

Abstract
The rescue of exhausted CD8+ cytolytic T-cells (CTLs) by anti-Programmed Cell Death-1 (anti-PD-1) blockade has been found to require CD28 expression. At the same time, we have shown that the inactivation of the serine/threonine kinase glycogen synthase kinase (GSK)-3α/β with small-interfering RNAs (siRNAs) and small molecule inhibitors (SMIs) specifically down-regulates PD-1 expression for enhanced CD8+ CTL function and clearance of tumors and viral infections. Despite this, it has been unclear whether the GSK-3α/β pathway accounts for CD28 costimulation of CD8+ CTL function. In this article, we show that inactivation of GSK-3α/β through siRNA or by SMIs during priming can substitute CD28 co-stimulation in the potentiation of cytotoxic CD8+ CTL function against the EL-4 lymphoma cells expressing OVA peptide. The effect was seen using several structurally distinct GSK-3 SMIs and was accompanied by an increase in Lamp-1 and GZMB expression. Conversely, CD28 crosslinking obviated the need for GSK-3α/β inhibition in its enhancement of CTL function. Our findings support a model where GSK-3 is the central cosignal for CD28 priming of CD8+ CTLs in anti-PD-1 immunotherapy.
Keywords: T-cells, glycogen synthase kinase-3, programmed cell death 1, Tbet, cancer
Introduction
Naive T-cells are cells that have not encountered cognate antigen are essential for responses to novel pathogens. In this instance, activation requires a combination of stimulatory signals (1). The first signal is provided by the T-cell receptor (TCR) upon lymphocyte interaction with major histocompatibility class (MHC) antigens on the antigen-presenting cells (APCs) within the immune synapse (2). The second signal for T-cell activation is provided by CD28 and other costimulatory coreceptors on T-cells (3–6). CD28 is a well-defined costimulatory molecule found on lymphocytes, which interacts with B7 (CD80 and CD86) proteins on the APC (7, 8). TCR signaling alone can result in the lymphocyte undergoing cell death, or becoming anergic and thus unable to respond to antigen (9). Simultaneous signaling through CD28 and the TCR gives rise to sustained activation characterized by interleukin (IL)-2 production and cell-cycle entry (8, 10–12). Anti-CD28 crosslinking using monoclonal antibodies (MAbs) that augment CD28 cosignaling, especially on CD4+ T-cells, leading to increased interleukin 2-receptor (IL-2R), CD69 expression and proliferation (13). Conversely, Fab fragments of antibodies in mice, inhibit T-cell responses and can induce long-term heart allograft survival (14), and ameliorate experimental autoimmune encephalomyelitis (15).
We and others have shown that CD28 can complement and amplify TCR signaling (8, 12, 16, 17). In addition, CD28 can generate signals independently of TCR engagement (6, 18–20). The Tyr-Met-Asn-Met (YMNM) motif in the cytoplasmic tail of CD28 binds the adaptor growth factor receptor-bound protein 2 (GRB-2) (5, 11, 21–25) and the p85 regulatory subunit of phosphoinositide 3-kinase (PI-3K) resulting in the activation of AKT (21, 26, 27). This, in turn, leads to optimal IL-2-gene activation (11, 28), the expression of the anti-apoptotic protein BCL-XL, and the induction of an antigen response in vivo (11, 24, 29, 30). In this context, CD28 is linked to the serine threonine kinase; glycogen synthase kinase-3 (GSK-3). GSK-3 is constitutively active in T-cells, facilitating the exit of nuclear factor of activated T-cells (NFAT-c1) from the nucleus (31). CD28 signaling via PI-3K leads to the phosphorylation and inactivation of GSK-3, thus increasing IL-2 production and T-cell proliferation (32, 33).
Programmed cell death 1 (PD-1; PDCD1) is a member of the CD28 supergene family which negatively regulates T-cell function (3, 34, 35). PD-1 is expressed in response to T-cell activation and contributes to the exhaustion of CD8+ T-cells during chronic infection (36, 37). The coreceptor binds to ligands, programmed cell death ligand 1 and 2 (PD-L1/L2), on lymphoid and non-lymphoid cells (38–40). Immune checkpoint blockade (ICB) with anti-PD-1 or anti-PD-L1 has also proven highly successful in the treatment of human cancers, alone or in combination with anti-CTLA-4 (41, 42). PD-1 expression on tumor-infiltrating CD8+ T-cells correlates with impaired effector cell function (3, 43). We recently showed that GSK-3 is a central regulator of PD-1 expression and that the inactivation of GSK-3 using small molecule inhibitors (SMIs) downregulates PD-1 expression resulting in enhanced clearance of viral infections and cancer (44, 45). Recently, it has also been shown that PD-1 check-point blockade requires CD28 expression (46–48).
Here, we show that inhibition of GSK-3α/β by either small-interfering RNAs (siRNAs) or SMIs can substitute CD28 stimulation in the potentiation of CD8+ cytolytic T-cell (CTL) function. We propose that GSK-3 is the key mediator that is responsible for CD28 priming of CD8+ CTLs in T-cell immunity and in response to anti-PD-1 ICB immunotherapy.
