

Abstract
Purpose of review
The current review aims to provide an update on the recent biomedical interest in oncogenic branched-chain amino acid (BCAA) metabolism, and discusses the advantages of using BCAAs and expression of BCAA-related enzymes in the treatment and diagnosis of cancers.
Recent findings
An accumulating body of evidence demonstrates that BCAAs are essential nutrients for cancer growth and are used by tumors in various biosynthetic pathways and as a source of energy. In addition, BCAA metabolic enzymes, such as the cytosolic branched-chain aminotransferase 1 (BCAT1) and mitochondrial branched-chain aminotransferase 2, have emerged as useful prognostic cancer markers. BCAT1 expression commonly correlates with more aggressive cancer growth and progression, and has attracted substantial scientific attention in the past few years. These studies have found the consequences of BCAT1 disruption to be heterogeneous; not all cancers share the same requirements for BCAA metabolites and the function of BCAT1 appears to vary between cancer types.
Summary
Both oncogenic mutations and cancer tissue-of-origin influence BCAA metabolism and expression of BCAA-associated metabolic enzymes. These new discoveries need to be taken into consideration during the development of new cancer therapies that target BCAA metabolism.
Keywords: branched-chain amino acids, branched-chain aminotransferase 1, branched-chain aminotransferase 2, cancer
INTRODUCTION
Cancer cells have unlimited potential to divide and sustain growth. This process is dependent on acquiring essential nutrients from the tumor microenvironment, which are used to maintain biomass and survival, even under conditions of poor nutrient and oxygen availability [1,2▪,3]. The metabolic flexibility of cancer cells is determined by their ability to reprogram anabolic and catabolic pathways, through altering gene expression programs as well as intercellular interactions within the tumor microenvironment [4].
The process of oncogenesis is dependent on amino acids, the building blocks for protein synthesis, and a source of energy and metabolites [3]. Many cancer types overexpress enzymes that function to degrade amino acids, which not only provide cellular energy and metabolites for anabolic processes but also serve as mechanisms of immune evasion by cancer cells [2▪,5]. For example, tumor overexpression of indoleamine-2,3, dioxygenase and arginase depletes the tumor microenvironment of tryptophan and arginine, respectively, which is beneficial for tumor growth but also suppresses local cytotoxic T-cell proliferation [6–8]. Thus, by using amino acid degrading enzymes as immunosuppressive factors, tumors increase their ability to survive.
In addition to arginine and tryptophan, tumors also preferentially uptake the branched-chain amino acids (BCAAs) leucine, isoleucine, and valine [5]. BCAAs can be used for protein synthesis or oxidized for energy purposes by tumors. BCAAs are essential amino acids; tumors must rely on dietary BCAA intake and their release from protein degradation [9] (Fig. 1). In recent years, it has become evident that the enzymes catalyzing the first step in BCAA degradation are overexpressed in many cancers [10,11,12▪]. These are the cytosolic [branched-chain aminotransferase 1 (BCAT1)] and mitochondrial [branched-chain aminotransferase 2 (BCAT2)] branched-chain aminotransferases, which convert BCAAs into their corresponding branched-chain α-keto acids by transferring the amino group onto α-ketoglutarate and thereby generating glutamate [13]. Of the two enzymes, BCAT1 is the major isoform implicated in cancer growth and has been proposed as a prognostic cancer cell marker [5,10,14–17,18▪▪]. The role of BCAT1 in cancer progression has become an intriguing but challenging topic to understand, with several different functions in tumor growth having been proposed [12▪,18▪▪]. In this review, we summarize the latest discoveries on the utility of BCAT1 expression as a prognostic cancer cell marker and the recent mechanistic insights into how BCAT1 contributes to the metabolic reprograming of cancer cells. Next, we address the most recent understanding of the role of BCAA metabolism in cancer growth and progression. Lastly, we discuss current and future opportunities to clinically target BCAA metabolism in the context of cancer therapies.
FIGURE 1.
