

Abstract
Tuberculosis (TB) is currently the number one killer among infectious diseases worldwide. Lipids are abundant molecules during the infectious cycle of Mycobacterium tuberculosis (Mtb) and studies better mimicking its actual metabolic state during pathogenesis are needed. Though most studies have focused on the mycobacterial lipid metabolism under standard culture conditions, little is known about the transcriptome of Mtb in a lipid environment. Here we determined the transcriptome of Mtb H37Rv in a lipid-rich environment (cholesterol and fatty acid) under aerobic and hypoxic conditions, using RNAseq. Lipids significantly induced the expression of 368 genes. A main core lipid response was observed involving efflux systems, iron caption and sulfur reduction. In co-expression with ncRNAs and other genes discussed below, may act coordinately to prepare the machinery conferring drug tolerance and increasing a persistent population. Our findings could be useful to tag relevant pathways for the development of new drugs, vaccines and new strategies to control TB.
Introduction
Mycobacterium tuberculosis (Mtb) remains a leading infectious agent since first described by Robert Koch as the cause of tuberculosis (TB) and is now the first killer among infectious diseases1. Mtb successfully disseminates based in part on its exceptional mechanisms of infection, survival, persistence and dormancy. Drug tolerance and drug resistance, metabolic reactivation from dormancy, and dissemination inside the host, all contribute to its successful pathogenesis and transmission. The World Health Organization declared TB as a global emergency in 1993 and TB research has greatly increased ever since1. However, we often study the virulence and survival factors of Mtb from liquid cultures under “in vitro standard conditions”, which usually implies dextrose as carbon source and an in vitro or in vivo TB model. Anyhow, studies have shown that it is important to control previous conditions of mycobacterial growth before setting up a TB model. For instance, implementation of a detergent-free medium to culture Mtb before setting a macrophage infection model, rather than using a medium containing Tween 80, significantly changed the transcriptome and pathways taken by murine macrophages2. Likewise, implementation of lipids in in vitro dormant models has yielded better mycobacterial recovery and different transcriptomic profiles during in vitro resuscitation, compared to dextrose as sole carbon source3,4. Lipids are important molecules during the life cycle of Mtb as well as during the metabolic dynamics within the host5,6. Mtb promotes and modulates its phagocytosis using complex lipids of the cell wall7 and enters the macrophage through lipid rafts, which mainly contain host cholesterol8. Also, Mtb recruits the protein TACO allocated in cholesterol-rich zones to avoid the phagosome-lysosome formation9. The granuloma formation is also promoted by released lipid microvesicles of Mtb, besides other virulence factors10,11, which led some authors to suggest the granuloma might be protecting the mycobacteria rather than the host12. Nevertheless, Mtb is not restricted to its own lipid homeostasis, it also influences host lipids by promoting the recruitment of low density lipoproteins and the formation of foamy macrophages13 producing cellular necrosis and caseous granuloma14 rich in cholesterol, triglycerides and lactosylceramides10. It is proposed that Mtb uses mainly lipids as carbon and energy source, rather than dextrose, during latency3,15,16. By using 14C-labeled lipids, transmission electron microscopy and auramine-nile staining, several studies have demonstrated that fatty acids and cholesterol consumption by Mtb lead to cytoplasmic lipid bodies accumulation, cell wall remodelling or synthesis of released lipoproteins11,16–19. Moreover, during active TB, Mtb evades the host immune cells by exchanging lipids in its cell wall and hiding its earlier identified pathogen-associated molecular patterns (PAMPs)20. Also, sputum from TB patients reflects the lipid environment from which mycobacteria is coming from, made up mainly of cholesterol, palmitic, stearic and oleic acids21,22. Sputum becomes in turn the microenvironment where the transmissible stage of Mtb resides, whose microscopic phenotype and transcriptional profile suggested there is a shift in the lipid metabolism. Mtb recovered from sputum showed cytoplasmic lipid bodies revealed with auramine-nile red staining, significant overexpression of the large Kst-R regulon of cholesterol catabolism, overpression of tgs1 gene required for triacylglycerol synthesis and down-regulation of genes involved in phthiocerol dimycocerosate and phenol glycolipid synthesis indicating cell wall remodeling23,24. There is also evidence that hypercholesterolemia increases susceptibility to TB in mice25 and a survey in Singapore found positive correlation between a cholesterol diet and an increased risk to active TB in Chinese population26. It is thus important to investigate not only the mycobacterial lipid metabolism but also, the mycobacterial response to lipids and the host lipid homeostasis during TB infection. Studies that better mimic the actual metabolic state of Mtb during pathogenesis are needed to fully tag therapeutic targets against TB. Although previous studies have focused on the mycobacterial lipid metabolism under different “standard” conditions (drugs, stress), less is known about the transcriptome of Mtb in a model that better mimics the lipid environment of the caseous granuloma, rich in cholesterol and fatty acids. This could uncover an unexplored field, potentially useful for the development of new drugs, vaccines and new strategies for controlling TB. In this study we determined the transcriptome of Mtb in a lipid-rich dormancy model through RNAseq technology, which has the advantage over qRT-PCR or microarrays, of producing the transcriptome of the organism with a higher resolution, including codifying and non-codifying genes, intergenic regions, small RNAs or antisense transcripts. Moreover, the overexpression of some genes is perfectly quantified without noise, which is usually found in other fluorescence-based systems. This study exposes the transcriptome landscape of Mtb H37Rv in the presence of the most abundant lipids inside the host, specifically, cholesterol and long chain fatty acids (C16:0, C18:0, C18:1), using dextrose carbon source as control, under aerobic and hypoxic conditions27.
Results
In vitro cultures of Mtb were grown in the presence of lipids (cholesterol and fatty acids) as previously described3 and parallel cultures with dextrose were used as control. Six conditions were studied: lipid exponential phase (LE), lipid stationary phase (LS), lipid NRP1 stage (LNRP1), dextrose exponential phase (DE), dextrose stationary phase (DS) and dextrose NRP1 (DNRP1). Mycobacterial RNA from all conditions was subjected to RNAseq analysis. Codifying genes (CDS) and non-coding RNAs (ncRNA) were classified according to functional categories listed in Tuberculist28, IGRs and sRNA candidates16,29, not listed in Tuberculist28, were added and featured as genes in the annotation file. After quality control evaluation and trimming of bad.