Results
Recently, we reported that the inactivation of GSK-3α/β with siRNAs and drug inhibitors specifically downregulate PD-1 expression for enhanced CD8+ CTL function and clearance of tumors and viral infections (44, 45). We also previously reported CD28 costimulation can induce the phosphorylation of GSK-3 and hence its inactivation (33, 49). To assess CD8+ CTL function in response to antigen-presentation, we utilized MHC class I-restricted OVA specific-TCR transgenic (OT-1) mice with a TCR specific for the SIINFEKL peptide of OVAlbumin (OVA257–264) as presented by H-2kb. Control samples showed an increase in killing targets concurrent with an increase in effector/target (E/T) ratios. As previously shown (44), inhibition of GSK-3 with the SMI, SB415286, increased killing of EL4 target cells loaded with OVA peptide as measured at day 6 (Figure 1A). We next assessed the role of CD28 in this process. To this end, cultures were coincubated with soluble CTLA-4 IgG to block the interaction between CD28 and CD80/86 on presenting cells. EL4 cells express CD80 (50) and were therefore used as target cells. CTLA-4-IgG effectively inhibited the level of CTL killing of target cells (left panels). Intriguingly, the addition of SMI SB415286 completely restored normal levels of high CTL killing of targets at all E/T ratios (right panels). This ability of a GSK-3 SMI to bypass CD28 blockade by CTLA-4-IgG indicated that the inhibition of GSK-3 can substitute for the signal that is normally provided by anti-CD28. Further to this, as expected from our previous work, SB415286 suppressed the expression of PD-1 under all conditions (Figure 1B).
Figure 1.

T-cell activation with anti-CD28 enhances cytolytic T-cell (CTL) killing of antigen specific target cells through glycogen synthase kinase 3 (GSK-3). (A) OT-1 CD8+ CTLs were activated with OVA peptide incubated in the presence (right panel) or absence (left panel) of SB415286 with or without anti-CD28 or blocking CD28 (CTLA-4 IgG fusion protein). After 5 days, CTLs were washed and counted before incubation with target (OVA-EL4) cells at the ratios shown for 4 h. Lactate dehydrogenase release was measured as an indication of target cell killing. Histogram depicts measurements normalized for background non-specific killing. OVA alone: light gray bars; anti-CD28: light blue bars; CTLA-4 IgG: dark blue bars (error bars based on triplicate values in individual experiments, data shown representative of four independent experiments). (B) Histogram showing MFI values of programmed cell death 1 (PD-1) expression as measured by flow cytometry. (C) Flow cytometry profiles of GZMB and Lamp-1 in the presence and absence of SB415286 alone, combined with anti-CD28 or CTLA-4 IgG. Error bars based on triplicate values in individual experiments; data shown representative of three independent experiments.
Anti-CD28 crosslinking has been found previously to augment CD28 signaling (13, 51). To assess this in the context of CD8+ CTLs, cultures were coincubated with anti-CD28 to crosslink the CD28 coreceptor for 7 days followed by an assessment of CTL function. Under these conditions, anti-CD28 greatly potentiated the killing potential of CTLs at all E/T ratios (left panel). Interesting, this level of enhanced killing was similar to that induced by GSK-3 SMI SB415286 (left panel). Further, the level of increased killing induced by anti-CD28 could not be further enhanced by SB415286 and vice versa. In the same vein, anti-CD28 coculture reduced the expression of PD-1 on CD8+ T-cells, similar to that seen with SB415286 (Figure 1B). Although it was originally assumed that CD28 would provide costimulation needed for the expression of PD-1 as in the case of CTLA-4 (52), we observed the opposite result. This was consistent with the generation of signals via GSK-3 whose inhibition also suppressed PD-1 expression. Consistent with this, CTLA-4-IgG blockade of CD28 was seen to increase PD-1 expression (left panel). This suggested that the normal engagement of CD28 by CD80/86 might also act to suppress PD-1 expression. Flow cytometry showed that SB415286 downregulated PD-1 expression on OVA peptide activated cells was accompanied by increased expression of Lamp-1 and GZMB in T-cells (Figure 1C).
In a related approach, anti-CD28 or CTLA-4 IgG was added to cells expressing siRNA for GSK-3α/β (Figure 2). In the scrambled control, anti-CD28 acted to increase the level of response. In addition, the knock-down of GSK-3α/β with siRNA increased the level of response to that of anti-CD28 such that the addition of anti-CD28 has no further effect. While CTLA-4-IgG markedly reduced the response of OT-1 T-cells expressing scrambled siRNA, it had no effect on cells expressing GSK-3α/β siRNA. Using a different approach, these data confirmed that GSK-3 inhibition could substitute for the signal provided by anti-CD28. In turn, the increased killing was reflected by a decrease in PD-1 expression (Figure 2B) and an increase in GZMB and Lamp-1 expression (Figure 2C).
Figure 2.

