chromatin condensation (5, 6).
The 18 mammalian Hdacs are classified into four groups (I–IV) based on their structure, function and localization in the cell (7, 8). Several class I and II Hdacs are known to contribute to bone and cartilage formation and remodeling (9, 10, 11). For example, Hdac4 (class II) is required for proper chondrocyte hypertrophy and bone lengthening (12). Despite this crucial role in development, Hdac4 is found an relatively low levels in adult bone and cartilage compared to most other Hdacs (9). Hdac5 polymorphisms may contribute to optimal bone mineral density and osteoporosis (13). Hdac5 deletion does not affect bone lengthening but is required for the accrual of bone mass and suppresses Sost expression (14, 15). Such temporal and spatial regulation contrasts the stable expression of class I Hdacs, particularly Hdac1 and Hdac3, which are the most abundant in bone and cartilage tissues (9). Class II Hdacs have little intrinsic enzymatic activity of their own and recruit Hdac3 and it unique co-repressors NCOR/SMRT to form functional deacetylase complexes (16). Thus, understanding how Hdac3 contributes to bone formation is essential for many aspects of skeletal biology.
Drugs that inhibit the activity of Hdac3 and other Zn-dependent Hdacs (Hdacs1-11) are approved as therapeutic treatments for some cancers and mood disorders in both children and adults, and are under evaluation for a variety of other pediatric and adult diseases (17, 18). These compounds are relatively safe in adults despite some side effects such as fatigue, thrombocytopenia, gastro-intestinal abnormalities and heightened fracture risk (7). However, these pan-Hdac inhibitors can have adverse effects during development as they are teratogenic to fetuses and raise fracture risk in children (19–21). Early Hdac inhibitors were non-selective, but newer molecules targeting specific Hdacs may improve therapeutic efficacy and reduce side-effects. Hdac3-selective inhibitors are potential treatments for Huntington’s disease and cancer (22, 23).
Hdac3 is detectable during early stages of skeletal development and is required for bone formation and maintenance. Somatic deletion of Hdac3 causes embryonic lethality (24), but conditional deletion of Hdac3 at various stages of skeletal development has provided insight into Hdac3 function in bone biology. Hdac3 deletion in mature osteoblasts using the OCN-Cre driver did not affect bone length or animal size but reduced bone strength and volume in adult mice (25). In contrast, deletion of Hdac3 in pre-osteoblasts and some hypertrophic chondrocytes using the Osx1-Cre driver stunted bone lengthening and reduced bone density and strength (26, 27). Deletion of Hdac3 in early chondrogenesis with Col2-Cre caused embryonic lethality and postnatal deletion of Hdac3 in chondro-progenitor cells using an inducible Col2a1-CreERT driver allowed for survival despite severe bone deformations including shorter limbs, disorganized trabecular structure and increased levels of matrix degrading proteins and inflammatory cytokines that promoted bone resorption (28, 29). In the present study, we deleted Hdac3 in mesenchymal cells expressing Prrx1-Cre. We found that Hdac3 expression in the mesenchymal progenitors is essential for both survival and limb development and that Hdac3 regulates a novel subset of genes in Prrx1-expressing limb bud cells. Notably, Fgf21 expression was elevated in Hdac3-deficient limbs and mesenchymal cells and may be one of many genes differentially controlled in progenitors by Hdac3 that affect limb development.
Materials and Methods
Animals
Mice harboring loxP sites flanking exon 7 of Hdac3 (Hdac3fl/fl mice (24) were bred with mice expressing the Cre recombinase under the Prrx1 promoter (30) to create Hdac3fl/fl;Prrx1-Cre+ conditional knockout (CKOPrrx1) mice and Hdac3fl/+;Prrx1-Cre+ conditional heterozygous (CHET) mice. All mice were maintained on C57BL/6 background. Mice were genotyped for normal or rearranged Hdac3 alleles and the Prrx1-Cre transgene with PCR using tail DNA as a template as previously described (26). All animal research was conducted in accordance with the guidelines provided by National Institutes of Health. The Mayo Clinic Institutional Animal Care and Use Committee approved all animal studies.
Whole Mount X-gal Staining
Prrx1-Cre;Rosa26-LacZ pups were euthanized on postnatal day 3 and processed for X-gal staining as previously described (31, 32). Briefly, neonates were carefully eviscerated and all internal organs were removed from the abdominal and thoracic cavities. The carcasses were rinsed with PBS (phosphate buffered saline, pH 7.4) and incubated overnight at 4°C in fixative (0.2% glutaraldehyde, 2 mM magnesium chloride, 5 mM EGTA, 0.1 M sodium phosphate buffer (pH 7.4)). Following fixation, the specimens were washed with PBS and incubated overnight at 37°C in the dark in X-gal reaction buffer (1 mg/ml 5-bromo-4-chloro-3-indolyl-β-δ-galactopyranoside, 5 mM potassium ferrocyanide, 2 mM magnesium chloride, 0.1% Triton X-100 in PBS (pH 7.4)). After X-gal incubation, neonate specimens were washed with PBS and incubated at 37°C in a series of solutions with increasing glycerol concentrations prepared in 1% (w/v) potassium hydroxide (KOH), starting with 20% (v/v) glycerol, 50% (v/v) glycerol, 80% (v/v) glycerol, and ending with 100% glycerol. Samples were incubated in each clearing solution for 7 days, after which the solution was changed to the next subsequent clearing solution. At the end of clearing, the X-gal stained neonates were stored in 100% glycerol until imaged.
Whole mount skeletal staining
Newborn pups were euthanized and skeletons fixed in 95% ethanol overnight. Cartilage elements were stained with a 30% Alcian blue dye following incubation in 2% KOH to dissolve the soft tissue. Bones were stained with 75μg/ml Alizarin red S (Sigma) in 1% KOH overnight and then destained in 20% glycerol, 1% KOH for 2 weeks with daily solution changes. Skeletons were stored in 50% glycerol, 50% ethanol solution. Images were obtained using a Wild M420 microscope (Wild Heerbrugg, Switzerland) and PreGres C3 camera (Jenoptik).
Histology and immunohistochemistry
Femura and humeri were harvested from 1 day-old mice, fixed in 10% neutral-buffered formalin (NBF) for 24 hours, and decalcified in 14% EDTA for 72 hours. Tissues were then embedded into paraffin and sectioned to a thickness 5μm. Alcian blue/hematoxylin/Orange G eosin staining was performed on sections for visualization of cartilage and bone.
For immunohistochemistry sections were deparaffinized and rehydrated. Antigen retrieval was performed in citric acid, pH 9.0, in a pressure chamber followed by quenching of endogenous peroxidase activity in 3% H2O2. Sections were incubated overnight with a 1:500 dilution of anti-Hdac3 antibody (Abcam, #ab7030), a 1:100 dilution of anti-Fgf21 antibody (R&D Systems # AF 3057A), or an anti-pERK antibody (Cell Signaling, #5683) and then rinsed in PBS. Images were developed using a polyvalent secondary HRP detection kit (Abcam #ab6464).
TUNEL assays
TUNEL staining was performed using the In Situ Cell Death Detection Kit (Fluorescein) according to the manufacturer’s specifications (Roche, Germany # 11684795910). DAPI staining (Sigma-Aldrich, # D9542) was used to measure cell numbers. To determine the percentage of TUNEL positive cells, the number of TUNEL-positive cells was divided by the number of DAPI positive cells in multiple regions.
BrdU assays
Newborn pups were intraperitoneally injected with BrdU labeling reagent (Invitrogen, #00-0103) at a dose of 1ml/100g three hours before euthanasia. BrdU incorporation was detected by IHC using anti-BrdU antibody (Cell Signaling, cat# 5292).