Qualitative reads, a mean of 54.8 ± 9.4 million paired reads per individual library were detected. 76.3 ± 3.9% of total reads mapped in the sense direction, while 4.3 ± 2.5% mapped in the anti-sense direction. It is worthy of notice that DS samples had slightly more anti-sense reads (9 ± 0.6%) than the rest of the conditions (Fig. 1A). Deep sequencing analysis of the sense reads showed that more ncRNAs were found in DS, LS and LNRP1, 32 ± 1.4%, 41 ± 0.8% and 28 ± 3.3% of total aligned sense-reads, respectively (Fig. 1B). Finally, the principal component analysis (PCA) and the clustering heatmap plots showed that the samples clustered by their biological replicates (Fig. 2).
Figure 1.

RNA-seq mapping results for every experimental condition and biological replicates. Sense and anti-sense mapping is figured in the chart (A). Grey bars show the reads mapped in the sense direction. Not mapped reads were mapped in the anti-sense direction and are represented by black bars. The chart (B) show stacked columns of total mapped reads in the sense direction. Gray bars with white dots show the percentage of reads aligned to ncRNA and light gray bars show the percentage of reads aligned to CDS. Detailed data of RNA-seq mapping can be seen in the Supplemental Material, Table S3.
Figure 2.

Plots of the principal component analysis (A) and the clustering-heatmap (B) of all genes after normalization using edge R. For the PCA plot, the data was rlog transformed. For the clustering heatmap, normalized counts were rescaled between −3 and 3, clustering was based on Pearson correlation. The six experimental conditions clustered with their biological replicates.
Differential gene expression analysis for lipid conditions
To investigate the impact of lipids during the different stages, differential gene expression (DGE) analysis for the first general lineal model was performed comparing lipid conditions vs dextrose (D vs L). Six replicates for each dextrose and lipid group were performed, ordered in three subgroups (exponential, stationary and NRP1). Differentially expressed genes were filtered using a false discovery rate (FDR) of <0.05 and a log2 fold change >1 or <−1 for biological significance. After filtering, 368 genes were found differentially expressed in the presence of lipids (Fig. 3 and Table S1 in the Supplemental Material) representing 7.8% of the total covered genes. From these, 183 genes decreased and 185 increased their expression (Fig. 3A). As shown in the PCA plot and in the heatmap visualization (Fig. 3B), the 368 genes clustered by the carbon source (lipid or dextrose) and by biological replicates. Regarding the functional categories, most of the differentially expressed genes belonged to the categories of conserved hypothetical proteins, cell wall and cell processes, intermediary metabolism and respiration and the appended category of IGR and sRNA candidates (Fig. 3C). These results were not unexpected given the substantial number of genes that are included in these four categories compared with the others in the whole genome of Mtb. For this reason, we proceeded to perform a differential functional category analysis, which took into account the category size. Results showed that the category of insertion sequences and phages as well as the PE/PPE category decreased their expression and, as expected, the presence of lipids prompted the expression of the lipid metabolism category, together with the intermediary metabolism and respiration category (Fig. 3D and Supplemental Material Table S4). Specifically, the ten genes with increased expression from the lipid metabolism were ltp2, pks2, tgs2, mutA, rv3087, rv1425, fadA5, fadD2, fadE7 and fadE9. Whilst the genes with diminished expression from the insertion sequences and phages category were rv2646, rv3427c, rv2647, rv1199c and rv2512c and from the PE/PPE family were PE_PGRS62, PE15, PE21, PE22, PE23, PE25, PE3, PE8, PPE1, PPE21, PPE22, PPE23, PPE24, PPE34, PPE57 and PPE67. It also draws our attention the MTS2823 ncRNA from the category of stable RNA, since it was the only one from this category with significant and differential increased expression in the presence of lipids, in the first general linear model (D vs L).
Figure 3.

Differential gene expression of Mtb H37Rv in presence of lipids. Conditions with dextrose were used as control and growth stages were used as batch effect. Counts were normalized using edge R, all genes with a CPM greater than 1 in at least 2 samples were used in the analysis. 368 significant differentially expressed genes were found. (A) Volcano plot (on the left) and its Venn diagram (on the right) where 185 genes with diminished expression and 183 genes with increased expression in presence of lipids are specified. (B) PCA and heatmap plots of the 368 differential expressed genes that were rescaled between −3 and 3, clustering was based on the Pearson correlation. (C) Number of differentially expressed genes by functional categories are shown, circle sizes are proportional to the number of genes with significant differential expression, total number of genes per functional category listed in tuberculist are indicated in parenthesis. (D) Differential functional categories analysis. Only significant expressed categories (FDR < 0.05) are indicated.
Additionally, a differential pathway analysis (DPA) was performed using the KEGG database30, to determine the impact of lipids over all mycobacterial phases. Again, dextrose was used as control, growth phases were set as a batch effect. This analysis predicted significantly induced or repressed pathways, either in a direct or indirect mode. The results of this analysis confirmed the induction of the steroid degradation and fatty acid degradation pathways (see Supplementary Material Table S5). Additionally, lipids promoted two-component systems, pyruvate metabolism, carbon metabolism, lysine degradation, valine, leucine and isoleucine degradation, tryptophan metabolism, butanoate metabolism and oxidative phosphorylation.
Differential gene expression analysis for every stage of growth of Mtb
Three linear models of DGE were made for the lipid growth stages LE, LS and LNRP1, with dextrose cultures (DE, DS and DNRP1) as controls. This analysis identified 1241 (557 down- and 684 up-regulated) differentially expressed genes in LE; 2043 (1196 down- and 847 up-regulated) differentially expressed genes in LS, while the LNRP1 stage showed the smallest number of genes, with 138 (43 down- and 95 up-regulated) differentially expressed (Fig. 4 and Supplementary Material Table S2). The number of genes differentially expressed per category can be seen in Fig. 5A. Again, the biggest categories: conserved hypothetical proteins, cell wall and cell processes, intermediary metabolism and respiration and the appended IGRs and sRNA candidates, had the higher number of significantly expressed genes. The differential functional categories analysis took into account the size of every category (Fig. 5B and Supplemental Material Tables S6, S7 and S8). Through this analysis, it was possible to define the categories up- and down-regulated in every phase of growth. The PE/PPE category was significantly down-regulated in the two models of dormancy in the presence of lipids (LS and LNRP1), while the lipid and the intermediary metabolism and respiration were significantly up-regulated in both stages. Among the differences between these two stages, we had two categories up-regulated in LS, information pathways and cell wall and cell processes categories, while the virulence and regulatory proteins categories were up-regulated during the lipid hypoxia LNRP1. On the other hand, the exponential stage only had the information pathways category significantly down-regulated.