CD28 activation is comparable to glycogen synthase kinase 3 (GSK-3) inactivation enhancing cytolytic function. (A) OT-1 CD8+ cytolytic T-cells (CTLs) were transfected with scrambled (left panel) or GSK-3 (right panel) small-interfering RNA (siRNA) prior to activation with OVA peptide and incubated with or without anti-CD28 or blocking CD28 (CTLA-4 IgG fusion protein). After 5 days CTLs were washed and counted before incubation with target (OVA-EL4) cells at the ratios shown for 4 h. Lactate dehydrogenase release was measured as an indication of target cell killing. Histogram depicts measurements normalized for background non-specific killing. OVA alone: light gray bars; anti-CD28: light blue bars; CTLA-4 IgG: dark blue bars (error bars based on triplicate values in individual experiments, data shown representative of four independent experiments). (B) Histogram showing MFI values of programmed cell death 1 (PD-1) expression as measured by flow cytometry. (C) Flow cytometry profiles of GZMB and Lamp-1 in either scrambled or GSK-3 siRNA transfected cells stimulated with Ova alone, or combined with anti-CD28 or CTLA-4 IgG. Error bars based on triplicate values in individual experiments; data shown representative of three independent experiments.
Importantly, the ability of GSK-3 inhibition to substitute for anti-CD28 in increasing CD8+ CTL function was seen with the use of different GSK-3 inhibitors; SB216763, CHIR99021, and L803-mts (Figure 3). Each have distinct structures but share a common target (53, 54). In each case, CD28 blockade by CTLA-4-IgG was reversed by the addition of any one of the four inhibitors used. Together, these data also support a key role for GSK-3 inhibition as a mediator of CD28 regulation of CD8+ T-cell killing.
Figure 3.

Anti-CD28 enhances cytolytic T-cell (CTL) killing of antigen-specific target cells to similar extent as glycogen synthase kinase 3 (GSK-3) inhibitors. OT-1 CD8+ CTLs were activated with OVA peptide incubated in the presence or absence of one of four small molecule inhibitors (from top to bottom; SB415286, CHIR99021, L803mts, SB216763) with or without anti-CD28 or blocking CD28 (CTLA-4 IgG fusion protein). After 5 days, CTLs were washed and counted before incubation with target (OVA-EL4) cells at the ratios shown for 4 h. Lactate dehydrogenase release was measured as an indication of target cell killing. Dark blue bars on left panel depicts background non-specific killing (non-pulsed target cell death). Error bars based on triplicate values in individual experiments; data shown representative of three independent experiments.
To assess the in vivo effect of CTL priming, OVA peptide in the presence or absence of SB415286 was injected intravenously into OT-1 transgenic mice followed by the harvest of spleens at day 7 (Figure 4). T-cells from extracted spleens were then subjected to further ex vivo stimulation for another 7 days in the presence or absence of SMI SB415286, anti-CD28, or CTLA-4-IgG followed by assessment of ex vivo killing of EL4-OVA targets. From this, it was observed that the in vivo administration of SMI enhanced cytolytic responses compared to OVA peptide alone (Figures 4A,B, left panel). This increase was also observed with OVA peptide alone primed cells when incubated with the GSK-3 SMI in vitro (Figure 4A, left panel). This finding showed that the cells were effectively primed in vivo with the SMI. In the case of cells primed with OVA peptide alone, the addition of anti-CD28 in vitro enhanced killing, whereas no additional effect was seen on cells primed with both OVA peptide and SMI. The addition of CTLA-4-IgG in vitro demonstrated the effects of priming with OVA peptide alone to be overcome by CD28 blockade. However, this was overcome by additional SMI in vitro (Figure 4B, left panel). Flow cytometry showed that priming with SMI, in addition to OVA peptide, slightly increased Lamp-1 and GZMB expression compared to OVA peptide alone. Further, anti-CD28 increased the numbers of CTLs expressing GZMB and Lamp-1, and this effect was reversed by CTLA-4-IgG (right panels). SMI had no further effect on anti-CD28-treated cells, but did overcome the CD28 blockade. Under both priming conditions, PD-1 expression was reduced in the presence of anti-CD28 to the same level as that seen with SMI. These data showed that GSK-3 inhibition in vivo augmented CTL function to a similar level as achieved in vitro with anti-CD28.
Figure 4.

Cytolytic potential of cytolytic T-cells (CTLs) primed in vivo can be enhanced in vitro with anti-CD28 antibody. OT-1 mice were injected intravenously with ova peptide alone (A) or in combination with SB415286 (B). Spleens were extracted on day 7. Ex vivo purified T-cells were then subjected to further stimulation with OVA peptide in the presence (right panel) or absence (left panel) of SB415286 with or without anti-CD28 or blocking CD28 (CTLA-4 IgG fusion protein). After 5 days, CTLs were washed and counted before incubation with target (OVA-EL4) cells at the ratios shown for 4 h. Lactate dehydrogenase release was measured as an indication of target cell killing. Histogram depicts measurements normalized for background non-specific killing. (Right panels) Flow cytometry profiles of GZMB, Lamp-1, and PD-1 in the presence and absence of SB415286, combined with anti-CD28 or CTLA-4 IgG (mean and SD of six mice per group). Error bars based on triplicate values in individual experiments; data shown representative of two independent experiments.