RNA extraction
Limbs were dissected from 1 day-old mice. Bones were cleaned of soft tissues and homogenized in TRIzol reagent (Invitrogen #15596026) using a high-speed homogenizer (Ultra-Turrax T25, IKA). RNA was isolated using Trizol and chloroform purification and then reverse transcribed into cDNA using the SuperScript III first strand synthesis system (Invitrogen). Relative expression of genes of interest was measured by real-time qPCR using the Bio-Rad iQ SYBR Green Supermix and the Bio-Rad MyiQ Single Color Real-Time PCR Detection System. Transcript levels were normalized to the house keeping gene tubulin and gene expression levels were quantified using the 2−ΔΔCt method. Primers used were: Hdac3: 5′-CCCGCATCGAGAATCAGAAC-3′ and 5′-TCAAAGATTGTCTGGCGGATCT-3′; Tubulin alpha: 5′-GGTTCCCAAAGATGTCAATGCT, and 5′-CAAACTGGATGGTACGCTTGGT′; Fgf21: 5′-AACAGCCATTCACTTTGCCTGAGC-3′ and 5′-GGCAGCTGGAATTGTGTTCTGACT-3′; Mmp3: 5′-GTCCCTCTATGGAACTCCCAC-3′ and 5′-AGTCCTGAGAGATTTGCGCC; and Mmp10: 5′ GACCCCAGACAAATGTGATCCT – 3′ and 5′-TTCAGGCTCGGGATTCCA; Sox9: 5′ -AGGAAGCTGGCAGACCAGTA-3′ and 5′-CGTTCTTCACCGACTTCCTC-3′; Aggrecan: 5′-CCGCTTGCCAGGGGGAGTTG-3′ and 5′-GATGATGGGCGCACGCCGTA-3′; Col2a1: 5′-ACTGGTAAGTGGGGCAAGAC-3′ and 5′-CCACACCAAATTCCTGTTCA-3′; Col10a1: 5′-CTTTTGTGTGCCTTTCAATCG-3′ and 5′-ACTGGTAAGTGGGGCAAGAC-5′. Col1a1: 5′-GCTTCACCTACAGCACCCTTGT-3′ and 5′-TGACTGTCTTGCCCCAAGTTC-3′; Runx2: 5′-GGCACAGACAGAAGCTTGATG-3′ and 5′- GAATGCGCCCTAAATCACTGA-3′.
High-throughput RNA sequencing (RNA-seq) and bioinformatics analysis
High-throughput RNA-seq and bioinformatics analyses were performed on RNA from three newborn WT and CKO mouse limbs as previously reported using the MAP-RSeq pipeline (29, 33–35). Briefly, after initial read alignment, paired-end reads were aligned by TopHat 2.0.6 (36) against the mm10 genome using the Bowtie1 aligner (37). Gene expression was expressed in reads per kilobase per million mapped reads (RPKM). Complete RNA-seq data sets are available at the Gene Expression Omnibus (GSE96907). Genes with RPKM values higher than 0.01, fold changes of more than 1.4, and p values less than 0.05 were considered differentially expressed in the CKO cells. The resulting gene lists were analyzed with DAVID (Database for Annotation, Visualization, and Integrated Discovery) v6.7 for functional annotation clustering of genes. To further identify molecular pathways, GSEA was performed with the C5 module (Gene Ontology derived gene sets) from the Molecular Signatures Database (MSigDB_v5.2; http://software.broadinstitute.org/gsea/msigdb/collections.jsp) (38, 39). Venn diagrams were generated in Venny2.1 (40) using data collected in this project and previously published datasets (29).
Protein expression analysis
Limbs were dissected from one day-old mice, cleaned of soft tissues, frozen in liquid nitrogen and ground with a mortar and pestle under liquid nitrogen. Specimens were lysed in radio-immunoprecipitation assay (RIPA) buffer (150 mM Tris, 160 mM NaCl, 0.1%SDS 1% NP-40) and stored at −80°C. Protein concentrations were determined with the DC Protein Assay (BioRad, # 500016) and 20μg protein were separated by SDS-PAGE (10% gels) and transferred to a polyvinylidene difluoride membrane. The membranes were blocked with 5% non-fat dry milk for 1 hour at room temperature and blotted with primary antibodies over night at 4°C. Primary antibodies used were: Hdac3 (1:5000, Abcam #ab63353), Fgf21 (1:100; R&D Systems, #AF3057A), tubulin (1:2000; Sigma Aldrich, #T8203); pERK (1:1000; Cell Signaling, #9010), total Erk (1:1000; Cell Signaling #9102,), beta-actin (1:5000, Sigma #A5316), and H3K27ac (1:1000; Abcam #ab177178). After washing with TBS-T (Tris-buffered saline, 0.05% Tween 20) the membranes were incubated with secondary antibodies 1:1000 and visualized using the SuperSignal West Femto Maximum Sensitivity substrate (Thermo Fisher, # 34095). Proteins were visualized and relative Hdac3 and Fgf21 protein levels were quantified based on band intensity with image analysis software (ImageJ).
Primary chondrocyte cultures
Chondrocytes were isolated from metatarsals of 1 day-old mice as previously described (41). The bones were cleaned from skin and muscles as much as possible, incubated in 0.2% trypsin versene (Thermo Fisher, # 15040066) for 2 hours with periodical vortexing, and then incubated with 0.2% collagenase for 3 hours with periodical vortexing. All incubations were done in 37°C. Cells were isolated using 70 micron strainer and seeded in monolayer at 5×104/cm2 plate for experiments. For Hdac inhibitor experiments, vorinostat (10μM, Sigma) and RGFP966 (10μM, Sigma) were added to immature mouse epiphyseal cell micromasses (29) every three to four days during a 17-day culture.
Calvarial cell isolation and osteogenic assays
Calvarial osteoblasts were isolated from newborn pups. Briefly, the pups were euthanized and bones were dissected and washed in sterile PBS. The bones were digested in digestion medium containing 2mg/ml collagenase and 3mg/ml trypsin versene (Worthington, #LS004182 and Invitrogen, #15040066, respectively). Three one hour-long digestions were performed. After the third digestion the cells were collected, strained through a 70μM mesh and plated in a dish and expanded. First and second passage cells were used for experiments.
Statistical analysis
Statistical analysis was performed with either paired or unpaired Student’s t-test and noted on the figures when applicable (*, p<0.05. **, p<0.01).
Results
Hdac3 deletion in mesenchymal progenitor delays bone lengthening
To determine how Hdac3 contributes to appendicular skeletal development we generated mice in which Hdac3 is deleted in mesenchymal progenitors under the regulation of the Prrx1 promoter, expression of which is largely restricted to mesenchymal tissues in the limbs and cranial bones (Figure 1A). The Hdac3-CKOPrrx1 mice died perinatally within 24 hours of birth and exhibited severe skeletal deficits, including shorter fore and hind limbs (Figure 1B). Mice retaining one copy of Hdac3 (CHETs) survived and did not show any gross phenotypes. Newborn Hdac3-CKOPrrx1 mice weighed 15% less than WT or CHET littermates (Figure 1C). Whole mount staining showed that skeletal patterning was normal and that ossification was initiated in the Hdac3-CKOPrrx1 animals but that limb lengths were shorter (Figure 1D).
Figure 1. Hdac3 deletion in mesenchymal progenitors results in severe bone defects.