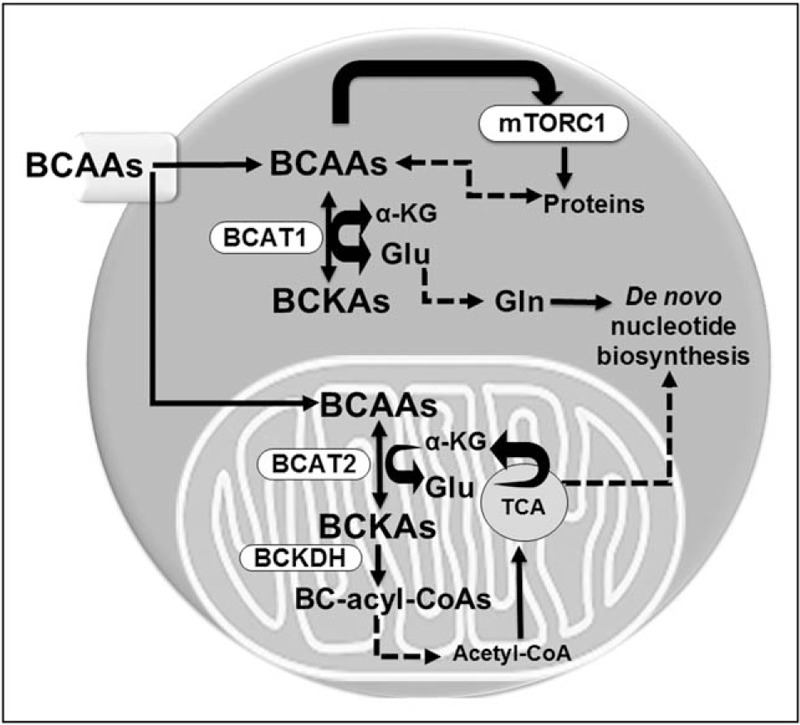
A model of branched-chain amino acid metabolism in cancer. As essential amino acids, cancer cells must obtain branched-chain amino acids from the tumor microenvironment or from protein degradation. Branched-chain amino acids are thought to play several roles in cancer cells: activate complex 1 of the mammalian target of rapamycin signaling, which stimulates protein translation, growth, and survival; serve as building blocks in protein synthesis; be metabolized into branched-chain α-keto acids in the cytosol (by branched-chain aminotransferase 1) and/or mitochondria (by branched-chain aminotransferase 2), a process involving conversion of α-ketoglutarate to glutamate; serve as indirect source of nitrogen for nucleotide (and nonessential amino acid) biosynthesis via the glutamate–glutamine axis; and become further catabolized to yield acetyl-CoA and succinyl-CoA (not shown) that feed into the cycle of tricarboxylic acids cycle and can contribute to energy production. Note that in some cancers (such as chronic myeloid leukemia), branched-chain aminotransferase 1 is proposed to convert branched-chain α-keto acids back to branched-chain amino acids. BCAA, branched-chain amino acid; BCKA, branched-chain α-keto acid; BCKDH, branched-chain keto acid dehydrogenase; BC-acyl-CoAs, branched-chain acyl-CoAs; α-KG, α-ketoglutarate; TCA, cycle of tricarboxylic acids; mTORC1, complex 1 of the mammalian target of rapamycin.
Box 1.

no caption available
BRANCHED-CHAIN AMINOTRANSFERASE 1 IS A PROGNOSTIC CANCER MARKER AND AN ATTRACTIVE TARGET FOR CANCER THERAPIES
The role of BCAT1 in cancer development was largely overlooked until recently. It was not until 2013, when Tonjes et al.[10] reported overexpression of BCAT1 in gliomas, that the scientific community became interested in this metabolic enzyme and its potential in cancer therapy. Since 2013, scientific knowledge about BCAT1 in cancer has been steadily accumulating with an average of seven publications/year (per ‘PubMed’ search). Current knowledge indicates that most cancer types express high levels of BCAT1 [5,14,17]. By contrast, BCAT1 expression in healthy humans is mainly limited to the nervous system and gonadal tissues [5], as well as activated T lymphocytes [13] and macrophages [19]. The cancer-specific expression of BCAT1 makes this gene an attractive target for therapeutic intervention. However, the biological functions of BCAT1 in cancer are not well understood, and recent evidence suggests it may be dependent on the cancer tissue-of-origin [9,12▪].
BCAT1 expression in glioblastoma tumors is specific to those carrying wild-type isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) [10]. Mutations in either IDH1 or IDH2, commonly seen in glioblastomas, contribute to downregulation of BCAT1 through DNA methylation of the BCAT1 promoter and the corresponding epigenetic silencing of BCAT1[10] (Table 1). Mutations in IDH1/2 are common in gliomas and acute myeloid leukemia (AML), whereas solid tumors rarely harbor IDH mutations [10,15,20,21]. Significantly, BCAT1 was recently found to be highly expressed in AML, where it contributed to growth in vitro[18▪▪]. On the other hand, an inverse relationship between BCAT1 and IDH1/2 was found in epithelial ovarian cancer (EOC), where BCAT1 silencing suppressed the expression of IDH1/2 genes [15]. Within the cycle of tricarboxylic acids (TCA) cycle, wild-type IDH enzymes convert isocitrate into α-ketoglutarate, whereas mutant IDH enzymes convert isocitrate into hydroxyglutarate [10]. Given α-ketoglutarate is used by BCAT1 for BCAA transamination, this points toward a possible metabolic link between IDH1/2 and BCAT1. Resulting perturbations to TCA cycle-associated metabolites and energy production may have contributed to the accelerated cellular proliferation, migration, and invasion observed in EOC [15] (Table 1).
Table 1.
