

Abstract
Recently, we reported a convergent cyclopropanation-Cope approach to the core of ineleganolide, which was the first disclosed synthesis of the core of the norditerpene natural product ineleganolide. In this complementary work, a model system for the core of ineleganolide has been prepared through a series of tandem cyclopropanation-Cope and translactonization-Cope rearrangements. Work with this model system has enriched our understanding of the cyclopropanation-Cope rearrangement sequence. Additionally, research into this model system has driven the development of tandem translactonization-Cope rearrangements.
Graphical Abstract
Introduction
In 1999, Duh and co-workers isolated a norditerpene from the soft coral Sinularia1 inelegans and named it ineleganolide (1, Figure 1).2,3 Ineleganolide demonstrates in vitro cytotoxicity against murine lymphocytic leukemia P388 cell lines (ED50 = 3.82 μg/mL),2 yet does not demonstrate activity against human oral epidermoid (KB) and liver (Hepa59T/VGH) carcinoma cells (ED50 > 20 μg/mL).3 The structure of ineleganolide (1) was elucidated by single crystal X-ray diffraction,2 revealing the relative configuration of nine stereocenters, six of which lined a central cycloheptanone core. The absolute configuration was established by Pattenden’s biomimetic semi-synthesis from 5-epi-sinuleptolide (3), which furnished the only disclosed laboratory preparation of ineleganolide.4 Owing to the novel structures and bioactivity of ineleganolide (1), Nicolaou, Moeller, Vanderwal, Frontier, Romo and co-workers have disclosed inventive and formidable approaches to its synthesis,5 none of which have provided access to its central [6,7,5,5]-core. We have accessed this core through a cyclopropanation-Cope cascade.6 Herein, we disclose model studies for that critical cyclopropanation-Cope sequence, as well as three complementary tandem transformations: translactonization-Cope, cyclopropanation-Cope-epoxidation and cyclopropanation-Cope-enolization cascades. Each transformation furnishes the [6,7,5,5]-core of ineleganolide.
Figure 1.
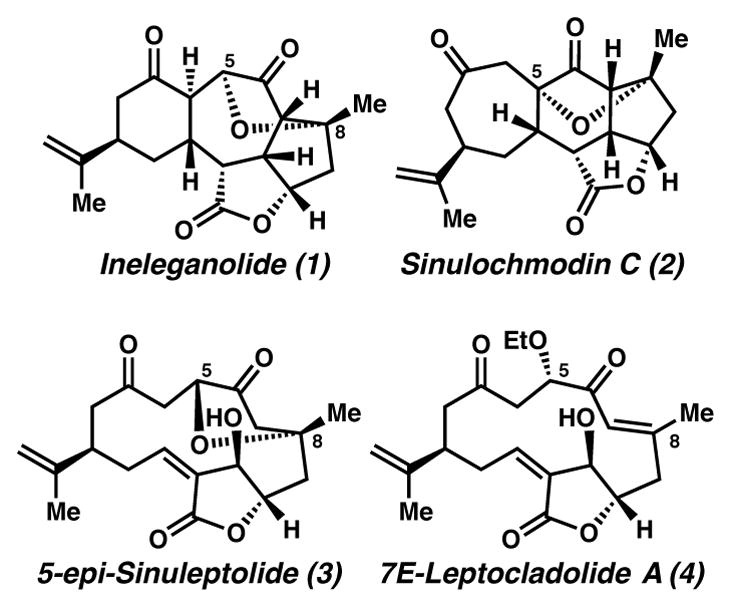
A subset of metabolites (1–4) isolated from plants of the genus Sinularia.
Results and Discussion
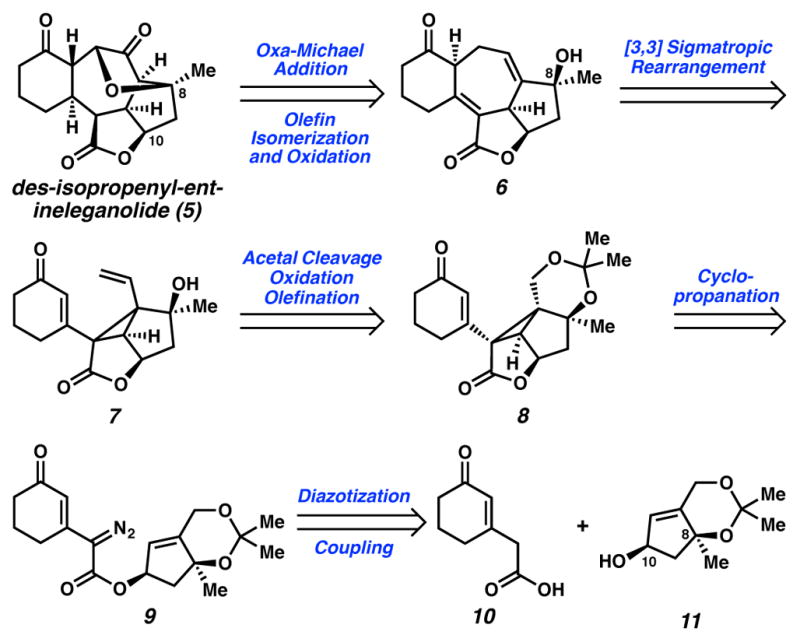
From the outset, we sought to overcome the challenges of the enantioselective installation of the C(8) tertiary ether and construction of the seven-membered ring core of ineleganolide (Scheme 1). To evaluate strategies, we targeted the simpler model, des-isopropenyl-ent-ineleganolide (5). Initial retrosynthetic simplification of the C(8)–C(5) ether and other oxidation state manipulations revealed cycloheptadienone 6. Access to model compound 6 would proceed through a strain-release Cope rearrangement of diene 7.7 The cyclopropane in diene 7 would arise from vinylcyclopropane 8. In turn, vinylcyclopropane 8 would be directly available from vinyldiazoester 9, which itself would be the coupling product of carboxylic acid 10 and alcohol 11. Embedded within alcohol 11 was a tertiary stereocenter at C(8), which we targeted through an enantioselective allylation strategy.8
Scheme 1.
Initial retrosynthetic analysis of des-isopropenyl-ent-ineleganolide (5)
In the forward direction, the first critical challenge was installation of the C(8) stereocenter. To this end, we found inspiration in work within our laboratory to establish quaternary stereogenic centers through the enantioselective decarboxylative allylation of cyclic enolates.9 If this method could be employed with a substrate bearing α-oxygenation,8b we would be able to access the C(8) center embedded within alcohol 11 (Scheme 2).8a At the time, there was limited evidence that α-heteroatoms might be tolerated in the reaction.10 Coincident with early model system investigations, we developed a general enantioselective alkylation of dioxanone-derived enol ethers,8a which could be used to prepare alcohol 118b by way of chloroallyl dioxanone 15. Treatment of triethylsilylether 12 with Pd(dmdba)2 (5 mol%), tris(CF3)-(S)-t-BuPHOX (14, 5.5 mol%),11 and chlorallyl me-sylate (13, 1.05 equiv) with an equivalent of TBAT in PhMe at 25 °C, yielded the desired tetrasubstituted ether 15 with good enantioselectivity (91% ee). Chloroolefin 15 was converted in a fickle two-step oxidative bromination/Wittig olefination sequence to volatile cyclopentenone 16.8a In turn, facile diastereoselective reduction furnished cyclopentenol 11, which contains both C(8) and C(10) stereogenic centers.
Scheme 2.

Synthesis of cyclopentenol 11
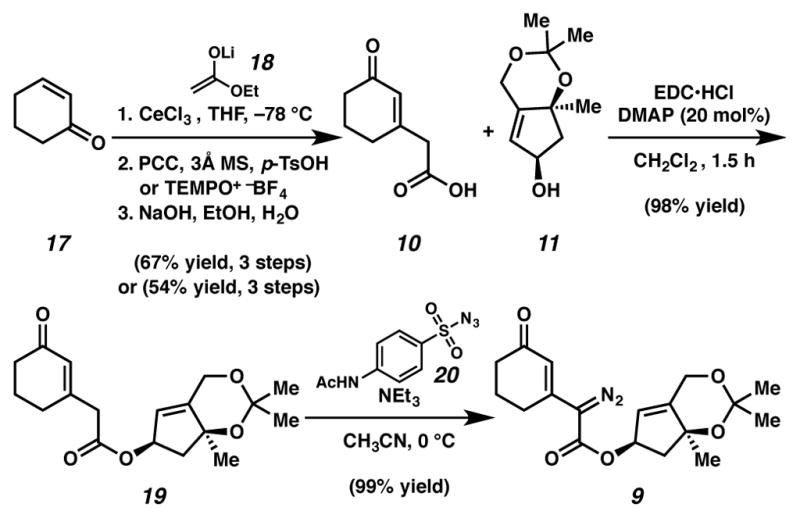
Having prepared enantioenriched cyclopentenol 11, we sought access to its coupling partner, carboxylic acid 10 (Scheme 3). Conversion of cyclohexenone (17) proceeded by an efficient CeCl3-mediated 1,2-addition of the lithium enolate of EtOAc.12 Oxidative rearrangement with allylic transposition13 and saponification generated carboxylic acid 10 in 67% overall yield. Notably, TEMPO+BF4− could be employed as an environmentally benign alternative to affect an oxidative rearrangement, albeit in slightly lower 54% overall yield.14 To prepare for the key cyclopropanation, we coupled alcohol 11 and carboxylic acid 10 with DCC. Subsequently, diazo transfer was accomplished upon treatment with p-ABSA (20) to provide the targeted cyclopropanation precursor (9) in excellent yield.
Scheme 3.
Toward the cyclopropanation substrate
Under standard cyclopropanation conditions, diazoester 9 did not give way to desired cyclopropane 8. Rather, a thermal rearrangement occurred to furnish pyrazole 21 in 59% yield (Scheme 4). This type of reaction had been previously reported,15 and Padwa had proposed that pyrazole formation proceeds through a 1,5-cyclization followed by a proton shift.15f Indeed, pyrazole 21 was generated cleanly when diazoester 9 was heated in benzene at reflux, in the absence of copper. The structure of pyrazole 21 was characterized by X-ray crystallographic diffraction, and this structure confirmed the syn-arrangement of C(8) and C(10) oxygenation.
Scheme 4.
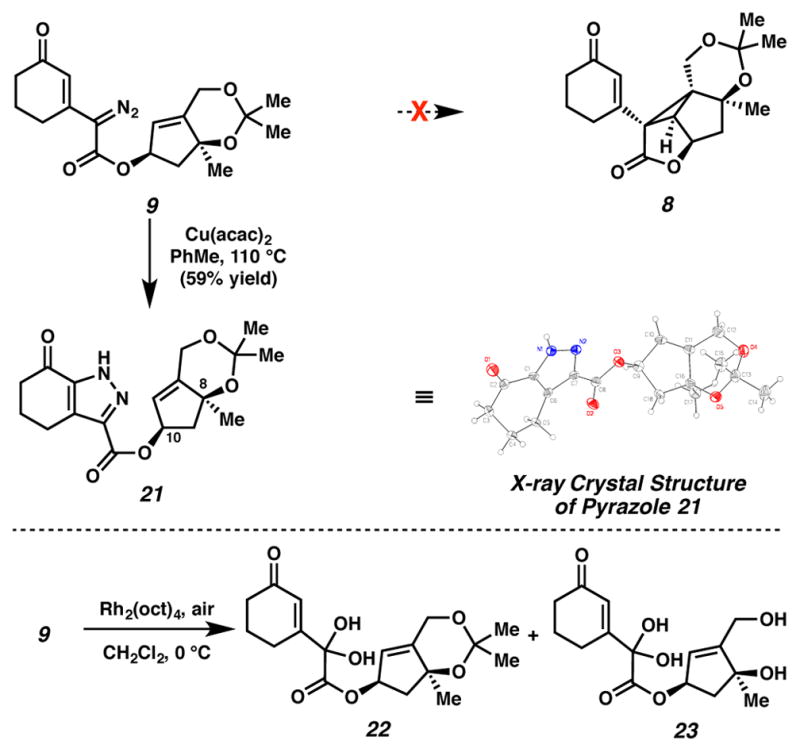
Pyrazole formation and oxidation plague initial cyclopropanation approach
At lower temperatures, neither cyclopropane 8 nor pyrazole 21 formed. One potential explanation for this lack of reactivity was that diazoester 9 did not generate the requisite metal carbenoid. To interrogate this hypothesis, we exposed diazoester 9 to Rh2(oct)4 and ambient air at 0 °C, anticipating that oxygen or water could intercept a formed metal carbenoid, ultimately furnishing oxidation products. Indeed, under these conditions, oxidized 22 formed along with its acetonide-cleaved analogue (23) suggesting that a carbenoid was generated upon exposure of diazoester 8 to Rh2(oct)4, but that this carbenoid failed to undergo cyclopropanation. Close examination of diazoester 9 and cyclopropane 8 revealed a 1,3-diaxial interaction between methyl groups at C(8) and on the acetonide that would likely disfavor cyclopropanation, as this 1,3-diaxial interaction was necessarily enhanced in cyclopropane 8, relative to diazoester 9.
To avoid this strain, we converted ester 19 to an alternative cyclopropanation precursor (Scheme 5). To this end, we turned to De Waard’s catalytic conditions for ketal cleavage,16 which enabled cleavage of acetonide 19 without olefin isomerization.16 Silylation provided ready access to an ether, which reacted with TsN3 to generate diazoester 24. We were pleased to find that copper tert-butyl salicylaldimine (Cu(tbs)2) affected cyclopropanation in 74% yield.
Scheme 5.

Successful cyclopropanation of silyl ether 24
With the cyclopropane successfully installed, we sought to advance silyl ether 25a to the requisite divinylcyclopropane Cope precursor (Scheme 6). Desilylation of cyclopropane 25a occurred readily; however, an array of products formed under a variety of oxidation conditions. At least one problematic side reaction was documented. Upon oxidation with Dess-Martin periodinane, ether 26 was observed as the major product (Scheme 6). Presumably, this product arises through conjugate addition of the primary alcohol into the enone functionality.
Scheme 6.

Byproduct formed in the desilylation–oxidation sequence
Having struggled with oxidation to provide the targeted Cope precursor (e.g., silyl ether 25a → divinyl cyclopropane 7, Scheme 1), we chose to mask the problematic C(8) tertiary alcohol and the C(3) ketone in cyclopropane 25a. Initially, we targeted formation of C(8) benzyl ether and acetyl ester (e.g., 25b and 25c, R=Bn and Ac respectively; Scheme 7). By standard analytical measurements (e.g., 1H- and 13C-NMR and IR spectroscopy, and HRMS), the formed products matched expectations for the C(8)-ester and C(8)-ether (e.g., 25b and 25c, R=Bn or Ac); however, careful analysis of gradient-heteronuclear multiple bond correlation (gHMBC) spectroscopy revealed that all efforts to alkylate the C(8) alcohol result in translactonization17 to furnish instead a C(8) lactone (e.g., 25a → 27). Specifically, benzylation of the C(10) alcohol was achieved on treatment of C(8) alcohol 25a with BnBr, KI, and Ag2O at room temperature forming benzylated ether 27a, while base-mediated acetylation (Ac2O, pyridine, DMAP) yielded the translactonized product 27b containing a C(8) lactone and a C(10) acetyl ester.18
Scheme 7.

Translactonization to form benzyl ether and acetyl ester
While these products were not originally targeted, we anticipated that acetyl-masked 27b could still be converted to the core of ineleganolide (i.e., 6) by way of a divinylcyclopropane (Scheme 8). Having previously recognized the problematic reactivity of the C(3) ketone (see Scheme 6), divinylcyclopropane 29 was targeted as a suitable precursor for this tandem transformation. We found that Luche reduction of ketone 27b furnished a 1:1.5 ratio of diastereomeric alcohols 28a and b. These diastereomers were separated and carried through subsequent reactions in parallel. Secondary alcohol 28b was masked as an acetyl ester, then the silyl ether was desilylated. The resultant alcohol was oxidized to an aldehyde, and subjected to methylenation to provide divinylcyclopropane 29b in 76% yield over four steps.
Scheme 8.

Access to divinylcyclopropane 29b
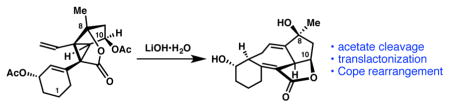
We suspected that bisacetyl-masked divinyl cyclopropane 29b could still be converted to the central [6,7,5,5]-fused system within ineleganolide through an acetate-cleavage/translactonization/Cope rearrangement (Scheme 9). We expected that both translactonization and Cope rearrangement would be thermodynamically governed transformations (Scheme 9). Consequently, when acetyl ester cleavage revealed alcohol 29c, it would enable the reaction to funnel to the lowest energy product, namely less-strained cycloheptadiene 32b via initial translactonization to lactone 31b and subsequent Cope rearrangement. A hypothetical and disfavored direct Cope rearrangement of the C(8) lactone would form highly-strained 30b, possessing two anti-Bredt, bridgehead alkenes.19 Were hypothetical intermediate 30b to form, it would presumably undergo translactonization to cycloheptadiene 32b. In the event, lithium hydroxide-mediated acetate cleavage of bis-acetyl ester 29b promoted a translactonization–Cope rearrangement cascade to deliver cycloheptadiene 32b in 55% yield, producing the carbocyclic core framework of ineganolide for the first time. To our knowledge, this is also the first example of a translactonization-triggered Cope rearrangement.
Scheme 9.

Translactonization-Cope approach to access cycloheptadiene 32b
An alternative translactonization–Cope approach was pursued concurrently, and also furnished the central [6,7,5,5]-fused system within inele-ganolide (Scheme 10). Efforts to mask the C(3) ketone through ketalization proved fruitless under many standard conditions, as ketones 25a and 27b either decomposed, or formed complex product mixtures. Moreover, these ketones were unreactive toward mild conditions designed to prevent alkene isomerization (TMSOTf, (TMSOCH2)2, CH2Cl2, −78 °C). At a higher temperature (0 °C), ketalization proceeded, albeit with undesired alkene isomerization. Subsequent desilylation and oxidation of the primary alcohol to an aldehyde followed by Wittig olefination delivers divinyl-cyclopropane 33, an appropriate Cope precursor.
Scheme 10.