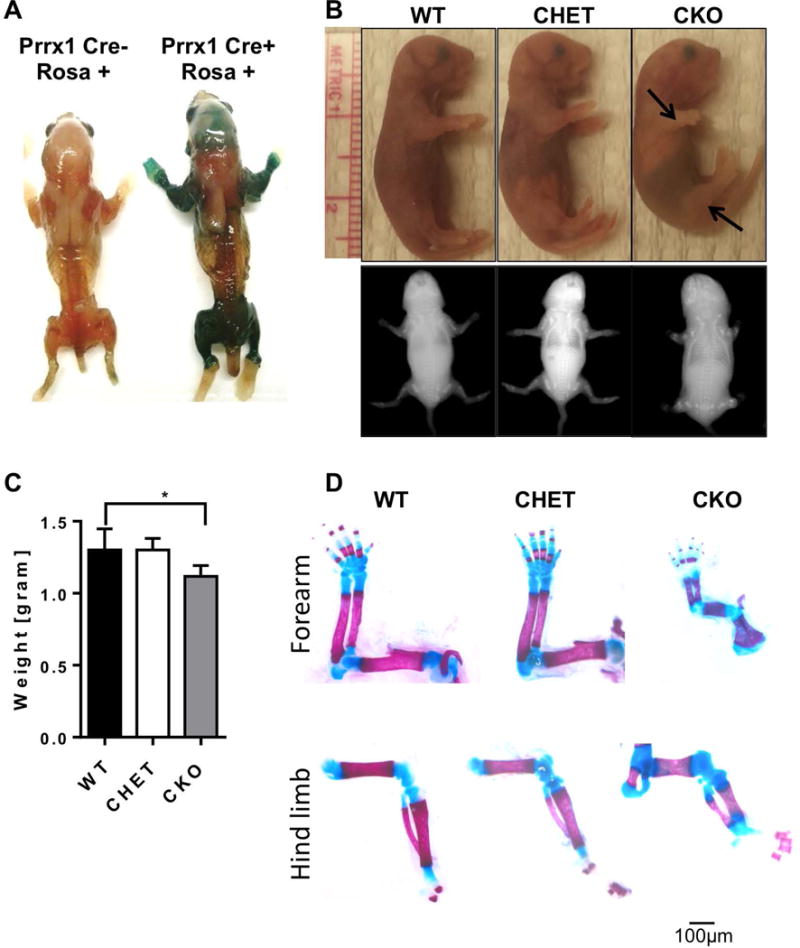
A. X-gal stains of Prrx1-Cre; Rosa26-LacZ mice showing expression patterns of the Prrx1-Cre. B. Photographs and x-ray images of 1 day-old pups. Arrows point to the shorter fore and hind limbs of Hdac3 CKO mice. C. Graphical representation of the mouse weights at one day of age. N = 8 *p<0.05. D. Whole mount Alizarin red and Alcian blue staining with representative images of the forearm and hind limbs of WT, Hdac3 CHET, and Hdac3 CKO mice. Scale bar represents 50 microns.
To gain further understanding of mechanisms governing shorter limb length of the Hdac3-CKOPrrx1 mice, femurs of the 1 day-old Hdac3-CKOPrrx1 and WT mice were examined histologically. Immunohistochemistry on WT mice confirmed our previous observations that Hdac3 is present throughout the growth plate and is notably abundant in the hypertrophic chondrocytes (25, 28). Hdac3 expression was markedly reduced in the growth plates of Hdac3-CKOPrrx1 mice, particularly in the hypertrophic zone, which was noticeably disorganized (Figure 2A). Overall, the hypertrophic zone was 70% longer and occupied a greater fraction of the bone, which was 40% shorter in the Hdac3-CKOPrrx1 mice as compared to WT and CHET littermates (Figure 2B–2C). More TUNEL positive cells (apoptotic cells) were also present in the hypertrophic chondrocytes and at the transition to primary spongiosa of Hdac3-CKOPrrx1 mice (Figure 3A–B). Hdac3-CKOPrrx1 mice also exhibited reduced BrdU labeling in the proliferative zone (Figure 3C). Aggrecan mRNA levels were modestly higher (p=0.09), but mRNA transcripts for Col2, Col10a1, Sox9, Pthrp, Ihh, Gli1, Gli2, Gli3, Ptch1, Ptch2, and Opn were not different between the Hdac3-CKOPrrx1 mice and controls (Figure 4A and data not shown). Together, these results indicate that defects in chondrocyte proliferation and survival contribute to the severe limb developmental delays in the Hdac3-CKOPrrx1 mice.
Figure 2. Hdac3 deletion in mesenchymal progenitors results in abnormal growth plate structure.
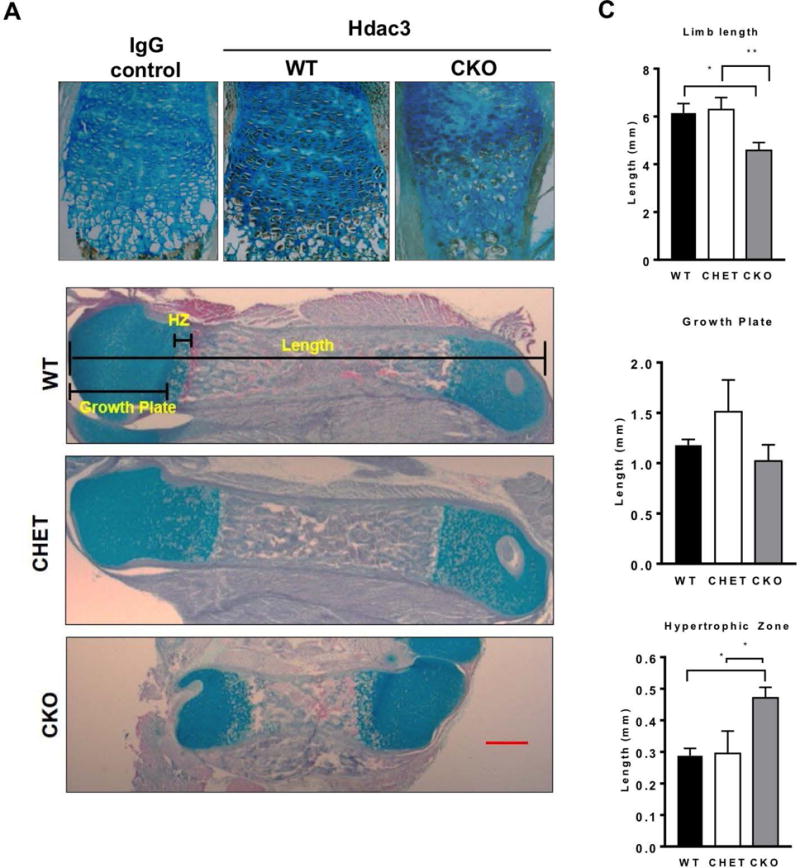
A. Immunohistochemistry for Hdac3 expression in 1 day-old WT and Hdac3 CKO mice. B. Alcian blue/Orange G staining of limbs from 1 day-old WT and CKO mice. C. Measurements of the limb length, growth plate length and hypertrophic length from WT and CKO limbs. N=5 *p<0.05. **p<0.01.
Figure 3. Hdac3 deletion in mesenchymal progenitors reduces proliferation and increase apoptosis in growth plates.