Figure 4.

Differential gene expression of Mtb H37Rv in every stage of growth in presence of lipids. Dextrose conditions were used as controls. The heatmap and volcano plots are shown in figure (A) for the 1241 differentially expressed genes in LE, figure (B) shows the 2043 differentially expressed genes in LS and figure (C) shows the 138 differentially expressed genes in LNRP1. Normalized counts in the heatmap were rescaled between −3 and 3 with clustering based on Pearson correlation and differentially expressed genes were considered significant using a false discovery rate of <0.05 and a log2 fold change >1 or <−1.
Figure 5.

Differential gene expression per functional categories of every stage of growth of Mtb. (A) The number of differentially expressed genes are shown by functional categories, where circle sizes are proportional to the number of genes with significant differential expression. Blue circles are used for genes with diminished expression and red circles are used for genes with increased expression. Total number of genes per functional category listed in tuberculist are indicated in parenthesis. (B) Differential functional Categories analysis are indicated in tables for every stage of growth, wherein the mean log2 fold change is significantly different from the general mean log2 fold change at a FDR <0.05.
The main core lipid response
Figure 6 shows the number of genes that belong to the core induced by the presence of lipids. It shows a Venn diagram with the set of significantly increased genes in lipids, from the first DGE analysis (D vs L), that overlaped with significantly increased genes in the subsequent DGE lineal models per stage of growth ([DE vs LE], [DS vs LS] and [DNRP1 vs LNRP1]). Although all of them were significantly overprexpressed in the presence of lipids, only six genes from the comparison D vs L overlapped in the three stages of Mtb, and they were called “the main core lipid response”, comprising Rv3161c, Rv3160c, Rv0678, Rv1217c, PPE53 and che1. Genes from the remain sets and detailed gene expression data can be checked in the Supplementary Material, Table S1 and S2.
Figure 6.
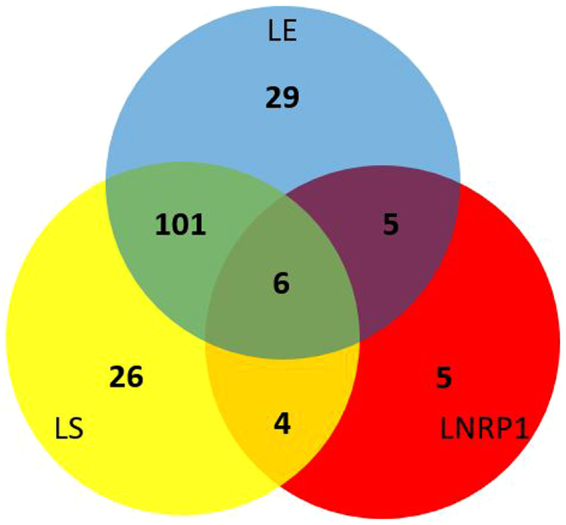
Core of genes induced by the presence of lipids.
qRT-PCR validation
Validation of RNAseq results was made through qRT-PCR, for which a set of ten significantly up-regulated genes in the presence of lipids were confirmed. Four technical replicates for each two biological replicates were tested for gene expression at every stage of growth in lipids or dextrose. Seven of the most significantly upregulated genes from the first linear DGE analysis (comparing L vs D) were chosen: rv3160c, rv3161c, PPE53, che1, mmpS5, hsd4A and usfY. Additionally, three significantly upregulated genes from the DGE analysis for every stage of growth were chosen based on lipid phase exclusivity: hrp1 was overexpressed in LE, rv1639c in LS and rv0560 in LNRP1. Absolute gene expression of the ten genes was determined and normalized to 16 S rRNA gene (Fig. 7A). For relative quantification, the absolute transcription level of genes in every lipid stage was compared with their respective dextrose control (LE/DE, LS/DS and LNRP1/DNRP1; Fig. 7B).
Figure 7.

Validation of ten differential expressed genes in lipid conditions by qRT-PCR. (A) Absolute gene expression during DE (gray bars), LE (black bars), DS (light blue bars), LS (dark blue bars), DNRP1 (light green bars) and LNRP1 (dark green bars) conditions. Data were normalized to the number of copies of rrs gene and results are expressed as log10 of the mean ± SD number of copies per microgram of RNA. Asterisks indicate significant diference between a given lipid stage compared to its respective dextrose stage (LE vs DE, LS vs DS and LNRP1 vs DNRP1), with P < 0.05 considered statistically significant. (B) The relative quantification is expressed as the ratio of [transcription in LE/transcripion in DE] (black bars), the ratio of [transcription in LS/transcription in DS] (blue bars) and the ratio of [transcription in LNRP1/transcription in DNRP1] (green bars).
Similar to the DGE results, the seven chosen genes from the comparison L vs D were up-regulated in the three lipid conditions (LE, LS and LNRP1) by qRT-PCR, though the upregulation of PPE53 in LS and che1 in LE were not statistically significant. Likewise, hrp1, rv1639c and rv0560 were the most expressed in LE, LS and LNRP1, respectively, although hrp1 was not exclusive of LE due to the given significant overexpression in LS as well (Fig. 7 and Supplemental Material S9 for statistics).
Discussion
Previous studies have shown a close link between persistent mycobacteria and drug tolerance31. Persistent mycobacteria are presumed to be the reason for many treatment failures, even in the presence of drug susceptible mycobacteria. The problem can reside in multiple mycobacterial phenotypes, including the persistent non-culturable mycobacteria, which cannot be subjected to DST. All these populations have in common the presence of host lipids, at several levels, from host membranes to caseous granuloma or sputum. In fact, Mtb not only modulates its own lipid metabolism but also influences the host lipid metabolism to be fully surrounded by lipids at one point of its life cycle5. Through RNAseq we confirmed that lipid accession is one more stimulus, besides other stress factors, by which Mtb increases their persistent population. The differences between the Rodriguez et al., study16 and ours are mainly based on the culture conditions and the bioinformatic analysis. Rodriguez et al. selected the even-length long-chain fatty acids (LC-FAs) as the sole carbon source. In the present study, to gain more insight into the M. tuberculosis adaptation to a fatty acid environment, we implemented cholesterol together with the fatty acids as carbon source, considering the propionyl-CoA pool, which is a central metabolite to produce methyl-branched lipids of the cell wall. Another difference is the hypoxic condition evaluated in the presence of lipids and dextrose of this study. Limited oxygen is a feature of granulomas and hypoxic models have been frequently used to study dormant mycobacteria. Additionally, the library construction, normalization of the data, DGE analysis and statistical analysis were completely different between both studies. Yet, the study of Rodríguez et al., must be considered since the authors confirmed that fatty acids prompted a phenotype of dormancy and the study highlighted relevant features of the transcriptome of Mtb during fatty acid consumption.