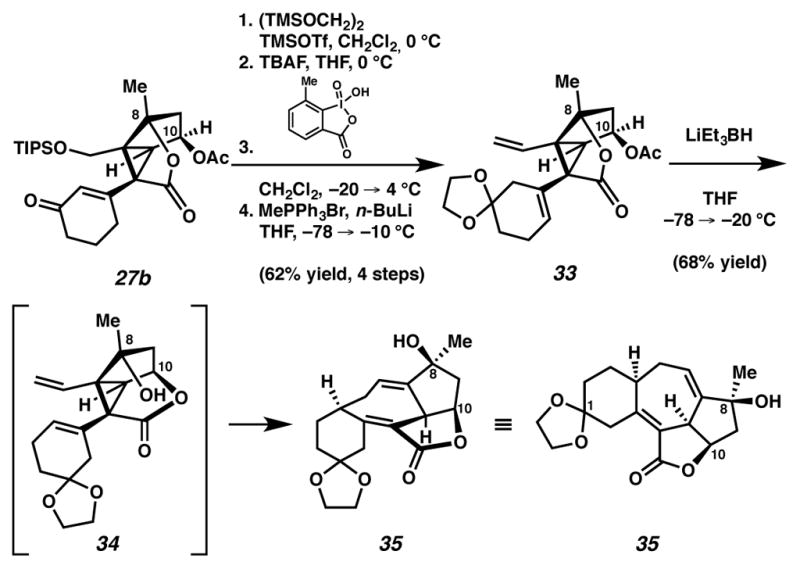
Ketalization with undesired olefin isomerization and subsequent translactonization-Cope rearrangement sequence
Although this olefin isomer was not originally designed into our synthetic approach to ineleganolide, we expected that ketal 33 would be an appropriate precursor for a tandem translactonization-Cope rearrangement. Acetyl ester 33 reacted with nucleophilic super-hydride to form desired cycloheptadiene 35, presumably by a translactonization-Cope process (i.e., 33 → 34 → 35). Importantly, this translactonization-Cope rearrangement again provided access to the [6,7,5,5]-fused core of ineleganolide, albeit with modified substitution on the cyclohexyl ring.
Having established a route to the carbon scaffold of ineleganolide (i.e., cycloheptadiene 35), we attended to C(6) oxygenation and reduction of the C(12)–C(13) tetrasubstituted alkene (Scheme 11). To our delight, the tertiary C(8) alcohol could direct vanadium-mediated diastereoselective epoxidation of the C(6)–C(7) alkene to generate epoxide 36. For subsequent functionalization, we anticipated that olefin reduction could prove useful. Expecting direct reduction of the C(12)–C(13) alkene to be challenging, we chose to pursue deketalization of cycloheptene 36, which could potentially induce alkene isomerization to furnish enone 37. We envisioned that the carbonyl in enone 37 could facilitate olefin reduction. Regrettably, selective deketalization of ketal 36 was not accomplished under a broad range of conditions.
Scheme 11.
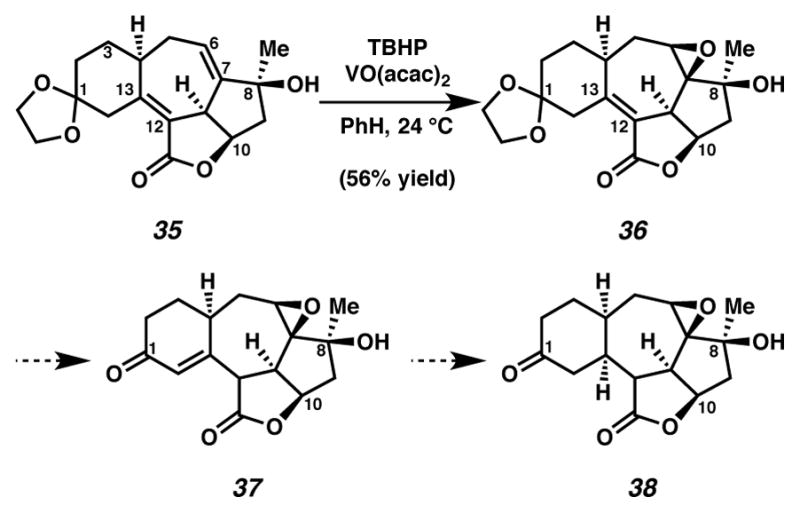
Olefin differentiation from cycloheptadiene 35
As a result of the aforementioned challenges, we continuted concurrent pursuit of a tandem cyclopropanation-Cope approach to form the [6,7,5,5]-fused core of ineleganolide. The tandem cyclopropanation-Cope20,21 rearrangement was developed by Davies and co-workers for application in diastereoselective intramolecular reactions.22,23,24,25 To investigate this reaction, we targeted diazoester 39 as the precursor to this key tandem cyclopropanation-Cope rearrangement (Scheme 12). Unfortunately, we were unable to directly convert acetonide 19 to diazoester 39 due to an inability to affect the requisite olefination. We speculated that deprotonation of the acidic γ-proton of the α,β-unsaturated ketone/δ-ester system interfered with the attempted Wittig methylenation. To access diazoester 39, we carried out the methylenation on the cyclopentene fragment prior to coupling it with the cyclohexenone carboxylic acid (Scheme 13). Thus, cyclopentenone 16 was converted to allylic benzoyl ester 40. Reduction and benzoylation furnished benzoyl ester 40 in 98% yield over two-steps. Benzoyl ester 40 was treated with acid to cleave the ketal, oxidized with Dess-Martin periodinane and olefinated to deliver diene 41. Alkoxide-mediated debenzoylation furnished alcohol 42, which was coupled with carboxylic acid 10. Subsequent diazotization provided targeted diazoester 39.
Scheme 12.
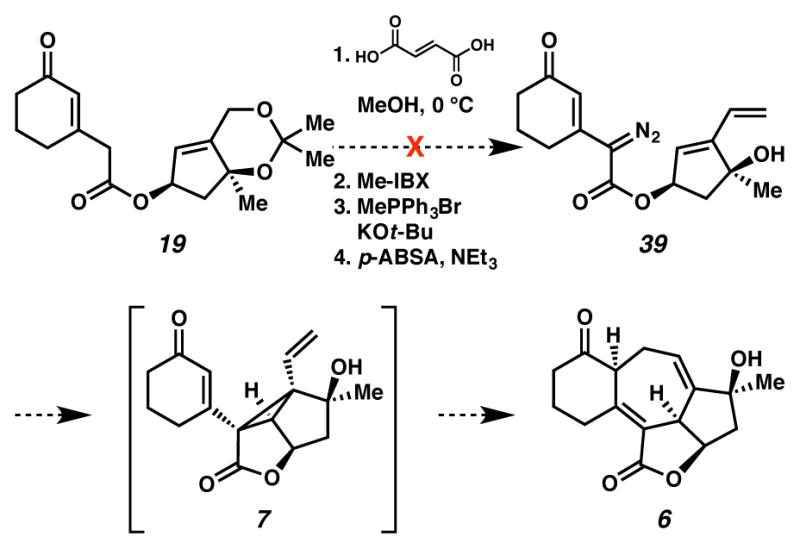
Alternative plan to access cycloheptadiene 6
Scheme 13.
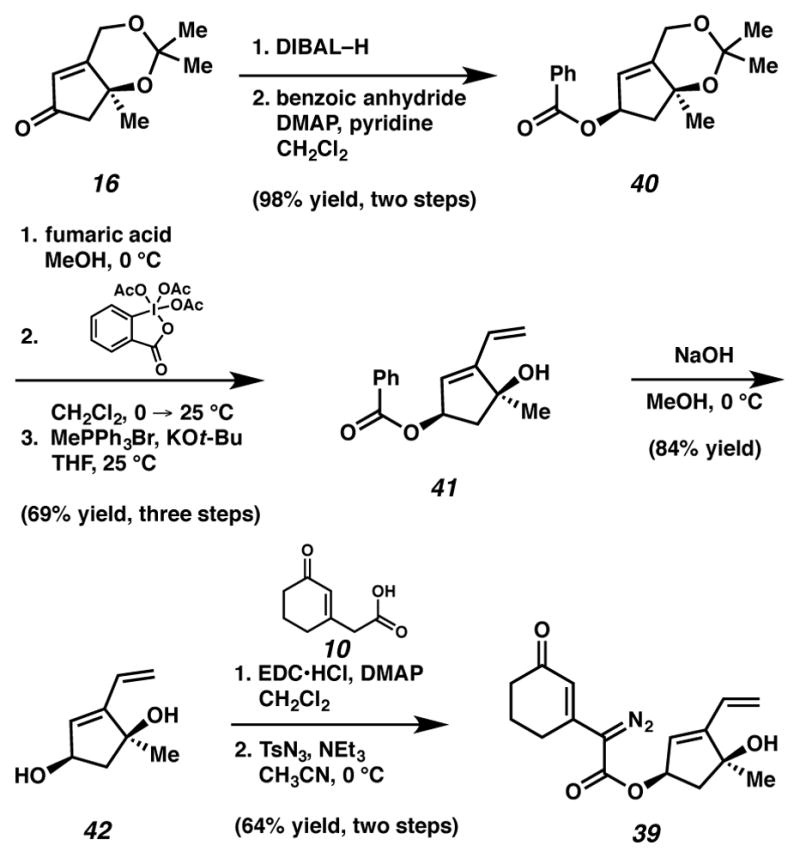
Alternative plan to access diazoester 39
This diazoester was poised for a tandem cyclopropanation-Cope cascade to generate cycloheptadiene 6 directly (Scheme 14). To our surprise, treatment of diazoester 39 with Cu(tbs)2 under N2-atmosphere in a glovebox resulted in the formation of a compound possessing three olefins, instead of two as in desired product 6. After careful analysis, we identified cycloheptatriene 44 as the major product of this reaction. Introduction of DBU into the reaction resulted in conversion of minor products, which were presumably olefin positional isomers of cycloheptatriene 44 to triene 44. Cycloheptatriene 44 likely arose from air-accelerated oxidation during work-up of cycloheptadiene 43. 1H NMR spectroscopic analysis of the crude reaction mixture prior to exposure to air revealed the presence of double-bond isomerized enone 43, instead of expected Cope product 6. Presumably, the undesired oxidation and isomerization reactions are facilitated by the acidic β- and δ-protons of the C(3) ketone.
Scheme 14.
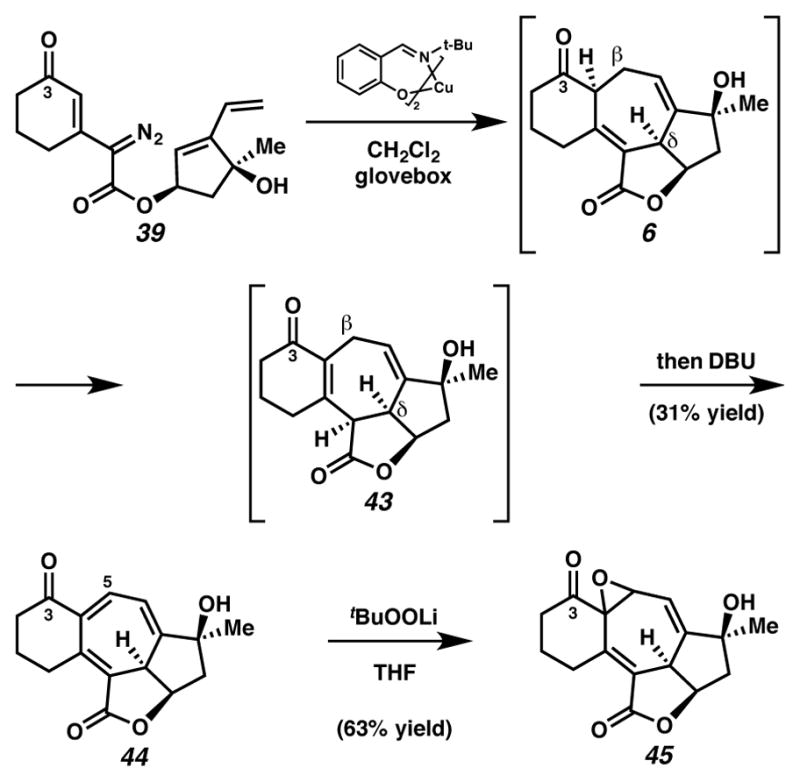
Cyclopropanation-Cope-oxidation sequence with nucleophilic epoxidation
With cycloheptatriene 44 in hand, we suspected that appropriate orbital overlap could render C(5) as the reactive electrophile in a conjugated acceptor, a property we would hope to use in a total synthesis of ineleganolide. Nucleophilic epoxidation of cycloheptatriene 44 with the lithium salt of tert-butyl hydroperoxide yielded epoxide 45, in a selective process that provided proof-of-concept for the anticipated reactivity of the C(4)–C(5) olefin.
Concurrently, alternative routes to the [6,7,5,5]-system were evaluated, with a focus on approaches that would avoid over-oxidation to the triene (Scheme 15). Formation of the triene could be avoided by employing lower Cu(tbs)2-loadings, and intercepting the intermediate cycloheptadiene 43 prior to exposure to air. For example, diastereoselective epoxidation of the C(6)–C(7) olefin of intermediate diene 43 with VO(acac)2 and TBHP afforded epoxide 46 in 32% yield in a formal cyclopropanation, Cope rearrangement, epoxidation sequence. Alternatively, acetylation of the intermediate C(3) ketone furnished enol ester 47 in 44% yield in a formal cyclopropanation, Cope rearrangement, enolate trapping sequence. In turn, triene 47 could undergo selective oxidation of the C(6)–C(7) olefin either by epoxidation catalyzed by VO(acac)2 and TBHP to generate epoxide 48 diastereoselectively, or by oxidative rearrangement with pyridinium chlorochromate (PCC) to install C(6) ketone 49, albeit with elimination of the critical C(8) alcohol. These additional transformations brought us tantalizingly close to a full synthesis of the des-isopropyl model system of ineleganolide (i.e., 5). Full details of the ensuing challenges have been described in a series of manuscripts detailing a related model system.6
Scheme 15.
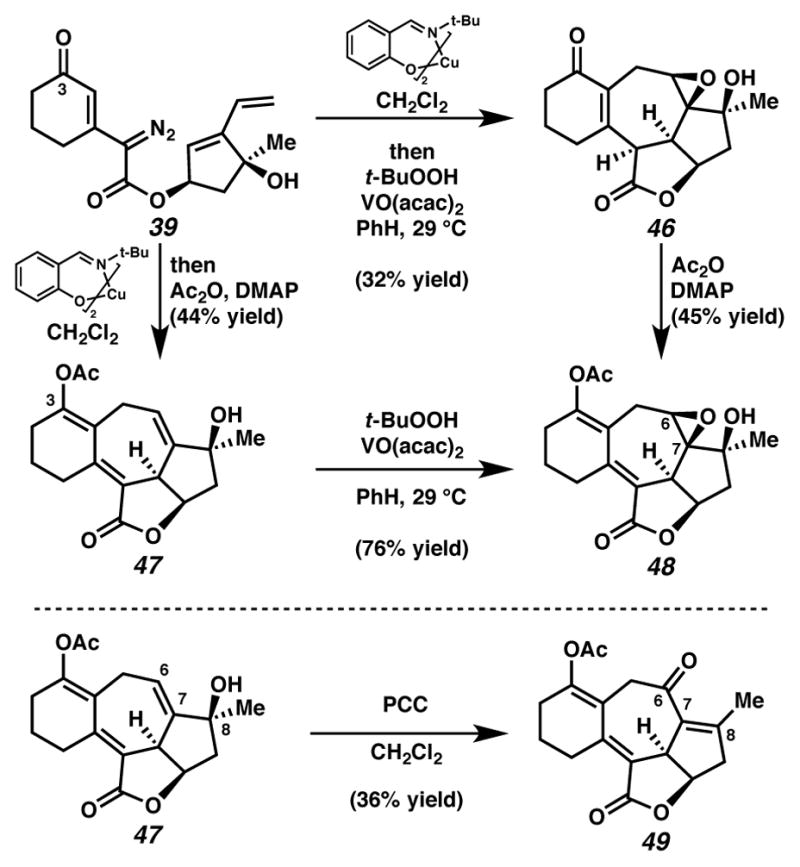
Cyclopropanation-Cope-epoxidation and cyclopropanation-Cope-enolate trapping sequences
Conclusion
The construction of the ring system of ineleganolide (1) has been demonstrated in a strategy that merges the asymmetric ketone alkylation and tandem translactonization-Cope rearrangements or formal cyclopropanation-Cope-epoxidation or cyclopropanation-Cope-enolate trapping sequences. This effort prompted the development of the previously unknown palladium-catalyzed asymmetric alkylation of dioxanones,8b and a mild oxidative bromination, Wittig olefination and reduction sequence8a to advance the resultant chloroalkene to enantioenriched cyclopentenol 11. This alcohol can be coupled with cyclohexenone acid 10, and the resultant vinylogous β-ketoesters can be advanced to rigid cyclopropanes. By necessity, we have created a rich body of chemistry exploring translactonizations in cis-substituted cyclopentane diols, including a translactonization-Cope cascade and a formal cyclopropanation-Cope-epoxidation sequence. This research complements a recently disclosed route to access the carbon framework of ineleganolide via a formal cyclopropanation–Cope sequence.6 All of these cascade sequences provide access to the rigid [6,7,5,5]-fused scaffold of ineleganolide (1).
EXPERIMENTAL SECTION
General Methods
Unless otherwise stated, reactions were performed in flame-dried glassware under an argon atmosphere using dry, deoxygenat-ed solvents. Solvents were dried by passage through an activated alumina column under argon. Tetrabutylammonium triphenyldifluorosilicate (TBAT) was purchased from Sigma-Aldrich Chemical Company and azeotropically dried five times from acetonitrile prior to use. Trimethylsilyl chloride (TMSCl), pyridine, and triethylamine (NEt3) were distilled from sodium hydride immediately prior to use. Sodium iodide was dried by heating at 90 °C (2 torr) for 12 h. TEMPO+BF4−,14 p-acetamidobenzenesulfonyl azide (pABSA), TsN3,26 bis(N-tert-butylsalicylaldehydiminato) copper (II, Cu(tbs)2),27 Dess-Martin periodinane (DMP),28 diazomethane,29 and Amberlyst A26 (S2O32−)30 were prepared by known methods. Other reagents were used without further purification. Molecular sieves were purchased from Sigma Aldrich Chemical Company as activated 5 μm powder and stored in a 120 °C drying oven until immediately prior to use unless otherwise noted. Rhodium octanoate was purchased from Strem. N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) and 4-(dimethyl amino)pyridine (DMAP) and N,N′-dicyclohexylcarbodiimide (DCC) were purchased from Sigma Aldrich Chemical Company. Reaction temperatures were controlled by an IKAmag temperature modulator. Thin-layer chromatography (TLC) was performed using E. Merck silica gel 60 F254 precoated plates (0.25 mm) and visualized by UV fluorescence quenching, anisaldehyde, or CAM staining. Florisil® (100–200 mesh) and ICN Silica gel (particle size 0.032–0.063 mm) were used for flash chromatography. Chiral HPLC analysis was performed with an Agilent 1100 Series HPLC utilizing chiralpak AD or chi-ralcel OD-H columns (4.6 mm × 25 cm) obtained from Daicel Chemical Industries, Ltd. with visualization at 254 nm or 220 nm. Chiral GC anaylsis was performed with an Agilent 6850 GC utilizing a G-TA (30 m × 0.25 cm) column (1.0 mL/min carrier gas flow). Optical rotations were measured with a Jasco P-1010 polarimeter at 589 nm. 1H and 13C NMR spectra were recorded on a Varian Mercury 300, 500, or 600 NMR spectrometer (at 300, 500, or 600 MHz, and 75, or 125 MHz, respectively), and are reported relative to Me4Si (δ 0.0). Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration). Data for 13C NMR spectra are reported in terms of chemical shift relative to Me4Si (δ 0.0). If carbons were not recorded in a particular spectrum, then the missing carbons were noted by italicizing the absent carbon in a partial formula at the beginning of the spectrum. IR spectra were recorded on a Perkin Elmer Paragon 1000 spectrometer and are reported in wavenumbers (cm−1). High resolution mass spectra were obtained from the Caltech Mass Spectral Facility, or acquired using an Agilent 6200 Series TOF with an Agilent G1978A Multimode source in electrospray ionization (ESI), atmospheric pressure chemical ionization (APCI), or mixed (MM) ionization mode.