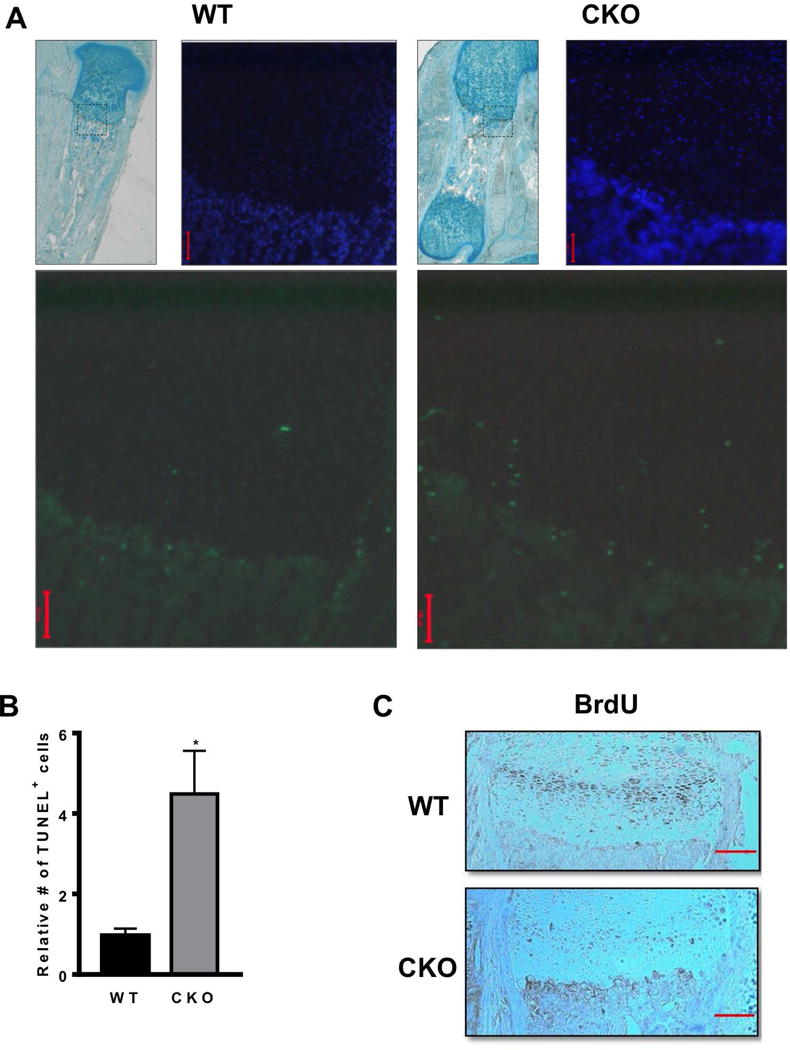
A-B. Images and quantification of TUNEL positive cells in the growth plates of WT and Hdac3-CKOPrrx1 mice (N=5). *p<0.05. C. BrdU staining in WT and Hdac3-CKOPrrx1 mice (N=3). Scale bars represent 100 microns.
Figure 4. Hdac3 deletion in the mesenchymal progenitor alters expression of genes that control matrix production and chondrocyte maturation.
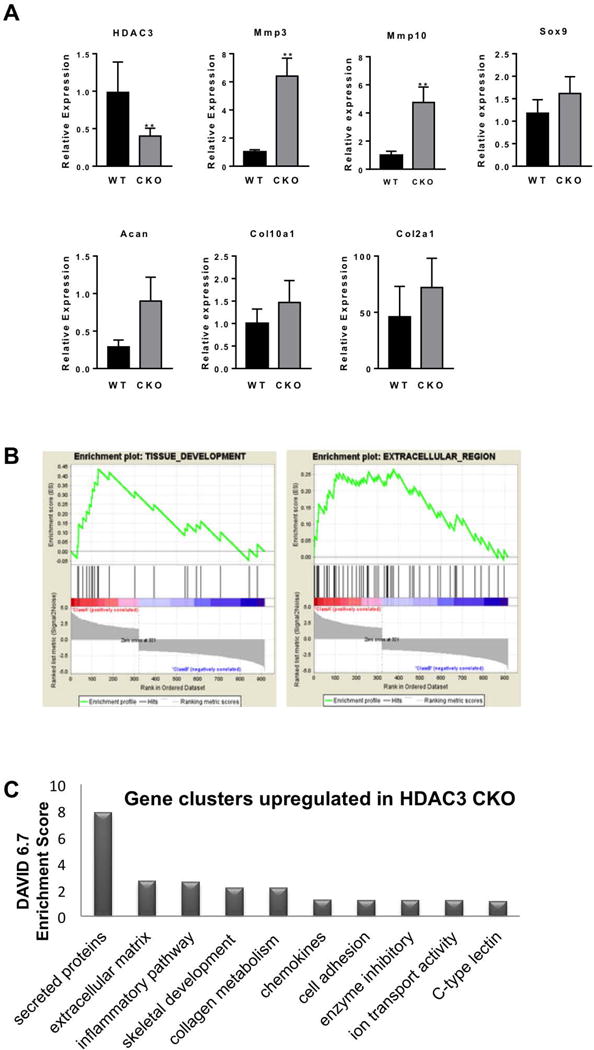
A. Gene expression analysis by qPCR to validate RNA-seq results. (N=3) B. Enrichment plots for Tissue Development and Extracellular Region data sets in GSEA. C. Gene clusters identified with DAVID analysis.
Hdac3 deletion in mesenchymal progenitor cells alters expression of genes important for bone development
To determine how Hdac3 controls appendicular skeletal development, a transcriptome analysis was performed on RNA isolated from newborn limbs of WT or Hdac3-CKOPrrx1 mice. Preliminary qPCR analysis showed a 55% reduction Hdac3 mRNA in this heterogeneous tissue (Figure 4). RNA-seq analysis identified 380 transcripts that were induced 1.4-fold or more and 1,213 transcripts that were suppressed 1.4-fold or more in limbs of Hdac3-CKOPrrx1 mice (p<0.05). The top 20 induced and suppressed transcripts are listed in Tables 1 and 2, respectively. Consistent with qPCR data, bioinformatics analyses of RNA-seq data did not reveal any changes in many chondrocyte related genes (e.g., Col2, Col10a1, Sox9, or aggrecan (Figure 4A), but several genes involved on bone biology were induced more then two-fold in Hdac3-CKOPrrx1 mice compared to WT mice (Table 3). For example, Mmp3 and Mmp10 were elevated in Hdac3-CKOPrrx1 limbs. Gene set enrichment analysis (GSEA) for gene ontogeny (GO) groups and pathways revealed that genes associated with tissue development processes and extracellular regions were significantly altered in Hdac3-CKOPrrx1 limbs (enrichment scores = 0.44 and 0.27, respectively; and FDR q-values = 0.05 and 0.14, respectively) (Figure 4B, Table 4, Supplementary Tables 1 and 2). DAVID pathway analysis of genes induced in the Hdac3-deficient limbs identified groups regulating extracellular matrix and matrix development (Figure 4C).
Table 1.
Transcripts that are significantly upregulated in limbs of the Hdac3-CKOPrrx1 mice
| Gene Name | Fold Change | p value |
|---|---|---|
| Tmem59l | 32.4 | 0.006 |
| Ccl22 | 16.8 | 0.000 |
| Fgf21 | 11.8 | 0.040 |
| Slc22a12 | 11.3 | 0.019 |
| Gm12946 | 11.1 | 0.027 |
| Got1l1 | 10.4 | 0.011 |
| RP23-235J5.14 | 10.2 | 0.024 |
| 1700013G24Rik | 10.2 | 0.027 |
| Msx3 | 10.0 | 0.044 |
| Saa3 | 8.69 | 0.009 |
| Ankdd1a | 8.67 | 0.045 |
| 2010308F09Rik | 7.59 | 0.020 |
| Gm27542 | 7.59 | 0.020 |
| Banf2os | 7.46 | 0.005 |
| Mir30d | 7.40 | 0.016 |
| Hist1h2br | 7.25 | 0.016 |
| Odam | 6.87 | 0.011 |
| Calm5 | 6.70 | 0.047 |
| Gm8909 | 6.59 | 0.040 |
| March10 | 6.57 | 0.003 |
| Mmp10 | 6.50 | 0.009 |
Table 2.
