

Abstract
Cholangiocytes, like most cells, express primary cilia extending from their membranes. These organelles function as antennae which detect stimuli from bile and transmit the information into cells regulating several signaling pathways involved in secretion, proliferation and apoptosis. The ability of primary cilia to detect different signals is provided by ciliary associated proteins which are expressed in its membrane. Defects in the structure and/or function of these organelles lead to cholangiociliopathies that result in cholangiocyte hyperproliferation, altered fluid secretion and absorption. Since primary cilia dysfunction has been observed in several epithelial tumors, including cholangiocarcinoma (CCA), primary cilia have been proposed as tumor suppressor organelles. In addition, the loss of cilia is associated with dysregulation of several molecular pathways resulting in CCA development and progression. Thus, restoration of the primary cilia may be a potential therapeutic approach for several ciliopathies and CCA.
CHOLANGIOCYTES
Cholangiocytes are the epithelial cells that line the biliary tree. Their major physiological function lies in modifying the hepatic canalicular bile secreted by hepatocytes via absorptive and secretory processes, while being transported along the biliary tree through transmembrane molecules expressed on the apical or basolateral membranes [1]. The most important and well-studied function of cholangiocytes is the release of bicarbonate into bile as induced by secretin, in a mechanism dependent on the increment of the levels of intracellular cyclic AMP (cAMP) and activation of protein kinase A (PKA) [2].
CHOLANGIOCYTE PRIMARY CILIA
Cholangiocytes, like many other cells, present primary cilia. In cholangiocytes, primary cilia extend from their apical plasma membranes into the ductal lumen, being ideally positioned to detect changes in bile flow, composition and osmolality [3]. The primary cilium or monocilium as each cell has only one, is composed by a non-motile microtubule-based core structure covered by the ciliary membrane called axoneme, which elongates from the basal body and extends from the cell surface (Fig. 1).
Figure 1. Primary cilium.
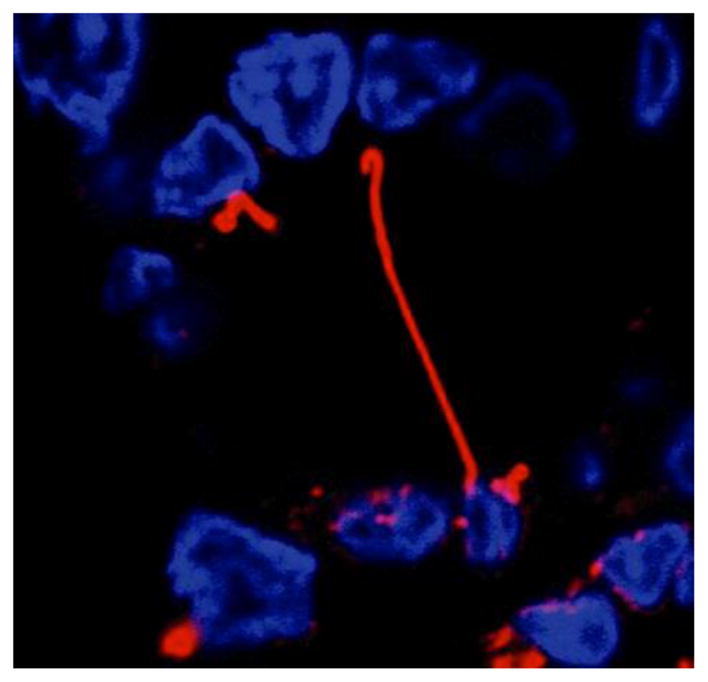
Confocal immunofluorescence showing a cross-section of a bile duct, where the nuclei of cholangiocytes lining the duct are stained in blue, and primary cilium extending into the ductal lumen in red.
Unlike most motile cilia or flagella that consist of axonemes assembled by nine peripheral microtubule doublets and two central single microtubules (9+2), primary cilia consist of axonemes lacking the two central microtubules, i.e. 9+0 structure [4]. Until recently, primary cilia were considered vestigial organelles with no physiologically relevant functions [5]. Nowadays, it is widely accepted that primary cilia are “the cellular antenna” and essential for development and cellular differentiation [6]. The ciliary membrane harbors many ciliary specific and ciliary associated receptors, ion channels and sensory signaling molecules (Fig. 2).
Figure 2. Cholangiocyte primary cilium regulates several signaling pathways.

The ability of primary cilia to detect different signals is a consequence of the expression of specific ciliary proteins and ciliary associated receptors. The activation of these receptors results in activation or inhibition of different signaling pathways. TGR5: G-protein-coupled bile acid receptor 1; PC-1: polycystin-1; PC-2: polycystin-2; TRPV4: Transient Receptor Potential Vanilloid 4; PTCH: patched receptor; SMO: Smoothened receptor; PDGFRα: Platelet-derived growth factor receptor α; P2Y12: Purinergic receptor 12; Inv: inversin; AMPK: AMP-activated protein kinase; LKB1: liver kinase B1.
There is no evidence that protein synthesis occurs in the cilium, but proteins and lipids are transported from the cytoplasm to the axoneme through the transition zone [7] followed by transport across the axoneme by the specialized system of transportation called intraflagellar transport (IFT). This specialized system can operate in the anterograde direction (from the basal body to the ciliary tip) and in the retrograde direction (towards the basal body) using different kind of motor proteins; i.e. Kinesin II is the main driver in the anterograde direction while Dynein II takes the lead in the retrograde direction [8]. This complex system allows primary cilia to function as an antenna, sensing extracellular signals acting as mechano-, chemo-, and osmo-sensory organelles (Fig. 3) [3, 9–12].
Figure 3. Primary cilia function as mechano-, chemo- and osmosensors.

PC-1 and PC-2 allows primary cilia to function as a mechanosensor. The bending of cholangiocytes primary cilia by luminal flow induces an increase in the intracellular ionic calcium through activation of PC-1 and PC-2, leading to inhibition of AC6, which is inhibited by calcium, resulting in a decrease in levels of cAMP and inhibition of PKA. TRPV4 participates in the osmosensory activity. This receptor is activated by extracellular hypotonicity and inhibited by extracellular hypertonicity, leading to an increase in intracellular calcium that results in PKA inhibition. The cilia chemosensory response is a consequence of purinergic receptors (P2Y12) and bile acid receptor (TGR5), which are activated causing changes in intracellular cAMP levels, possibly via AC6, that induces PKA activation or inhibition. PC-1: polycystin-1; PC-2: polycystin-2; AC6: adenylyl cyclase 6.
The mechanosensory capacity of primary cilia is provided by the ciliary associated proteins polycystin-1 (PC-1), a cell surface receptor, and polycystin-2 (PC-2), a calcium channel. Thus, the bending of cholangiocyte primary cilia by luminal flow induces an increase in the intracellular ionic calcium, which depends on the activation of PC-1 and PC-2 receptors [3]. The osmosensory activity is provided by the Transient Receptor Potential Vanilloid 4 (TRPV4) that is activated by extracellular hypotonicity and inhibited by hypertonicity [9, 13] leading to an increase in intracellular calcium, which depends on extracellular calcium sources. Finally, the chemosensory function is a consequence of several proteins expressed in the primary cilia that recognize appropriate ligands from the environment and are able to transduce those signals into the cell. Among the specific receptors with chemosensory functions, primary cilia present important purinergic receptors such as P2Y12, which is activated by nucleotides causing changes in intracellular cAMP levels. Other proteins associated with the chemosensory functions include the AKAP (A-kinase anchoring protein) signaling complex, AKAP150, PKA, one isoform of EPAC and the G-protein-coupled bile acid receptor 1 (TGR5) among others [12]. Interestingly, cholangiocyte primary cilia are enriched in TGR5 coupled to Gαi in contrast to Gαs when it is localized on the apical domain of the plasma membrane [14]. Since all of these proteins are involved in the cAMP signaling cascade, it is suggested that the chemosensory function modulates cAMP signaling [10].