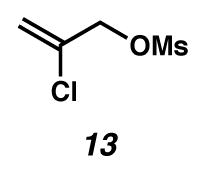
2-Chloroallyl mesylate (13)
A flask was charged with 2-chloroallyl alcohol (1.0 g, 10.8 mmol, 1 equiv), THF (20 mL, 0.54 M), and NEt3 (3.0 mL, 21.5 mmol, 2 equiv) at 0 °C (ice water bath). The solution was treated dropwise with mesyl chloride (1.26 mL, 16.2 mmol, 1.5 equiv). After 2 h at 0 °C, the reaction was quenched by addition of NaHCO3, and the mixture was extracted with Et2O. The organic layer was washed successively with 1N HCl (20 mL), aq NaHCO3 (20 mL), and brine (20 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2 (ca. 50 g) with 1:4 EtOAc:hexanes as the eluent) to provide a colorless oil (1.716 g, 93% yield). Rf 0.34 (1:2 EtOAc:hexanes); 1H NMR (300 MHz, CDCl3) δ 5.65 (d, 1H, J = 0.5 Hz), 5.55 (d, 1H, J = 0.5 Hz), 4.77 (s, 2H), 3.10 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 134.1, 117.7, 70.9, 38.4; IR (thin film/NaCl) 3033, 2943, 1639, 1360, 1175, 1010 cm−1; HRMS (EI) m/z: [M·+]+ Calcd for C4H7O3SCl 169.9804; Found 169.9811.
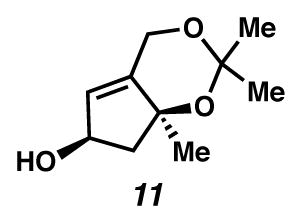
(6R,7αS)-2,2,7α-Trimethyl-4,6,7,7α-tetrahydrocyclopenta[d][1,3]dioxin-6-ol (11)
A flask was charged with a colorless solution of ketone 16 (0.110 g, 0.60 mmol, 1 equiv), MeOH (7.4 mL, 0.082 M) and CeCl3·7H2O (0.292 g, 0.78 mmol, 1.3 equiv). The solution was cooled to 0 °C (ice water bath), and treated with NaBH4 (55 mg). Bubbles evolved. The solution was treated with a 1.5 M NaOH solution, extracted with EtOAc (to 150 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure, and immediately diluted with CH2Cl2. Alcohol 11 was purified by silica gel chromatography (SiO2 (ca. 16 mL)) with 1:1 pentane:Et2O as the eluent to give pure alcohol 11 as a colorless oil (75 mg, 67% yield). The alcohol was immediately diluted in Et2O. Rf 0.11 (50% Et2O in hexanes; visualized with anisaldehyde); 1H NMR (300 MHz, CDCl3) δ 5.57 (q, J = 1.7, 1H), 4.77–4.70 (m, 1H), 4.70–4.61 (m, 2H), 2.57 (dd, J = 12.4, 6.5, 1H), 1.82 (ddd, J = 12.5, 6.8, 1.1, 1H), 1.63 (dt, J = 6.8, 3.3, 1H), 1.48 (s, 3H), 1.41 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 144.7, 125.7, 105.0, 100.2, 81.1, 74.0, 60.4, 53.5, 30.0, 28.3, 26.0; IR (NaCl) 3400 (b), 1197, 1156, 1107, 1088, 1061, 1037, 847; HRMS (EI) m/z: [M–Me]+ Calcd for C9H13O3 169.0865; Found 169.0858; [α]D21.6 −2.7° (c 0.235, CHCl3, 91% ee).
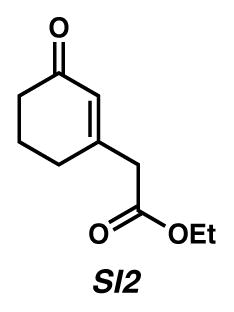
Ethyl 2-(3-oxocyclohex-1-enyl)acetate (SI2)12
To a 0.17 M acetonitrile solution of known allylic alcohol 5012 (1.14 g, 6.19 mmol) at ambient temperature was added TEMPO+BF4− (1.49 g, 6.15 mmol). After stirring for 1 hour, the orange solution was poured onto H2O (30 mL) and Et2O (100 mL). The organic and aqueous phases were separated and the aqueous layer further extracted with Et2O (2 × 100 mL). The combined organic layers were dried with Na2SO4, filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (1:10→1:4→1:1 EtOAc:hexanes) to yield known12 cyclohexenone 51 (0.77 g, 69% yield) as a colorless liquid. 1H NMR of this compound was as reported in the literature.12
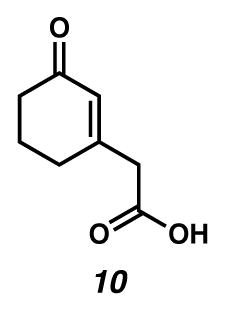
2-(3-Oxocyclohex-1-enyl)acetic acid (10)
To a 1:1 EtOH/H2O solution (0.14 M) of ester 5112 (0.76 g, 4.2 mmol) cooled in an ice water bath was added NaOH (aq) (1.8 mL, 3.04 M, 5.47 mmol) dropwise. The bright yellow solution was allowed to warm to room temperature and stirred for 5 hours. The solution was quenched with 10% HCl (aq) (15 mL) and extracted with EtOAc (5 × 100 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude oil was purified by flash chromatography (3:1→1:1→0:1 hexanes/EtOAc eluent) to yield carboxylic acid 10 (0.57 g, 89% yield) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 6.02–6.00 (m, 1H), 3.28 (d, J = 0.7, 2H), 2.43–2.39 (m, 4H), 2.06–2.00 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 200.1, 174.5, 157.1, 129.1, 43.1, 37.0, 29.8, 22.6; IR (thin film/NaCl) 3445, 1722, 1652, 1260 cm−1; HRMS (ESI–APCI) m/z: [M+H]+ Calcd for C8H10O3 155.0703; Found 155.0704.
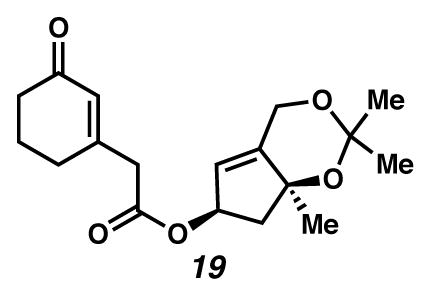
Ester 19
A concentration flask was charged with a yellow mixture of acid 10 (1.303 g, 8.4 mmol, 3 equiv), alcohol 11 (from 2.95 mmol ketone 16, 1 equiv) and EDC·HCl (1.131g, 5.9 mmol, 2 equiv) in CH2Cl2 (29.5 mL, 0.1 M). The suspension was cooled to 0 °C (ice water bath) and treated with DMAP (72 mg, 0.59 mmol, 0.2 equiv). The suspension was allowed to gradually warm to room temperature (ca. 25 °C) as the ice bath melted. After 3.5 hours, the reaction was treated with additional EDC·HCl (1.131g, 5.9 mmol, 2 equiv). After 1.5 hours, the solution was cooled to 0 °C (ice water bath) and treated with 0.1 N HCl (6 mL). The layers were separated, and the organics were further rinsed with 0.1 N HCl (6 mL), 5% K-2CO3 (aq, 6 mL × 2), and brine (6 mL) in succession. Each aqueous layer was further extracted with EtOAc (12 mL × 10). The organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure to give ester 19 as a yellow oil (0.923 g, 98% yield). Rf 0.72 (EtOAc; UV active; visualized with anisaldehyde stain); 1H NMR (300 MHz, CDCl3) δ 5.93 (d, J = 0.7, 1H), 5.59–5.56 (m, 1H), 5.54–5.53 (m, 1H), 4.67–4.63 (m, 2H), 3.24 (s, 2H), 2.58 (ddd, J = 12.9, 6.7, 1.2, 1H), 2.40–2.32 (m, 4H), 2.03 (dt, J = 12.7, 6.5, 2H), 1.94 (dd, J = 12.8, 6.6, 1H), 1.46 (s, 3H), 1.44 (s, 3H), 1.41 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 199.5, 169.4, 157.1, 147.2, 129.1, 121.3, 100.4, 81.0, 77.0, 60.4, 49.3, 43.6, 37.3, 30.1, 29.8, 28.6, 25.8, 22.8; IR (thin film/NaCl) 1732, 1667, 1169, 1127, 974 cm−1; HRMS (EI) m/z: [M+]+ Calcd for C18H24O5 320.1624; Found 320.1613; [α]D24.1 28.4° (c 0.11, CHCl3, derived from ketone 16 with 91% ee).
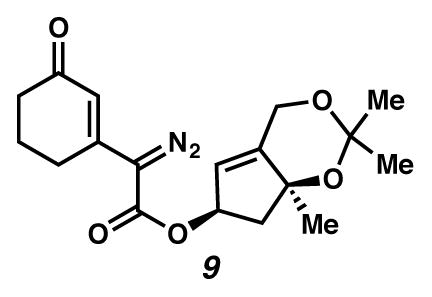
Diazoester 9
A vial beneath N2(g) was charged with ester 19 (32 mg, 0.1 mmol, 1 equiv), CH3CN (0.5 mL, 0.2 M) and pABSA (31 mg, 0.13 mmol, 3 equiv).31 The solution turned deep yellow upon dropwise addition of NEt3 (0.04 mL, 0.3 mmol, 3 equiv). After six hours, the yellow mixture was treated with Et2O and filtered through a plug of SiO2 (ca. 1 mL) with copious elution. The yellow solution was concentrated under reduced pressure. The residue was diluted with EtOAc and purified by flash chromatography (SiO2 (ca. 14 mL), 1:3 → 1:2 EtOAc:hexanes eluent) to give diazoester 9 as a yellow oil (37 mg, >99% yield). Rf 0.38 (1:1 EtOAc:hexanes; visualized with UV); 1H NMR (500 MHz, CDCl3) δ 6.37 (s, 1H), 5.67 (dddd, J = 8.5, 6.7, 3.4, 1.9, 1H), 5.54 (dd, J = 3.5, 1.5, 1H), 4.69–4.65 (m, 1H), 4.65–4.61 (m, 1H), 2.58 (dd, J = 12.8, 6.9, 1H), 2.54–2.49 (m, 2H), 2.41–2.36 (m, 2H), 2.06 (dt, J = 12.7, 6.3, 2H), 1.97 (dd, J = 12.9, 6.5, 1H), 1.43 (s, 3H), 1.43 (s, 3H), 1.39 (s, 3H); 13C NMR (125 MHz, CDCl3, –C=N2 or –C(CH3)2) δ 197.4, 162.7, 147.5, 147.2, 121.3, 120.9, 100.5, 81.0, 77.5, 60.4, 49.4, 37.0, 30.0, 28.6, 26.8, 25.9, 22.5; IR (CH2Cl2) 2101, 1708, 1580, 1191, 1137, 1110, 1064, 994 cm−1; HRMS (EI) m/z: [M+]+ Calcd for C18H22O5N2 346.1529; Found 346.1524; [α]D24.4 47.9° (c 0.11, CHCl3, derived from ketone 16 with 91% ee).
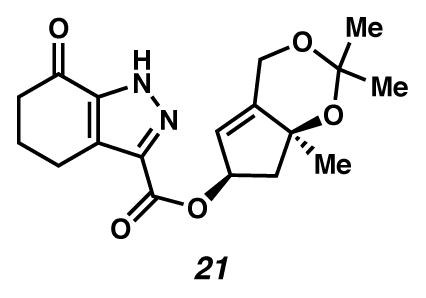
Pyrazole 21
A flask fitted with a reflux condenser was charged with diazoester 9 (20 mg, 0.054 mmol, 1 equiv) and PhMe (8 mL, 0.00675 M). The yellow solution was treated with a warm (previously refluxing) solution of copper(II) acetylacetonate (2.8 mg, 0.011 mmol, 0.2 equiv) in PhMe (2 mL, with 10 mL wash, 0.054 M in diazoester 9). The solution was heated in an oil bath at 110 °C. After 40 minutes, the solution was removed from heat and concentrated under reduced pressure. The residue was diluted with 1:1 EtOAc:hexanes and purified by flash chromatography (SiO2 ~ 8 mL; 1:1 EtOAc:hexanes eluent) to give pyrazole 21 as a white solid (10 mg, 59% yield). Colorless crystals could be obtained by slow diffusion of PhH into a solution of pyrazole 21 in CHCl3. Melting point analysis resulted in crystals yellowing at 149–150 °C and then converting to a red liquid as gas evolved at 170–172 °C. Rf 0.26 (1:1 EtOAc:hexanes; visualized with UV or anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 11.40 (s, 1H), 5.83 (ddtd, J = 6.8, 5.0, 3.4, 1.9, 1H), 5.70 (dt, J = 3.0, 1.5, 1H), 4.73 (ddd, J = 15.7, 3.3, 2.4, 1H), 4.70–4.68 (m, 1H), 3.04 (t, J = 6.1, 2H), 2.71 (dd, J = 12.7, 6.8, 1H), 2.63 (dd, J = 7.3, 5.6, 2H), 2.23–2.13 (m, 3H), 1.66 (s, 2H), 1.49 (s, 3H), 1.48 (s, 3H), 1.40 (s, 3H); 13C NMR (125 MHz, CDCl3, –C=O, –CC(O)OR) δ 147.2, 131.8, 121.4, 110.2, 100.4, 81.0, 77.2, 60.4, 49.5, 38.7, 30.1, 29.9, 28.6, 25.9, 24.6, 21.5; IR (thin film/NaCl) 3286, 1716, 1689, 1188, 1172, 1094, 1042, 994 cm−1; HRMS (EI) m/z: [M+H]+ Calcd for C18H23O5N2 347.1607; Found 347.1596; [α]D27.4 +45.6° (c 0.59, acetone, derived from ketone 16 with 91% ee).
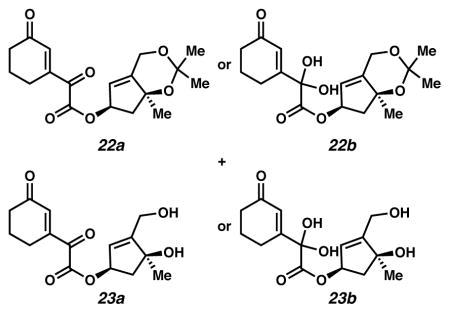
Oxidized 22a, or 22b, and 23a, or 23b17a
As a note of clarification, one set of data has been acquired for compound 22, and another for compound 23. 13C and IR spectra support their assignment as ketones 22a and 23a, respectively, while HRMS data supports their assignment as hydrates 22b and 23b, respectively. It is likely that these hydrates form under the conditions used to obtain HRMS data.
A vial was charged with rhodium octanoate dimer (Rh2(oct)4, 0.6 mg, 0.0007 mmol, 0.01 equiv) and CH2Cl2 (0.2 mL, 0.375 M relative to diazoester 9). The pale green solution was cooled to 0 °C (ice water bath). To it was added dropwise diazoester 9 (28 mg, 0.075 mmol, 1 equiv) in CH2Cl2 (0.22 mL, 0.341 M). After 25 minutes, the yellow solution was treated with additional Rh2(oct)4 (0.4 mg, 0.0005 mmol, 0.007 equiv). After an additional 18 hours, the yellow solution was treated with a third portion of Rh2(oct)4 (0.4 mg, 0.0005 mmol, 0.007 equiv). After an additional two hours, the reaction was diluted with EtOAc (10 mL), filtered through a plug of celite and concentrated under reduced pressure. The residue was diluted with EtOAc (10 mL), filtered and concentrated. This residue was purified by flash chromatography (SiO2 ~ 16 mL; 1:40 → 1:20 → 1:5 → 1:0 EtOAc:CH2Cl2 then 1:20 MeOH:EtOAc elution) to give oxidized 22a (9.8 mg, 41% yield). Rf 0.61 (1:5 EtOAc:CH2Cl2; UV/Vis); 1H NMR (500 MHz, CDCl3) δ 6.62 (t, J = 1.7, 1H), 5.75–5.70 (m, 1H), 5.58 (dd, J = 3.7, 1.7, 1H), 4.70–4.62 (m, 2H), 2.64 (dd, J = 12.9, 6.9, 1H), 2.54 (td, J = 6.1, 1.8, 2H), 2.52–2.47 (m, 2H), 2.11–2.02 (m, 3H), 1.45 (d, J = 0.5, 3H), 1.42 (s, 3H), 1.38 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 200.0, 188.7, 162.7, 150.0, 148.6, 137.0, 120.2, 100.6, 81.0, 79.0, 60.3, 49.0, 38.2, 30.1, 28.7, 25.7, 23.0, 21.9; IR (thin film/NaCl) 3384, 2934, 2871, 1732, 1682, 1455, 1429, 1416, 1373, 1350, 1317, 1257, 1195, 1148, 1110, 1065, 1030, 978, 949, 902, 849, 735 cm−1; HRMS (ES) m/z: [M+H]+ Calcd for 22 C18H24O7 353.1600; Found 353.1631; [α]D26.6 +30.4° (c 0.15, CHCl3, derived from ketone 16 with 91% ee).
Chromatography also furnished slightly impure acetonide-cleaved 23a or 23b. Acetonide-cleaved 23a or 23b was purified by flash chromatography (SiO2 ~ 2 mL; 4:1 EtOAc:hexanes elution) to give acetonide-cleaved 23a (4.8 mg, 23% yield). Rf 0.40 (EtOAc; UV active, visualized with anisaldehyde stain); 1H NMR (300 MHz, CDCl3) δ 6.65 (t, J = 1.7, 1H), 5.84 (q, J = 1.7, 1H), 5.70–5.64 (m, 1H), 4.45–4.35 (m, 2H), 2.60 (dd, J = 14.7, 7.2, 2H), 2.54 (ddd, J = 6.0, 6.0, 1.8, 2H), 2.51–2.43 (m, 2H), 2.44 (s, 1H), 2.16 (dd, J = 14.6, 3.3, 1H), 2.13–2.03 (m, 3H), 1.44 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 200.2, 188.6, 162.6, 155.2, 150.1, 137.1, 124.4, 81.4, 78.8, 59.2, 47.9, 38.2, 26.6, 23.0, 21.9; IR (thin film/NaCl) 3382, 2968, 2935, 1732, 1682, 1193, 1110, 1092, 1065, 1030, cm−1; HRMS (ES+) m/z Calcd for 23 C15H18O6 [M+H]+: 295.1182; Found 295.1195; HRMS (ES) m/z: [M+H]+ Calcd for 23 C15H20O7 313.1287; Found 313.1286; [α]D24.9 +48.8° (c 0.09, CHCl3, derived from ketone 16 with 91% ee).