Here, Mtb differentially expressed a core of 368 genes in the presence of lipids (183 down- and 185 up-regulated). Our study included lipids in aerobic exponential growth, aerobic stationary phase and hypoxic stage (NRP1), the last two better mimicking dormancy27,32. Since lipids are present and involved in most stages of Mtb during TB pathogenesis6, these 368 genes may be clues in the mycobacterial metabolism during its survival. The genes Rv3161c, Rv3160c, Rv0678, Rv1217c, PPE53 and che1, form what we call “the main core lipid response”; and have been linked to processes of drug detoxification, iron acquisition and efflux and transport systems also co-expressed with the mmpL5-mmpS5 operon. The correlated overexpression of Rv3160c-RV3161c and PPE53 have also been found in the presence of cell wall and respiration inhibitors (Fig. 8)33,34. Thus, it is proposed that this operon is required for detoxification of drugs35, although there is no evidence of its direct involvement in drug resistance36. This operon is co-expressed with mmpS5-mmpL5 in the presence of drugs (Fig. 9)33 as it happened in the presence of lipids of our study. The MmpS5-mmpL5 efflux pump is not only related to drug resistance but siderophore transportation37,38. Another gene from the mean core lipid response related to iron utilization was che1, whose codified product is a ferrochelatase. This gene resides in a sulfur reduction operon, constituted by sirA, cysH, che1, ggtB 39,40. The role of Che1 is to insert iron into a siroheme.
Figure 8.

Correlated gene expression among PPE53, Rv3160c and Rv3161c. Plots show data reported by Boshoff and colleagues through microarray assay, which is deposited in the correlation catalog of the TBDB data base41,42. (A). Comparison between PPE53 and Rv3160c; (B). Comparison between PPE53 and Rv3161C; (C). Comparison between Rv3161c and Rv3161c. Black frames and circles highlight positive correlation between two genes in samples of Mycobacterium tuberculosis under diferent conditions. Gene expression responses of Mtb to 76 treatment groups are represseented by red dots. Plots resumed experimental categories: 1) samples of Mtb under inhibitors of cell wall synthesis (left), 2) samples of Mtb under diferent inhibitors of respiration (middle) and 3) “others” (on the right) inlcludes 8 categories more, such as acidified médium, agents that affect DNA integrty or topology, aromatic amides that can be hydrolyzed intracellylary, growth associated with dosR regulon, inhibitors of protein synthesis, minimal medium with palmitate or succinate as carbon, starvation and transcriptional inhibitors. It can be seen that PPE53, Rv3160c and Rv3161c are mainly co-expressed in compounds that affect cell wall synthesis and respiration. On the other hand, these genes are not induced by compounds from the cathegory “others”.
Figure 9.

Correlated gene expression between Rv3161c and mmpL5. Plots show data reported by Boshoff and colleagues33 through microarray assay, which is deposited in the correlation catalog of the TBDB data base41,53. Black frames highlight positive correlation between Rv3161c and mmpL5 (>1 for both) in samples of Mycobacterium tuberculosis under diferent conditions. Gene expression responses of Mtb to 76 treatment groups are represseented by red dots. Plots resumed experimental categories: 1) samples of Mtb under inhibitors of cell wall synthesis (upper left), 2) samples of Mtb under diferent inhibitors of respiration (upper right), 3) in presence of aromatic amides (lower left) and 4) “others” (lower right) inlcludes 7 categories more, such as acidified médium, agents that affect DNA integrty or topology, growth associated with dosR regulon, inhibitors of protein synthesis, minimal medium with palmitate or succinate as carbon, starvation and transcriptional inhibitors. DCCD: Dicyclohexylcarbodiimide, TRZ: Thioridazine, CPZ: Chlorpromazine, CFZ: Clofazimine, KCN: Potassium cyanide, PZA: Pyrazinamide33,54. It can be seen that Rv3161c and mmpL5 are mainly co-expressed in compounds that affect cell wall synthesis and respiration as well as in presence of aromatic amides. Besides, these genes are not co-expressed by compounds from the cathegory “others”.
Rv1217c (from the mean core lipid response) is predicted to be part of the operon integrated by Rv1216c, Rv1217c, Rv1218c and Rv1219c 39. All were differentially overexpressed in the presence of lipids in our study (Table S1). Likewise, this operon is co-expressed with mmpS5-mmpL5 in the presence of the drugs41 as it happened in the presence of lipids of our study.
It is also important to mention the overexpression of an efflux pump codified by mmr gene and a sulfur reduction operon (Rv2391 – Rv2394). All these genes are linked somehow and were significantly overexpressed in the presence of lipids in this study (Table S1). Mmr is an efflux pump involved in transport of multidrugs and codified next to Rv3066 that encodes a transcriptional regulator belonging to the deoR-family. They belong to the same operon39 and Rv3066 is predicted to repress mmr 42. Rv3066 was not up-regulated under lipid conditions; in fact, its transcription was significantly diminished in LS stage allowing transcription of mmr. Hence, we hypothesize that all genes mentioned above are coordinated to act not only at the level of intracellular redox balance but also preparing the machinery that confers drug tolerance to Mtb.
According to the differential functional category analysis (DFCA), it is not surprising that lipids mostly induced the expression of the lipid metabolism category (Fig. 3D). This was also confirmed by a differential pathway analysis using KEGG30, which showed activation of fatty acid degradation and steroid degradation pathways. Specifically, essential genes for in vitro growth in cholesterol43 such as cyp125, kstD, hsaB, hsaC, fadA5, ltp2 and pks2 were induced, as well as genes for lipid degradation, including fadE9, fadD2, fadE7. Additionally, there were genes involved in lipid biosynthesis, such as for triacylglycerol (TAG) (tgs2, Rv3087 and Rv1425) and mutA, necessary for multimethyl-branched lipid biosynthesis through the methylmalonyl pathway. Based on this, we can assume that Mtb used cholesterol and fatty acids as carbon source and synthetized new macro-lipids, either for lipid deposition as TAG or for cell wall remodeling.