Transcripts that are significantly downregulated in limbs of the Hdac3-CKOPrrx1 mice
| Gene Name | Fold Change | p value |
|---|---|---|
| Grpr | 30.64 | 0.043 |
| RP23-243H19.2 | 20.07 | 0.006 |
| Daw1 | 16.99 | 0.001 |
| Gm28712 | 16.79 | 0.005 |
| Gm12159 | 12.61 | 0.001 |
| Gm3617 | 11.38 | 0.043 |
| RP23-92D17.3 | 10.37 | 0.036 |
| BC117090 | 10.29 | 0.030 |
| Gm26641 | 10.06 | 0.032 |
| Zic3 | 10.02 | 0.044 |
| Slco4c1 | 9.51 | 0.036 |
| RP23-114N13.2 | 9.39 | 0.004 |
| RP23-87P16.4 | 9.24 | 0.026 |
| Gm26347 | 9.24 | 0.026 |
| Pcp4 | 9.13 | 0.004 |
| Gm13230 | 9.10 | 0.008 |
| Aqp9 | 9.00 | 0.004 |
| Rpl21-ps3 | 8.76 | 0.028 |
| Gm11439 | 8.59 | 0.048 |
| Dio3 | 8.30 | 0.018 |
Table 3.
Genes associated with bone formation that upregulated in the limbs of Hdac3-CKOPrrx1
| Gene Name | Fold Change | p value |
|---|---|---|
| Msx3 | 10.0 | 0.040 |
| Saa3 | 8.70 | 0.008 |
| Mmp10 | 6.75 | 0.002 |
| Clip | 3.25 | 0.007 |
| Mmp3 | 2.75 | 0.020 |
| Mgp | 2.50 | 0.006 |
| Ibsp | 2.00 | 0.040 |
Table 4.
Pathways found in C5 module
| NAME | ES | NOM p-val | FDR q-val |
|---|---|---|---|
| TISSUE_DEVELOPMENT | 0.44 | 0.00 | 0.05 |
| EXTRACELLULAR_REGION | 0.27 | 0.00 | 0.14 |
| TRANSPORT | 0.28 | 0.01 | 0.19 |
| ESTABLISHMENT_OF_LOCALIZATION | 0.26 | 0.02 | 0.19 |
| STRUCTURAL_MOLECULE_ACTIVITY | 0.37 | 0.01 | 0.23 |
Hdac3 deletion in the mesenchyme increases Fgf21 expression
Elevated levels of Mmp3 were previously observed in chondrocytes lacking Hdac3 (29). However, a Venn diagram comparison of differentially expressed genes in Hdac3-CKOPrrx1 limb cells versus Hdac3-CKOCol2ERT chondrocytes (29) revealed that only a small number of genes were similarly affected by Hdac3 deletion in the two experiments (Figure 5A). Only 62 genes (2.1%) were induced by Hdac3 deletion in both chondrocytes and limb progenitors and 37 genes (1.2%) were suppressed in both cell types. This suggests that Hdac3 plays distinct roles in these cell populations during development.
Figure 5. Hdac3 CKO limbs show elevated levels of Fgf21 expression.
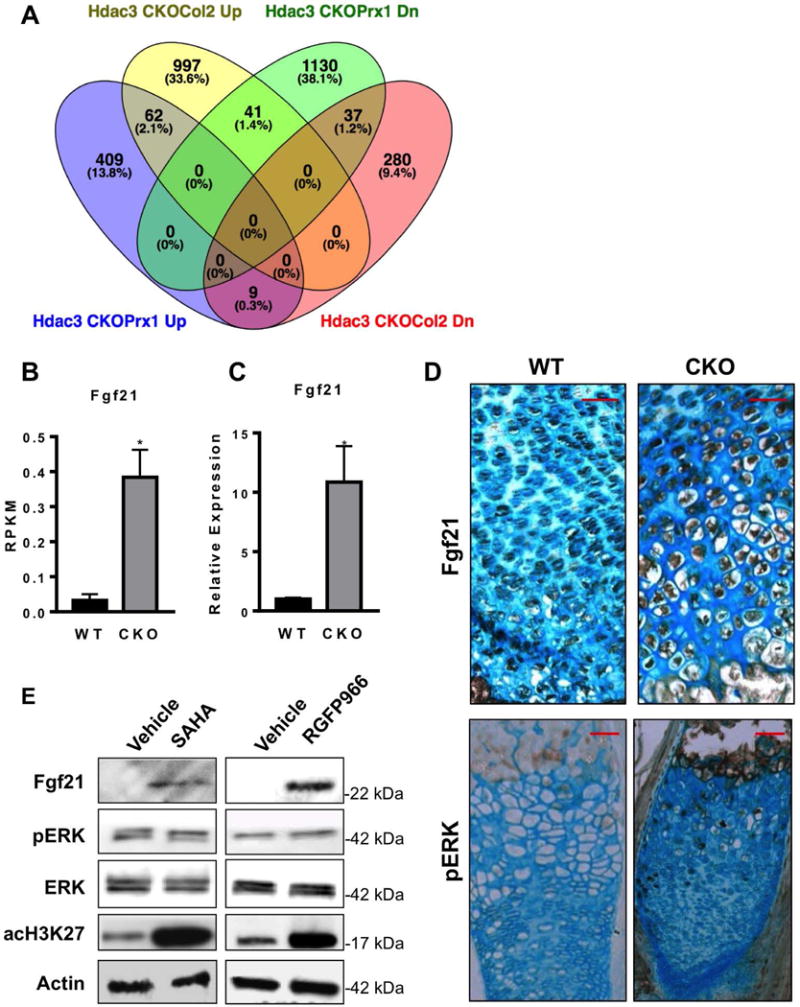
A. Venn Diagram comparing RNA-seq results from this study using Hdac3-CKOPrrx1 limb cells and Hdac3-CKOCol2ERT chondrocytes (29). B. Fgf21 RPKMs values from RNA-seq analysis of WT and Hdac3 CKO limbs (N=3). C. QPCR analysis for Fgf21 expression in WT and Hdac3 CKO limbs (N=8). D. Immunohistochemistry staining for Fgf21 in hypertrophic zone of WT and Hdac3 CKO mice. E. A representative image from one of at least three western blots of Fgf21 and other proteins in immature mouse articular chondrocyte micromass cultures incubated with SAHA/vorinostat or RGFP966 for 17 days.
One of the most enriched genes in the Hdac3-CKOPrxx1 limb cells was Fgf21. It was suppressed 2.2 fold in previous RNA-seq studies of Hdac3-CKOCol2ERT chondrocytes (29), but the high levels of Fgf21 mRNA that were observed in Hdac3-CKOPrrx1 limb cells by RNA-seq (Figure 5B) were confirmed by qPCR analyses of primary limb mRNA (Figure 5C) and were also detected by immunohistochemistry of growth plates of Hdac3-CKOPrrx1 mice (Figure 5D). In accordance with increased Fgf21 levels, elevated pERK was also detected in Hdac3-CKOPrrx1 bones though it was sporadic. Similarly, treatment of immature epiphyseal cells from mouse limbs with SAHA/vorinostat (a broad acting Hdac inhibitor that targets Hdac3 as well as other Hdacs) or RGFP966 (a Hdac3 selective inhibitor) induced Fgf21 expression and had modest effects on pERK (Figure 5E and data not shown). These data demonstrate that Hdac3 suppression increases Fgf21 expression though the functional consequences are yet to be realized.