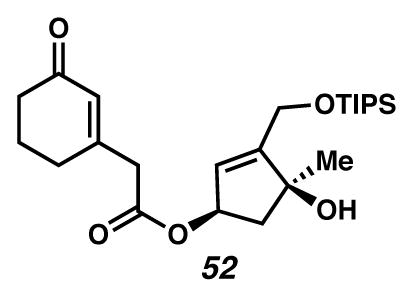
Silyl ether 52
A flask was charged with acetonide 19 (0.451 g, 1.41 mmol, 1 equiv) in MeOH (30 mL, 0.047 M), and the solution was cooled to 0 °C (ice water bath). The solution was treated dropwise with a solution of fumaric acid (59 mg, 0.50 mmol, 0.36 equiv) in MeOH (10 mL, 0.14 M total relative to acetonide). After 3 days, the reaction was quenched by addition of a 1:1 solution of H2O and saturated aq NaHCO3 (40 mL each), and extracted with EtOAc (300 mL, and then 200 mL × 5). The organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure to give a golden/orange oil.
The resultant oil was dissolved in CH2Cl2 (35 mL, 0.040 M), placed beneath an Ar(g) atmosphere, and cooled to 0 °C (ice water bath). The solution was treated dropwise with imidazole (0.303 g, 4.46 mmol, 3.16 equiv) in CH2Cl2, followed by TIPSCl (0.91 mL, 4.25 mmol, 3.01 equiv) and DMAP (20 mg, 0.16 mmol, 0.11 equiv) in CH2Cl2. After 17 hours, the solution was quenched with saturated aq NH4Cl (50 mL), and extracted with EtOAc (150 mL × 3). The organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The reaction was purified by flash chromatography (1:19 → 1:9 EtOAc:CH2Cl2 eluent) to give silyl ether 52 (0.371 g, 59% yield over two steps) as a yellow oil. Rf 0.76 (EtOAc, UV/Vis, visualized with anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 5.95 (t, J = 1.0, 1H), 5.73 (dd, J = 3.5, 1.7, 1H), 5.51 (dtd, J = 7.2, 3.6, 1.8, 1H), 4.59–4.38 (m, 2H), 3.24 (s, 2H), 2.82 (s, 1H), 2.52 (dd, J = 14.4, 7.3, 1H), 2.42–2.35 (m, 4H), 2.11–1.91 (m, 3H), 1.41 (s, 3H), 1.21–1.10 (m, 3H), 1.09–1.05 (m, 18H); 13C NMR (125 MHz, CDCl3) δ 199.6, 169.5, 157.3, 153.6, 129.1, 124.5, 81.1, 76.9, 60.4, 48.0, 43.7, 37.4, 29.8, 26.8, 22.8, 18.2, 12.0; IR (thin film/NaCl) 3440, 1733, 1668, 1192, 1165, 1127, 1107, 1055, cm−1; HRMS (mixed EIC) m/z: [M+Na]+ Calcd for C24H40O5Si 459.2537; Found 459.2539; [α]D18.9 +44.8° (c 0.93, CHCl3, derived from ketone 16 with 91% ee).
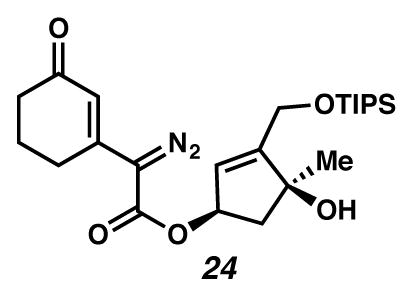
Diazoester 24
A flask was charged with a pale yellow solution of ester 52 (53 mg, 0.121 mmol, 1 equiv) in CH3CN (4.7 mL, 0.026 M) with TsN3 (0.600 g, 0.535 mmol, 4.4 equiv), and cooled to 0 °C (ice water bath). CAUTION!!! TsN3 is SHOCK SENSITIVE AND POTENTIALLY EXPLOSIVE. The solution was treated with Et3N (0.03 mL, 0.22 mmol, 1.8 equiv) dropwise, and immediately turned deeper yellow in color. After 24 hours, the reaction was concentrated under reduced pressure. The crude yellow mixture was purified by flash chromatography (SiO2 ~ 16 mL; 1:5 EtOAc:PhH eluent) to give diazoester 24 (48.9 mg, 87% yield) as a yellow oil. Rf 0.48 (1:2 EtOAc:PhH; visualized with UV); 1H NMR (500 MHz, CDCl3) δ 6.37 (s, 1H), 5.71 (dd, J = 3.3, 1.6, 1H), 5.59 (ddd, J = 7.3, 3.7, 1.9, 1H), 2.82 (s, 1H), 2.58–2.49 (m, 3H), 2.41–2.35 (m, 2H), 2.10–2.00 (m, 3H), 1.40 (s, 3H), 1.13 (tt, J = 12.2, 6.9, 3H), 1.08–1.03 (m, 21H); 13C NMR (125 MHz, CDCl3, –C=N2) δ 197.5, 162.7, 153.7, 147.5, 124.4, 120.6, 81.0, 77.4, 60.4, 48.2, 37.0, 27.0, 26.8, 22.5, 18.2, 12.0; IR (thin film/NaCl) 3407, 2101, 1709, 1645, 1191, 1138, 1106, 1061, 993 cm−1; HRMS (ES) m/z: [M+H]+ Calcd for C24H39N2O5Si 463.2628; Found 463.2640; [α]D23.4 +48.5° (c 0.175, CHCl3, derived from ketone 16 with 91% ee).
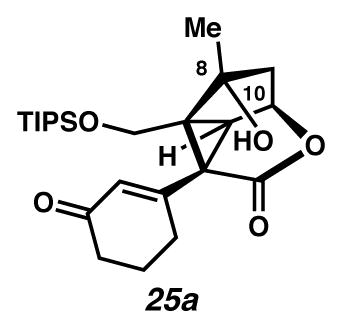
Cyclopropane 25a
In the glovebox, a flask was charged with Cu(tbs)2 (92.2 mg, 0.22 mmol, 1.1 equiv) and CH2Cl2 (20 mL, 0.01 M). This solution was treated with diazoester 24 (93.2 mg, 0.20 mmol, 1 equiv) in CH2Cl2 (8 mL, 0.025 M). After 7 days, the reaction was removed from the glovebox and concentrated under reduced pressure to yield a brown residue, which was purified by flash chromatography (SiO2 ~ 16 mL; 1:1 EtOAc:hexanes eluent) to give cyclopropane 25a as a yellow oil (62.8 mg, 74% yield). Rf 0.18 (1:1 EtOAc:hexanes; UV active; visualized with anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 5.87 (s, 1H), 4.87 (td, J = 6.2, 2.9, 1H), 4.07 (d, J = 11.5, 1H), 3.49 (d, J = 11.5, 1H), 2.68–2.60 (m, 1H), 2.59 (d, J = 5.9, 1H), 2.45–2.23 (m, 2H), 2.17–1.97 (m, 3H), 1.93–1.83 (m, 1H), 1.74 (s, 1H), 1.49 (s, 3H), 1.11–0.76 (m, 21H); 13C NMR (125 MHz, CDCl3) δ 199.7, 173.1, 156.4, 130.0, 93.8, 70.6, 59.9, 59.6, 53.8, 48.0, 42.8, 37.6, 29.5, 22.8, 19.1, 18.2, 12.1; IR (thin film/NaCl) 3427, 1759, 1668, 1626, 1192, 1175, 1122, 1085, 1065 cm−1; HRMS (FAB) m/z: [M+H]+ Calcd for C24H39O5Si 435.2567; Found 435.2586; [α]D21.2 −12.6° (c 0.125, CHCl3, derived from ketone 16 with 91% ee).
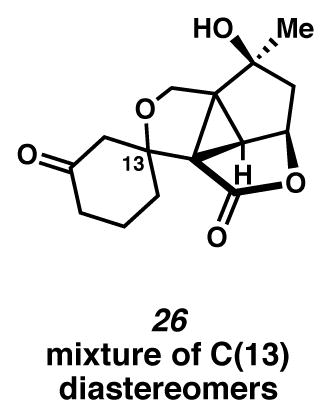
Dihydropyran 26
A flask was charged with a colorless solution of silyl ether 25a (1 equiv) in THF (0.00825 M) at 0 °C (ice water bath). The solution was treated with TBAF (1M in THF, 1.2 equiv), and the solution turned yellow in color. After 30 min, the reaction was filtered through SiO2 (ca. 1 mL; EtOAc elution), and concentrated under reduced pressure to give a crude white paste, assigned as crude diol 53. Rf 0.14 (EtOAc; UV/Vis active); 1H NMR (500 MHz, CDCl3) δ 5.93 (s, 1H), 4.93 (t, J = 6.3, 1H), 3.94 (d, J = 11.9, 1H), 3.66 (d, J = 12.0, 1H), 2.74–2.65 (m, 2H), 2.69 (d, J = 6.0, 1H), 2.47–2.34 (m, 3H), 2.31–2.23 (m, 2H), 2.19 (dd, J = 14.0, 7.0, 1H), 2.13 (d, J = 13.5, 1H), 2.11–2.01 (m, 2H), 1.99–1.89 (m, 2H), 1.69 (s, 3H); HRMS (EI) m/z: [M+·]+ Calcd for C15H18O5 278.1154; Found 278.1143.
A flask was charged with a solution of crude diol 53, CH2Cl2 (1.1 mL, 0.012 M) and pyridine (0.02 mL, 0.159 mmol, 12 equiv) at 0 °C (ice water bath), which was subsequently treated with DMP (7.9 mg, 0.018 mmol, 1.4 equiv). CAUTION! DMP is a HEAT- and SHOCK-SENSITIVE COMPOUND, showing exotherms when heated (>130 °C). ALL OPERATIONS SHOULD BE CONDUCTED BEHIND A BLAST SHIELD. After 2 days at this temperature, the mixture was diluted with EtOAc (ca. 10 mL), and concentrated under reduced pressure. The resultant semi-solid was treated with CH2Cl2 (ca. 1 mL) and Amberlyst A26 (S2O32−) (19.6 mg). After 6.5 h, the mixture was treated with JandaJel (2.3 mmol/g, 17.2 mg, 0.0396 mmol, 3.0 equiv). After an additional 17.5 h, the mixture was filtered through SiO2 (ca. 0.2 mL; EtOAc elution), and concentrated under reduced pressure. The diastereomeric mixture was purified by thin layer chromatography on a 250 μm silica gel plate (10 cm wide, 20 cm tall; Et2O × 2 elution). The desired diastereomeric mixture was removed from the plate at Rf 0.10. The diastereomeric mixture of dihydropyrans (26) was further purified by thin layer chromatography on a 250 μm silica gel plate (10 cm wide, 20 cm tall; 1:1 EtOAc:PhH elution). The diastereomeric mixture was removed from the plate at Rf 0.27, as a white solid (<20% yield). Major diastereomer: 1H NMR (500 MHz, CDCl3) δ 4.94 (app. q, J = 3.7, 1H), 4.02 (d, J = 9.4, 1H), 3.90 (d, J = 9.4, 1H), 3.18 (d, J = 4.8, 1H), 2.77 (ddd, J = 14.2, 11.6, 5.1, 1H), 2.71 (d, J = 14.1, 1H), 2.43–2.28 (m, 2H), 2.27 (dt, J = 14.1, 2.3, 1H), 2.18 (dd, J = 14.3, 2.0, 1H), 2.08–2.03 (m, 1H), 2.04–1.86 (m, 3H), 1.76–1.70 (m, 1H), 1.52 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 208.5, 172.9, 84.7, 80.5, 78.0, 64.7, 55.7, 53.0, 46.2, 45.9, 43.7, 40.9, 31.5, 27.8, 20.2. Minor dia-stereomer: 1H NMR (500 MHz, CDCl3) δ 4.94 (app. q, J = 3.7, 1H), 4.11 (d, J = 9.6, 1H), 3.88 (d, J = 9.6, 1H), 3.61 (d, J = 15.1, 1H), 3.18 (d, J = 4.8, 1H), 2.77 (ddd, J = 14.2, 11.6, 5.1, 1H), 2.59 (d, J = 15.1, 1H), 2.43–2.28 (m, 2H), 2.18 (dd, J = 14.3, 2.0, 1H), 2.08–2.03 (m, 1H), 2.04–1.86 (m, 3H), 1.76–1.70 (m, 1H), 1.51 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 209.7, 173.0, 84.7, 80.5, 78.1, 64.7, 56.6, 52.8, 48.5, 45.6, 43.9, 40.6, 31.2, 27.8, 21.5. Diastereomeric Mixture: IR (thin film/NaCl) 3460, 1756, 1712, 1172, 1109, 1073, 1047, 1012 cm−1; HRMS (ESI–APCI) m/z: [M+Na]+ Calcd for C15H18O5 301.1046; Found 301.1067.
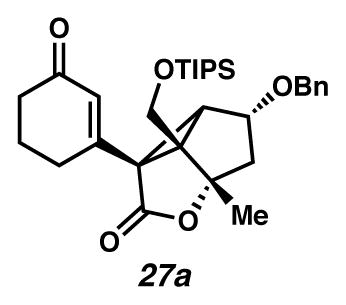
Benzyl ether 27a32
A flask was charged with a solution of silyl ether 25a (1.1 mg, 0.0026 mmol, 1 equiv) in PhMe (0.2 mL, 0.013 M) beneath an Ar(g) atmosphere at room temperature (22.5 °C). The solution was treated with KI (0.9 mg, 0.0054 mmol, 2.1 equiv) and Ag2O (0.9 mg, 0.0039 mmol, 1.5 equiv), followed by BnBr (10 μL, 0.084 mmol, 32 equiv). After 2 days, the yellow mixture was heated in an oil bath at 40 °C for 22 hours, at which point additional BnBr (10 μL, 0.084 mmol, 32 equiv) was added to the reaction. After 24 h, the suspension was treated with Ag2O (3.0 mg, 0.013 mmol, 5 equiv) and KI (4.6 mg, 0.028 mmol, 10.6 equiv). After an additional 27 h, additional BnBr (10 μL, 0.084 mmol, 32 equiv) was added to the reaction. After an additional 27 h, the reaction was diluted with EtOAc (10 mL), filtered through SiO2, and concentrated under reduced pressure. The mixture was purified by thin layer chromatography on a 250 μm silica gel plate (10 cm wide, 20 cm tall; 1:10 acetone:CH2Cl2 elution) to give benzoyl ester 27a (0.5 mg, 37% yield, Rf 0.69). Rf 0.22 (1:1 Et2O:heptanes, UV/Vis, visualized with anisaldehyde stain); 1H NMR (600 MHz, CDCl3) δ 7.36–7.32 (m, 4H), 7.29–7.26 (m, 1H), 5.93 (t, J = 1.4, 1H), 4.63 (d, J = 11.7, 1H), 4.53 (dd, J = 7.5, 5.6, 1H), 4.38 (d, J = 11.6, 1H), 4.17 (d, J = 11.5, 1H), 3.57 (d, J = 11.5, 1H), 2.79–2.72 (m, 1H), 2.61 (d, J = 5.6, 1H), 2.47 (ddd, J = 16.6, 9.2, 4.7, 1H), 2.39 (ddd, J = 12.1, 7.9, 4.7, 1H), 2.29 (d, J = 13.9, 1H), 2.21–2.08 (m, 2H), 2.14 (dd, J = 13.9, 7.6, 1H), 1.95 (tdd, J = 13.8, 9.3, 4.6, 1H), 1.70 (s, 3H), 1.12–0.90 (m, 21H); 13C NMR (125 MHz, C6D6) δ 199.2, 172.5, 156.2, 137.5, 129.7, 128.7, 128.2, 128.0, 93.1, 76.9, 71.4, 59.9, 59.4, 52.1, 48.1, 40.3, 37.6, 29.5, 22.9, 19.0, 18.2, 12.1; IR (thin film/NaCl) 1760, 1674, 1190, 1177, 1125, 1086, 1039 cm−1; HRMS-MM (ESI–APCI) m/z: [M+H]+ Calcd for C31H44O5Si 525.3031; Found 525.3023; [α]D22.9 +5.9° (c 0.20, CHCl3, derived from ketone 16 with 91% ee).
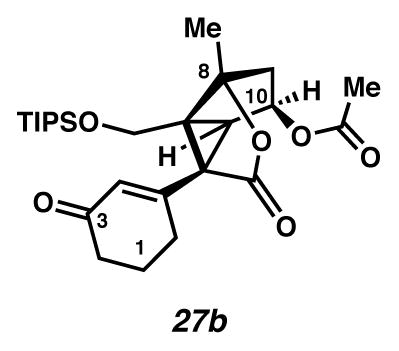
Secondary acetate 27b
A flask charged with CH2Cl2 (0.96 mL, 0.027 M), tertiary alcohol 25a (11.2 mg, 0.0258 mmol, 1 equiv), Ac2O (0.03 mL, 0.33 mmol, 13 equiv) and pyridine (0.03 mL, 0.38 mmol, 15 equiv) at room temperature (23 °C) was treated with DMAP (0.4 mg, 0.003 mmol, 0.13 equiv). After 1 h 10 m, the solution was extracted with EtOAc (10 mL × 4), and rinsed successively with 1N HCl (3 mL) and brine (3 mL × 2). The organic extracts were dried over MgSO4, filtered, and concentrated under reduced pressure to yield a secondary acetate 27b (11.7 mg, 87.3% yield) as a pale yellow oil. Rf 0.64 (1:1 EtOAc:hexanes, UV/Vis, visualized with anisaldehyde stain); 1H NMR (500 MHz, C6D6) δ 6.17 (t, J = 1.5, 1H), 5.11 (dd, J = 7.7, 5.9, 1H), 3.77 (d, J = 11.6, 1H), 3.40 (d, J = 11.6, 1H), 2.62 (d, J = 5.9, 1H), 2.46 (dddd, J = 9.4, 7.3, 5.0, 1.7, 1H), 2.11–1.96 (m, 4H), 1.80–1.72 (m, 4H), 1.58–1.44 (m, 2H), 1.42 (s, 3H), 0.93 (d, J = 1.9, 18H), 0.95–0.87 (m, 3H); 1H NMR (500 MHz, CDCl3) δ 5.89 (t, J = 1.5, 1H), 5.40 (td, J = 6.0, 2.1, 1H), 4.18 (d, J = 11.4, 1H), 3.52 (d, J = 11.5, 1H), 2.80 (d, J = 5.9, 1H), 2.54 (dddd, J = 18.1, 7.3, 4.9, 1.5 1H), 2.45–2.29 (m, 2H), 2.23 (d, J = 0.9, 1H), 2.23 (d, J = 5.0, 1H), 2.21–2.13 (m, 1H), 2.09–2.01 (m, 1H), 2.00 (s, 3H), 1.99–1.89 (m, 1H), 1.69 (s, 3H), 1.10–0.92 (m, 3H), 1.01 (d, J = 3.8, 18H); 13C NMR (125 MHz, C6D6) δ 196.5, 171.2, 170.1, 153.9, 130.1, 91.8, 73.1, 59.7, 57.9, 50.2, 47.6, 41.3, 37.1, 28.8, 22.5, 19.9, 18.5, 17.7, 11.8; IR (film) 1769, 1740, 1675, 1191, 1173, 1127, 1082, 1064 cm−1; HRMS (ESI–APCI) m/z: [M+H]+ Calcd for C26H40O6Si 477.2667; Found 477.2667; [α]D17.9 −11.6° (c 0.35, CH3CN, derived from ketone 16 with 91% ee).