Discussion
Both CD28 and the serine/threonine kinase GSK-3α/β have been found to play important roles in the activation of T-cells (4, 5, 44). The PI-3K/3-phosphoinositide-dependent protein kinase 1 (PDK1)/AKT signaling axis is central to cellular homeostasis, cell growth and proliferation (55, 56). We previously showed that GSK-3α/β inactivation with siRNAs and SMIs specifically downregulates PD-1 expression which leads to enhanced CD8+ CTL function and clearance of viral infections and cancer (44, 45). Despite this, it has been unclear how the GSK-3 pathway is linked to CD28 costimulation in the generation of CD8+ CTL function. We previously showed that CD28 has a cytoplasmic YMNM motif for binding to PI-3K, and that the pathway promotes the phosphorylation and inactivation of GSK-3 (21, 27, 33). The binding motif for PI-3K is phosphorylated by the src kinases, p56lck and p59fyn (22). Here, we show that GSK-3 inactivation substitutes for CD28 in the priming of cytotoxic CD8+ T-cells, while the enhanced cytotoxic function induced by anti-CD28 Mab crosslinking obviates the effects of GSK-3 SMIs.
Our first observation was that GSK-3 inactivation, using either siRNAs or SMIs, could substitute for CD28 in providing cosignals for enhanced cytotoxicity. GSK-3 inactivation reversed the effects of CD28 blockade with CTLA-4-IgG in the cytotoxic response OT-1 CTLs against EL4 cells expressing the OVA peptide. This was seen at all effector to target ratios studied. In each case this enhanced function was accompanied by an increase in Lamp-1 and GZMB expression. The efficacy of SMIs indicated that the inhibition of the catalytic activity of GSK-3, and not its potential role as a molecular scaffold for the binding of other proteins, was primarily responsible for increased function. Further, the effects were seen with four different SMIs with distinct structures whose shared property is the inhibition of GSK-3. These included ATP-competitive inhibitors SB216763, CHIR99021, and L803-mts, where SB216763 has a greater preference of inhibition for the GSK-3α isoform, while CHIR99021 and L803-mts preferentially inhibits GSK-3β (54, 57). Our previous work assessed longevity of the effectiveness of the SMIs by monitoring PD-1 expression in mice coinjected with EL4 tumors and a single injection of SMI. These data indicate that the effects of SB415286 were sustained for over 7–10 days (44).
The close relationship between CD28 and GSK-3 was also observed by the ability of anti-CD28 MAb crosslinking to over-ride or substitute for GSK-3 SMI inhibition in the potentiation of CTL function. While anti-CD28 blocks the interaction between CD28 and CD80/86, it also crosslinks the coreceptor in the generation of cosignals. CD28 crosslinking by CD80/86 is generally thought to be suboptimal, while the higher concentration of anti-CD28 can be more effective in occupying and crosslinking the coreceptor. Consistent with this, anti-CD28 MAb PVI greatly enhanced the killing function of OT-1 CTLs against OVA-EL4 targets. The level of increased killing was identical to the level observed with the addition of GSK-3 SMIs. The addition of GSK-3 SMI SB415286 to cultures that had been incubated with anti-CD28 provided no further potentiation of the CTL response and vice versa. This was confirmed in both in vitro and in vivo assays. This is reminiscent of the similarity in the effects of GSK-3 SMIs and anti-PD-1 blockade (44). Whether a similar relationship between GSK-3 and CD28 exists in CD4+ T-cells and operates in response to activating CD28 superagonists (58) remains to be studied.
Overall, we propose a model where GSK-3 is the center of effects mediated via CD28 (Figure 5). Recently, it was reported that the rescue of exhausted CD8+ T-cells by anti-PD-1 blockade requires CD28 expression (46, 47). One proposed mechanism was the de-phosphorylation of CD28 by PD-1-associated Src homology region 2 domain-containing phosphatase (SHP)-2 (48). By connecting these observations to our findings, we propose a new model for the mechanism by which anti-PD-1 ICB operates in immunotherapy (see Figure 5). In the absence of anti-PD-1 ICB, PD-1-associated phosphatases SHP-1 and SHP-2 would dephosphorylate the CD28 YMNM motif for the activation of PI-3K. In the presence of anti-PD-1 ICB, the activation of SHP-1/2 is blocked, allowing for the phosphorylation of the CD28 YMNM motif and the recruitment of PI-3K (4, 5). PI-3K produces phosphatidylinositol (3,4,5) trisphosphates (PIP3) which serve as plasma membrane docking sites for proteins with pleckstrin-homology (PH) domains. CD28 induced PI-3K would promote PDK1 to the membrane where it would activate serine/threonine kinase AKT (also known as protein kinase B or PKB). AKT would in turn inhibit GSK-3 by phosphorylation of sites of human GSK-3α (Ser21) and GSK-3β (Ser9). As we have shown (44, 45), GSK-3 inhibition up-regulates the transcription of the transcription factor Tbx21 (Tbet) that inhibits PD-1 expression. We propose that CD28 regulation of GSK-3 accounts for the requirement for CD28 in the rescue of the response of CD8+ T-cells to anti-PD-1 blockade (46, 47). Further studies are needed to assess the full range of targets of the CD28-GSK-3-Tbet-PD-1 axis in T-cell biology.
Figure 5.