Hdac3 deletion in the mesenchyme reduces chondrocyte and osteoblast differentiation
We next sought to examine the differentiation potential of Hdac3-CKOPrrx1 chondrocytes and osteoblasts in culture. After 6 days in chondrogenic differentiation medium, Hdac3-CKOPrrx1 primary metatarsal chondrocyte cultures showed less Alcian blue staining than wildtype chondrocytes. (Figure 6A). In accordance with the RNA-seq data, the cultured chondrocytes from Hdac3-CKOPrrx1 mice expressed lower levels of Hdac3 and higher levels of Fgf21 (Figure 6B–C). Higher levels of Mmp3 and Mmp10 were also detected in the Hdac3-CKOPrrx1 primary metatarsal chondrocyte cultures (Figure 6B). Since transcript levels of aggrecan and Col2 were largely unchanged, these results suggest that the increased matrix degradation in the Hdac3-CKOPrrx1 cultures is the major contributing factor to the reduced proteoglycan content and Alcian blue staining of these cultures. Osteoblast differentiation was also delayed as shown by reduced matrix calcification and reduced expression of Col1a1 and Runx2 in Hdac3 CKOPrrx1 primary calvarial osteoblast cultures (Figure 7). Together, these data demonstrate that Hdac3 is required for limb formation because it regulates the expression of many genes involved in bone development and extracellular matrix structure.
Figure 6. Hdac3 CKO chondrocyte cultures have fewer proteoglycans.
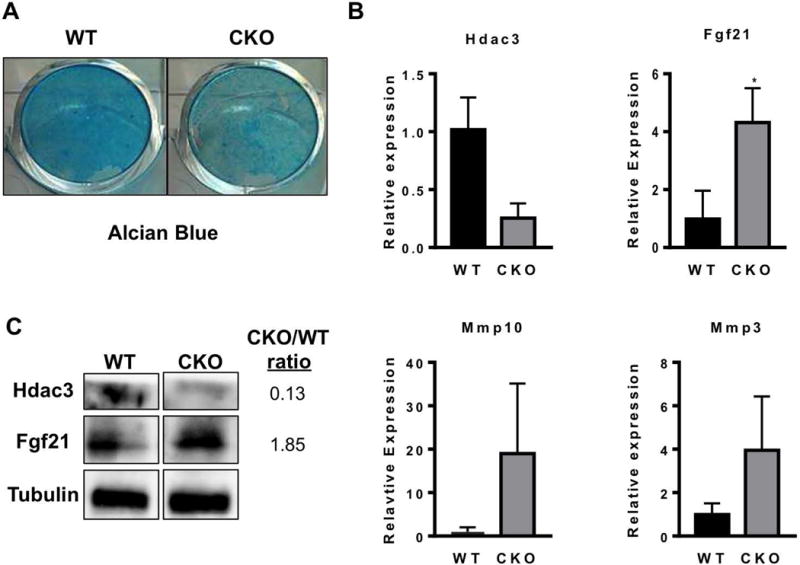
A. Chondrocyte cultures from Hdac3 CKO mice stain poorly with Alcian blue indicating that fewer proteoglycans are present. B. Gene expression analysis from chondrogenic cultures of WT and Hdac3 CKO pups. N=3 *p<0.05.
Figure 7. Hdac3 CKO calvarial osteoblast cultures show delayed maturation.
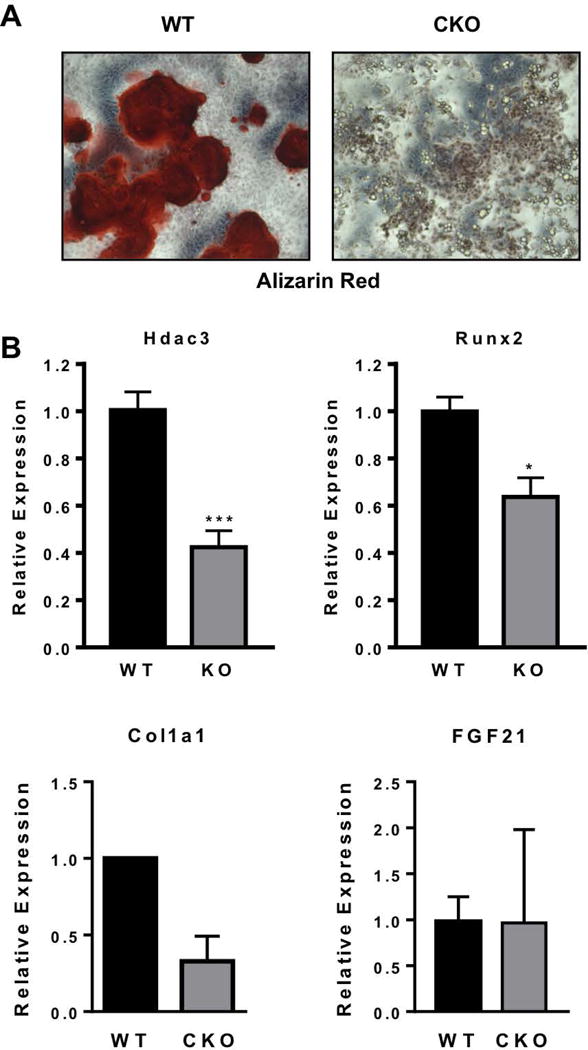
A. Primary calvarial osteoblast cultures from Hdac3 CKO mice stain poorly with Alizarin red indicating poor calcification. The image is representative of at least three experiments. B. Gene expression analysis from osteogenic cultures of WT and Hdac3 CKO pups. N: WT=6, CKO=7 *p<0.05.
Discussion
Hdac inhibitors are emerging clinical therapies for numerous conditions but preclinical data show that they interfere with skeletal development and remodeling. The objective of this study was to investigate the role of Hdac3, one of the enzymes blocked by the broad acting Hdac inhibitors, in mesenchymal progenitor cells responsible for early formation of the appendicular skeleton. We focused on appendicular skeletal development to evaluate the effects of Hdac3 depletion on mesenchymal proliferation, commitment and differentiation and to avoid embryonic lethality that was observed when Hdac3 was depleted in chondrocyte progenitors of both the axial and appendicular skeleton (28). Our results demonstrate that Hdac3 expression in Prrx1-expressing mesenchymal progenitor cells is essential for limb development. Deletion of Hdac3 in the Prrx1-expressing cells caused mice to be born with significantly shorter limbs and to die within one day of birth. Interestingly, similar phenotypes are also seen when Ezh2, an Hdac3-interacting protein, was ablated in Prrx1-expressing cells (42). The cause of death is unknown but may be related to defects in the diaphragm, where Prrx1 is also expressed (30), since mice had difficulty breathing. The bone defects seem to be caused by a variety of factors including reduced proliferation of chondrocytes in the proliferative zone of the growth plate, increased apoptosis in the hypertrophic zone, and increased expression of matrix degrading genes that may contribute to a highly disorganized cell structure within the growth plate. High-throughput RNA-seq analysis revealed enrichment of genes regulating tissue development, chondrocytic matrix degradation (such as Mmp3 and Mmp10) and chondrocyte maturation (Fgf21) in the limbs of the Hdac3-CKOPrrx1 mice. Together with our previous studies, this project emphasizes the essential role that Hdac3 plays in early endochondral ossification, as well as bone maintenance (25, 26, 28, 29).