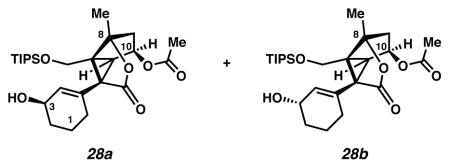
Alcohols 28
Alcohol 25a (57.2 mg, 0.131 mmol, 1 equiv) was converted to acetate 27b, as described above. The crude 27b as a pale yellow oil was dissolved in MeOH (12.0 mL, 0.011 M), cooled to 0 °C (ice water bath) and treated with NaBH4 (18.6 mg, 0.491 mmol, 3.75 equiv). After 24 m, the yellow solution was treated with a saturated aq solution of NH4Cl (0.70 mL). The mixture was diluted with EtOAc (125 mL), filtered through SiO2 (ca. 2 mL) and concentrated under reduced pressure to an off-white solid. The solid was purified by preparative thin layer chromatography on a 250 μm analytical plate (2:1 CH2Cl2:EtOAc × 2 elution) to give alcohol 28a (21.4 mg, 34% yield) as a colorless oil and alcohol 28b (32.2 mg, 51% yield) as a colorless oil. Alcohol 28a: Rf 0.65 (1:1 EtOAc:hexanes, visualized by UV/Vis, or with anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 5.73 (app dt, J = 3.1, 1.7, 1H), 5.41 (dt, J = 5.9, 4.1, 1H), 4.27 (d, J = 11.4, 2H), 3.45 (dd, J = 11.4, 8.7, 1H), 2.79 (d, J = 5.9, 1H), 2.26–2.21 (m, 2H), 2.19–2.11 (m, 1H), 2.00 (s, 3H), 1.91 (tdd, J = 7.8, 4.8, 3.1, 1H), 1.79–1.74 (m, 1H), 1.73 (ddd, J = 7.6, 4.3, 2.2, 1H), 1.70 (s, 3H), 1.68–1.61 (m, 1H), 1.50 (dddd, J = 12.5, 9.4, 6.5, 3.2, 1H), 1.44 (s, 1H), 1.14–0.94 (m, 21H); 13C NMR (125 MHz, CDCl3) δ 174.2, 171.3, 133.7, 133.0, 93.0, 73.8, 65.9, 60.0, 57.5, 51.0, 48.3, 41.9, 31.6, 28.1, 20.9, 19.3, 18.9, 18.2, 12.1; IR (film) 3482, 2941, 2866, 1760, 1740, 1462, 1374, 1242, 1080, 1065, 882, 683 cm−1; HRMS (ESI–APCI) m/z: [M+NH4]+ Calcd for C26H42O6Si 496.3089; Found 496.3071; [α]D27.2 +8.7° (c 1.52, CHCl3, derived from ketone 16 with 91% ee). Alcohol 28b: Rf 0.61 (1:1 EtOAc:hexanes, UV/Vis, visualized with anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 5.74 (dt, J = 3.3, 1.6, 1H), 5.42 (dt, J = 5.9, 4.1, 1H), 4.26 (d, J = 11.4, 1H), 4.17 (app d, 1H), 3.43 (d, J = 11.4, 1H), 2.77 (d, J = 5.9, 1H), 2.23 (d, J = 4.1, 1H), 2.19–2.14 (m, 2H), 2.02 (s, 3H), 1.82–1.72 (m, 3H), 1.69 (s, 3H), 1.66–1.51 (m, 2H), 1.11–0.95 (m, 21H); 13C NMR (125 MHz, CDCl3) δ 174.1, 171.2, 133.9, 132.6, 93.0, 73.7, 65.8, 59.9, 57.2, 51.0, 48.3, 41.7, 31.7, 28.1, 20.9, 19.3, 18.9, 18.2, 12.1; IR (thin film/NaCl) 3447, 1763, 1740, 882 cm−1; HRMS (EI) m/z: [M+NH4]+ Calcd for C26H42O6Si 496.3089; Found 496.3099; [α]D26.8 +33.1° (c 1.01, CHCl3, derived from ketone 16 with 91% ee).
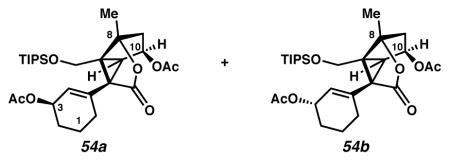
Bisacetates 54
For representative acetylation and reduction procedures, see acetylation of tertiary alcohol 25a to 27c, and reduction of 27c to 28. Alcohols 28 can be acetylated without attempting their purification or separation. Bisacetates 54 can be separated by preparative thin layer chromatography on a 250 μm analytical plate (eluent: 20:1 CH2Cl2:EtOAc × 2) to give bisacetate 54b as a colorless oil (6.3 mg, 51% yield over three steps) and bisacetate 54a as a white solid (2.6 mg, 21% yield over three steps). Alternatively, bisacetates 54 can be formed by acylation in parallel. Alcohol 28b (20.0 mg, 0.042 mmol, 1 equiv) furnished bisacetate 54b (21.7 mg, 99% yield), while alcohol 28a (13.9 mg, 0.029 mmol, 1 equiv) provided bisacetate 54a (12.1 mg, 80% yield). In this case, bisacetates were carried on without further purification. Bisacetate 54a: Rf 0.41 (1:20 EtOAc:CH2Cl2 × 2, visualized with anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 5.68 (app dt, J = 3.5, 1.6, 1H), 5.43 (dd, J = 10.0, 4.1, 1H), 5.34–5.29 (m, 1H), 4.27 (d, J = 11.4, 1H), 3.45 (d, J = 11.4, 1H), 2.81 (d, J = 6.0, 1H), 2.28–2.21 (m, 3H), 2.02 (d, J = 5.7, 6H), 1.93–1.86 (m, 1H), 1.80–1.70 (m, 1H), 1.69 (s, 3H), 1.69–1.65 (m, 2H), 1.65–1.60 (m, 1H), 1.12–0.94 (m, 21H); 13C NMR (125 MHz, CDCl3) δ 173.8, 171.3, 170.6, 135.9, 128.3, 93.0, 73.7, 68.0, 59.8, 57.6, 51.1, 48.4, 41.5, 28.1 (2C), 21.5, 20.9, 19.1, 18.9, 18.2, 12.1; IR (thin film/NaCl) 2944, 2867, 1770, 1738, 1732, 1667, 1463, 1433, 1373, 1321, 1297, 1241, 1200, 1172, 1129, 1100, 1080, 1065, 1045, 1025, 965, 947, 918, 901, 883, 792, 748, 730 cm−1; HRMS (ESI–APCI) m/z: [M–OAc]+ Calcd for C28H44O7Si 461.2718; Found 461.2740; [α]D25.8 +72.4° (c 0.19, CHCl3, derived from ketone 16 with 91% ee). Bisacetate 54b: Rf 0.56 (1:20 EtOAc:CH2Cl2 × 2, visualized with anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 5.62 (app dt, J = 3.5, 1.8, 1H), 5.43 (ddd, J = 5.9, 4.9, 3.4, 1H), 5.23–5.18 (m, 1H), 4.26 (d, J = 11.4, 1H), 3.46 (d, J = 11.4, 1H), 2.71 (d, J = 6.0, 1H), 2.28–2.21 (m, 3H), 2.04 (s, 3H), 2.03 (s, 3H), 1.86–1.76 (m, 3H), 1.73–1.63 (m, 1H), 1.69 (s, 3H), 1.63–1.55 (m, 1H), 1.14–0.82 (m, 21H); 13C NMR (125 MHz, CDCl3) δ 173.8, 171.2, 171.0, 136.1, 128.1, 92.9, 73.7, 68.1, 60.1, 57.3, 51.1, 48.1, 41.6, 28.1, 27.9, 21.5, 20.9, 19.4, 18.9, 18.2, 12.1; IR (film) 1770, 1738, 883 cm−1; HRMS (ESI–APCI) m/z: [M–OAc]+ Calcd forC28H44O7Si 461.2718; Found 461.2732; [α]D 26.0 −28.5° (c 0.16, CHCl3, derived from ketone 16 with 91% ee).
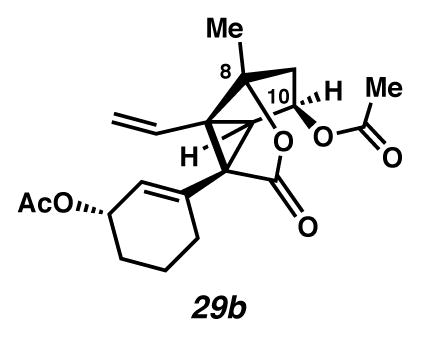
Divinylcyclopropane 29b
Desilylation
A flask charged with THF (4.8 mL, 0.012 M) and bisacetate 54b (30.0 mg, 0.0576 mmol, 1 equiv) was cooled to 0 °C (ice-water bath) and treated dropwise with TBAF (1 M in THF, 0.09 mL, 0.09 mmol, 1.56 equiv). After 30 m, the yellow solution was diluted with EtOAc (to 25 mL), filtered through SiO2 (ca. 0.3 mL) and concentrated under reduced pressure to give desilylated compound as an oil (Rf 0.11 (1:1 EtOAc:hexanes, visualized with anisaldehyde stain)).
Oxidation
The crude oil was taken up in CH2Cl2 (30 mL), cooled to −20 °C (cryocool) beneath an Ar atmosphere, and treated with crushed Me-IBX (34 mg, 0.12 mmol, 2.0 equiv). After 10 m, the mixture was allowed to gradually warm to 4 °C (cold room). Two additional portions of crushed Me-IBX (34.6 and 29.5 mg, 0.12 and 0.10 mmol, 2.0 and 1.7 equiv) were added to the mixture ca. 6 and ca. 9 hours after the initial addition. On completion of the reaction (ca. 21 h), the mixture was diluted with EtOAc (to 100 mL) and filtered through SiO2 (ca. 2 mL). The organics were concentrated under reduced pressure to give aldehyde as a cream-colored solid (Rf 0.31 (1:1 EtOAc:hexanes, visualized with anisaldehyde stain)). The solid was placed under an argon atmosphere and quickly dissolved in THF (0.5 mL).
Wittig olefination
A room temperature (ca. 24 °C) flask charged with a mixture of MePPh3Br (0.230 g, 0.643 mmol, 11.1 equiv) and THF (30 mL, 0.0019 M) beneath an Ar atmosphere was treated dropwise with n-BuLi (2.5 M in hexanes, 0.40 mL, 1 mmol, 17 equiv). The yellow solution was stirred for 30 min, and then cooled to −65 °C (i-PrOH/dry ice bath). The yellow solution was treated dropwise with aldehyde in THF (0.5 mL × 2). The solution was allowed to warm to −10 °C over ca. 1 h, at which point it was quenched by addition of acetone (1 mL). The white mixture was rinsed with 1:1 H2O: brine (ca. 5 mL) and extracted with EtOAc (to 75 mL). The extracts were dried over MgSO4, filtered, and concentrated under reduced pressure. The resulting solid was dissolved in EtOAc (10 mL), filtered through SiO2 (ca. 0.3 mL), and concentrated under reduced pressure. The semi-solid was purified by preparatory thin layer chromatography on a 250 μm plate (1:15 EtOAc:CH2Cl2 × 2 eluent) to give divinylcyclopropane 29b as a colorless oil (16.0 mg, 77% yield). Rf 0.17 (1:15 EtOAc:CH2Cl2, visualized with anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 6.09 (dd, J = 17.0, 10.6, 1H), 5.67–5.62 (m, 1H), 5.44 (t, J = 6.6, 1H), 5.30–5.23 (m, 1H), 5.17 (dd, J = 10.5, 1.2, 1H), 5.01 (dd, J = 17.0, 1.2, 1H), 3.03 (d, J = 5.9, 1H), 2.23 (d, J = 14.1, 2H), 2.17 (dd, J = 14.4, 7.5, 2H), 2.06 (s, 3H), 2.04 (s, 3H), 1.83–1.62 (m, 4H), 1.59 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 173.4, 171.2, 171.1, 135.4, 128.7, 127.0, 118.7, 92.6, 73.2, 68.2, 58.3, 50.8, 49.8, 40.0, 28.1, 27.7, 21.6, 20.9, 20.0, 19.4; IR (film) 2937, 1760, 1738, 1733, 915 cm-1; HRMS (ESI–APCI) m/z: [M–H]− Calcd for C20H24O6 359.1500; Found 359.1513; [α]D22.8 −48.0° (c 0.13, CHCl3, derived from ketone 16 with 91% ee).
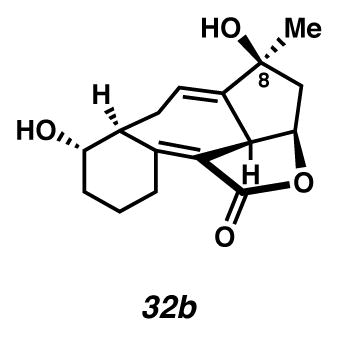
Cycloheptadiene 32b
A flask charged with bisacetate 29b (7.3 mg, 0.020 mmol, 1 equiv) in THF (5.2 mL, 0.0039 M) at 0 °C (ice water bath) was treated dropwise with a solution of LiOH·H2O (9.1 mg, 0.22 mmol, 10.8 equiv) in H2O (0.91 mL, 0.022 M). After 9 h, the solution was diluted with EtOAc (to 40 mL), filtered through SiO2 (ca. 0.4 mL) and concentrated under reduced pressure. The semi-solid was purified by preparatory thin layer chromatography on a 250 μm plate (1:3 acetone:CH2Cl2 elution) to give cycloheptadiene 32b (3.1 mg, 55% yield, Rf 0.14) as a white solid. Rf 0.45 (EtOAc, UV/Vis, stained blue in anysaldehyde); 1H NMR (600 MHz, CD2Cl2) δ 5.71 (app td, J = 5.2, 2.7, 1H), 4.82 (dt, J = 9.2, 7.5, 1H), 4.23 (d, J = 9.0, 1H), 3.69 (ddd, J = 8.5, 7.0, 1H), 3.13–3.05 (m, 1H), 2.84–2.75 (m, 1H), 2.66 (t, J = 8.0, 1H), 2.57–2.50 (m, 1H), 2.44 (dd, J = 12.8, 7.1, 1H), 2.32 (dddd, J = 16.3, 9.2, 4.9, 2.2, 1H), 1.91 (dtd, J = 12.8, 5.4, 3.6, 1H), 1.84–1.76 (m, 1H), 1.74 (dd, J = 12.8, 7.9, 1H), 1.60–1.52 (m, 3H), 1.51–1.44 (m, 1H), 1.25 (s, 3H); 13C NMR (125 MHz, CD2Cl2, –lactone carbon, –disubstituted carbon of trisubstituted olefin) δ 158.4, 123.3, 118.9, 78.8, 76.1, 72.0, 49.0, 48.7, 43.7, 32.6, 28.7, 27.1, 26.1, 21.0; IR (film) 3393, 2929, 1719, 1653, 966 cm−1; HRMS (ESI–APCI) m/z: [M+H]+ Calcd for C16H20O4 277.1434; Found 277.1427; [α]D28.1 +33.5° (c 0.17, CH2Cl2, derived from ketone 16 with 91% ee).
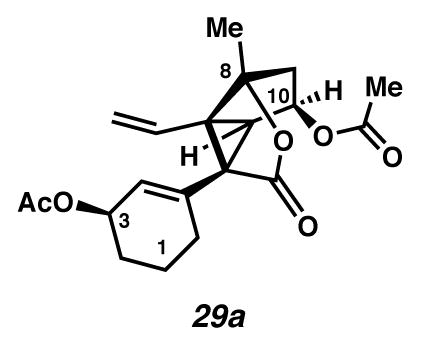
Divinylcyclopropane 29a
For a representative procedure, see silyl ether 54b → divinylcyclopropane 29b. 59% yield over three steps. Rf 0.35 (1:20 acetone:CH2Cl2, visualized with anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 6.13 (dd, J = 17.1, 10.5, 1H), 5.67 (dt, J = 3.5, 1.7, 1H), 5.42 (dd, J = 7.1, 6.1, 1H), 5.31 (dddd, J = 7.3, 5.5, 3.7, 1.8, 1H), 5.19 (dd, J = 10.5, 1.1, 1H), 5.02 (dd, J = 17.1, 1.1, 1H), 3.06 (d, J = 5.9, 1H), 2.24 (d, J = 14.1, 1H), 2.18 (dd, J = 14.4, 7.5, 1H), 2.14–2.08 (m, 1H), 2.04 (s, 3H), 2.03 (s, 3H), 1.89 (ddd, J = 17.8, 9.6, 5.6, 1H), 1.81–1.74 (m, 1H), 1.65 (dt, J = 12.4, 6.2, 2H), 1.60 (s, 3H), 1.57–1.50 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 173.4, 171.2, 170.7, 135.3, 129.3, 127.1, 118.5, 92.6, 73.3, 68.4, 58.5, 50.8, 49.7, 40.6, 28.0, 27.7, 21.5, 20.9, 20.0, 19.2; IR (film) 2937, 1759, 1738, 1733, 915 cm−1; HRMS (ESI–APCI) m/z: [M – H]− Calcd for C20H24O6 359.1500; Found 359.1488; [α]D23.7 +30.4° (c 0.18, CHCl3, derived from ketone 16 with 91% ee).
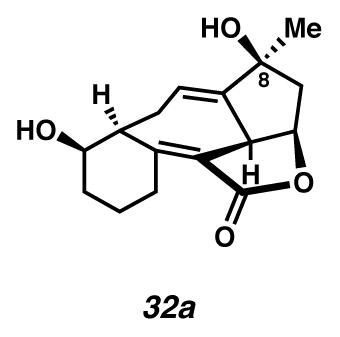
Cycloheptadiene 32a (white solid)
For a representative procedure see, divinylcyclopropane 29b → cycloheptadiene 32b. 51% yield. Rf 0.44 (EtOAc, visualized by UV/Vis or with anisaldehyde stain); 1H NMR (500 MHz, C6D6) δ 5.55 (s, 1H), 4.22 (dd, J = 16.1, 7.8, 1H), 3.50 (dd, J = 12.2, 6.7, 1H), 3.43 (d, J = 7.7, 1H), 3.32–3.09 (m, 1H), 2.91 (app s, 1H), 2.56 (dd, J = 10.0, 4.6, 1H), 2.44 (ddd, J = 17.3, 8.6, 4.2, 1H), 1.99 (dd, J = 12.6, 7.3, 1H), 1.76–1.63 (m, 1H), 1.56 (dq, J = 19.2, 6.4, 1H), 1.46–1.11 (m, 6H), 0.93 (s, 3H).; 13C NMR (125 MHz, C6D6) δ 168.7, 157.1, 148.5, 125.4, 119.4, 78.6, 75.7, 71.6, 48.9, 44.8, 43.3, 30.7, 28.3, 25.2, 24.4, 21.6; IR (film) 3369, 2929, 1724, 1654 cm−1; HRMS (ESI–APCI) m/z [M+H]+ Calcd for C16H20O4 277.1434; Found 277.1445; [α]D26.6 +100.0° (c 0.16, acetone, derived from ketone 16 with 91% ee).