Primary cilia may react to exosomes, which are vesicles implicated in several physiological and pathological processes. Recent studies suggest that the principal function of exosomes is associated with intercellular communication [15]. Cholangiocyte primary cilia can deliver and receive exosomes which induces changes in intracellular signaling pathways, miRNA expression levels and cholangiocytes proliferation [11]. Specifically, cholangiocytes are exosome-releasing cells and cholangiocyte primary cilia are able to recognize the presence of biliary exosomes and transduce this signal into modifications in ERK1/2 signaling that result in the regulation of proliferation [11]. Interestingly, the exposition of cholangiocytes expressing primary cilia to biliary exosomes results in a decrease in ERK signaling and inhibition of proliferation while this effect is abolished by deciliation [11], suggesting a major role of primary cilia in the regulation of proliferation via ciliary-exosomes dependent mechanisms.
In general, the physiological function of primary cilia in cholangiocytes seems to be the fine-tuning regulation of bile secretion by feedback loops where variations in bile flow and/or composition are recognized by ciliary mechanisms, which in turn send regulatory signals inside the cells. For example, a bile flow increase by the hormone secretin would be detected by the bending of primary cilia, which in turn would generate a signal to cease bicarbonate secretion [16]. On the other hand, when changes in osmolarity or nucleotides are detected, a signal to induce bicarbonate secretion is generated by the primary cilia [9, 17]. Thus, proper function of these organelles is essential for the normal physiology of cholangiocytes and, in general, to normal function of the cells and tissues that compose different organs.
PRIMARY CILIA AND CHOLANGIOCILIOPATHIES
Since cholangiocyte primary cilia are important for normal physiology, defects in its structure and/or function lead to cholangiociliopathies, which are a diverse group of biliary disorders where cholangiocytes are the target cells. Generally, these alterations result in decreased intracellular ionic calcium and increased cAMP, leading to cholangiocyte hyperproliferation, altered fluid secretion and bile absorption. Cholangiociliopathies can be genetic or acquired and have in common the presence of cholestasis and chronic liver injury. They are caused by mutations in genes that encode proteins localized in primary cilia, proteins involved with ciliary functions or participate in ciliogenesis. The most studied cholangiociliopathy is polycystic liver disease (PCLD), which is caused by the presence of mutations that can occur in a dominant or recessive mode, and can also occur alone or in association with polycystic kidney disease (PKD). There is a form of isolated PCLD called autosomal dominant polycystic liver disease (ADPLD), which is characterized by the presence of cysts exclusively in the liver. Primary cilia in the liver are expressed by cholangiocytes but not hepatocytes. Liver cysts are derived from cholangiocytes and are associated with hyperproliferation, cell cycle dysregulation and alteration in the secreted bile flow [18]. The prevalence of this disease is estimated to be 1:1100000 [18] and is caused by mutations in the PRKCSH or in SEC63 genes. PRKCSH encodes the protein called hepatocystin (also known as protein kinase C substrate 80k-H) [19] which is suggested to act as the regulatory subunit of glucosidase II and is involved in the folding and quality control of glycoproteins [19]. Hepatocystin is required for the normal construction of the bile ducts and experimental evidence has shown that the absence of hepatocystin leads to cyst formation. SEC63 gene encodes the protein Sec63p that is involved in oligosaccharide processing of glycoproteins, and participates in the transport of proteins into/out of the endoplasmic reticulum [20, 21]. In contrast to hepatocystin, Sec63p is expressed in patients with PLCD suggesting that mutations result in abnormal function of the protein [22].
Diseases that occur when PCLD is associated with PKD includes autosomal dominant PKD (ADPKD), autosomal recessive PKD (ARPKD), Meckel-Gruber syndrome (MKS), Bardet-Biedl syndrome (BBS), nephronophthisis (NPHP) and Joubert syndrome (JBTS). ADPKD is particularly caused by mutations in polycystic kidney disease 1 (PKD1) and 2 (PKD2). PKD1 encodes the transmembrane protein PC-1 which is mutated in 85% of ADPKD cases while PKD2 encodes the transmembrane protein PC-2 which is found to be mutated in 15% of cases. Both proteins form a functional complex in primary cilia that participates not only in the mechanosensation but also in the control of several cell functions such as cell cycling and signaling pathways such as Wnt [23]. ARPKD is less frequent than the dominant mode and is caused by mutations in the PKHD1 gene that encodes the integral transmembrane protein fibrocystin expressed at subcellular levels in primary cilia [18]. All of these proteins are localized in primary cilia, thus their mutations lead to the absence or malfunction of primary cilia resulting in defective ciliogenesis, ciliary structure and function along with cyst formation. Therefore, the loss of ciliary structure is associated with an impairment in the transducing function leading to decreased intracellular calcium and increased cAMP which in turn results in cholangiocytes proliferation, altered fluid secretion and absorption [12].
Another example of ciliopathy is MSK, an autosomal recessive disease where biliary dysgenesis and hepatic fibrosis are common features in patients. Three genes are involved in the development of this disease, MSK1, MSK2 and MSK3. The MSK protein, encoded by MSK1, is predicted to be a cytoplasmic protein while meckelin, encoded by MSK3, is predicted to be a receptor that is localized in primary cilia [18].
NPHP is a rare autosomal recessive disease in which at least 9 mutated genes are candidates in causing this disease. NPHP genes encode nephrocystins proteins, which are expressed in primary cilia and basal bodies [18]. JBTS is another cholangiociliopathy and is considered a genetically heterogeneous disorder where several genes such as JBTS1, JTBS2 and JTBS3 among others are implicated in the development of the disease. These genes encode transport proteins localized in primary cilia, cell junctions and centrosome. Finally, BBS cases are caused by multiorgan genetic disorders where BBS genes are involved and all of the variants encode proteins localized to primary cilia and basal bodies.
Although primary sclerosing cholangitis (PSC), which is a chronic and progressive cholestatic disease whose cause and pathogenesis are still unknown and is not yet considered as a cholangiociliopathy, some authors suggest its inclusion as a novel member because cholangiocytes in this disease possess longer primary cilia compared to normal cholangiocytes, and the abnormal elongation of cilia have a consequential impairment of its functions as previously described [24]. This disease is characterized by inflammation and fibrosis of intrahepatic and/or extrahepatic bile ducts, and it is suggested that the autoimmune mediated process is involved. Most cases of PSC are associated with inflammatory bowel disease, colorectal dysplasia and colorectal cancer [25] and have an increased risk of developing cholangiocarcinoma (CCA) [26]. Although the mechanism that links PCS and CCA is unclear, it is suggested that bile acids, which are molecules synthesized from cholesterol and are essential for cholesterol excretion and dietary lipid absorption, could be involved. Consistent with this suggestion, secondary bile acids such as deoxycholic acid have been found to have carcinogenic properties in rat models [27], and bile acid exposures in humans are associated with increasing incidence of gastrointestinal cancers and progression. Thus, bile acids can induce cancer cell proliferation via different receptors such as epidermal growth factor receptors (EGFR), farnesoid X receptors (FXR), pregnane X receptor (PXR) and TGR5 among others, some of which are primary cilia associated receptors.