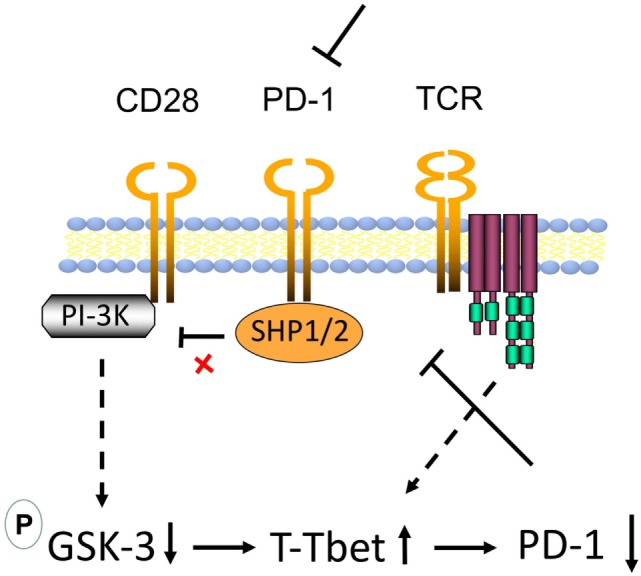
Model of CD28 mediated upregulation of CD8+ cytolytic T-cell (CTL) function via glycogen synthase kinase 3 (GSK-3) in the context of antiprogrammed cell death 1 (anti-PD-1) immune checkpoint blockade (ICB). In the absence of anti-PD-1 ICB, PD-1-associated phosphatases Src homology region 2 domain-containing phosphatase (SHP)-1 and SHP-2 dephosphorylate the CD28 phosphoinositide 3-kinase (PI-3K)-binding site Tyr-Met-Asn-Met (YMNM), thereby preventing the binding and engagement of PI-3K by CD28. In the presence of anti-PD-1, the activation of SHP-1/2 is blocked, allowing for the phosphorylation of the YMNM motif and the recruitment of PI-3K. PI-3K produces phosphatidylinositol (3,4,5) trisphosphates (PIP3) which serve as plasma membrane docking sites for proteins with pleckstrin-homology (PH) domains, including of the serine/threonine kinase AKT (also known as protein kinase B or PKB) and its upstream activator of the 3-phosphoinositide-dependent protein kinase 1 (PDK1). In our model, in T-cells, CD28 ligation by CD80/86 allows for the activation of PI-3K leading to the activation of PDK1 and the phosphorylation and activation of AKT. Phosphorylation of AKT at Ser473 by mTORC2 can also stimulate its full enzymatic activity. AKT in turn inhibits GSK-3 by phosphorylation [GSK-3α (Ser21) or GSK-3β (Ser9)]. We have shown that GSK-3 inhibition in turn upregulates the transcription of the transcription factor Tbx21 (Tbet) which in turn binds and inhibits transcription of PD-1. This pathway could downregulate PD-1 leading to more effective anti-PD-1 immunotherapy.
Materials and Methods
Mice
C57BL/6–OT-1Tg and wt mice were used throughout the majority of the study. The research on mice was regulated under the Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 following ethical review by the University of Cambridge Animal Welfare and Ethical Review Body Home Office UK PPL No. 70/7544.
Cells and Cultures
OVA specific CD8+ cytolytic T-cells were generated by incubating isolated splenocytes from OT-1 mice with SIINFEKL peptide of OVA (OVA257–264) at 10 ng/mL for 5–7 days. In certain cases, naive OT-1 T-cells were isolated from spleens using T-cell enrichment columns (R&D) and subjected to nuclear transfection (see method below). In the case of purified naive T-cells, the thymoma EL4 cell line was used to present OVA257–264 to primary T-cells. EL4 cells were incubated with 10 nM OVA257–264 peptide (Bachem) for 1 h at 37°C and treated with mitomycin C (Sigma-Aldrich, St. Louis, MO, USA) (final concentration of 10 µg/mL) prior to mixing with primary T-cells by coculturing at a ratio of 1:5 of EL4 and T-cells to generate cytotoxic T-cells. In either case, CTLs were generated in the presence or absence of SMI and/or anti-CD28 or CTLA-4-Ig (inhibitors/Abs added simultaneously with OVA-stimulation for 5–7 days) prior to washing and analysis by FACs, PCR, or cytotoxicity assays. Cells were cultured in RPMI 1640 medium supplemented with 10% FCS, 50 mM beta-mercaptoethanol, sodium pyruvate, 2 mM l-glutamine, 100 U/ml penicillin, and streptomycin (GIBCO).
Antibodies/Reagents
Stimulations were performed using 10 nM OVA257–264 peptide (Bachem), anti-CD28 (clone PV1, bioXpress), and CTLA-4 IgG Fusion Protein (BD Pharmingen) where stated. SMI (GSK-3 inhibitor) was obtained from Abcam plc. and suspended in DMSO to give a stock solution of 25 mM and diluted to a concentration of 10 μM in vitro. Fluorescently labeled Abs to GZMB, PD-1, and Lamp-1 (CD107a) were obtained from Biolegend.
Cytotoxicity Assays
Cytotoxicity was assayed using a Cytotox 96 nonradioactive kit (Promega) following the instructions provided. In brief, purified T-cells were plated in 96-well plates at the effector/target ratios shown using 104 EL4 (ova peptide-pulsed) target cells per well in a final volume of 200 µl per well using RPMI lacking phenol red. Lactate dehydrogenase release was assayed after 4 h incubation at 37°C by removal of 50 µl supernatant from each well and incubation with substrate provided for 30 min and the absorbance read at 490 nm using the Thermomax plate reader (Molecular Devices). Percentage cytotoxicity = [(experimental effectorspontaneous − target spontaneous)/(targetmaximum − target spontaneous)] × 100. All cytotoxicity assays were reproducible in at least three independent assays (59).