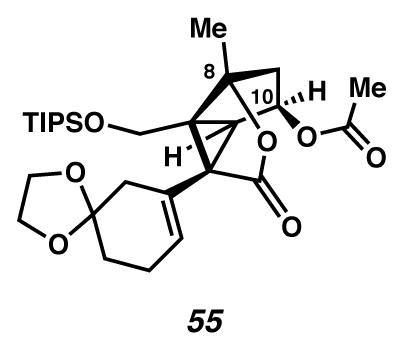
Ketal 55
A flask was charged with a colorless solution of ketone (7.0 mg, 0.015 mmol, 1 equiv), CH2Cl2 (2 mL, 0.0073 M), and 1,2-bis(trimethylsilyloxy)ethane (0.10 mL, 0.41 mmol, 28 equiv). The solution was cooled to 0 °C (ice water bath), and treated dropwise with a 1% v/v solution of TMSOTf in CH2Cl2 (150 μL, 0.000008 mmol, 0.0005 equiv). After two days, the solution was treated sequentially with CH2Cl2 (0.5 mL, 0.0294 M), 1,2-bis(trimethylsilyloxy)ethane (0.10 mL, 0.41 mmol, 28 equiv) and TMSOTf (150 μL of a solution containing TMSOTf (10 μL) in CH2Cl2 (1 mL), 0.000008 mmol, 0.0005 equiv). After an additional 12 h, the reaction was quenched through addition of pyridine (0.3 mL, 0.0037 mmol, 0.25 equiv), and then concentrated under reduced pressure. The resultant yellow oil was purified by thin layer chromatography on a 250 μm plate (12 cm × 20 cm; 1:1 Et2O:hexanes, then 2:1 Et2O:pentane elution) to furnish ketal 55 (5.1 mg, 67% yield) as a colorless oil. Rf 0.64 (1:2 EtOAc:PhH, visualized with anisaldehyde stain); 1H NMR (600 MHz, CDCl3) δ 5.71 (tt, J = 3.5, 1.6, 1H), 5.42 (ddd, J = 5.6, 5.6, 2.8, 1H), 4.33 (d, J = 11.4, 1H), 3.99–3.88 (m, 3H), 3.88–3.82 (m, 1H), 3.39 (d, J = 11.5, 1H), 2.81 (d, J = 6.0, 1H), 2.49 (d, J = 16.8, 1H), 2.36–2.26 (m, 1H), 2.26–2.18 (m, 3H), 2.02 (s, 3H), 1.77 (dddd, J = 16.8, 9.2, 4.8, 1.2, 2H), 1.70–1.60 (m, 4H), 1.56–1.51 (m, 3H), 1.10–0.98 (m, 21H); 13C NMR (125 MHz, CDCl3) δ 174.1, 171.4, 129.5, 127.9, 107.9, 92.8, 73.9, 64.7, 64.5, 59.6, 57.2, 51.2, 48.5, 42.2, 38.4, 30.6, 24.6, 21.0, 18.9, 18.2, 18.2, 12.1; IR (thin film/NaCl) 2943, 2892, 1763, 1739, 903, 852 cm−1; HRMS (ESI–APCI) m/z: [M+H]+ Calcd for C28H44O7Si 521.2925; Found 521.2929; [α]D21.0 +16.8° (c 0.37, CHCl3, derived from ketone 16 with 91% ee).
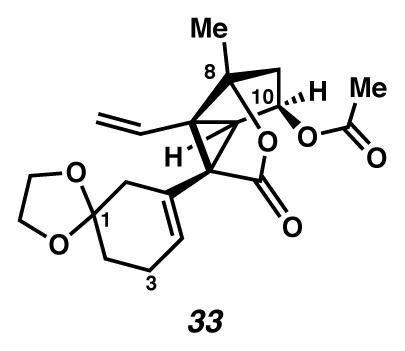
Divinylcyclopropane 33
A flask was charged with silyl ether 55 (15.0 mg, 0.027 mmol, 1 equiv) in THF (2.3 mL, 0.012 M) at 0 °C (ice water bath). The colorless solution was treated dropwise with TBAF (1M in THF, 40 μL, 0.040 mmol, 1.5 equiv), generating a yellow solution. After 1.5 h, the solution was diluted with EtOAc (to 40 mL), filtered through SiO2 (ca. 0.1 mL) and concentrated under reduced pressure to give primary alcohol 56 as a yellow oil. This yellow oil had already been characterized analytically as a byproduct in the ketalization reaction. Rf 0.07 (1:1 EtOAc:hexanes, visualized with anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 5.67 (dd, J = 5.9, 3.3, 1H), 5.40 (td, J = 6.7, 1.6, 1H), 4.02 (dd, J = 6.1, 2.2, 1H), 4.01 (d, J = 6.1, 1H), 3.94 (d, J = 6.1, 1H), 3.92 (dd, J = 6.1, 1.3, 1H), 3.85 (d, J = 12.9, 1H), 3.82 (d, J = 13.0, 1H), 2.95 (d, J = 6.0, 1H), 2.75–2.68 (m, 1H), 2.40–2.32 (m, 1H), 2.27–2.18 (m, 3H), 2.02 (s, 3H), 1.97 (app d, J = 16.7, 1H), 1.78 (dd, J = 9.3, 6.6, 1H), 1.83–1.73 (m, 2H), 1.69 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 173.9, 171.3, 128.1, 127.1, 108.3, 92.7, 73.5, 64.8, 64.6, 58.6, 57.8, 50.9, 47.8, 41.0, 38.8, 30.0, 24.6, 20.9, 18.7; IR (thin film/NaCl) 2932, 1738 cm−1; HRMS (ESI–APCI) m/z: [M+H]+ Calcd for C19H24O7 365.1595; Found 365.1598; [α]D22.5 +36.7° (c 0.13, CHCl3, derived from ketone 16 with 91% ee).
The crude alcohol 56 was thrice concentrated from PhH (0.15 mL each), and backfilled with Ar(g). The diol 56 was diluted with CH2Cl2 (16 mL, 0.0017 M), cooled to −15 °C (cryocool temperature), and treated with crushed Me-IBX (15 mg, 0.051 mmol, 1.9 equiv). After 5 min, the solution was allowed to warm gradually to 4 °C (cold room temperature). After 18, 24, 30, 45, and 55 h, additional portions were added of crushed Me-IBX (67.4 mg total, 0.23 mmol, 8.5 equiv). After 65 h, the reaction was diluted with EtOAc (to ca. 25 mL), filtered through SiO2 (ca. 0.2 mL), and concentrated under reduced pressure to give crude aldehyde 57.
A flask was charged with a mixture of Ph3PMeBr (0.129 g, 0.36 mmol, 13 equiv) in THF (9 mL, 0.003 M) at room temperature (ca. 24 °C). The mixture was treated dropwise with n-BuLi (2.5 M in hexanes, 0.13 mL, 0.325 mmol, 12.0 equiv), generating an orange solution. After 55 min, the orange solution had been cooled to −78 °C (acetone/dry ice) and was treated dropwise with crude aldehyde 57 in THF (0.3 mL × 2). The solution was allowed to gradually warm to −10 °C (over 1.33 h), at which point it was treated with acetone (0.3 mL). The reaction was rinsed with 1:1 brine:H2O (ca. 1 mL), and extracted with EtOAc (to ca. 50 mL). Extracts were dried over MgSO4, filtered and concentrated under reduced pressure. The mixture was purified by thin layer chromatography on 250 μm plates (10 cm × 20 cm; EtOAc × 2 elution) to furnish the desired divinyl-cyclopropane (33, Rf 0.79, 7.9 mg including ca. 5% other, 91% yield of slightly impure material) as an oil. 1H NMR (500 MHz, CDCl3) δ 6.09 (ddd, J = 17.1, 10.5, 0.5, 1H), 5.74 (tt, J = 3.5, 1.7, 1H), 5.40 (dd, J = 6.9, 5.9, 1H), 5.17 (dd, J = 10.5, 1.1, 1H), 5.03 (dd, J = 17.1, 1.1, 1H), 4.02–3.91 (m, 4H), 3.04 (d, J = 5.9, 1H), 2.38–2.30 (m, 2H), 2.24 (app dd, J = 14.3, 0.6, 1H), 2.26–2.19 (m, 1H), 2.17 (dd, J = 14.4, 7.6, 1H), 2.03 (s, 3H), 1.95–1.90 (m, 1H), 1.75 (ddd, J = 14.0, 6.9, 3.9, 1H), 1.64–1.59 (m, 1H), 1.59 (s, 3H); 13C NMR (125 MHz, CDCl3, –CO2R) δ 171.4, 130.1, 127.8, 127.0, 118.5, 107.9, 92.4, 73.4, 64.6, 64.5, 58.1, 50.6, 49.8, 41.0, 37.6, 30.7, 24.5, 21.0, 20.0; IR (thin film/NaCl) 2932, 1759, 1738, 958, 851 cm−1; HRMS (ESI–APCI) m/z: [M+Na]+ Calcd for C20H24O6 383.1465; Found 383.1466; [α]D20.4 −11.2° (c 0.17, CHCl3, derived from ketone 16 with 91% ee).
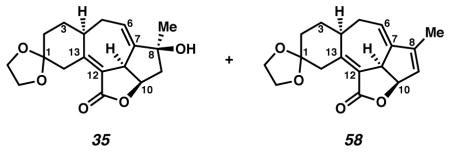
Cycloheptadiene 35
A flask was charged with acetate 33 (2.0 mg, 0.0055 mmol, 1 equiv) in THF (2.0 mL, 0.0028 M) at −78 °C (acetone, dry ice bath). The colorless solution was treated dropwise with LiEt3BH (0.5 M solution in THF, 24 μL, 2.2 equiv). After 10 min, the solution was allowed to warm to −20 °C (cryocool temperature). After an additional 6 h, the reaction was treated with H2O (0.02 mL), allowed to warm to room temperature (ca. 24 °C), diluted with EtOAc (to 10 mL), filtered through SiO2 (ca. 0.2 mL), and concentrated under reduced pressure. The product was combined with product from two equivalent reactions that had been run with acetate 33 (0.4 and 1.0 mg, respectively). The reactions were purified by thin layer chromatography on a 250 μm plate (10 cm × 20 cm; EtOAc elution) to furnish the desired cycloheptadiene (35, Rf 0.34, slightly impure material). The material was further purified by thin layer chromatography on a 250 μm plate (10 cm × 20 cm; 1:5 acetone:CH2Cl2 elution) to furnish the desired cycloheptadiene (35, Rf 0.29, 1.8 mg, 68% yield). Rf 0.56 (EtOAc, UV/Vis, visualized with anisaldehyde stain); 1H NMR (600 MHz, C6D6) δ 5.37 (ddd, J = 2.8, 5.0, 5.0 Hz, 1H), 4.17 (ddd, J = 7.1, 8.0, 9.2 Hz, 1H), 3.71 (ddd, J = 6.2, 6.8, 6.8 Hz, 1H), 3.68 (ddd, J = 5.1, 6.9, 6.9 Hz, 1H), 3.57 (br d, J = 15 Hz, 1H), 3.52 (ddd, J = 4.8, 6.9, 6.9 Hz, 1H), 3.48 (ddd, J = 5.9, 6.9, 6.9 Hz, 1H), 3.32 (br d, J = 9 Hz, 1H), 2.21 (dddd, J = 4.5, 4.5, 8.9, 8.9 Hz, 1H), 1.96 (dd, J = 7.1, 12.8 Hz, 1H), 1.93 (dddd, J = 3.6, 3.6, 5.4, 16.7 Hz, 1H), 1.89 (dddd, J = 1.0, 4.3, 6.0, 13.7 Hz, 1H), 1.73 (ddd, J = 4.9, 10.8, 13.9 Hz, 1H), 1.67 (dddd, J = 2.4, 4.8, 9.5, 16.7, 1H), 1.43 (dddd, J = 5.4, 5.4, 5.4, 13.6 Hz, 1H), 1.37 (dddd, J = 4.2, 9.0, 10.6, 13.5 Hz, 1H), 1.34 (dd, J = 7.9, 12.8 Hz, 1H), 0.89 (d, J = 0.9 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 169.6, 156.0, 149.9, 124.5, 119.1, 109.8, 78.8, 76.3, 65.0, 64.8, 48.9, 43.6, 39.6, 35.6, 34.3, 30.8, 28.5, 28.5; IR (thin film/NaCl) 3446, 2927, 1738, 1647, 955 cm−1; HRMS (ESI–APCI) m/z: [M+H]+ Calcd for C18H22O5 319.1540; Found 319.1530; [α]D23.8 +23.3° (c 0.18, CH2Cl2, derived from ketone 16 with 91% ee).
Dehydration product 58. Rf 0.68 (1:5 acetone:PhH; visualized with anisaldehyde stain); 1H NMR (600 MHz, CD2Cl2) δ 5.88 (s, 1H), 5.58 (dt, J = 2.2, 6.0, 1H), 5.36–5.33 (m, 1H), 4.32–4.30 (m, 1H) 3.96–3.91 (m, 2H), 3.90–3.83 (m, 2H), 2.87 (ddd, J = 16.1, 5.0, 5.0, 1H), 2.68 (m, 1H), 2.47–2.42 (m, 1H), 2.17 (dddd, J = 15.9, 6.5, 6.5 1.1, 1H), 1.85–1.73 (m, 6H), 1.63–1.71 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 170.1, 154.0, 150.8, 145.7, 130.0, 123.1, 117.0, 109.3, 81.6, 64.6, 64.3, 43.6, 40.1, 36.0, 34.8, 30.8, 29.1, 12.1; IR (thin film/NaCl) 3456 (br), 2934, 1738, 1733, 1645 cm−1; HRMS (ESI–APCI) m/z: [M+H]+ Calcd for C18H20O4 301.1434; Found 301.1439; [α]D25.8 −25.5° (c 0.22, CH2Cl2, derived from ketone 16 with 91% ee).
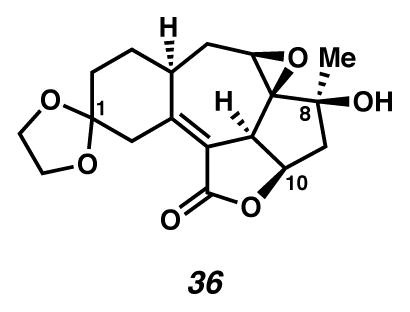
Epoxide 36
A flask was charged with cycloheptadiene 35 (0.9 mg, 0.0028 mmol, 1 equiv) and VO(acac)2 (0.2 mg, 0.00075 mmol, 0.27 equiv) in PhH (0.4 mL). The pale green solution was treated with TBHP, 5.5 M in decane (ca. 10 μL, 1 drop), and then turned burgundy in color. After 12 minutes, the solution was treated with saturated aq Na2SO3 (0.04 mL), diluted with EtOAc (to 10 mL), filtered through SiO2 (ca. 0.2 mL), and concentrated under reduced pressure. This compound was combined with the crude product from an analogous reaction carried out with cyclohep-tadiene 35 (1.8 mg, 0.0056 mmol). The reactions were purified by thin layer chromatography on a 250 μm plate (10 cm × 20 cm; 1:5 acetone:CH2Cl2 elution) to furnish epoxide 36 (1.6 mg, 56% yield, Rf 0.34) as a colorless oil. Rf 0.40 (EtOAc, visualized by UV/Vis, or with anisaldehyde stain); 1H NMR (500 MHz, CD2Cl2) δ 4.83 (ddd, J = 8.7, 7.5, 6.6, 1H), 4.03–3.85 (m, 5H), 3.34 (t, J = 3.0, 1H), 3.28 (d, J = 15.0, 1H), 3.07 (d, J = 15.1, 1H), 2.70–2.62 (m, 1H), 2.52 (dd, J = 13.5, 7.5, 1H), 2.27 (s, 1H), 2.07–2.03 (m, 1H), 2.05 (d, J = 2.6, 1H), 1.90–1.77 (m, 3H), 1.73–1.67 (m, 2H), 1.29 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 169.3, 156.4, 120.0, 109.5, 75.8, 74.8, 68.7, 65.1, 64.8, 55.9, 47.6, 42.8, 39.4, 35.3, 34.0, 29.2, 29.1, 23.7; IR (thin film/NaCl) 3473, 1739, 1662, 1268, 1120 cm−1; HRMS (ESI–APCI) m/z: [M+H]+ Calcd for C18H22O6 335.1489; Found 335.1483; [α]D22.1 +28.6° (c 0.16, CH2Cl2, derived from ketone 16 with 91% ee).
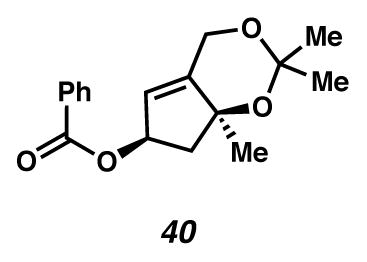
Benzoyl ester 40
A dilute solution of enone 16 in Et2O was concentrated at 150 torr to 0.645 g of a 30% (w/w) solution as determined by 1H NMR (0.19 g enone, 1.04 mmol) and dissolved in THF (8 mL, 0.13 M). [The volatility of the starting enone precluded preparation of fully concentrated samples without significant loss of material.] The solution was cooled in a dry ice/acetone bath and to it was added DIBALH (2.3 mL, 1 M toluene, 2.3 mmol). The starting material was consumed within 10 minutes according to TLC. The solution was warmed to room temperature and quenched with saturated aq sodium potassium tartrate (20 mL) and saturated aq NH4Cl (20 mL) and stirred vigorously for 2 h. The solution was extracted with Et2O (6 × 50 mL) and the combined organic phases dried with Na2SO4, filtered and concentrated at 150 torr. The crude material was purified by flash chromatography (1:1 pentane:Et2O), concentrated at 150 torr and dissolved immediately in CH2Cl2 (17 mL, 0.06 M) for use in the next step. The CH2Cl2 solution of allylic alcohol was cooled in an ice water bath and to it was added DMAP (0.27 g, 2.2 mmol) and Et3N (1.3 mL, 9.38 mmol) followed by benzoic anhydride (0.472 g, 2.1 mmol). The solution was stirred for 4 hr with warming to ambient temperature, then quenched with saturated aq NH4Cl (20 mL) and extracted with EtOAc (3 × 75 mL). The combined organic phases were dried with Na2SO4, filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (2:1 hexanes:CH2Cl2 with 2% Et3N) to yield benzoate ester 40 (0.294 g, 98% yield, 2 steps) as a colorless oil. Rf 0.66 (1:1 EtOAc:hexanes, visualized by UV/Vis, or with anisaldehyde stain); 1H NMR (300 MHz, CDCl3) δ 8.08–8.03 (m, 2H), 7.60–7.53 (m, 1H), 7.48–7.41 (m, 2H), 5.83–5.75 (m, 1H), 5.69–5.66 (m, 1H), 4.77–4.64 (m, 2H), 2.69 (dd, J = 6.9, 12.8, 1H), 2.13 (dd, J = 6.9, 12.9, 1H), 1.50 (s, 3H), 1.50 (s, 3H), 1.42 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 166.6, 146.5, 133.2, 130.4, 129.8, 128.5, 122.0, 100.3, 81.0, 76.6, 60.4, 49.4, 30.0, 28.6, 25.9; IR (thin film/NaCl) 2990, 1715, 1602, 1268, 1158 cm−1; HRMS (EI) m/z: [M+H]+ Calcd for C17H20O4 288.1362; Found 288.1366; [α]D24.7 +74.2° (c 1.0, CHCl3, derived from ketone with 91% ee).