Bile acids also act as signaling molecules activating different nuclear receptors such as FXR and membrane receptors such as TGR5 [28–30]. Since both bile acid receptors FXR and PXR are found in low levels or are absent in cholangiocytes, it is suggested that TGR5, which transmits bile acid signaling via cAMP pathway, is presumably the major receptor for bile acid signaling in these cells. TGR5 is localized in different non-parenchymal cells of the liver, cholangiocytes, sinusoidal endothelial cell and gallbladder epithelial cells. The activation of TGR5 has opposite effects depending on whether it is coupled to the Gαs or the Gαi protein. When coupled to Gαs, the activation leads to an increase of cAMP through activation of adenylate cyclase, resulting in PKA activation. In contrast, when coupled to Gαi, the activation leads to a decrease of cAMP through inhibition of adenylate cyclase, resulting in PKA inhibition. TGR5 is activated by unconjugated and conjugated bile acids in the order of taurolithocholic acid (TLCA) > lithocholic acid (LCA) > deoxycholic acid (DCA) > chenodeoxycholic acid (CDCA) > cholic acid (CA) and participates in the regulation of the bile acid pool, bile composition, modulation of hepatic inflammation and improvement of insulin sensitivity and regulation of glucose metabolism among other functions [29, 31]. Using in vitro studies, Masyuk et al., showed that in ciliated cholangiocytes TGR5 is localized in the apical membrane coupled to Gαs and in primary cilia coupled to Gαi, while TGR5 coupled to Gαi is absent in non-ciliated cholangiocytes. Therefore, the localization of TGR5 determines the cholangiocyte functional response to bile acid signaling. In ciliated cholangiocytes, activation of TGR5 leads to a decrease in cAMP levels and an increase in ERK signaling that results in inhibition of proliferation, while activation in non-ciliated cholangiocytes leads to an increase in cAMP levels and a decrease in ERK signaling resulting in cell proliferation [14].
Since patients with PSC have reduced TGR5 mRNA and proteins levels in the liver and bile ducts, it is suggested that TGR5 could be involved in the pathogenesis of PSC. In contrast, high expression of TGR5 has been found in human cholangiocarcinoma (CCA) suggesting that overexpression would be involved in CCA development [32].
CHOLANGIOCARCINOMA
CCA is a rare tumor originating from cholangiocytes. Anatomically, and according to its localization, CCA can be classified as intrahepatic (originating from the intrahepatic biliary tree) or extrahepatic (arising between the ampulla of Vater and the hepatic hilum) [33–37]. The prevalence of CCA shows a wide geographic variability [38]. In the United States, the incidence of CCA has been reported to be 0.95/100,000 for intrahepatic forms and 0.82/100,000 for extrahepatic forms of the disease. Its prevalence in different racial and ethnic groups is heterogeneously distributed, with the highest age-adjusted prevalence in Hispanics (1.22/100,000) and the lowest in African Americans (0.17-0.5/100,000) [34]. Due to its late clinical presentation in addition to the lack of effective non-surgical therapeutic modalities, patients diagnosed with CCA have a poor overall survival rate [39, 40].
The causes for CCA development in most cases are unknown. However, there are several conditions associated with biliary tract inflammation and cholestasis that are recognized as risk factors, such as liver infection by parasites, mainly of the Opisthorchis viverrini and Clonorchis sinensis species, and congenital abnormalities of the bile ducts, such as the Caroli’s disease. Infections with hepatitis B and C have also recently been considered as risk factors [34, 36, 41].
A wide range of oncogenic mutations have been identified in CCA and several studies have demonstrated that mutations or abnormal expression of KRAS and TP53 are associated with CCA development while other tumor suppressors genes may be inactivated such as P16INK4a, P14ARF, DPC4/SMAD4 and APC [42–44]. Patients with CCA have a high serum concentration of Interleukin-6 (IL-6) that seems to be a critical signaling molecule in the pathogenesis of CCA. As a matter of fact, CCA cells produce and release IL-6 leading to an increased expression of antiapoptotic protein MCL-1 making these cells resistant to therapies [45]. On the other hand, IL-6 induces an increase in telomerase activity resulting in evasion of senescence, activating the p44/42 and p38 MAPKs that decreases the levels of the cell cycle negative regulator cyclin-dependent kinase inhibitor p21. Activation of p44/42 and p38 MAPKs induces an increase in COX-2 expression which has an important role in the carcinogenesis of CCA by inhibition of apoptosis and stimulation of cell growth [46].
PRIMARY CILIA AND CHOLANGIOCARCINOMA
The fact that primary cilia dysfunction has been observed in several epithelial and non-epithelial derived tumors such as pancreatic, renal, breast, and prostate cancer [47–51] and the fact that malformation or loss of the primary cilia has been described in several cholangiopathies [52–55] suggests that the loss of primary cilia has an essential role in these diseases. Moreover, we recently found reduced expression of primary cilia in CCA that strengthens the association between loss of cilia and cancer [56, 57]. Therefore, ciliary dysfunction may be associated with cholangiocarcinoma development and/or progression. The mechanisms leading to deciliation in these tumor cells as well as the consequences of such a loss remain understudied. We found that the dysregulation of microRNAs (miRNAs) in CCA cells induces the overexpression of Histone deacetylase 6 (HDAC6) resulting in ciliary resorption. HDAC6 is a unique histone deacetylase that is localized exclusively in the cytoplasm and functions as a tubulin deacetylase [58], decreasing the level of acetylated α-tubulin that is one of the main structural components of the primary cilia and is required for ciliary assembly.
Since primary cilia regulate several signaling pathways, including the platelet-derived growth factor receptor α (PDGFRα), Hedgehog, Wnt, Notch, mTOR and bile acids signaling pathways (Fig. 4–8), primary cilia dysfunction might result in tumorigenesis through different mechanisms, which includes loss of cell cycle checkpoint control [59, 60]. The loss of primary cilia results in the dysregulation of the aforementioned pathways, alteration of the proteasomal activity, dysregulation of autophagy and suppression of p53 expression, therefore loss of the cholangiocytes primary cilia may be involved in CCA development and progression [61–63].
Figure 4. PDGRF signaling pathway.

Quiescent cells express PDGFRα in primary cilia. Its activation induces ERK1/2 phosphorylation by induction of MEK1/2 and activation of AKT through PI3K resulting in cell cycle entry.
Figure 8.

Cholangiocyte primary cilia regulate PKA activation trough TGR5. In the apical membrane TGR5 is coupled to Gαs, while in primary cilia TGR5 is coupled to Gαi. The activation of apical TGR5 leads to adenylyl cyclase activation and increase in the level of cAMP resulting in the activation of PKA and increase in ERK1/2 signaling, while the activation of ciliary TGR5 has an opposite effect.