Nuclear Transfection
The 3.0 μg GSK-3α/β siRNA was added to 1 × 106 PBMC that had been washed in PBS and resuspended in 100 µl of Nucleofector™ solution for T-cells (Amaxa Biosystems, Cologne, Germany). Cells were transferred into a cuvette and electroporated using program X-01 of the Nucleofector™ (Amaxa Biosystems), and then immediately transferred into prewarmed cRPMI medium supplemented as recommended. GSK-3α/β specific and control siRNA were synthesized by Cell Signaling Technology. Control cells were transfected with 3.0 μg siRNA using the same protocol. Transfected cells were rested 24 h, before assays commenced.
Priming OT-1Tg Cells In Vivo
Ova peptide (1 µg) was injected intravenously into OT-1Tg mice with and without SB415286 (100 µg) in 100 µl of PBS. Spleens were harvested after 7 days and T-cells purified before further stimulation in vitro for 5 days with the indicated antibodies.
Statistical Analysis
The mean and SE of each treatment group were calculated for all experiments. The number of samples is indicated in the figure legends. Unpaired Student’s t-tests or ANOVA tests were performed using the InStat 3.0 software (GraphPad).*P < 0.05, **P < 0.01, and ***P < 0.001.
Ethics Statement
The research was regulated under the Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 following ethical review by the University of Cambridge Animal Welfare and Ethical Review Body (AWERB) Home Office UK PPL No. 70/7544.
Author Contributions
CR supervised, contributed conceptionally, and helped to write the article. AT contributed conceptionally, conducted experiments, and helped to write the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. CR and AT were supported by CRUK grant A20105. CR was also supported by Wellcome Trust 092627/Z/10/Z and a Foundation Award from the Centre de Recherche Hopital Maisonneuve-Rosemont.
References
- 1.Bretscher P, Cohn M. A theory of self-nonself discrimination. Science (1970) 169:1042–9. 10.1126/science.169.3950.1042 [DOI] [PubMed] [Google Scholar]
- 2.Dustin ML. A dynamic view of the immunological synapse. Semin Immunol (2005) 17:400–10. 10.1016/j.smim.2005.09.002 [DOI] [PubMed] [Google Scholar]
- 3.Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol (2016) 34:539–73. 10.1146/annurev-immunol-032414-112049 [DOI] [PubMed] [Google Scholar]
- 4.Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev (2009) 229:12–26. 10.1111/j.1600-065X.2009.00770.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rudd CE, Schneider H. Unifying concepts in CD28, ICOS and CTLA-4 co-receptor signalling. Nat Rev Immunol (2003) 3:544–56. 10.1038/nri1131 [DOI] [PubMed] [Google Scholar]
- 6.Rudd CE, Raab M. Independent CD28 signaling via VAV and SLP-76: a model for in trans costimulation. Immunol Rev (2003) 192:32–41. 10.1034/j.1600-065X.2003.00005.x [DOI] [PubMed] [Google Scholar]
- 7.van der Merwe PA, Bodian DL, Daenke S, Linsley P, Davis SJ. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J Exp Med (1997) 185:393–403. 10.1084/jem.185.3.393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.June CH, Ledbetter JA, Linsley PS, Thompson CB. Role of the CD28 receptor in T-cell activation. Immunol Today (1990) 11:211–6. 10.1016/0167-5699(90)90085-N [DOI] [PubMed] [Google Scholar]
- 9.Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science (1990) 248:1349–56. 10.1126/science.2113314 [DOI] [PubMed] [Google Scholar]
- 10.Lanier LL, O’Fallon S, Somoza C, Phillips JH, Linsley PS, Okumura K, et al. CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL. J Immunol (1995) 154:97–105. [PubMed] [Google Scholar]
- 11.Cai YC, Cefai D, Schneider H, Raab M, Nabavi N, Rudd CE. Selective CD28pYMNM mutations implicate phosphatidylinositol 3-kinase in CD86-CD28-mediated costimulation. Immunity (1995) 3:417–26. 10.1016/1074-7613(95)90171-X [DOI] [PubMed] [Google Scholar]
- 12.Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol (2001) 1:220–8. 10.1038/35105024 [DOI] [PubMed] [Google Scholar]
- 13.Abe R, Vandenberghe P, Craighead N, Smoot DS, Lee KP, June CH. Distinct signal transduction in mouse CD4+ and CD8+ splenic T cells after CD28 receptor ligation. J Immunol (1995) 154:985–97. [PubMed] [Google Scholar]
- 14.Jang MS, Pan F, Erickson LM, Fisniku O, Crews G, Wynn C, et al. A blocking anti-CD28-specific antibody induces long-term heart allograft survival by suppression of the PKC theta-JNK signal pathway. Transplantation (2008) 85:1051–5. 10.1097/TP.0b013e31816846f6 [DOI] [PubMed] [Google Scholar]