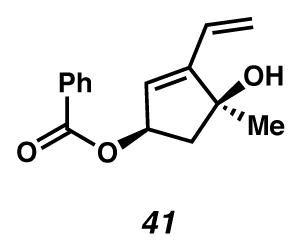
Allylic benzoyl ester 41
To a MeOH (3 mL, 0.037 M) solution of acetonide 40 (0.031 g, 0.11 mmol), cooled in an ice water bath, was added trimethyl orthoformate (0.16 mL, 0.96 mmol) followed by a MeOH (1.5 mL) solution of fumaric acid (0.032 g, 0.28 mmol). The solution was allowed to gradually warm to ambient temperature and stirred an additional 10 hr. The solution was quenched with saturated aq NaHCO3 (10 mL) and extracted with EtOAc (3×20 mL). The combined organic phases were dried with MgSO4, filtered and concentrated in vacuo to yield crude diol 59 (0.028 g, >99% yield) as a colorless oil, which was used immediately.
Crude diol 59 (0.028, 0.11 mmol) was dissolved in CH2Cl2 (2 mL, 0.055 M) and cooled in an ice water bath. To the solution was added DMP (0.11 g, 0.27 mmol). CAUTION! DMP is a HEAT- and SHOCK-SENSITIVE COMPOUND, showing exotherms when heated (>130 °C). All operations should be carried out behind a blast shield. The solution was stirred for an additional two hr (with warming to ambient temperature) and quenched with saturated aq NaHCO3 (3 mL) and saturated aq Na2S2O3 (6 ml). The two phases were stirred vigorously for 5 min and then extracted with EtOAc (6 × 25 mL). The combined organic phases were dried with MgSO4, filtered and concentrated in vacuo. The crude aldehyde was used immediately as is. A solution of methylenetriphenylphosphorane was prepared in THF (8 mL, 0.06 M) from triphenylphosphonium bromide (0.17 g, 0.48 mmol) and potassium t-butoxide (0.053 g, 0.47 mmol). The yellow solution was cooled in an ice water bath after stirring for 30 min at ambient temperature, and to it added crude aldehyde dropwise as a solution in THF (4 mL). The reaction was complete within 10 min according to TLC. The brown solution was quenched with H2O (20 mL) and extracted with CH2Cl2 (3 × 30 mL). The combined organic phases were dried with Na2SO4, filtered and concentrated in vacuo. The crude material was purified by flash chromatography (1:20 EtOAc:hexanes elution) to yield allylic benzoyl ester 41 as a pale yellow oil (0.019 g, 69% yield, 3 steps). Rf 0.62 (1:2 EtOAc:hexanes, visualized with anisaldehyde stain); 1H NMR (300 MHz, CDCl3) δ 8.03 (dt, J = 8.4, 1.5, 2H), 7.56 (app t, J = 7.4, 1H), 7.44 (app t, J = 7.7, 2H), 6.36 (dd, J = 17.9, 11.3, 1H), 5.94 (d, J = 2.2, 1H), 5.84–5.77 (m, 1H), 5.75 (ddd, J = 7.0, 4.7, 2.0, 1H), 5.34 (dd, J = 11.3, 1.6, 1H), 2.75 (dd, J = 14.1, 7.3, 1H), 2.16 (dd, J = 14.1, 4.7, 1H), 1.91 (s, 1H), 1.50 (s, 3H); 13C NMR (75 MHz, CDCl3, –CO2Ph) δ 151.3, 133.2, 130.4, 129.8, 129.2, 128.6, 127.2, 119.4, 81.3, 76.1, 49.5, 27.0; IR (thin film/NaCl) 3436 (br), 3063, 2974, 2931, 2865, 1715, 1602, 924, 859, 713 cm−1; HRMS (EI) m/z: [M+·]+ Calcd for C15H16O3 244.1100; Found 244.1101; [α]D27.3 +150.0° (c 0.13, CHCl3, derived from ketone 16 with 91% ee).
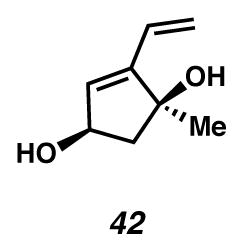
Alcohol 42
To a MeOH (5 mL, 0.05 M) solution of allylic benzoyl ester 41 (0.062 g, 0.25 mmol) was added a MeOH solution of sodium hydroxide (0.85 mL, 0.64 M, 0.54 mmol) dropwise. The solution was stirred at ambient temperature for 3 hours, quenched with H2O (10 mL) and extracted with EtOAc (6 × 25 mL). The combined organic phases were dried with Na2SO4, filtered and concentrated in vacuo. The crude material was purified by flash chromatography (4:1 CH2Cl2:EtOAc → 1:1 hexanes/EtOAc elution) to yield alcohol 42 as a white solid (0.030 g, 84% yield). Rf 0.13 (1:5 EtOAc:CH2Cl2, visualized with anisaldehyde stain); 1H NMR (300 MHz, CDCl3) δ 6.33 (ddt, J = 17.7, 11.2, 0.7, 1H), 5.82 (d, J = 2.1, 1H), 5.75 (dd, J = 17.9, 1.7, 1H), 5.29 (dd, J = 11.2, 1.7, 1H), 4.68 (dd, J = 11.5, 5.1, 1H), 2.57 (dd, J = 13.6, 6.8, 1H), 1.88 (app dd, J = 13.6, 4.8, 2H), 1.71 (d, J = 6.6, 1H), 1.43 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 149.5, 131.3, 129.5, 118.6, 81.3, 73.0, 53.1, 26.8; IR (thin film/NaCl) 3287, 3252, 2968, 2930, 2873, 926 cm−1; HRMS (EI) m/z: [M+·]+ Calcd for C8H12O2, 140.0837; Found 140.0859; [α]D24.2 +100.0° (c 0.085, MeOH, derived from ketone 16 with 91% ee).
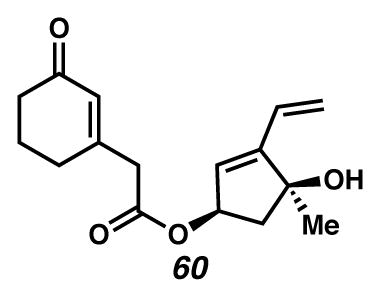
Ester 60
A CH2Cl2 (15 mL, 0.026 M) solution of alcohol 42 (0.054 g, 0.39 mmol) with carboxylic acid 10 (0.16 g, 1.1 mmol) was cooled in an ice water bath. EDC·HCl (0.21 g, 1.1 mmol) was added followed by DMAP (0.011 g, 0.09 mmol). The deep yellow solution was allowed to warm to ambient temperature and stirred an additional 1 hour. The solution was quenched with aq HCl (20 mL, 0.12 M) and extracted with EtOAc (2 × 50 mL). The combined organic phases were washed with aq HCl (20 mL, 0.12 M), aq K2CO3 (2 × 20 mL 5 % w/v), brine (20 mL), and saturated aq NH4Cl (20 mL). The organic phase was dried with Na2SO4, filtered and concentrated to provide crude ester 60 (0.125 g, 117% crude yield) as an orange oil. The crude ester was used without further purification. An analytical sample was obtained at another point, upon purification by preparatory thin layer chromatography with Et2O × 2 as the eluent. Rf 0.21 (1:5 EtOAc:CH2Cl2, visualized with anisaldehyde stain); 1H NMR (300 MHz, CDCl3) δ 6.30 (dd, J = 17.9, 11.3, 1H), 5.94–5.91 (m, 1H), 5.80 (d, J = 2.2, 1H), 5.83–5.75 (m, 1H), 5.53–5.47 (m, 1H), 5.36 (dq, J = 1.6, 0.5, 1H), 5.32 (dq, J = 1.7, 0.6, 1H), 3.23 (s, 2H), 2.64 (dd, J = 14.1, 7.3, 1H), 2.42–2.38 (m, 4H), 2.06–1.93 (m, 3H), 1.84 (s, 1H), 1.46 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 199.6, 169.4, 157.2, 151.7, 129.1, 129.0, 126.5, 119.7, 81.1, 76.5, 49.2, 43.7, 37.3, 29.8, 27.0, 22.7; IR (thin film/NaCl) 3417, 2925, 2853, 1732, 1661, 959, 887 cm−1; HRMS (EI) m/z: [M·]+ Calcd for C16H20O4 276.1362; Found 276.1363; [α]D23.2 +45.6° (c 0.20, CHCl3, derived from ketone 16 with 91% ee).
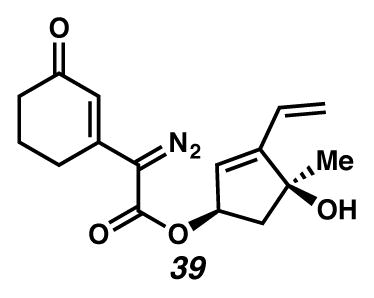
Diazoester 39
To a CH3CN (42 mL, 0.01 M) solution of ester 60 (≤0.125 g, 0.385 mmol) was added TsN3 (0.670 g, 3.4 mmol) dropwise using a flame-dulled pipette. CAUTION!!! TsN3 is SHOCK SENSITIVE AND POTENTIALLY EXPLOSIVE. The flask was equipped with an argon balloon and cooled in an ice water bath. Et3N (0.8 mL, 5.8 mmol) was added dropwise causing the solution to become deep orange in color. The solution was allowed to warm to ambient temperature and stirred an additional 11 hr. The solution was concentrated in vacuo and purified using flash chromatography (4:1 CH2Cl2:EtOAc) to yield diazoester 39 (0.746 g, 64% yield, 2 steps) as a bright yellow oil. Rf 0.18 (1:5 EtOAc:CH2Cl2, UV/Vis, visualized with anisaldehyde stain); 1H NMR (500 MHz, C6D6) 6.75 (t, J = 1.3, 1H), 6.15 (dd, J = 17.8, 11.3, 1H), 5.72 (dd, J = 17.8, 1.8, 1H), 5.60 (d, J = 2.0, 1H), 5.46–5.24 (m, 1H), 5.11 (dd, J = 11.3, 1.8, 1H), 2.32 (dd, J = 13.7, 7.3, 1H), 2.16–2.07 (m, 2H), 1.82 (dd, J = 13.7, 5.2, 1H), 1.64 (m, 2H), 1.39 (dt, J = 12.4, 6.2, 2H), 1.37–1.20 (m, 1H), 1.17 (s, 3H); 13C NMR (125 MHz, C6D6, –CN2) δ 195.0, 162.4, 151.7, 144.6, 129.3, 126.2, 121.6, 119.0, 80.3, 76.7, 49.2, 36.8, 26.9, 26.0, 22.1; IR (thin film/NaCl) 3407, 2930, 2103, 1707, 1644, 992 cm−1; HRMS (EI) m/z: [M+H]+ Calcd for C16H18N2O4 303.1345; Found 303.1339; [α]D20.4 +67.0° (c 0.24, CHCl3, derived from ketone 16 with 91% ee).
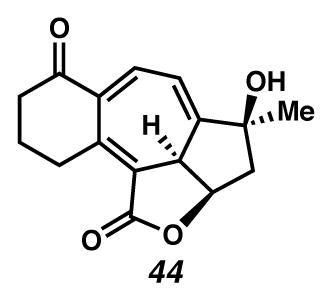
Cycloheptatriene 44
To a CH2Cl2 (2.4 mL) solution of Cu(tbs)2 (0.035 g, 0.084 mmol, 3.2 eq.) in a moisture-free, oxygen-free glovebox was added diazoester 39 (0.008 g, 0.026 mmol) as a solution in CH2Cl2 (5.6 mL). The deep maroon solution was stirred for 5 days at 29 °C and removed from the glove box. DBN (0.01 mL, 0.08 mmol) was added and the solution stirred for 2 hr. The solution was diluted with EtOAc, filtered over a sort silica plug, concentrated in vacuo and purified by preparatory thin layer chromatography (2:1 EtOAc/benzene) to yield cycloheptatriene 44 (0.0022 g, 31% yield) as a pale yellow solid. Rf 0.50 (1:2 EtOAc:PhH, UV/Vis, visualized with anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 7.72 (d, J = 6.4, 1H), 6.54 (dd, J = 6.4, 2.2, 1H), 5.17 (ddd, J = 7.9, 6.6, 4.5, 1H), 3.59 (m, 1H), 3.21 (d, J = 7.9, 1H), 2.94 (dt, J = 17.5, 5.7, 1H), 2.61 (m, 2H), 2.52 (dd, J = 14.5, 6.6, 1H), 2.27 (dd, J = 14.5, 4.4, 1H), 2.03–1.92 (m, 2H), 1.77 (s, 1H), 1.54 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 199.9, 168.4, 156.3, 144.8, 139.8, 139.2, 117.3, 115.3, 79.2, 78.2, 48.3, 46.4, 39.6, 28.1, 26.5, 20.9; IR (thin film/NaCl) 3416 (br), 2956, 2925, 2853, 1739, 1734, 802 cm−1; HRMS (TOF) m/z: [M+H]+ Calcd for C16H17O4 273.1121; Found 273.1131; [α]D25 −86.9 (c 0.43, CH2Cl2, derived from ketone 16 with 91% ee).
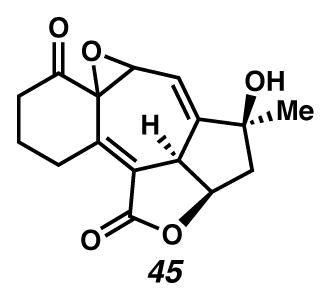
Epoxide 45
In a flame-dried, argon purged RBF was added THF (4 mL) and t-BuOOH (0.07 mL, 5–6 M/decane, 0.35–0.42 mmol). The solution was cooled in a dry ice/acetone bath and to it added n-BuLi (0.1 mL, 2.3 M/hexanes, 0.25 mmol). The solution was allowed to warm to room temperature. A separate RBF was prepared with triene 44 (2.1 mg, 0.0077 mmol) in THF (0.8 mL) and cooled in a dry ice/acetone bath. The t-BuOOLi/THF solution (0.25 mL, 0.058 M, 0.015 mmol) was added dropwise. The reaction was allowed to warm to room temperature, then quenched with Na2S2O3 (aq) and stirred vigorously for 10 min. The solution was extracted with EtOAc (3 × 5 mL), dried with Na2SO4 and filtered. Purification by thin-layer chromatography with 2:1 EtOAc:PhH eluent yielded the epoxide (1.4 mg, 64% yield). 1H NMR (500 MHz, CDCl3): δ 5.95 (dd, J = 4.3, 2.6 Hz, 1H), 4.99 (ddd, J = 8.6, 6.5, 5.7 Hz, 1H), 4.59 (dd, J = 8.6, 2.5 Hz, 1H), 3.90 (dd, J = 4.2, 0.5 Hz, 1H), 3.62 (dm, J = 17.2, 1H), 3.14 (dm, J = 17.2 Hz, 1H), 2.73 (dt, J = 17.8, 6.6 Hz, 1H), 2.64 (dt, J = 17.8, 6.6 Hz, 1H), 2.40 (dd, J = 13.9, 6.5 Hz, 1H), 2.06–1.97 (m, 2H), 1.99 (dd, J = 13.9, 5.7 Hz, 1H), 1.68 (s, 1H), 1.41 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 202.9, 169.0, 155.2, 146.9, 122.4, 113.5, 79.3, 78.1, 62.7, 62.5, 47.3, 43.2, 40.6, 28.5, 28.4, 20.0; IR (Neat film, NaCl) 3446 (br), 2965, 2928, 2872, 1732, 1738, 1262, 806, 735 cm−1; HRMS (TOF) m/z: [M+H]+ Calcd for C16H17O5 289.1071; Found 289.1085; [α]D25 = +35.1 (c 0.14, CH2Cl2)
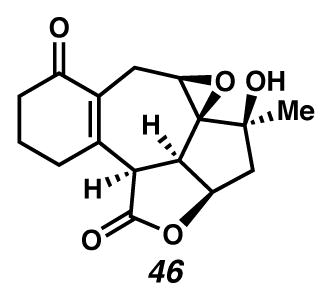
Epoxide 46
To a CH2Cl2 (2 mL) solution of Cu(tbs)2 (0.0062 g, 0.015 mmol, 0.4 equiv) in a moisture-free, oxygen-free glove box was added diazoester 39 (0.0108 g, 0.036 mmol) as a solution in CH2Cl2 (2.5 mL). The deep maroon solution was stirred for 5 days at 30–31 °C and then concentrated in vacuo (predominantly diene, Rf 0.36 in 2:1 EtOAc:benzene, visualized by UV/Vis, or with anisaldehyde stain). PhH (3 mL) was added and the solution removed from the glovebox. VO(acac)2 (0.010 g, 0.038 mmol) followed by t-BuOOH (0.05 mL, 5.5 M decane, 0.275 mmol). Upon addition of t-BuOOH the solution turns an even deeper maroon and over the course of 20 minutes, after which time starting material has been consume by TLC, the solution gradually becomes a light tan color. The solution was diluted with EtOAc, filtered over a short silica plug, concentrated in vacuo and purified by preparatory TLC (2:1 EtOAc:PhH) to yield epoxide 46 (3.3 mg, 32% yield) as an off-white solid. Rf 0.23 (1:2 EtOAc:PhH, UV/Vis, visualized with KMnO4 stain); 1H NMR (600 MHz, CDCl3) δ 4.84 (ddd, J = 6.0, 4.6, 1.6, 1H), 3.82 (dd, J = 18.4, 6.1, 1H), 3.52 (d, J = 6, 1H), 3.41 (dd, J = 6.3, 4.6, 1H), 3.38 (d, J = 6.4, 1H), 3.26–3.18 (m, 1H), 2.54 (ddd, J = 16.9, 7.5, 4.8, 1H), 2.52 (s, 1H), 2.46–2.36 (m, 3H), 2.24–2.14 (m, 1H), 2.12–2.04 (m, 1H), 1.97–1.90 (m, 1H), 1.37 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 198.7, 172.2, 150.0, 129.9, 80.0, 75.0, 70.3, 54.3, 51.0, 46.1, 43.7, 37.9, 32.7, 26.9, 22.0, 21.8; IR (thin film/NaCl) 3472 (br), 2960, 2933, 2857, 1766, 1661, 1266, 919, 785 cm−1; HRMS (ESI–APCI) m/z: [M+H]+ Calcd for C16H18O5 291.1227; Found 291.1228; [M–H]−: Calcd for C16H18O5 289.1081; Found 289.1086; [α]D25.4 +86.2 (c 0.16, CH2Cl2, derived from ketone 16 with 91% ee).