PDGFRα signaling plays a major role in cell growth, survival and migration. In quiescent NIH3T3 fibroblasts, PDGFRα is localized in primary cilia and its activation induces ERK1/2 signaling by activation of MEK1/2 and activation of AKT resulting in cell cycle entry (Fig. 4) [64]. These events are coordinated by the primary cilia where not only the activation but also up-regulation of PDGFRα is required for controlling the cell cycling. Moreover, the expression of this receptor in primary cilia is required for sensing chemotactic gradients that result in cell migration. Thus, the loss of cholangiocytes primary cilia would affect the PDGFRα ciliary expression and function. However, Boonjaraspinyo et al. showed that this receptor is overexpressed during the tumorigenesis of CCA induced by O. viverrini infection in a hamster model [65].
Primary cilia are also involved in Sonic hedgehog (Hh) and Wnt signaling, which are the pathways most intensively studied over the last decades [66]. In mammalians, there are three Hh proteins, Sonic (SHH), Indian (IHH) and Desert (DHH) that participate in the signaling pathway. However, SHH and IHH are the most important and have overlapping functions in many tissues. In comparison, DHH, which is suggested to depend on primary cilia, appears to be restricted to the gonads [67]. These ligands bind and activate the receptor patched (PTCH), leading to the attenuation of the inhibition of Smoothened (SMO). Activation of SMO converts glioblastoma proteins transcription factors (Gli) from transcriptional repressors to activators, targeting the expression of genes. Primary cilium is essential for proper Shh signaling due to the involvement of the ciliary structure and IFTs proteins in this process [68]. In the absence of ligands, PTCH is located in the basal body and prevents entry of SMO to the cilia, while Suppressor of Fused (SUFU) binds Gli, facilitating its proteasomal degradation. Thus, SUFU negatively regulates the pathway. (26). In this context, PKA, regulated by ciliary Gpr161 through the activation of adenylyl cyclase, together with other kinases such as glycogen synthase kinase-3 (GSK-3β) and Casein kinase I (CKI), localized in the basal body, prevent the accumulation of the complex Gli2/3-SUFU in cilia by phosphorylation. This promotes partial proteolysis of Gli3 by β-TRCP originating the Gli3 repressor (Gli3R) that moves to the nucleus repressing the expression of Hh targets genes. When the ligands bind to PTCH, the receptor leaves the cilia, allowing SMO translocation to the ciliary membrane. SMO is phosphorylated mainly by CKI and GRK2, a G protein-coupled receptor kinase, and at the same time, Gpr161 is removed from the cilia. Unstimulated PKA is unable to phosphorylate Gli2/3-SUFU, thus this complex is accumulated at the ciliary tip where Gli2/3 is released from SUFU and activated, generating the active form of GLI2 (Gli2A) that results in the transcriptional activation of Shh targets (Fig 5). Since primary cilia structure and the SMO receptor translocation to the cilia are required for correct Shh signaling, loss of primary cilia leads to dysregulation of this pathway.
Figure 5. Hedgehog signaling pathways.

Canonical Hedgehog signaling in mammals is ligand dependent. In the absence of Hh ligands, PTCH accumulates in the primary cilium and inhibits the function of Smo. PTCH facilitates the activation of several kinases (CK1, PKA, and GSK3) at the base of the primary cilium, which differentially phosphorylates the Gli protein. This can lead to complete degradation of the Gli protein by the proteasome, as well as partial cleavage of Gli2 and Gli3. Partially cleaved Gli3 (Gli3R) is translocated to the nucleus and functions as a transcriptional repressor for Hh target genes. In the presence of Hh ligands, Shh, Ihh, or Dhh binds to PTCH, which induces its lysosomal degradation. This relieves its inhibitory effect on Smo, which accumulates in the primary cilium and prevents degradation of Gli proteins. Activated Gli protein is translocated to the nucleus and functions as a transcriptional activator for Hh target genes.
Cholangiocytes have a very low proliferative index, however, following biliary injury they start to proliferate and acquire a reactive phenotype responding to the SHH and IHH ligands. Reactive cholangiocytes release chemokines and cytokines generating a pro-inflammatory environment. The sustained activation of the hedgehog signaling can result in the blockage of epithelial differentiation, the induction of epithelial mesenchymal transition (EMT), and can even be associated with the pathogenesis of CCA [69]. The association between CCA and Hh signaling is controversial and unclear. On one hand, it was demonstrated that Gli3 regulates the expression of the death receptor 4 (DR4) in some CCA cells, which is required for the apoptosis induced by the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) [70]. On the other hand, a recent study showed that the canonical Hh pathway does not play a major role in CCA cells [57].
Furthermore a non-canonical pathway for Shh signaling, which was identified in drosophila, shows cells that lack primary cilia participating in cytoskeleton remodeling and cell migration through a mechanism independent of SMO localized on primary cilia but dependent of the small Rho GTPases Rac1 and RhoA activation [71]. This novel non-canonical pathway is activated in CCA and was found to contribute to CCA progression [57]. Thus, dysregulation of both Shh signaling pathways is associated with CCA development and progression.
Wnt signaling is another important pathway regulated by primary cilia (Fig 6). Wnt signaling is a well-known studied pathway involved in many cancers. In the canonical pathway and in absence of ligand, two scaffolding proteins (Apc and Axin), which compose the destruction complex, bind β-catenin and induce its phosphorylation by GSK-3β and CKI. Once phosphorylated, β-catenin becomes a substrate of the ubiquitin E3 ligase, which results in its degradation via proteasome. When Wnt ligands bind to the Frizzled receptor, this induces inactivation of the destruction complex resulting in stabilization of β-catenin in the cytoplasm and its subsequent translocation to nucleus acting as a transcription factor in coordination with TCF/LEF [72]. This process appears to be regulated by Dishevelled (Dvl) due to the interaction between Dvl and Axin required to stop GSK-3β from phosphorylating β-catenin.
Figure 6. Wnt signalling pathways.

In the canonical pathway, when the Frizzled receptor is not activated, the destruction complex (Apc and Axin) binds β-catenin inducing its phosphorylation as mediated by GSK-3β and CKI, which results in β-catenin degradation. When the Frizzled receptor is activated, the destruction complex is inactivated resulting in the stabilization of β-catenin that translocates to the nucleus and induces target gene expression in coordination with TCF/LEF. In the non-canonical pathway, the activation of Frizzled receptors lead to activation of the small GTPases Rho and Rac. Inversin, localized in the basal body, functions as a switch between both pathways. DVL: Dishevelled; APC: Adenomatous polyposis coli, Inv: inversin.
In the non-canonical pathway, Wnt signaling stimulates the planar cell polarity through activation of the small GTPases Rho and Rac that induces cytoskeletal rearrangements [72]. Inversin (Inv), which is localized in the basal body, interacts with cytoplasmic Dvl but not with membranous Dvl promoting its degradation. Thus, Inv functions as a switch between the canonical and non-canonical pathways. In a recent study, Boulter et al. showed that Wnt ligands are overexpressed in CCA and the number of activated β-catenin targets increases suggesting that canonical Wnt signaling is upregulated in human CCA [73]. In accordance with this, inhibition of canonical Wnt signaling by pharmacological blockage reduced CCA formation in a rat model by stimulation of apoptosis and reduced cell cycle entry [74]. Thus, Wnt signaling is important for proliferation and survival of CCA cells.