There were eight PE/PPE genes overexpressed in the DGE core (PPE53, PE_PGRS54, PE_PGRS5, PE_PGRS10, PE_PGRS57, PE_PGRS15, PE_PGRS53, PE_PGRS33). From these, only PPE53 was strongly up-regulated in the three lipid phases of this study. The role of PE/PPE proteins remains uncertain but evidence suggests that they confer virulence variability to mycobacteria. A system of oxidative stress was significantly overexpressed in the lipid stationary phase (LS) compared to cultures in dextrose (DS). This system includes the enzymes KatG, SodA and AhpC44, all essential for virulence44. Also, during stationary phase, lipids induced expression of vapC1, vapC22, vapB1, vapB10, vapB48 and vapB46, while during exponential phase (LE) and hypoxia (LNRP1), there was overexpression of complete modules vapB22/vapC22 and vapB9/vapC9, respectively (Table S2), suggesting that lipids promote a higher survival through vapBC components in exponential and hypoxic phases and may contribute to persistence population appearance.
Lipids also induced the overexpression of non-codifying genes in this study. At least one third of the Mtb transcriptome, including rRNA, belongs to non-codifying RNAs (ncRNAs)45. Our results confirmed that the transcriptome of Mtb drifts towards a greater proportion of non-codifying transcripts, out of rRNA, under different in vitro stress conditions as previously reported16,45,46. In this study, those ncRNA transcripts were more abundant in the stationary phases and under lipid hypoxia (Fig. 1B; DS, LS and LNRP1), confirming previous studies that suggest that ncRNA are strongly involved in mycobacterial survival. The small ncRNA MTS2823 was the only significantly overexpressed ncRNA found from the stable RNA’s category (Fig. 3C). The DGE analysis showed that up-regulation of MTS2823 was co-expressed with sigE which was up-regulated in the lipid exponential phase with significant down regulation of prpC/prpD genes, needed for the methylcitrate cycle (Table S2). There was also overexpression of desA3 in the lipid hypoxic condition (LNRP1).
In summary, using RNAseq we determined the transcriptomic profile induced by a mixture of cholesterol, palmitic, stearic and oleic acids, in Mtb H37Rv, over three metabolic stages, exponential and stationary stages, as well as for the non-replicating persistence stage 1 of hypoxia (NRP1). The transcriptomic profile suggested that cholesterol and fatty acids were used as carbon source and induced what we have called the mean core lipid response. This mainly induced a machinery that confers drug tolerance to a vast variety of drugs but also ensures the maintenance of iron caption and sulfide production, necessary for enzymes and coenzymes needed for redox balance and production of central metabolites, such as acetyl-CoA and methyl-malonyl-CoA, needed for de novo lipid biosynthesis (Fig. 10). As additional studies confirm the importance of lipids during the TB pathology, the core lipid response may give us some clues to target relevant pathways in the Mtb metabolism and find better therapeutic targets, for antibiotic and vaccine development as well as for better therapeutic regimens of TB patients.
Figure 10.

Principal metabolic pathways activated by the core lipid response.
Methods
Bacterial strain and growth conditions
Mtb cultures were handled under the specific biosafety recommendations established by WHO47. Mtb H37Rv was cultured in Dubos broth (Difco), without glycerol, containing 0.5% albumin (Sigma Fraction V), supplemented with either 0.2% dextrose or a lipid mixture (oleic acid, palmitic acid, stearic acid, at final concentration of 0.001% each, plus 0.01% cholesterol). Three growth phases of Mtb were cultivated for each carbon source (dextrose or lipids), as it was previously described by Soto-Ramirez et al., 2017. Specifically, Mtb was grown until reaching 1) its aerobic exponential phase (DE: dextrose exponential phase and LE: lipid exponential phase), 2) its aerobic stationary phase (DS: dextrose stationary phase and LS: lipid stationary phase) and 3) its NRP1 stage of dormancy (DNRP1: dextrose NRP1 stage and LNRP1: lipid NRP1 stage), for which, a parallel culture supplemented with methylene-blue (1.5 mg mL) was used as an indicator of oxygen depletion. A total of six experimental conditions were assessed and at least two biological replicates were done for all conditions. For lipid stages Mtb was grown to exponential stage containing 2.4 × 106 ± 9 × 105 CFU/mL, OD600 between 0.3 to 0.5, at day 7 of incubation, stationary stage containing 2.9 × 106 ± 2 × 106 CFU/mL, OD600 between 0.7 to 0.9, at day 15 of incubation and NRP1 stage containing 2.7 × 106 ± 8 × 105 CFU/mL, OD600 between 0.3 to 0.4, at day 8 of hypoxia. For dextrose stages Mtb was grown to exponential stage containing 8.3 × 106 ± 1.86 × 106 CFU/mL, OD600 = 0.3, at day 7 of incubation, stationary stage containing 2.9 × 108 ± 9.43 × 106 CFU/mL, OD600 = 0.6, at day 15 of incubation and NRP1 stage containing 5.3 × 106 ± 1.1 × 106 CFU/mL, OD600 = 0.3, at day 5 of hypoxia.
RNA isolation
Total RNA was isolated from all Mtb cultures as previously described48. All work involving viable Mtb was performed in a dedicated Biosafety Level 3 (BSL 3) laboratory. In short, each culture (200 mL) was harvested by centrifugation and pellets were resuspended in 2 mL of lysis buffer (6 M guanidinium chloride, 0.1% Tween 80, 1 mM 2-mercaptoethanol, 10 mM EDTA). Cells were lysed mechanically in a FastPrep cell disrupting equipment (Thermo Scientific) with 150 μm–to 200 μm glass beads (Sigma-Aldrich) by performing three lysis cycles of 45 s, at 4.0 m/s followed by two lysis cycles of 15 s at 6.5 m/s. Nucleic acids were extracted with phenol-chloroform-isoamyl alcohol (25:24:1) and RNA was precipitated with 0.4 volume of absolute ethanol. Total RNA was purified three times with Trizol reagent (Invitrogen), RNA integrity was analyzed with a Bioanalyzer (Agilent Technologies), quantified by spectrophotometry with the NanoDrop ND-1000 (Thermo Scientific) and DNA absence was evaluated by qPCR, using primers for amplification of the gene rpsL (see Supplementary Material, Table S10). Finally, RNA was dissolved in DEPC-treated water and stored at −70 °C until further use.