- 15.Perrin PJ, June CH, Maldonado JH, Ratts RB, Racke MK. Blockade of CD28 during in vitro activation of encephalitogenic T cells or after disease onset ameliorates experimental autoimmune encephalomyelitis. J Immunol (1999) 163:1704–10. [PubMed] [Google Scholar]
- 16.Michel F, Mangino G, Attal-Bonnefoy G, Tuosto L, Alcover A, Roumier A, et al. CD28 utilizes Vav-1 to enhance TCR-proximal signaling and NF-AT activation. J Immunol (2000) 165:3820–9. 10.4049/jimmunol.165.7.3820 [DOI] [PubMed] [Google Scholar]
- 17.Alegre M, Fallarino F, Zhou P, Frauwirth K, Thistlethwaite J, Newell K, et al. Transplantation and the CD28/CTLA-4/B7 pathway. Transplant Proc (2001) 33:209–11. 10.1016/S0041-1345(00)01977-1 [DOI] [PubMed] [Google Scholar]
- 18.Raab M, da Silva AJ, Findell PR, Rudd CE. Regulation of Vav-SLP-76 binding by ZAP-70 and its relevance to TCR zeta/CD3 induction of interleukin-2. Immunity (1997) 6:155–64. 10.1016/S1074-7613(00)80422-7 [DOI] [PubMed] [Google Scholar]
- 19.Marinari B, Costanzo A, Marzano V, Piccolella E, Tuosto L. CD28 delivers a unique signal leading to the selective recruitment of RelA and p52 NF-kappaB subunits on IL-8 and Bcl-xL gene promoters. Proc Natl Acad Sci U S A (2004) 101:6098–103. 10.1073/pnas.0308688101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thaker YR, Schneider H, Rudd CE. TCR and CD28 activate the transcription factor NF-kappaB in T-cells via distinct adaptor signaling complexes. Immunol Lett (2015) 163:113–9. 10.1016/j.imlet.2014.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prasad KV, Cai YC, Raab M, Duckworth B, Cantley L, Shoelson SE, et al. T-cell antigen CD28 interacts with the lipid kinase phosphatidylinositol 3-kinase by a cytoplasmic Tyr(P)-Met-Xaa-Met motif. Proc Natl Acad Sci U S A (1994) 91:2834–8. 10.1073/pnas.91.7.2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raab M, Cai YC, Bunnell SC, Heyeck SD, Berg LJ, Rudd CE. p56Lck and p59Fyn regulate CD28 binding to phosphatidylinositol 3-kinase, growth factor receptor-bound protein GRB-2, and T cell-specific protein-tyrosine kinase ITK: implications for T-cell costimulation. Proc Natl Acad Sci U S A (1995) 92:8891–5. 10.1073/pnas.92.19.8891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider H, Cai YC, Prasad KV, Shoelson SE, Rudd CE. T cell antigen CD28 binds to the GRB-2/SOS complex, regulators of p21ras. Eur J Immunol (1995) 25:1044–50. 10.1002/eji.1830250428 [DOI] [PubMed] [Google Scholar]
- 24.Kim HH, Tharayil M, Rudd CE. Growth factor receptor-bound protein 2 SH2/SH3 domain binding to CD28 and its role in co-signaling. J Biol Chem (1998) 273:296–301. 10.1074/jbc.273.1.296 [DOI] [PubMed] [Google Scholar]
- 25.Raab M, Pfister S, Rudd CE. CD28 signaling via VAV/SLP-76 adaptors: regulation of cytokine transcription independent of TCR ligation. Immunity (2001) 15:921–33. 10.1016/S1074-7613(01)00248-5 [DOI] [PubMed] [Google Scholar]
- 26.Okkenhaug K, Wu L, Garza KM, La Rose J, Khoo W, Odermatt B, et al. A point mutation in CD28 distinguishes proliferative signals from survival signals. Nat Immunol (2001) 2:325–32. 10.1038/35073061 [DOI] [PubMed] [Google Scholar]
- 27.Rudd CE. Upstream-downstream: CD28 cosignaling pathways and T cell function. Immunity (1996) 4:527–34. 10.1016/S1074-7613(00)80479-3 [DOI] [PubMed] [Google Scholar]
- 28.Burr JS, Savage ND, Messah GE, Kimzey SL, Shaw AS, Arch RH, et al. Cutting edge: distinct motifs within CD28 regulate T cell proliferation and induction of Bcl-XL. J Immunol (2001) 166:5331–5. 10.4049/jimmunol.166.9.5331 [DOI] [PubMed] [Google Scholar]
- 29.Harada Y, Ohgai D, Watanabe R, Okano K, Koiwai O, Tanabe K, et al. A single amino acid alteration in cytoplasmic domain determines IL-2 promoter activation by ligation of CD28 but not inducible costimulator (ICOS). J Exp Med (2003) 197:257–62. 10.1084/jem.20021305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harada Y, Tanabe E, Watanabe R, Weiss BD, Matsumoto A, Ariga H, et al. Novel role of phosphatidylinositol 3-kinase in CD28-mediated costimulation. J Biol Chem (2001) 276:9003–8. 10.1074/jbc.M005051200 [DOI] [PubMed] [Google Scholar]
- 31.Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science (1997) 275:1930–4. 10.1126/science.275.5308.1930 [DOI] [PubMed] [Google Scholar]
- 32.Diehn M, Alizadeh AA, Rando OJ, Liu CL, Stankunas K, Botstein D, et al. Genomic expression programs and the integration of the CD28 costimulatory signal in T cell activation. Proc Natl Acad Sci U S A (2002) 99:11796–801. 10.1073/pnas.092284399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wood JE, Schneider H, Rudd CE. TcR and TcR-CD28 engagement of protein kinase B (PKB/AKT) and glycogen synthase kinase-3 (GSK-3) operates independently of guanine nucleotide exchange factor VAV-1. J Biol Chem (2006) 281:32385–94. 10.1074/jbc.M604878200 [DOI] [PubMed] [Google Scholar]