Another important pathway related to primary cilia is the Notch pathway. Notch is a transmembrane receptor bound and activated by ligands of the delta jagged family proteins. Its activation results in proteolitic cleavages of the Notch receptor generating a Notch intracellular domain fragment that translocates to the nucleus to drive transcription targets genes associated with suppression of cell differentiation and proliferation [75, 76]. Ciliary regulation of Notch is unclear but there is strong evidence that Notch and cilia participate in an intricate mechanism of feedback where primary cilia regulate the activation of Notch, which in turn regulates the length of the cilia. In fact, in basal corneal epithelial cells the ablation of the cilia diminishes Notch pathway activity and induces hyperproliferation [77]. Since loss of primary cilia induces dysregulation of the Notch pathway whose activation is necessary for the proliferation of cholangiocarcinoma, Notch signaling dysregulation upon cholangiocyte ciliary loss may be another driver of carcinogenesis in CCA [78, 79].
Primary cilia also participate in regulation of the mammalian target of rapamycin (mTOR) activity via liver kinase B1 (LKB1) activation in MDCK cells (Fig. 7) [80]. mTOR is a serine/threonine kinase that forms two different types of multiprotein complexes, mTORC1 and mTORC2 which participate in a number of cellular functions including cell size regulation, cell metabolism, cell growth, proliferation and survival. mTORC1 controls multiple anabolic pathways such as protein synthesis, ribosome production, lipogenesis, nucleotides synthesis, all pathways that are related to cell and tissue growth, and also suppresses the production of lysosomes that participates in autophagy [81, 82]. mTORC1 activation results in phosphorylation of the translation repressor protein 4E-BP1 (4EBP1) and ribosomal protein S6 kinase (S6K1) regulating cell proliferation while mTORC2 regulates distinct proteins target including AKT, which is involved in anabolism and controls actin cytoskeleton and cell migration via PKC [83, 84]. Interestingly, the PI3K/Akt/mTOR signaling, which is up-regulated in CCA [85], has a central role in cell survival, apoptosis, metabolism, motility, and angiogenesis [86]. However, loss of primary cilia seems to particularly affect mTORC1 activity in MDCK cells [87]. Since mechanical stimulation of primary cilia induces inhibition of mTOR via AMP-activated protein kinase (AMPK) activation in a LKB1 dependent mechanism in MDCK cells in vitro [80], the loss of cilia observed in CCA may lead to dysregulation of mTOR activity contributing to CCA development.
Figure 7. Primary cilia regulate mTOR.

Changes in the bile flow would induce activation of LKB1, which is localized along the entire length of the cilia. Once activated LKB1 phosphorylates and activates AMPK, which is recruited in the basal body. TSC is phosphorylated by AMPK leading to inhibition of Rheb that normally activates mTOR. As result, mTOR and its downstream proteins are inhibited. Thus, primary cilia participate in the regulation of cell size, cell metabolism, cell growth, proliferation and survival.
Finally, since TGR5 is expressed in primary cilia and apical membrane and the fact that TGR5 activation has opposite effects in ciliated and non-ciliated cells (Fig 8), loss of primary cilia would result in dysregulation of bile acid signaling involved in CCA development. Indeed, activation of TGR5 promotes proliferation in non-ciliated cholangiocytes and CCA and induces resistance to apoptosis [88, 89].
CILIOTHERAPHY
The fact that primary cilia are absent or malfunctional in most ciliopathies, and the fact that both molecular and chemical deciliation in vitro of normal cholangiocytes induce a malignant-like phenotype resulting in several dysregulated pathways (Figure 9) lead to the proposed therapeutical approaches based on cilia restoration.
Figure 9. Ciliotherapy.

Primary cilia function as tumor suppressor organelles by controlling several signaling pathways involved in metabolic process, cell growth, proliferation and survival. In CCA, the loss of primary cilia, which is associated with HDAC6 overexpression, generates malignant cholangiocytes unable to properly regulate these signaling pathways. Therefore, ciliotherapies, which are based on ciliary restoration, might have a potential value for cancer prevention and treatment.
Different drugs such as clofibratem, gefitinib, imexon, ciprofloxacin and dexamethasone have the ability to restore primary cilia in cancer cells [90]. This ability is associated with induction of growth arrest and, in some cases, is accompanied by apoptosis [90]. Since HDAC6 overexpression in cholangiocytes results in ciliary resorption, HDAC6 represents a novel target for the treatment of ciliopathies. HDAC6 is phosphorylated and activated by a mechanism dependent on the Aurora A kinase. Once activated, HDAC6 promotes α-tubulin deacetylation that destabilizes the axoneme resulting in ciliary resorption. HDAC6 inhibitors represent very attractive drugs for treatments because these small molecules are not toxic to normal tissues and have minimal adverse effects. Recent findings show a relationship between high expression of HDAC6 and loss of primary cilia in CCA [56]. Therefore, the use of HDAC6 inhibitor as an approach to block the malignant phenotype in CCA cells seems promising. In line with this, treatment with an HDAC6 inhibitor decreases the tumorigenic phenotype with significant decrease in tumor growth in a ciliary re-expression dependent manner in in vitro and in vivo models of CCA [56]. Thus, the restoration of the primary cilia may be a potential therapeutic approach for CCA.
The use of HDAC6 inhibitors is promising for several ciliopathies. Recent studies using ACY 1215 treatment, a specific HDAC6 inhibitor, have shown reduced cell proliferation and cyst growth in in vitro and in animal models of PLC [91]. Thus, HDAC6 inhibitors may be important for the treatment of ciliopathies.
Another potential drug for use in ciliotherapy is fenoldopan. A recent study showed that fenoldopan, which is usually used to decrease blood pressure by activating peripheral D1 receptors, has the ability to increase ciliary length in vascular endothelial cells [92]. The use of fenoldopan resulted in increased levels of nitric oxide in serum and reduced blood pressure in a PKD mouse model. This result suggests the use of fenoldopan to address the hypertension associated with PKD.
Since the loss of primary cilia results in dysregulation of several molecular pathways resulting in abnormal functions of cholangiocytes, ciliotherapy might have great potential value in cancer prevention and treatment.
CONCLUSION
Cholangiocyte primary cilia function as mechano- chemo- and osmosensors organelles where several signals in the bile flow are detected and information is transmitted into the nucleus regulating bile secretion, cell proliferation, and apoptosis among others. Defects in the structure and/or function of cholangiocytes primary cilia lead to dysregulation of several signaling pathways resulting in cholangiociliopathies. Since primary cilia are absent in CCA and other tumors, loss of cilia may be associated with dysregulation of several molecular pathways resulting in CCA development and progression. Thus, primary cilia would function as a tumor suppressor organelle. Therapies based on ciliary restoration such as treatment with HDAC6 inhibitors might have potential value in cholangiociliopathies and in CCA prevention and treatment.
Acknowledgments
This work was supported by National Institutes of Health Grant R01CA183764 (to S.A.G), The Randy Shaver Cancer Research and Community Fund Award (to S.A.G.), and The Hormel Foundation.