Hdac3 acts as a transcriptional repressor and controls numerous cellular processes including cell growth and metabolism. Due to its repressor functions, genes directly affected by Hdac3 should be induced upon its deletion. The many genes that are down-regulated upon Hdac suppression are typically indirect targets. Gene set enrichment analysis of transcripts induced by Hdac3 deletion in mesenchymal progenitors indicated disruption of tissue development processes and extracellular proteins. Only a small fraction (~3%) of differentially expressed genes in Hdac3-CKOPrrx1 cells were regulated similarly in Hdac3-CKOCol2ERT mice (29). There are several limitations to the comparisons including the cell isolation and culturing procedures. The new experiments in this report used a heterogeneous mix of limb mesenchymal cells due to more careful dissection of the diaphysis was impossible due to the disorganization, softness, and small size of the Hdac3-CKOPrxx1 limbs. In contrast, previous experiments used more homogenous immature mouse chondrocytes. Despite these differences in the models used, the low similarity between the transcriptomic results was unexpected and points to unique functions of Hdac3 in the developing limb.
Of the most highly induced genes, Fgf21 was studied further because it is a regulator of endochondral ossification. Fgf21 is an atypical member of the FGF family as it can act locally as well as distantly in an endocrine fashion (43). Fgf21 is highly expressed in pancreas and liver, where it regulates insulin and lipid metabolism and may be a novel therapeutic for the treatment of hyperlipidemia. Fgf21 was also previously shown to play a role in bone development. Mice overexpressing Fgf21 are significantly smaller with shorter bones due to inhibition of chondrocyte function in the growth plate (44). In culture, Fgf21 reduced chondrocyte differentiation and maturation in a concentration dependent matter (45). Fgf21 transgenic mice exhibit severe bone loss while genetic deletion of Fgf21 increased bone mass (44). Similarly, in our study we observed that Hdac3 deletion in mesenchymal cells reduced their differentiation capacity and also increased Fgf21 expression in chondrogenic conditions in vitro and in vivo. However, the mechanisms and pathways by which Fgf21 controls limb lengthening require further study as we only observed sporadic and minor changes in pERK levels. It is possible that Fgf21 activities are temporal and could require Klotho.
Fgf21 expression can be regulated by variety of factors including hormones, nutrients and insulin (43). Leng and colleagues showed that histone deacetylase inhibitors such valproic acid and SAHA induce Fgf21 expression in glial cells (46). Similar results were achieved with in vitro suppression of either Hdac2 or Hdac3 (46). Moreover, in human hepatocytes HDAC3 suppression was associated with elevated levels of FGF21 and improved hepatic function (47). As such, we propose that Hdac3 deletion in Prrx1-expressing cells increases Fgf21 levels, which inhibits bone development. Interestingly, we did not observe changes in Fgf21 expression when Hdac3 was deleted in Col2-Cre expressing cells, but increases in Mmp3 expression were detected in both models (28). This suggests that Hdac3 influences maturation of mesenchymal progenitors and controls bone growth by regulating Fgf21, but it affects a different set of gene targets in more committed chondrocytes. Additional studies are needed to understand the mechanisms by which Hdac3 and Hdac inhibitors control Fgf21 expression.
Fgf21 may also be an important regulator of lipid and glucose metabolism (48, 49). Our previous studies showed that Hdac3 deletion in bone progenitor using the Osx1-Cre promotes lipid accumulation in bone marrow (50). In the current study, pathways controlling lipid metabolism were also enriched in Hdac3-CKOPrrx1 limbs though no evidence for lipid accumulation was observed in Hdac3-CKOPrrx1 limbs or in the limbs from Hdac3 conditionally heterozygotic (CHET) mice. More studies will be required to contribution of Hdac3 deletion and Fgf21 interactions and metabolic activity.
In summary, our work demonstrates that Hdac3 is essential for proper long bone growth and development. The severity of the bone phenotype, as well as perinatal lethality of these mice, emphasizes the importance of Hdac3 in bone biology. Together with our previously published work, it is clear that Hdac3 regulates distinct genes depending on the cell type and stage of skeletal formation. Our work adds to the notion of the complexity of Hdac3 biology and regulation of variety of genes and pathways.
Supplementary Material
Acknowledgments
The authors thank Xiaodong Li and David Razidlo for technical expertise. We thank the molecular biology core at Mayo Clinic and Chanana Pritha for the help with RNA-seq data analysis. This work was supported in part by grants from the National Institutes of Health (AR65397 (EWB)).
Author roles: MF- study design, data collection, data interpretation manuscript drafting and approval of the final manuscript version. LCS-data collection, data analysis, data interpretation and revising manuscript content. ETC, SR, AJvW- data analysis, data interpretation and revising manuscript content. EWB-revising manuscript content, JJW-study design, data interpretation and manuscript drafting. MF takes responsibility for the integrity of data analysis.
Footnotes
Disclosures
The authors have no conflicts of interest to report.
References
- 1.McGee-Lawrence ME, Carpio LR, Bradley EW, Dudakovic A, Lian JB, van Wijnen AJ, et al. Runx2 is required for early stages of endochondral bone formation but delays final stages of bone repair in Axin2-deficient mice. Bone. 2014;66:277–86. doi: 10.1016/j.bone.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kronenberg HM. PTHrP and skeletal development. Annals of the New York Academy of Sciences. 2006;1068:1–13. doi: 10.1196/annals.1346.002. [DOI] [PubMed] [Google Scholar]
- 3.Salazar VS, Gamer LW, Rosen V. BMP signalling in skeletal development, disease and repair. Nature reviews Endocrinology. 2016;12(4):203–21. doi: 10.1038/nrendo.2016.12. [DOI] [PubMed] [Google Scholar]
- 4.Long F, Ornitz DM. Development of the endochondral skeleton. Cold Spring Harbor perspectives in biology. 2013;5(1):a008334. doi: 10.1101/cshperspect.a008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389(6649):349–52. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 6.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annual review of biochemistry. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 7.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Longworth MS, Laimins LA. Histone deacetylase 3 localizes to the plasma membrane and is a substrate of Src. Oncogene. 2006;25(32):4495–500. doi: 10.1038/sj.onc.1209473. [DOI] [PubMed] [Google Scholar]
- 9.Bradley EW, Carpio LR, van Wijnen AJ, McGee-Lawrence ME, Westendorf JJ. Histone Deacetylases in Bone Development and Skeletal Disorders. Physiol Rev. 2015;95(4):1359–81. doi: 10.1152/physrev.00004.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGee-Lawrence ME, Westendorf JJ. Histone deacetylases in skeletal development and bone mass maintenance. Gene. 2011;474(1–2):1–11. doi: 10.1016/j.gene.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carpio LR, Westendorf JJ. Histone Deacetylases in Cartilage Homeostasis and Osteoarthritis. Curr Rheumatol Rep. 2016;18(8):52. doi: 10.1007/s11926-016-0602-z. [DOI] [PubMed] [Google Scholar]
- 12.Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119(4):555–66. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 13.Rivadeneira F, Styrkarsdottir U, Estrada K, Halldorsson BV, Hsu YH, Richards JB, et al. Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nature genetics. 2009;41(11):1199–206. doi: 10.1038/ng.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Molecular and cellular biology. 2004;24(19):8467–76. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wein MN, Spatz J, Nishimori S, Doench J, Root D, Babij P, et al. HDAC5 controls MEF2C-driven sclerostin expression in osteocytes. J Bone Miner Res. 2015;30(3):400–11. doi: 10.1002/jbmr.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, et al. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 2002;9(1):45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 17.Lakshmaiah KC, Jacob LA, Aparna S, Lokanatha D, Saldanha SC. Epigenetic therapy of cancer with histone deacetylase inhibitors. Journal of cancer research and therapeutics. 2014;10(3):469–78. doi: 10.4103/0973-1482.137937. [DOI] [PubMed] [Google Scholar]