RNA amplification and sequencing library preparation
Concentration and quality of the total extracted RNA was checked by using the ‘Quant-it ribogreen RNA assay’ (Life Technologies, Grand island, NY, US) and the RNA 6000 pico chip (Agilent Technologies, Santa Clara, CA, USA), respectively. One µg of RNA was used to start the library preparation. Ribosomal RNA was removed from the total RNA using the ‘Ribo-zero Magnetic Bacteria kit’ (Epicentre), according to manufacturer’s protocol. Subsequently, RNA cleanup was performed with the ‘Rneasy MinElute Cleanup kit’ (Qiagen). After fragmentation and random priming, first and second strand cDNA was synthesized using the ‘NEBNextUltra Directional RNA Library Prep Kit for Illumina’ (New England Biolabs), according to manufacturer’s protocol. After adapter ligation, the library was cleaned up twice using the Agencourt AMPure XP system (Beckman Coulter Inc.), followed by a 14 cycles PCR amplification, according to manufacturer’s instructions. Then, the library underwent a final cleanup using the Agencourt AMPure XP system (Beckman Coulter Inc.) after which the libraries’ size distribution and quality was assessed using a high sensitivity DNA chip (Agilent Technologies). Libraries were subsequently quantified by qPCR, according to Illumina’s protocol ‘Sequencing Library qPCR Quantification protocol guide’, version February 2011. Finally, equimolar quantities (1.9 pM) of all libraries were sequenced by a high throughput run on the Illumina NextSeq. 500 sequencer using 2 × 150 bp paired-end reads and a 1% Phix spike-in control.
Read alignment and differential gene-expression analysis
A custom.gff annotation file was made for reference genome NC_000962.3 of Mtb H37Rv. The original RefSeq annotation file was appended with all sRNA candidate and IGR’s described in the Supplementary data Miotto et al., 2012 and Rodriguez et al., 2014. Annotations on the same strand with more than 50% overlap with another annotation were considered duplicates. After filtering by origin, where RefSeq annotations were retained over Rodriguez et al., which in turn were retained over Miotto et al., a single duplicate with maximal length remained. All annotated features: genes, IGRs and candidates were annotated as genes for gene expression analysis.
To quantify gene expression, the RNA-seq Analysis tool from the CLC Genomics Workbench 9.0.1 software (CLC Bio, Aarhus, Denmark) was used to map the reads to gene regions only with strand-specificity set to reverse and default settings. Count tables were made by counting the reads that mapped against the genes and exported from CLC.
To explore if the samples from different treatment groups clustered together and to detect outlier samples, a Principal Component Analysis (PCA) was performed in R on the rlog49 normalized and transformed counts of the genes expressed at cpm above 1 in at least 2 samples.
Differential gene expression analysis was performed using edgeR50. Genes were only retained if they were expressed at a cpm above 1 in at least 2 samples. Four different models were built. The first general linear model was made with 2 treatments “dextrose” vs “lipid”, defining the effects from the growth phases “exponential”, “stationary” and “NRP1” as batch effect. The last three general linear models were made with 2 treatments each “dextrose exponential” vs “lipid exponential”, “dextrose stationary” vs “lipid stationary” and “dextrose NRP1” vs “lipid NRP1”. A differential functional categories analysis was done for the mentioned four comparisons, using dextrose groups as control, lipids as treatment and growth stages as batch effect. Likewise, a differential pathways analysis was performed using KEGG30 for the general linear model D vs L.
The set of genes with significant increased expression obtained from the first general linear model (DGE: D vs L), that overlapped with significant increased expression in the subsequent DGE lineal models per stage of growth ([DE vs LE], [DS vs LS] and [DNRP1 vs LNRP1]) were grouped in a Venn diagram. This set of genes was defined as the “core lipid response”. Additionally, only those genes that overlapped in the three stages of Mtb, were called “the mean core lipid response”.
The data is available at the GEO database of NCBI. The accession number is GSE100097.
qRT-PCR
The total transcripts of selected genes were measured by real-time qRT-PCR. cDNA was synthetized from 1 µg of total RNA using the “SuperScriptTM First-Strand Synthesis System for RT-PCR” kit (Invitrogen). Quantification was performed with gene-specific primers (see Supplemental Material Table S10), which were designed by the Primer-Blast tool51 and based on the genome sequence for Mtb H37Rv with Gene bank access number NC_000962.3. qPCR was performed by using the start SYBR Green I Master mix and the LightCycler® 480 system (Life Science Roche). Quantification was carried out in the six experimental conditions (DE, LE, DS, LS, DNRP1, LNRP1), using four technical replicates for each two biological replicates. The qPCR was subjected to 40 cycles of amplification and a melting curve analysis was performed for ensuring that the fluorescence levels detected were due to the amplification of a specific product. To determine the absolute gene expression, a standard curve was obtained for each set of primers by using 10-fold dilutions of known amounts of Mtb H37RV chromosomal DNA (100, 1 000, 10 000, 100 000, and 1 000 000 theoretical copies). Threshold cycle values of each RNA quantitation were interpolated to standard curve to obtain gene expression (number of gene copies per microgram of RNA). Normalization of these data was performed by using rrs gene (16 S rRNA) expression levels. The statistical analysis was carried out in SIGMASTAT software version 3.5, through a Two-way ANOVA and Turkey’s multiple-comparison, P value < 0.05 was considered significant.
Statistical analysis
Statistical testing for differential gene expression (DGE) was done using the empirical Bayes quasi-likelihood F-test. For all DGE analysis performed in this work, genes, functional categories and KEGG pathways, having a false discovery rate (FDR) <0.05 and a fold change >2 were considered significantly differential. PCA was performed in R on rlog49 transformed cpm of the differentially expressed genes. To produce the heat map52, the normalized count data of the differentially expressed genes was rescaled between −3 and 3 and clustered based on Pearson correlation.
The statistical analysis for qRT-PCR results was carried out in SIGMASTAT software version 3.5, through a Two-way ANOVA and Turkey’s multiple-comparison, to determine the significance of differences of gene expression between lipid and dextrose conditions. P value < 0.05 was considered significant, with one degree of freedom.