Footnotes
Conflicts of interest
The authors confirm that there no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Masyuk AI, Marinelli RA, LaRusso NF. Water transport by epithelia of the digestive tract. Gastroenterology. 2002;122:545–562. doi: 10.1053/gast.2002.31035. [DOI] [PubMed] [Google Scholar]
- 2.Afroze S, Meng F, Jensen K, McDaniel K, Rahal K, Onori P, Gaudio E, Alpini G, Glaser SS. The physiological roles of secretin and its receptor. Annals of translational medicine. 2013;1:29. doi: 10.3978/j.issn.2305-5839.2012.12.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, Stroope AJ, LaRusso NF. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology. 2006;131:911–920. doi: 10.1053/j.gastro.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abou Alaiwi WA, Lo ST, Nauli SM. Primary cilia: highly sophisticated biological sensors. Sensors (Basel, Switzerland) 2009;9:7003–7020. doi: 10.3390/s90907003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christensen ST, Pedersen LB, Schneider L, Satir P. Sensory cilia and integration of signal transduction in human health and disease. Traffic (Copenhagen, Denmark) 2007;8:97–109. doi: 10.1111/j.1600-0854.2006.00516.x. [DOI] [PubMed] [Google Scholar]
- 6.Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nature reviews Genetics. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Czarnecki PG, Shah JV. The ciliary transition zone: from morphology and molecules to medicine. Trends in cell biology. 2012;22:201–210. doi: 10.1016/j.tcb.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JE, Gleeson JG. A systems-biology approach to understanding the ciliopathy disorders. Genome medicine. 2011;3:59. doi: 10.1186/gm275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gradilone SA, Masyuk AI, Splinter PL, Banales JM, Huang BQ, Tietz PS, Masyuk TV, Larusso NF. Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19138–19143. doi: 10.1073/pnas.0705964104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masyuk AI, Gradilone SA, Banales JM, Huang BQ, Masyuk TV, Lee SO, Splinter PL, Stroope AJ, Larusso NF. Cholangiocyte primary cilia are chemosensory organelles that detect biliary nucleotides via P2Y12 purinergic receptors. American journal of physiology Gastrointestinal and liver physiology. 2008;295:G725–734. doi: 10.1152/ajpgi.90265.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masyuk AI, Huang BQ, Ward CJ, Gradilone SA, Banales JM, Masyuk TV, Radtke B, Splinter PL, LaRusso NF. Biliary exosomes influence cholangiocyte regulatory mechanisms and proliferation through interaction with primary cilia. American journal of physiology Gastrointestinal and liver physiology. 2010;299:G990–999. doi: 10.1152/ajpgi.00093.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Masyuk AI, Masyuk TV, LaRusso NF. Cholangiocyte primary cilia in liver health and disease. Developmental dynamics: an official publication of the American Association of Anatomists. 2008;237:2007–2012. doi: 10.1002/dvdy.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liedtke W. Transient receptor potential vanilloid channels functioning in transduction of osmotic stimuli. The Journal of endocrinology. 2006;191:515–523. doi: 10.1677/joe.1.07000. [DOI] [PubMed] [Google Scholar]
- 14.Masyuk AI, Huang BQ, Radtke BN, Gajdos GB, Splinter PL, Masyuk TV, Gradilone SA, LaRusso NF. Ciliary subcellular localization of TGR5 determines the cholangiocyte functional response to bile acid signaling. American journal of physiology Gastrointestinal and liver physiology. 2013;304:G1013–1024. doi: 10.1152/ajpgi.00383.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simpson RJ, Jensen SS, Lim JW. Proteomic profiling of exosomes: current perspectives. Proteomics. 2008;8:4083–4099. doi: 10.1002/pmic.200800109. [DOI] [PubMed] [Google Scholar]
- 16.Maroni L, Haibo B, Ray D, Zhou T, Wan Y, Meng F, Marzioni M, Alpini G. Functional and structural features of cholangiocytes in health and disease. Cellular and molecular gastroenterology and hepatology. 2015;1:368–380. doi: 10.1016/j.jcmgh.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larusso NF, Masyuk TV. The role of cilia in the regulation of bile flow. Digestive diseases (Basel, Switzerland) 2011;29:6–12. doi: 10.1159/000324121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masyuk T, Masyuk A, LaRusso N. Cholangiociliopathies: genetics, molecular mechanisms and potential therapies. Current opinion in gastroenterology. 2009;25:265–271. doi: 10.1097/MOG.0b013e328328f4ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drenth JP, Martina JA, Te Morsche RH, Jansen JB, Bonifacino JS. Molecular characterization of hepatocystin, the protein that is defective in autosomal dominant polycystic liver disease. Gastroenterology. 2004;126:1819–1827. doi: 10.1053/j.gastro.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 20.Ponting CP. Proteins of the endoplasmic-reticulum-associated degradation pathway: domain detection and function prediction. The Biochemical journal. 2000;351(Pt 2):527–535. [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer HA, Grau H, Kraft R, Kostka S, Prehn S, Kalies KU, Hartmann E. Mammalian Sec61 is associated with Sec62 and Sec63. The Journal of biological chemistry. 2000;275:14550–14557. doi: 10.1074/jbc.275.19.14550. [DOI] [PubMed] [Google Scholar]
- 22.Waanders E, Croes HJ, Maass CN, te Morsche RH, van Geffen HJ, van Krieken JH, Fransen JA, Drenth JP. Cysts of PRKCSH mutated polycystic liver disease patients lack hepatocystin but express Sec63p. Histochemistry and cell biology. 2008;129:301–310. doi: 10.1007/s00418-008-0381-3. [DOI] [PubMed] [Google Scholar]
- 23.Ong AC, Harris PC. Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney international. 2005;67:1234–1247. doi: 10.1111/j.1523-1755.2005.00201.x. [DOI] [PubMed] [Google Scholar]
- 24.Masyuk TV, Masyuk AI, LaRusso NF. TGR5 in the Cholangiociliopathies. Digestive diseases (Basel, Switzerland) 2015;33:420–425. doi: 10.1159/000371696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palmela C, Peerani F, Castaneda D, Torres J, Itzkowitz SH. Inflammatory Bowel Disease and Primary Sclerosing Cholangitis: A Review of the Phenotype and Associated Specific Features. Gut and liver. 2017 doi: 10.5009/gnl16510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Razumilava N, Gores GJ, Lindor KD. Cancer surveillance in patients with primary sclerosing cholangitis. Hepatology (Baltimore, Md) 2011;54:1842–1852. doi: 10.1002/hep.24570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B, Bernstein H. Carcinogenicity of deoxycholate, a secondary bile acid. Archives of toxicology. 2011;85:863–871. doi: 10.1007/s00204-011-0648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li T, Chiang JYL. Nuclear receptors in bile acid metabolism. Drug metabolism reviews. 2013;45:145–155. doi: 10.3109/03602532.2012.740048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan L, Bambha K. Bile acid receptors and nonalcoholic fatty liver disease. World Journal of Hepatology. 2015;7:2811–2818. doi: 10.4254/wjh.v7.i28.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reich M, Klindt C, Deutschmann K, Spomer L, Haussinger D, Keitel V. Role of the G Protein-Coupled Bile Acid Receptor TGR5 in Liver Damage. Digestive diseases (Basel, Switzerland) 2017;35:235–240. doi: 10.1159/000450917. [DOI] [PubMed] [Google Scholar]