- 18.Dinarello CA, Fossati G, Mascagni P. Histone Deacetylase Inhibitors for Treating a Spectrum of Diseases Not Related to Cancer. Mol Med. 2011;17(5–6):333–52. doi: 10.2119/molmed.2011.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paradis FH, Hales BF. The Effects of Class-Specific Histone Deacetylase Inhibitors on the Development of Limbs During Organogenesis. Toxicological sciences: an official journal of the Society of Toxicology. 2015;148(1):220–8. doi: 10.1093/toxsci/kfv174. [DOI] [PubMed] [Google Scholar]
- 20.Rakitin A, Koks S, Reimann E, Prans E, Haldre S. Changes in the Peripheral Blood Gene Expression Profile Induced by 3 Months of Valproate Treatment in Patients with Newly Diagnosed Epilepsy. Frontiers in neurology. 2015;6:188. doi: 10.3389/fneur.2015.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fathe K, Palacios A, Finnell RH. Brief report novel mechanism for valproate-induced teratogenicity. Birth defects research Part A, Clinical and molecular teratology. 2014;100(8):592–7. doi: 10.1002/bdra.23277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jia H, Kast RJ, Steffan JS, Thomas EA. Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington’s disease mice: implications for the ubiquitin-proteasomal and autophagy systems. Human molecular genetics. 2012;21(24):5280–93. doi: 10.1093/hmg/dds379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minami J, Suzuki R, Mazitschek R, Gorgun G, Ghosh B, Cirstea D, et al. Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia. 2014;28(3):680–9. doi: 10.1038/leu.2013.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer cell. 2010;18(5):436–47. doi: 10.1016/j.ccr.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGee-Lawrence ME, Bradley EW, Dudakovic A, Carlson SW, Ryan ZC, Kumar R, et al. Histone deacetylase 3 is required for maintenance of bone mass during aging. Bone. 2013;52(1):296–307. doi: 10.1016/j.bone.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Razidlo DF, Whitney TJ, Casper ME, McGee-Lawrence ME, Stensgard BA, Li X, et al. Histone deacetylase 3 depletion in osteo/chondroprogenitor cells decreases bone density and increases marrow fat. PLoS One. 2010;5(7):e11492. doi: 10.1371/journal.pone.0011492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradley EW, Carpio LR, Westendorf JJ. Histone deacetylase 3 suppression increases PH domain and leucine-rich repeat phosphatase (Phlpp)1 expression in chondrocytes to suppress Akt signaling and matrix secretion. J Biol Chem. 2013;288(14):9572–82. doi: 10.1074/jbc.M112.423723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradley EW, Carpio LR, McGee-Lawrence ME, Castillejo Becerra C, Amanatullah DF, Ta LE, et al. Phlpp1 facilitates post-traumatic osteoarthritis and is induced by inflammation and promoter demethylation in human osteoarthritis. Osteoarthritis Cartilage. 2016;24(6):1021–8. doi: 10.1016/j.joca.2015.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carpio LR, Bradley EW, McGee-Lawrence ME, Weivoda MM, Poston DD, Dudakovic A, et al. Histone deacetylase 3 supports endochondral bone formation by controlling cytokine signaling and matrix remodeling. Sci Signal. 2016;9(440):ra79. doi: 10.1126/scisignal.aaf3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33(2):77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 31.Kawaguchi J, Wilson V, Mee PJ. Visualization of whole-mount skeletal expression patterns of LacZ reporters using a tissue clearing protocol. BioTechniques. 2002;32(1):66, 8–70, 2–3. doi: 10.2144/02321st02. [DOI] [PubMed] [Google Scholar]
- 32.Schatz O, Golenser E, Ben-Arie N. Clearing and photography of whole mount X-gal stained mouse embryos. BioTechniques. 2005;39(5):650. doi: 10.2144/000112034. 2, 4 passim. [DOI] [PubMed] [Google Scholar]
- 33.Kremer KN, Dudakovic A, McGee-Lawrence ME, Philips RL, Hess AD, Smith BD, et al. Osteoblasts protect AML cells from SDF-1-induced apoptosis. Journal of cellular biochemistry. 2014;115(6):1128–37. doi: 10.1002/jcb.24755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dudakovic A, Camilleri E, Riester SM, Lewallen EA, Kvasha S, Chen X, et al. High-resolution molecular validation of self-renewal and spontaneous differentiation in clinical-grade adipose-tissue derived human mesenchymal stem cells. Journal of cellular biochemistry. 2014;115(10):1816–28. doi: 10.1002/jcb.24852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalari KR, Nair AA, Bhavsar JD, O’Brien DR, Davila JI, Bockol MA, et al. MAP-RSeq: Mayo Analysis Pipeline for RNA sequencing. BMC bioinformatics. 2014;15:224. doi: 10.1186/1471-2105-15-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28(16):2184–5. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- 37.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell systems. 2015;1(6):417–25. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oliveros JC. Venny. An interactive tool for comparing lists with Venn’s diagrams. 2007–2015 [Available from: http://bioinfogp.cnb.csic.es/tools/venny/index.html.]
- 41.Dao DY, Jonason JH, Zhang Y, Hsu W, Chen D, Hilton MJ, et al. Cartilage-specific beta-catenin signaling regulates chondrocyte maturation, generation of ossification centers, and perichondrial bone formation during skeletal development. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2012;27(8):1680–94. doi: 10.1002/jbmr.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dudakovic A, Camilleri ET, Xu F, Riester SM, McGee-Lawrence ME, Bradley EW, et al. Epigenetic Control of Skeletal Development by the Histone Methyltransferase Ezh2. The Journal of biological chemistry. 2015;290(46):27604–17. doi: 10.1074/jbc.M115.672345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fisher FM, Maratos-Flier E. Understanding the Physiology of FGF21. Annual review of physiology. 2016;78:223–41. doi: 10.1146/annurev-physiol-021115-105339. [DOI] [PubMed] [Google Scholar]
- 44.Wei W, Dutchak PA, Wang X, Ding X, Bookout AL, Goetz R, et al. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor gamma. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(8):3143–8. doi: 10.1073/pnas.1200797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu S, Levenson A, Kharitonenkov A, De Luca F. Fibroblast growth factor 21 (FGF21) inhibits chondrocyte function and growth hormone action directly at the growth plate. The Journal of biological chemistry. 2012;287(31):26060–7. doi: 10.1074/jbc.M112.343707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leng Y, Wang J, Wang Z, Liao HM, Wei M, Leeds P, et al. Valproic Acid and Other HDAC Inhibitors Upregulate FGF21 Gene Expression and Promote Process Elongation in Glia by Inhibiting HDAC2 and 3. The international journal of neuropsychopharmacology. 2016;19(8) doi: 10.1093/ijnp/pyw035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Gao Z, Zhang J, Ye X, Xu A, Ye J, et al. Sodium butyrate stimulates expression of fibroblast growth factor 21 in liver by inhibition of histone deacetylase 3. Diabetes. 2012;61(4):797–806. doi: 10.2337/db11-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Canto C, Auwerx J. Cell biology. FGF21 takes a fat bite. Science. 2012;336(6082):675–6. doi: 10.1126/science.1222646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dutchak PA, Katafuchi T, Bookout AL, Choi JH, Yu RT, Mangelsdorf DJ, et al. Fibroblast growth factor-21 regulates PPARgamma activity and the antidiabetic actions of thiazolidinediones. Cell. 2012;148(3):556–67. doi: 10.1016/j.cell.2011.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGee-Lawrence ME, Carpio LR, Schulze RJ, Pierce JL, McNiven MA, Farr JN, et al. Hdac3 Deficiency Increases Marrow Adiposity and Induces Lipid Storage and Glucocorticoid Metabolism in Osteochondroprogenitor Cells. J Bone Miner Res. 2016;31(1):116–28. doi: 10.1002/jbmr.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.