Electronic supplementary material
Acknowledgements
This study was partially funded by a Research Grant 2015 given by the European Society of Clinical Microbiology and Infectious Diseases (ESCMID) to DAAA. We acknowledge the support of the BOF fellowship (“Bijzonder Onderzoeksfonds” register number 01W01514, special foundation for research of Ghent University, Belgium) to DAA. It is gratefully acknowledged the support of the CONACyT (Mexico) scholarship to DAA and CONACYT grant, CB-2016-283984 to JAG-y-M. Jorge A. Gonzalez-y-Merchand is fellow of COFAA and EDI, IPN. The authors acknowledge Sander Willems for his preliminary bioinformatic data processing contribution.
Author Contributions
D.A.A.A. conducted the experimental work, analyzed the results, designed and compiled the figures and tables, and wrote the complete first draft of the manuscript. L.T., F.V.N. and D.D. carried out the bioinformatic and static analysis related to RNAseq. P.V. supervised and guided the research project, ensured resolution of financial issues and equipment availability. J.C.P. contributed with advices and constructive critiques, reviewed results and corrected language style in the final manuscript. J.A.G.Y.M. and A.M. jointly and equally supervised the experimental work and results, guided the research project, ensured resolution of financial issues and equipment availability, contributed with advices and constructive critiques.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-17751-x.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.WHO. Global tuberculosis report 2016. Geneva: World Health Organization Available at: http://www.who.int/tb/about/ (2016).
- 2.Leisching G, et al. The host response to a clinical MDR mycobacterial strain cultured in a detergent- free environment: A global transcriptomics approach. PLoS One. 2016;11:1–16. doi: 10.1371/journal.pone.0153079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soto-Ramirez MD, et al. Cholesterol plays a larger role during Mycobacterium tuberculosis in vitro dormancy and reactivation than previously suspected. Tuberculosis. 2017;103:1–9. doi: 10.1016/j.tube.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Shleeva, M. et al. Cyclic AMP-dependent resuscitation of dormant mycobacteria by exogenous free fatty acids. PLoS One8, (2013). [DOI] [PMC free article] [PubMed]
- 5.Lovewell RR, Sassetti CM, VanderVen BC. Chewing the fat: lipid metabolism and homeostasis during M. tuberculosis infection. Curr. Opin. Microbiol. 2016;29:30–36. doi: 10.1016/j.mib.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Aguilar-Ayala DA, Palomino JC, Vandamme P, Martin A, Gonzalez-y-Merchand JA. ‘Genetic regulation of Mycobacterium tuberculosis in a lipid-rich environment’. Infect. Genet. Evol. 2017;55:392–402. doi: 10.1016/j.meegid.2016.10.015. [DOI] [PubMed] [Google Scholar]
- 7.Siegrist MS, Bertozzi CR. Mycobacterial lipid logic. Cell Host Microbe. 2014;15:1–2. doi: 10.1016/j.chom.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muñoz S, Rivas-Santiago B, Enciso JA. Mycobacterium tuberculosis entry into mast cells through cholesterol-rich membrane microdomains. Scand. J. Immunol. 2009;70:256–263. doi: 10.1111/j.1365-3083.2009.02295.x. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen L, Pieters J. The Trojan horse: Survival tactics of pathogenic mycobacteria in macrophages. Trends Cell Biol. 2005;15:269–276. doi: 10.1016/j.tcb.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Kim M-J, et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2010;2:258–274. doi: 10.1002/emmm.201000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rezwan M, Grau T, Tschumi A, Sander P. Lipoprotein synthesis in mycobacteria. Microbiology. 2007;153:652–658. doi: 10.1099/mic.0.2006/000216-0. [DOI] [PubMed] [Google Scholar]
- 12.Pagán AJ, Ramakrishnan L. Immunity and immunopathology in the tuberculous granuloma. Cold Spring Harb. Perspect. Med. 2014;5:1–19. doi: 10.1101/cshperspect.a018499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahajan S, et al. Mycobacterium tuberculosis modulates macrophage lipid-sensing nuclear receptors PPARγ and TR4 for survival. J. Immunol. 2012;188:5593–603. doi: 10.4049/jimmunol.1103038. [DOI] [PubMed] [Google Scholar]
- 14.Dietzold J, Gopalakrishnan A, Salgame P. Duality of lipid mediators in host response against Mycobacterium tuberculosis: good cop, bad cop. 2015;8:1–8. doi: 10.12703/P7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cantrell SA, et al. Free mycolic acid accumulation in the cell wall of the mce1 operon mutant strain of Mycobacterium tuberculosis. J. Microbiol. 2013;51:619–626. doi: 10.1007/s12275-013-3092-y. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez JG, et al. Global Adaptation to a Lipid Environment Triggers the Dormancy-Related Phenotype of Mycobacterium tuberculosis. MBio. 2014;5:e01125–14. doi: 10.1128/mBio.01125-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. USA. 2008;105:4376–80. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee W, VanderVen BC, Fahey RJ, Russell DG. Intracellular Mycobacterium tuberculosis Exploits Host-derived Fatty Acids to Limit Metabolic Stress. J. Biol. Chem. 2013;288:6788–6800. doi: 10.1074/jbc.M112.445056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daniel, J., Maamar, H., Deb, C., Sirakova, T. D. & Kolattukudy, P. E. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 7, (2011). [DOI] [PMC free article] [PubMed]
- 20.Arbues A, Lugo-Villarino G, Neyrolles O, Guilhot C, Astarie-Dequeker C. Playing hide-and-seek with host macrophages through the use of mycobacterial cell envelope phthiocerol dimycocerosates and phenolic glycolipids. Front. Cell. Infect. Microbiol. 2014;4:173. doi: 10.3389/fcimb.2014.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cha D, et al. Analysis of fatty acids in sputum from patients with pulmonary tuberculosis using gas chromatography-mass spectrometry preceded by solid-phase microextraction and post-derivatization on the fiber. J. Chromatogr. A. 2009;1216:1450–1457. doi: 10.1016/j.chroma.2008.12.039. [DOI] [PubMed] [Google Scholar]
- 22.Mourão MPB, Denekamp I, Kuijper S, Kolk AHJ, Janssen H-G. Hyphenated and comprehensive liquid chromatography × gas chromatography–mass spectrometry for the identification of Mycobacterium tuberculosis. J. Chromatogr. A. 2016;1439:152–160. doi: 10.1016/j.chroma.2015.10.054. [DOI] [PubMed] [Google Scholar]