- 34.Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity (2016) 44:955–72. 10.1016/j.immuni.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A (2002) 99:12293–7. 10.1073/pnas.192461099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2006) 439:682–7. 10.1038/nature04444 [DOI] [PubMed] [Google Scholar]
- 37.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443:350–4. 10.1038/nature05115 [DOI] [PubMed] [Google Scholar]
- 38.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192:1027–34. 10.1084/jem.192.7.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okazaki T, Iwai Y, Honjo T. New regulatory co-receptors: inducible co-stimulator and PD-1. Curr Opin Immunol (2002) 14:779–82. 10.1016/S0952-7915(02)00398-9 [DOI] [PubMed] [Google Scholar]
- 40.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol (2001) 2:261–8. 10.1038/85330 [DOI] [PubMed] [Google Scholar]
- 41.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366:2443–54. 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369:122–33. 10.1056/NEJMoa1302369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114:1537–44. 10.1182/blood-2008-12-195792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor A, Harker JA, Chanthong K, Stevenson PG, Zuniga EI, Rudd CE. Glycogen synthase kinase 3 inactivation drives T-bet-mediated downregulation of co-receptor PD-1 to enhance CD8(+) cytolytic T cell responses. Immunity (2016) 44:274–86. 10.1016/j.immuni.2016.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor A, Rothstein D, Rudd CE. Small molecule drug inhibition of PD-1 transcription is an effective alternative to antibody blockade in cancer therapy. Cancer Res (2017):0491. 10.1158/0008-5472.CAN-17-0491 [DOI] [PubMed] [Google Scholar]
- 46.Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science (2017) 355:1423–7. 10.1126/science.aaf0683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krueger J, Rudd CE. Two strings in one bow: PD-1 negatively regulates via co-receptor CD28 on T cells. Immunity (2017) 46:529–31. 10.1016/j.immuni.2017.04.003 [DOI] [PubMed] [Google Scholar]
- 48.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (2017) 355:1428–33. 10.1126/science.aaf1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schneider H, Valk E, Leung R, Rudd CE. CTLA-4 activation of phosphatidylinositol 3-kinase (PI 3-K) and protein kinase B (PKB/AKT) sustains T-cell anergy without cell death. PLoS One (2008) 3:e3842. 10.1371/journal.pone.0003842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng P, Wu Y, Guo Y, Lee C, Liu Y. B7-CTLA-4 interaction enhances both production of antitumor cytotoxic T lymphocytes and resistance to tumor challenge. Proc Natl Acad Sci U S A (1998) 95:6284–9. 10.1073/pnas.95.11.6284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.June CH, Bluestone JA, Nadler LM, Thompson CB. The B7 and CD28 receptor families. Immunol Today (1994) 15:321–31. 10.1016/0167-5699(94)90080-9 [DOI] [PubMed] [Google Scholar]
- 52.Linsley PS, Ledbetter JA. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol (1993) 11:191–212. 10.1146/annurev.iy.11.040193.001203 [DOI] [PubMed] [Google Scholar]
- 53.Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov (2004) 3:479–87. 10.1038/nrd1415 [DOI] [PubMed] [Google Scholar]
- 54.Martinez A, Castro A, Dorronsoro I, Alonso M. Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neurodegeneration, cancer, and inflammation. Med Res Rev (2002) 22:373–84. 10.1002/med.10011 [DOI] [PubMed] [Google Scholar]
- 55.Dieterle AM, Bohler P, Keppeler H, Alers S, Berleth N, Driessen S, et al. PDK1 controls upstream PI3K expression and PIP3 generation. Oncogene (2014) 33:3043–53. 10.1038/onc.2013.266 [DOI] [PubMed] [Google Scholar]
- 56.Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment. Annu Rev Med (2016) 67:11–28. 10.1146/annurev-med-062913-051343 [DOI] [PubMed] [Google Scholar]
- 57.Kaidanovich-Beilin O, Eldar-Finkelman H. Peptides targeting protein kinases: strategies and implications. Physiology (2006) 21:411–8. 10.1152/physiol.00022.2006 [DOI] [PubMed] [Google Scholar]
- 58.Hunig T, Dennehy K. CD28 superagonists: mode of action and therapeutic potential. Immunol Lett (2005) 100:21–8. 10.1016/j.imlet.2005.06.012 [DOI] [PubMed] [Google Scholar]
- 59.Jenkins MR, Tsun A, Stinchcombe JC, Griffiths GM. The strength of T cell receptor signal controls the polarization of cytotoxic machinery to the immunological synapse. Immunity (2009) 31:621–31. 10.1016/j.immuni.2009.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]