- 31.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, Hinuma S, Fujisawa Y, Fujino M. A G protein-coupled receptor responsive to bile acids. The Journal of biological chemistry. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 32.Keitel V, Reinehr R, Reich M, Sommerfeld A, Cupisti K, Knoefel WT, Häussinger D. The membrane-bound bile acid receptor TGR5 (GPBAR-1) is highly expressed in intrahepatic cholangiocarcinoma. Hepatology (Baltimore, Md) 2011;54:769A. [Google Scholar]
- 33.Mao K, Jiang W, Liu J, Wang J. Incidence of subsequent cholangiocarcinomas after another malignancy: trends in a population-based study. Medicine. 2015;94:e596. doi: 10.1097/MD.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blechacz B, Gores GJ. Cholangiocarcinoma: advances in pathogenesis, diagnosis, and treatment. Hepatology (Baltimore, Md) 2008;48:308–321. doi: 10.1002/hep.22310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cardinale V, Bragazzi MC, Carpino G, Torrice A, Fraveto A, Gentile R, Pasqualino V, Melandro F, Aliberti C, Bastianelli C, Brunelli R, Berloco PB, Gaudio E, Alvaro D. Cholangiocarcinoma: increasing burden of classifications. Hepatobiliary surgery and nutrition. 2013;2:272–280. doi: 10.3978/j.issn.2304-3881.2013.10.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson CD, Pinson CW, Berlin J, Chari RS. Diagnosis and treatment of cholangiocarcinoma. The oncologist. 2004;9:43–57. doi: 10.1634/theoncologist.9-1-43. [DOI] [PubMed] [Google Scholar]
- 37.Goodman ZD. Neoplasms of the liver. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2007;20(Suppl 1):S49–60. doi: 10.1038/modpathol.3800682. [DOI] [PubMed] [Google Scholar]
- 38.Gatto M, Alvaro D. New insights on cholangiocarcinoma. World journal of gastrointestinal oncology. 2010;2:136–145. doi: 10.4251/wjgo.v2.i3.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishak KG, Anthony PP, SOBIN L. Histological typing of tumours of the liver. Springer Science & Business Media; 2012. [Google Scholar]
- 40.Shaib Y, El-Serag HB. The epidemiology of cholangiocarcinoma. Seminars in liver disease. 2004;24:115–125. doi: 10.1055/s-2004-828889. [DOI] [PubMed] [Google Scholar]
- 41.Friman S. Cholangiocarcinoma--current treatment options. Scandinavian journal of surgery: SJS: official organ for the Finnish Surgical Society and the Scandinavian Surgical Society. 2011;100:30–34. doi: 10.1177/145749691110000106. [DOI] [PubMed] [Google Scholar]
- 42.Kang YK, Kim WH, Lee HW, Lee HK, Kim YI. Mutation of p53 and K-ras, and loss of heterozygosity of APC in intrahepatic cholangiocarcinoma. Laboratory investigation; a journal of technical methods and pathology. 1999;79:477–483. [PubMed] [Google Scholar]
- 43.Hahn SA, Bartsch D, Schroers A, Galehdari H, Becker M, Ramaswamy A, Schwarte-Waldhoff I, Maschek H, Schmiegel W. Mutations of the DPC4/Smad4 gene in biliary tract carcinoma. Cancer Res. 1998;58:1124–1126. [PubMed] [Google Scholar]
- 44.Taniai M, Higuchi H, Burgart LJ, Gores GJ. p16INK4a promoter mutations are frequent in primary sclerosing cholangitis (PSC) and PSC-associated cholangiocarcinoma. Gastroenterology. 2002;123:1090–1098. doi: 10.1053/gast.2002.36021. [DOI] [PubMed] [Google Scholar]
- 45.Wehbe H, Henson R, Meng F, Mize-Berge J, Patel T. Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res. 2006;66:10517–10524. doi: 10.1158/0008-5472.CAN-06-2130. [DOI] [PubMed] [Google Scholar]
- 46.Brito AF, Abrantes AM, Encarnacao JC, Tralhao JG, Botelho MF. Cholangiocarcinoma: from molecular biology to treatment. Medical oncology (Northwood, London, England) 2015;32:245. doi: 10.1007/s12032-015-0692-x. [DOI] [PubMed] [Google Scholar]
- 47.Seeley ES, Carriere C, Goetze T, Longnecker DS, Korc M. Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res. 2009;69:422–430. doi: 10.1158/0008-5472.CAN-08-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Basten SG, Willekers S, Vermaat JS, Slaats GG, Voest EE, van Diest PJ, Giles RH. Reduced cilia frequencies in human renal cell carcinomas versus neighboring parenchymal tissue. Cilia. 2013;2:2. doi: 10.1186/2046-2530-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan K, Frolova N, Xie Y, Wang D, Cook L, Kwon YJ, Steg AD, Serra R, Frost AR. Primary cilia are decreased in breast cancer: analysis of a collection of human breast cancer cell lines and tissues. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society. 2010;58:857–870. doi: 10.1369/jhc.2010.955856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hassounah NB, Nagle R, Saboda K, Roe DJ, Dalkin BL, McDermott KM. Primary cilia are lost in preinvasive and invasive prostate cancer. PloS one. 2013;8:e68521. doi: 10.1371/journal.pone.0068521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nobutani K, Shimono Y, Yoshida M, Mizutani K, Minami A, Kono S, Mukohara T, Yamasaki T, Itoh T, Takao S, Minami H, Azuma T, Takai Y. Absence of primary cilia in cell cycle-arrested human breast cancer cells. Genes to cells: devoted to molecular & cellular mechanisms. 2014;19:141–152. doi: 10.1111/gtc.12122. [DOI] [PubMed] [Google Scholar]
- 52.Masyuk TV, Huang BQ, Ward CJ, Masyuk AI, Yuan D, Splinter PL, Punyashthiti R, Ritman EL, Torres VE, Harris PC, LaRusso NF. Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat. Gastroenterology. 2003;125:1303–1310. doi: 10.1016/j.gastro.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 53.Alvaro D, Onori P, Alpini G, Franchitto A, Jefferson DM, Torrice A, Cardinale V, Stefanelli F, Mancino MG, Strazzabosco M, Angelico M, Attili A, Gaudio E. Morphological and functional features of hepatic cyst epithelium in autosomal dominant polycystic kidney disease. The American journal of pathology. 2008;172:321–332. doi: 10.2353/ajpath.2008.070293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chu AS, Russo PA, Wells RG. Cholangiocyte cilia are abnormal in syndromic and non-syndromic biliary atresia. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2012;25:751–757. doi: 10.1038/modpathol.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karjoo S, Hand NJ, Loarca L, Russo PA, Friedman JR, Wells RG. Extrahepatic cholangiocyte cilia are abnormal in biliary atresia. Journal of pediatric gastroenterology and nutrition. 2013;57:96–101. doi: 10.1097/MPG.0b013e318296e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gradilone SA, Radtke BN, Bogert PS, Huang BQ, Gajdos GB, LaRusso NF. HDAC6 inhibition restores ciliary expression and decreases tumor growth. Cancer Res. 2013;73:2259–2270. doi: 10.1158/0008-5472.CAN-12-2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Razumilava N, Gradilone SA, Smoot RL, Mertens JC, Bronk SF, Sirica AE, Gores GJ. Non-canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. Journal of hepatology. 2014;60:599–605. doi: 10.1016/j.jhep.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 59.Mans DA, Voest EE, Giles RH. All along the watchtower: is the cilium a tumor suppressor organelle? Biochimica et biophysica acta. 2008;1786:114–125. doi: 10.1016/j.bbcan.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 60.Izawa I, Goto H, Kasahara K, Inagaki M. Current topics of functional links between primary cilia and cell cycle. Cilia. 2015;4:12. doi: 10.1186/s13630-015-0021-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wong SY, Seol AD, So PL, Ermilov AN, Bichakjian CK, Epstein EH, Jr, Dlugosz AA, Reiter JF. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nature medicine. 2009;15:1055–1061. doi: 10.1038/nm.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gerhardt C, Leu T, Lier JM, Ruther U. The cilia-regulated proteasome and its role in the development of ciliopathies and cancer. Cilia. 2016;5:14. doi: 10.1186/s13630-016-0035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cao M, Zhong Q. Cilia in autophagy and cancer. Cilia. 2015;5:4. doi: 10.1186/s13630-016-0027-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Current biology: CB. 2005;15:1861–1866. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 65.Boonjaraspinyo S, Wu Z, Boonmars T, Kaewkes S, Loilome W, Sithithaworn P, Nagano I, Takahashi Y, Yongvanit P, Bhudhisawasdi V. Overexpression of PDGFA and its receptor during carcinogenesis of Opisthorchis viverrini-associated cholangiocarcinoma. Parasitology international. 2012;61:145–150. doi: 10.1016/j.parint.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 66.Oh EC, Katsanis N. Context-dependent regulation of Wnt signaling through the primary cilium. Journal of the American Society of Nephrology: JASN. 2013;24:10–18. doi: 10.1681/ASN.2012050526. [DOI] [PubMed] [Google Scholar]
- 67.Nygaard MB, Almstrup K, Lindbaek L, Christensen ST, Svingen T. Cell context-specific expression of primary cilia in the human testis and ciliary coordination of Hedgehog signalling in mouse Leydig cells. Scientific reports. 2015;5:10364. doi: 10.1038/srep10364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ocbina PJ, Anderson KV. Intraflagellar transport, cilia, and mammalian Hedgehog signaling: analysis in mouse embryonic fibroblasts. Developmental dynamics: an official publication of the American Association of Anatomists. 2008;237:2030–2038. doi: 10.1002/dvdy.21551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Omenetti A, Diehl AM. Hedgehog signaling in cholangiocytes. Current opinion in gastroenterology. 2011;27:268–275. doi: 10.1097/MOG.0b013e32834550b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kurita S, Mott JL, Almada LL, Bronk SF, Werneburg NW, Sun SY, Roberts LR, Fernandez-Zapico ME, Gores GJ. GLI3-dependent repression of DR4 mediates hedgehog antagonism of TRAIL-induced apoptosis. Oncogene. 2010;29:4848–4858. doi: 10.1038/onc.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Polizio AH, Chinchilla P, Chen X, Kim S, Manning DR, Riobo NA. Heterotrimeric Gi proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. The Journal of biological chemistry. 2011;286:19589–19596. doi: 10.1074/jbc.M110.197111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nusse R. Wnt signaling. Cold Spring Harbor perspectives in biology. 2012;4 doi: 10.1101/cshperspect.a011163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Boulter L, Guest RV, Kendall TJ, Wilson DH, Wojtacha D, Robson AJ, Ridgway RA, Samuel K, Van Rooijen N, Barry ST, Wigmore SJ, Sansom OJ, Forbes SJ. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. The Journal of clinical investigation. 2015;125:1269–1285. doi: 10.1172/JCI76452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Noll AT, Cramer T, Olde Damink SW, Schaap FG. Cholangiocarcinoma, gone without the Wnt? World J Hepatol. 2016;8:1093–1096. doi: 10.4254/wjh.v8.i26.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ahnfelt-Ronne J, Hald J, Bodker A, Yassin H, Serup P, Hecksher-Sorensen J. Preservation of proliferating pancreatic progenitor cells by Delta-Notch signaling in the embryonic chicken pancreas. BMC developmental biology. 2007;7:63. doi: 10.1186/1471-213X-7-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schweisguth F. Regulation of notch signaling activity. Current biology: CB. 2004;14:R129–138. [PubMed] [Google Scholar]
- 77.Grisanti L, Revenkova E, Gordon RE, Iomini C. Primary cilia maintain corneal epithelial homeostasis by regulation of the Notch signaling pathway. Development (Cambridge, England) 2016;143:2160–2171. doi: 10.1242/dev.132704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cigliano A, Wang J, Chen X, Calvisi DF. Role of the Notch signaling in cholangiocarcinoma. Expert opinion on therapeutic targets. 2017;21:471–483. doi: 10.1080/14728222.2017.1310842. [DOI] [PubMed] [Google Scholar]
- 79.El Khatib M, Bozko P, Palagani V, Malek NP, Wilkens L, Plentz RR. Activation of Notch signaling is required for cholangiocarcinoma progression and is enhanced by inactivation of p53 in vivo. PloS one. 2013;8:e77433. doi: 10.1371/journal.pone.0077433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boehlke C, Kotsis F, Patel V, Braeg S, Voelker H, Bredt S, Beyer T, Janusch H, Hamann C, Godel M, Muller K, Herbst M, Hornung M, Doerken M, Kottgen M, Nitschke R, Igarashi P, Walz G, Kuehn EW. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nature cell biology. 2010;12:1115–1122. doi: 10.1038/ncb2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xie J, Wang X, Proud CG. mTOR inhibitors in cancer therapy. F1000Research. 2016;5 doi: 10.12688/f1000research.9207.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. The Journal of clinical investigation. 2015;125:25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nature reviews Molecular cell biology. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 84.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Current opinion in cell biology. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 85.Riener MO, Bawohl M, Clavien PA, Jochum W. Rare PIK3CA hotspot mutations in carcinomas of the biliary tract, Genes. chromosomes & cancer. 2008;47:363–367. doi: 10.1002/gcc.20540. [DOI] [PubMed] [Google Scholar]
- 86.Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway--beyond rapalogs. Oncotarget. 2010;1:530–543. doi: 10.18632/oncotarget.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.(!!! INVALID CITATION !!! {}).
- 88.Reich M, Deutschmann K, Sommerfeld A, Klindt C, Kluge S, Kubitz R, Ullmer C, Knoefel WT, Herebian D, Mayatepek E, Haussinger D, Keitel V. TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut. 2016;65:487–501. doi: 10.1136/gutjnl-2015-309458. [DOI] [PubMed] [Google Scholar]
- 89.Keitel V, Häussinger D. TGR5 in cholangiocytes. Current opinion in gastroenterology. 2013;29:299–304. doi: 10.1097/MOG.0b013e32835f3f14. [DOI] [PubMed] [Google Scholar]
- 90.Khan NA, Willemarck N, Talebi A, Marchand A, Binda MM, Dehairs J, Rueda-Rincon N, Daniels VW, Bagadi M, Thimiri Govinda Raj DB, Vanderhoydonc F, Munck S, Chaltin P, Swinnen JV. Identification of drugs that restore primary cilium expression in cancer cells. Oncotarget. 2016;7:9975–9992. doi: 10.18632/oncotarget.7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gradilone SA, Habringer S, Masyuk TV, Howard BN, Masyuk AI, Larusso NF. HDAC6 is overexpressed in cystic cholangiocytes and its inhibition reduces cystogenesis. The American journal of pathology. 2014;184:600–608. doi: 10.1016/j.ajpath.2013.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kathem SH, Mohieldin AM, Abdul-Majeed S, Ismail SH, Altaei QH, Alshimmari IK, Alsaidi MM, Khammas H, Nauli AM, Joe B, Nauli SM. Ciliotherapy: a novel intervention in polycystic kidney disease. Journal of geriatric cardiology: JGC. 2014;11:63–73. doi: 10.3969/j.issn.1671-5411.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]