- 23.Sharma S, et al. Transcriptome analysis of mycobacteria in sputum samples of pulmonary tuberculosis patients. PLoS One. 2017;12:e0173508. doi: 10.1371/journal.pone.0173508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garton NJ, et al. Cytological and Transcript Analyses Reveal Fat and Lazy Persister-Like Bacilli in Tuberculous Sputum. PLoS Med. 2008;5:e75. doi: 10.1371/journal.pmed.0050075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martens GW, et al. Hypercholesterolemia impairs immunity to tuberculosis. Infect. Immun. 2008;76:3464–3472. doi: 10.1128/IAI.00037-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soh, A. Z., Wang, Y., Yuan, J. & Koh, W. Dietary Cholesterol Increases the Risk whereas PUFAs Reduce the Risk of Active Tuberculosis. J. Nutr. 1–8 10.3945/jn.115.228049.The (2016). [DOI] [PMC free article] [PubMed]
- 27.Wayne LG, Hayes LG. An In vitro Model for Sequential Study of Shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun. 1996;64:2062–2069. doi: 10.1128/iai.64.6.2062-2069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lew JM, Kapopoulou A, Jones LM, Cole ST. TubercuList – 10 years after. Tuberculosis. 2011;91:1–7. doi: 10.1016/j.tube.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 29.Miotto P, et al. Genome-Wide Discovery of Small RNAs in Mycobacterium tuberculosis. PLoS One. 2012;7:e51950. doi: 10.1371/journal.pone.0051950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogata H, et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ayrapetyan M, Williams TC, Oliver JD. Bridging the gap between viable but non- culturable and antibiotic persistent bacteria. Trends Microbiol. 2014;23:7–13. doi: 10.1016/j.tim.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 32.Keren, I., Minami, S., Rubin, E. & Lewis, K. C. −3119538. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. MBio 2, e00100–11 ST–Characterization and transcriptome (2011). [DOI] [PMC free article] [PubMed]
- 33.Boshoff HIM, et al. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism. Novel insights into drug mechanisms of action. J. Biol. Chem. 2004;279:40174–40184. doi: 10.1074/jbc.M406796200. [DOI] [PubMed] [Google Scholar]
- 34.Betts JC, et al. Signature gene expression profiles discrimate between isoniazid- thiolactomycin-, and triclosan-treated Mycobacterium tuberculosis. Antimicrob.Agents Chemother. 2003;47:2903–2913. doi: 10.1128/AAC.47.9.2903-2913.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dutta NK, Mazumdar K, Dastidar SG, Amaral PCK, L New Patentable Use of an Old Neuroleptic Compound Thioridazine to Combat Tuberculosis: A Gene RegulationPerspective. Recent Patents on Anti-Infective Drug Discovery. 2011;6:128–138. doi: 10.2174/157489111796064597. [DOI] [PubMed] [Google Scholar]
- 36.Gomez A, Andreu N, Ferrer-Navarro M, Yero D, Gibert I. Triclosan-induced genes Rv1686c-Rv1687c and Rv3161c are not involved in triclosan resistance in Mycobacterium tuberculosis. Sci. Rep. 2016;6:1–7. doi: 10.1038/srep26221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bailo R, Bhatt A, Aínsa JA. Lipid transport in Mycobacterium tuberculosis and its implications in virulence and drug development. Biochem. Pharmacol. 2015;96:159–167. doi: 10.1016/j.bcp.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 38.Almeida Da Silva, P. E. et al. Antimicrobial Resistance in Bacteria. 10.1007/978-3-319-39658-3 (2007).
- 39.Roback P, et al. A predicted operon map for Mycobacterium tuberculosis. Nucleic Acids Res. 2007;35:5085–5095. doi: 10.1093/nar/gkm518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pinto R, Harrison JS, Hsu T, Jacobs WR, Leyh TS. Sulfite reduction in mycobacteria. J. Bacteriol. 2007;189:6714–6722. doi: 10.1128/JB.00487-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boshoff, H. I. M. et al. Correlation Catalog for M. tuberculosis H37Rv. Available at: http://tuberculosis.bu.edu/tbdb_sysbio/CC/Rv1217c.html. (Accessed: 1st May 2017)
- 42.Reddy TBK, et al. TB database: An integrated platform for tuberculosis research. Nucleic Acids Res. 2009;37:499–508. doi: 10.1093/nar/gkn652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Griffin JE, et al. High-Resolution Phenotypic Profiling Defines Genes Essential for Mycobacterial Growth and Cholesterol Catabolism. PLoS Pathog. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forrellad MA, et al. Virulence factors of the Mycobacterium tuberculosis complex. Virulence. 2013;4:3–66. doi: 10.4161/viru.22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arnvig KB, et al. Sequence-Based Analysis Uncovers an Abundance of Non-Coding RNA in the Total Transcriptome of Mycobacterium tuberculosis. PLoS Pathog. 2011;7:e1002342. doi: 10.1371/journal.ppat.1002342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rienksma RA, et al. Comprehensive insights into transcriptional adaptation of intracellular mycobacteria by microbe-enriched dual RNA sequencing. BMC Genomics. 2015;16:34. doi: 10.1186/s12864-014-1197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.WHO. World Health Organization. Tuberculosis Laboratory Biosafety Manual. (2012). [PubMed]
- 48.Gonzalez-y-Merchand JA, Colstonl MJ, Cox RA. The rRNA operons of Mycobacterium smegmatis and Mycobacterium tuberculosis: comparison of promoter elements and of neighbouring upstream genes. Microbiology. 1996;142:667–674. doi: 10.1099/13500872-142-3-667. [DOI] [PubMed] [Google Scholar]
- 49.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson MD, McCarthy DJ, Smyth G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ye J, et al. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Culhane AC, Thioulouse J, Perrière G, Higgins DG. MADE4: An R package for multivariate analysis of gene expression data. Bioinformatics. 2005;21:2789–2790. doi: 10.1093/bioinformatics/bti394. [DOI] [PubMed] [Google Scholar]
- 53.McGuire A, et al. Comparative analysis of mycobacterium and related actinomycetes yields insight into the evolution of Mycobacterium tuberculosis pathogenesis. BMC Genomics. 2012;13:120. doi: 10.1186/1471-2164-13-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boshoff HIM, Mizrahi V. Expression of Mycobacterium smegmatis pyrazinamidase in Mycobacterium tuberculosis confers hypersensitivity to pyrazinamide and related amides. J. Bacteriol. 2000;182:5479–5485. doi: 10.1128/JB.182.19.5479-5485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.