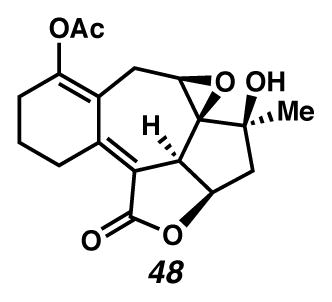
Epoxide 48
To CH2Cl2 (0.6 mL) solution of epoxide 46 (2.9 mg, 0.01 mmol) was added DMAP (3.3 mg, 0.027 mmol) followed by Ac2O (0.005 mL, 0.053 mmol), added in two equal batches. The solution was purified directly by thin-layer chromatography (EtOAc) to yield epoxide 48 (1.5 mg, 45% yield) as a white solid. 1H NMR (500 MHz, CDCl3) δ 4.83 (ddd, J = 9.4, 7.6 Hz, 1H), 3.84 (d, J = 9.4 Hz, 1H), 3.51–3.45 (m, 1H), 3.47 (d, J = 6.1 Hz, 1H), 3.29 (dd, J = 16.5, 6.3 Hz, 1H), 2.94–2.87 (m, 1H), 2.60 (dd, J = 13.0, 7.1 Hz, 1H), 2.49 (dt, J = 16.5, 2.8 Hz, 1H), 2.40–2.37 (m, 2H), 2.34 (s, OH), 2.25 (s, 3H), 1.95 (dd, J = 13.0, 7.9 Hz, 1H), 1.89–1.82 (m, 1H), 1.78–1.71 (m, 1H), 1.31 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 170.4, 168.2, 156.7, 152.5, 122.6, 115.0, 74.0, 73.3, 70.9, 53.1, 48.6, 42.2, 29.3, 25.7, 23.8, 23.6, 21.3, 21.1; IR (neat film, NaCl) 3491 (br), 2972, 2935, 1757, 1732, 1274, 934, 788 cm−1; HRMS (TOF) m/z: [M+H]+ Calcd for C18H21O6 333.1333; Found 333.1333; [α]D25 = +265.6 (c 0.15, CH2Cl2).
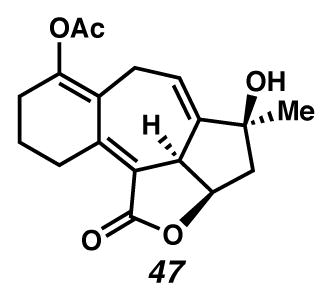
Cycloheptadiene 47
To a CH2Cl2 (1 mL) solution of Cu(tbs)2 (0.0055 g, 0.013 mmol) in a moisture-free, oxygen-free glove box was added diazoester 39 (0.0087 g, 0.029 mmol) as a solution in CH2Cl2 (1.9 mL). The deep maroon solution was stirred for 2 days at 35–36 °C. The solution was then removed from the glove box and to it added acetic anhydride (0.02 mL, 0.21 mmol) followed by DMAP (0.0056 g, 0.046 mmol) as a solution in 0.4 mL. The solution turned to a light green solution and was loaded directly onto a single analytical TLC plate and purified using 2:1 EtOAc:PhH eluent to provide enol acetate 47 (0.004 g, 0.013 mmol, 44% yield) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 5.82 (ddd, J = 8, 6, 2.7 Hz, 1H), 4.85 (ddd, J = 8.5, 6.7, 1H), 4.35 (d, J = 9 Hz, 1H), 3.18 (dd, J = 14.8, 8 Hz, 1H), 3.15–3.04 (m, 2H), 2.94 (ddt, J = 14.6, 3.3 Hz, 1H), 2.44 (dd, J = 13.1, 6.8 Hz, 1H), 2.40–2.30 (m, 1H), 2.24 (s, 3H), 2.11 (s, 1H), 1.97 (dd, J = 12.8, 8 Hz, 1H), 1.82–1.76 (m, 2H), 1.33 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 170.4, 168.6, 155.2, 154.1, 149.9, 121.4, 119.0, 117.3, 77.4, 75.5, 49.3, 46.2, 29.1, 28.5, 26.0, 25.9, 21.5, 21.1; IR (Neat film, NaCl) 3444 (br), 2970, 2933, 2868, 1738, 1626 cm−1; HRMS (TOF) m/z: [M+H]+ Calcd for C18H21O5 317.1384; Found 317.1388; [α]D25.4 = +65.6 (c 0.38, CH2Cl2).
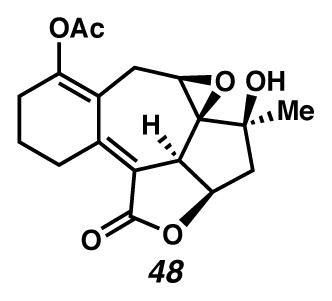
Epoxide 48
Enol acetate 47 (4 mg, 0.013 mmol) was dissolved in benzene (2 mL) and to it added excess t-BuOOH followed by VO(acac)2 (3.4 mg, 0.013 mmol). The deep maroon solution was purified directly by thin-layer chromatography (EtOAc) to yield epoxide (3.3 mg, 76% yield) as a white solid.
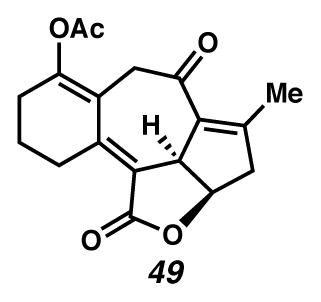
Ketone 49
To a CH2Cl2 (1.8 mL) solution of allylic alcohol 47 (0.0028 g, 0.0089 mmol) was added PCC (0.031 g, 0.15 mmol). The solution was stirred for 45 minutes, diluted with ethyl acetate and filtered through a plug of silica. The crude reaction mixture was purified by preparatory thin-layer chromatography with 2:1 EtOAc/PhH eluent to provide the rearranged enone (0.001 g, 0.0032 mmol, 36% yield) as a colorless oil. 1H NMR (500 Mhz, CDCl3) δ 5.13 (ddd, J = 8.3, 8.3, 3.2 Hz, 1H), 4.64 (d, J = 8.3, 1H), 3.57 (dt, J = 13.7, 2.4 Hz, 1H), 3.52 (d, J = 13.7 Hz, 1H), 3.36–3.28 (m, 1H), 3.07 (dd, J = 20.1, 8.3 Hz, 1H), 3.01–2.92 (m, 1H), 2.76 (d, J = 20.1 Hz, 1H), 2.43–2.39 (m, 2H), 2.26 (s, 3H), 2.15 (app q, J ≤ 1.1 Hz, 3H), 1.86–1.76 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 193.8, 170.5, 168.2, 156.8, 152.6, 148.2, 133.9, 120.6, 119.6, 75.8, 51.6, 47.6, 43.6, 29.3, 25.6, 21.3, 21.1, 16.1; IR (neat film, NaCl) 2931, 1762, 1737, 1687 cm-1; HRMS (TOF) m/z: [M+H]+ Calcd for C18H19O5 315.1227; Found 315.1233; [α]D25 = +122.56 (c 0.16, CH2Cl2).
Supplementary Material
Acknowledgments
The authors wish to thank the NIH-NIGMS (R01GM080269), Amgen, the Gordon and Betty Moore Foundation, and Caltech for financial support. Additionally, the authors gratefully acknowledge Larry Henling and the late Dr. Michael Day (Caltech) for X-ray crystallographic structural determination, Dr. Mona Shahgholi and Naseem Torian (Caltech) for mass spectrometric analysis, Dr. David VanderVelde (Caltech) for NMR experimental assistance and helpful discussions, Dr. Masaki Seto, Dr. N. Sherden, and Dr. D. E. White for helpful discussions, and Dr. R. A. Craig, II for editorial suggestions. J.L.R. thanks the California Tobacco-Related Disease Research Program of the University of California, Grant Number 14DT-0004 for a predoctoral fellowship. A.C.J. thanks the NIH (F32GM082000) for a postdoctoral fellowship.
Footnotes
Notes
The authors declare no competing financial interest.
Spectra (1H NMR, 13C NMR, and IR). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For a review on the terpenoids that have been isolated from Sinularia, see Kamel HN, Slattery M. Pharmaceutical Biology. 2005;43:253.. The taxonomy of Sinularia has been well reviewed in Coll JC. Chem Rev. 1992;92:613.
- 2.Duh CY, Wang SK, Chia MC, Chiang MY. Tetrahedron Lett. 1999;40:6033. [Google Scholar]
- 3.(a) Sheu JH, Ahmed AF, Shiue RT, Dai CF, Kuo YH. J Nat Prod. 2002;65:1904. doi: 10.1021/np020280r. [DOI] [PubMed] [Google Scholar]; (b) Ahmed AF, Shiue RT, Wang GH, Dai CF, Kuo YH, Sheu JH. Tetrahedron. 2003;59:7337. [Google Scholar]; (c) Tseng YJ, Ahmed AF, Hsu CH, Su JH, Dai CF, Sheu JH. J Chinese Chem Soc. 2007;54:1041. [Google Scholar]; (d) Cheng SY, Chuang CT, Wen ZH, Wang SK, Chiou SF, Hsu CH, Dai CF, Duh CY. Bioorg Med Chem. 2010;18:3379. doi: 10.1016/j.bmc.2010.04.012. [DOI] [PubMed] [Google Scholar]; (e) Thao NP, Nam NH, Cuong NX, Quang TH, Tung PT, Dat LD, Chae D, Kim S, Koh YS, Kiem PV, Minh CV, Kim YH. Bioorg Med Chem Lett. 2013;23:228. doi: 10.1016/j.bmcl.2012.10.129. [DOI] [PubMed] [Google Scholar]; (f) Lillsunde KE, Festa C, Adel H, De Marino S, Lombardi V, Tilvi S, Nawrot DA, Zampella A, D’Souza L, D’Auria MV, Tammela P. Mar Drugs. 2014;12:4045. doi: 10.3390/md12074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Li Y, Pattenden G. Tetrahedron. 2011;67:10045. [Google Scholar]; (b) Li Y, Pattenden G. Nat Prod Rep. 2011;28:1269. doi: 10.1039/c1np00023c. [DOI] [PubMed] [Google Scholar]; (c) Li Y, Pattenden G. Nat Prod Rep. 2011;28:429. doi: 10.1039/c0np00029a. [DOI] [PubMed] [Google Scholar]
- 5.(a) O’Connell C. PhD dissertation. Queen’s Universtiy of Belfast; Belfast, Northern Ireland, U.K: 2006. [Google Scholar]; (b) O’Connell CE, Frontier AJ. Abstracts of Papers, 32nd Northeast Regional Meeting of the American Chemical Society; Rochester, NY. Oct 31–Nov 3, 2004; American Chemical Society; 2004. GEN-088. [Google Scholar]; (c) Pratt BA. PhD dissertation. Scripps Research Institute; La Jolla, California: 2008. [Google Scholar]; (d) Liu G. PhD dissertation. Texas A&M University; College Station, Texas: 2011. [Google Scholar]; (e) Liu G, Romo D. Abstracts of Papers, 237th American Chemical Society National Meeting; Salt Lake City, UT. Mar 22–26, 2009; American Chemical Society; 2009. ORGN-083. [Google Scholar]; (f) Tang F. PhD dissertation. Washington University in St. Louis; St. Louis, Missouri: 2009. [Google Scholar]; (g) Tang F, Moeller KD. Tetrahedron. 2009;65:10863. [Google Scholar]; (h) Tang F, Moeller KD. J Am Chem Soc. 2007;129:12414. doi: 10.1021/ja076172e. [DOI] [PubMed] [Google Scholar]; (i) Horn EJ, Silverston JS, Vanderwal CD. J Org Chem. 2016;81:1819. doi: 10.1021/acs.joc.5b02550. [DOI] [PubMed] [Google Scholar]; (j) Horn EJ. PhD dissertation. University of California at Irvine; Irvine, CA: 2014. [Google Scholar]
- 6.Craig RA, II, Roizen JL, Smith RC, Jones AC, Virgil SC, Stoltz BM. Chem Sci. 2017;8:507. doi: 10.1039/c6sc03347d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Krüger S, Gaich T. Beilstein J Org Chem. 2014;10:183. doi: 10.3762/bjoc.10.14. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Davies HML, Stafford DG, Doan BD, Houser JH. J Am Chem Soc. 1998;120:3326. [Google Scholar]; (c) Davies HML. Tetrahedron. 1993;49:5203. [Google Scholar]; (d) Vogel E. Angew Chem, Int Ed. 1963;2:1. [Google Scholar]
- 8.(a) Craig RA, II, Roizen JL, Smith RC, Jones AC, Stoltz BM. Org Lett. 2012;14:5716. doi: 10.1021/ol3027297. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Seto M, Roizen JL, Stoltz BM. Angew Chem Int Ed. 2008;47:6873. doi: 10.1002/anie.200801424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Behenna DC, Mohr JT, Sherden NH, Marinescu SC, Harned AM, Tani K, Seto M, Ma S, Novák Z, Krout MR, McFadden RM, Roizen JL, Enquist JA, Jr, White DE, Levine SR, Petrova KV, Iwashita A, Virgil SC, Stoltz BM. Chem – Eur J. 2011;17:14199. doi: 10.1002/chem.201003383. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mohr JT, Stoltz BM. Chem–Asian J. 2007;2:1476. doi: 10.1002/asia.200700183. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew Chem Int Ed. 2005;44:6924. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]; (d) Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]
- 10.Trost BM, Xu J, Reichle M. J Am Chem Soc. 2007;129:282. doi: 10.1021/ja067342a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Tani K, Behenna DC, McFadden RM, Stoltz BM. Org Lett. 2007;9:2529. doi: 10.1021/ol070884s. [DOI] [PubMed] [Google Scholar]; (b) Krout MR, Mohr JT, Stoltz BM. Org Synth. 2009;86:181. doi: 10.15227/orgsyn.086.0181. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Helmchen G, Pfaltz A. Acc Chem Res. 2000;33:336. doi: 10.1021/ar9900865. [DOI] [PubMed] [Google Scholar]; (d) Williams JMJ. Synlett. 1996:705. and references therein. [Google Scholar]
- 12.Liu HJ, Zhu BY. Can J Chem. 1991;69:2008. [Google Scholar]
- 13.Herscovici J, Egron M-J, Antonakis K. J Chem Soc, Perkin Trans 1. 1982:1967. [Google Scholar]
- 14.For similar transformations, see: Shibuya M, Tomizawa M, Iwabuchi Y. J Org Chem. 2008;73:4750. doi: 10.1021/jo800634r.
- 15.Pyrazole formation has been previously reported in thermolysis of vinyl diazo compounds: Doyle MP, Yan M, Gao HM, Blossey EC. Org Lett. 2003;5:561. doi: 10.1021/ol027475a.Doyle MP, Yan M. J Org Chem. 2002;67:602. doi: 10.1021/jo016135k.Hutchinson IS, Matlin SA, Mete A. Tetrahedron Lett. 2001;42:1773.Pellicciari R, Natalini B, Sadeghpour BM, Marinozzi M, Snyder JP, Williamson BL, Kuethe JT, Padwa A. J Am Chem Soc. 1993;118:1.Davies HML, Huby NJS, Cantrell WR, Jr, Olive JL. J Am Chem Soc. 1993;115:9468.Padwa A, Kulkarni YS, Zhang Z. J Org Chem. 1990;55:4144.Bailey RJ, Card PJ, Shechter H. J Am Chem Soc. 1983;105:6096.Hurd CD, Lui SC. J Am Chem Soc. 1935;57:2656.
- 16.De Leeuw JW, De Waard ER, Beetz T, Huisman HO. Recl Trav Chim Pays-Bas. 1973;92:1047. [Google Scholar]
- 17.For related examples involving intramolecular translactonization of γ-lactones, see: Raffaelli B, Pohjoispää M, Hase T, Cardin CJ, Gan Y, Wähälä K. Org Biomol Chem. 2008;6:2619. doi: 10.1039/b801593g.Mulzer J, Giester G, Gilbert M. Helv Chim Acta. 2005;88:1560.Duret P, Figadère B, Hocquemiller R, Cavé A. Tetrahedron Lett. 1997;38:8849.Clardy J, Springer JP, Buechi G, Matsuo K, Wightman R. J Am Chem Soc. 1975;97:663. doi: 10.1021/ja00836a045.
- 18.The immediate product of acetylation does not demonstrate the necessary gHMBC correlations; however, ketone reduction generates two diastereomers, one of which exhibits diagnostic gHMBC correlations.
- 19.Bredt J. Justus Liebigs Annalen der Chemie. 1924;437:1. [Google Scholar]
- 20.(a) Tei T, Sugimura T, Katagiri T, Tai A, Okuyama T. Tetrahedron: Asymmetry. 2001;12:2727. [Google Scholar]; (b) Davies HML, Calvo RL, Townsend RJ, Ren P, Churchill RM. J Org Chem. 2000;65:4261. doi: 10.1021/jo991959b. [DOI] [PubMed] [Google Scholar]; (c) Davies HML. Tetrahedron. 1993;49:5203. [Google Scholar]; (d) Davies HML, Clark TJ, Smith HD. J Org Chem. 1991;56:3817. [Google Scholar]; (e) Cantrell WR, Jr, Davies HML. J Org Chem. 1991;56:723. [Google Scholar]; (f) Davies HML, Clark DM, Smith TK. Tetrahedron Lett. 1985;26:5659. [Google Scholar]
- 21.Davies HML, Smith HD, Korkor O. Tetrahedron Lett. 1987;28:1853. [Google Scholar]
- 22.(a) Davies HML, McAfee MJ, Oldenburg CEM. J Org Chem. 1989;54:930. [Google Scholar]; (b) Davies HML, Oldenburg CEM, McAfee MJ, Nordahl JG, Henretta JP, Romines KR. Tetrahedron Lett. 1988;29:975. [Google Scholar]
- 23.For intermolecular cascades, see: Davies HML, Stafford DG, Doan BD, Houser JH. J Am Chem Soc. 1998;120:3326.Davies HML, Peng ZQ, Houser JH. Tetrahedron Lett. 1994;35:8939.
- 24.Davies HML, Doan BD. J Org Chem. 1999;64:8501. [Google Scholar]
- 25.For asymmetric intramolecular cascades, see: Davies HML, Ahmed G, Churchill MR. J Am Chem Soc. 1996;118:10774.. For related uses of chiral auxiliaries in cyclopropanation/Cope reactions, see: Davies HML, Matasi JJ, Hodges LM, Huby NJS, Thornley C, Kong N, Houser JH. J Org Chem. 1997;62:1095.
- 26.Danheiser RL, Miller RF, Brisbois RG, Park SZ. J Org Chem. 1990;55:1959. [Google Scholar]
- 27.Nozaki H, Takaya H, Moriuti S, Noyori R. Tetrahedron. 1968;24:3655. [Google Scholar]
- 28.(a) Frigerio M, Santagostino M, Sputore S. J Org Chem. 1999;64:4537. [Google Scholar]; (b) Boeckman RK, Jr, Shao P, Mullins JJ. Org Syn. 2000;77:141. [Google Scholar]
- 29.de Boer Th J, Backer HJ. Org Syn. 1963 [Google Scholar]; Coll. 4:250–253. [Google Scholar]
- 30.Parlow JJ, Case BL, South MS. Tetrahedron. 1999;55:6785. [Google Scholar]
- 31.Baum JS, Shook DA, Davies HML, Smith HD. Synth Comm. 1987;17:1709. [Google Scholar]
- 32.This procedure has been influenced by: Bouzide A, Sauvé G. Tetrahedron Lett. 1997;38:5945.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.