Cancer type-dependent expression of branched-chain aminotransferase and their downstream effects
| Cancer type | BCAT expression | Upstream regulators of BCAT | Metabolites (cancer tissue) | Metabolites (plasma) | Downstream targets of BCAT | Downstream effects | References |
| Glioblastoma | BCAT1 overexpression in IDHwt BCAT1 epigenetic silencing in IDHmut glioblastomas | IDHmut-dependent silencing of BCAT1 | In Bcat1 knockdown:Increased BCAAs Decreased Glu excretion | N/A | Bcat1 knockdown led to:HADH suppression | Bcat1 suppression led to:Smaller tumors in mice | [10] |
| PDAC | ME3 dependent suppression of BCAT2Low BCAT1 expressionHigh BCAT2 expression | ME3, AMPK and SREBP1 regulate BCAT2N/A | In ME3 depleted PATU8988T cells:Increased BCAAsDecreased BCAA uptakeSlightly decreased BCAAsIncreased citrate | N/AIncreased BCAAs | BCAT2-dependent nucleotide biosynthesisN/A | Bcat2 overexpression led to:Aggressive tumor growth in miceBcat1 suppression led to:No change in tumor growth | [11,12▪] |
| NSCLC | High BCAT1 expressionHigh BCAT2 expression | N/A | High BCAAsHigh Glu + GlnHigh nucleotides | Decreased BCAAs | N/A | Bcat1 suppression led to:Impaired ability to form tumors in mice | [12▪] |
| Ovarian cancer | High BCAT1 expression in EOC | Epigenetic hypomethylation of BCAT1Activation of BCAT1 via c-Myc | BCAT1 knockdown in SKOV3 cells led to:Low Leu and IleLow sphingolipidsLow glycerophospholipid | N/A | Bcat1 knockdown led to:suppression of IDH1, IDH2, AKR1C1, and PHGDH | Bcat1 suppression led to:No effect on tumor burden Increased survival rate in mice | [15] |
| Breast cancer | High BCAT1 expression in unspecified breast cancer tissuesHigh BCAT1 expression in:ERα−, HER2, and TNBC | N/AEpigenetic hypomethylation of BCAT1 in ERα− | High BCAAsBCAT1 knockdown in TamR8 and MDA-MB231 led to: High Glu, Ala, Pro, and BCAAs | High BCAAsLow Glu, GlnN/A | BCAT1 knockdown led to:Inhibition of mTOR Suppression of PGC-1a, NRF-1, TFAM, SOD, Catalase, and Gpx1p27kip1 (inhibition) by BCAT1 | BCAT1 overexpression in MCF-7 and T47D led to:Increased growth and colony formationBcat1 suppression led to:Decreased tumor size in miceReduced migration and invasion | [16,30] |
| Liver cancer | High expression in HCC | Activation of BCAT1 via c-Myc | N/A | N/A | Bcat1 knockdown led to:Suppression of G2/M phaseLC3A/B, p62 (inhibition) | Bcat1 overexpression led to:Accelerated tumor growth Increased tumor size in mice | [17] |
| CML | High BCAT1 expression in BC-CML, AML | MSI2 binds BCAT1mRNA | In Bcat1 knockdown: Low BCAAsLow GluNo change in BCAA uptake | N/A | mTOR (activation) | Bcat1 and BCR-ABL1 overexpression in HSPCs led to:Splenomegaly and increased mortality in mice | [18▪▪] |
AKR1C1, aldo-keto reductase family 1 member C1; Ala, alanine; AML, acute myeloid leukemia; AMPK, AMP-dependent protein kinase; BCAA, branched-chain amino acid; BCAT1, branched-chain aminotransferase 1; BC-CML, blast crisis of chronic myeloid leukemia; EOC, epithelial ovarian cancer; ERα−, estrogen receptor negative breast cancer; Gln, glutamine; Glu, glutamate; Gpx1, glutathione peroxidase 1; HADH, hydroxyacyl-CoA dehydrogenase; HCC, hepatocellular carcinoma; HER2, human epidermal growth factor receptor 2 triggered breast cancer; HSPC, hematopoietic stem and progenitor cell; IDHmut, tissue specific knock-in of isocitrate dehydrogenase; IDHwt, wild-type isocitrate dehydrogenase; Ile, isoleucine; LC3A/B, autophagy marker light chain 3, isoforms A and B; Leu, leucine; MCF-7, breast cancer cell line; MDA-MB231, human breast adenocarcinoma cell line; ME3,malic enzyme 3; MSI2, musashi RNA binding protein 2; mTOR, mammalian target of rapamycin; NRF-1, nuclear respiratory factor 1; NSCLC, nonsmall cell lung cancer; p62, ubiquitin-binding scaffold protein 62; PDAC, pancreatic ductal adenocarcinoma; PGC-1a, peroxisome proliferator-activated receptor-gamma coactivator 1alpha; PHGDH, phosphoglycerate dehydrogenase; SKOV3, ovarian carcinoma cell line; SOD, superoxide dismutase; SREBP-1, sterol regulatory element-binding protein 1; T47D, human breast cancer cell line; TFAM, mitochondrial transcription factor A; TNBC, triple negative breast cancer.
Most, but not all, reports indicate that BCAT1 overexpression correlates with enhanced cancer growth, whereas suppression of BCAT1 limits proliferation. For example, suppression of BCAT1 in U-87MG, a human primary glioblastoma cell line, produced smaller tumors in mice [10]. Similarly, when Bcat1-null nonsmall lung carcinoma (NSCLC) cells were implanted subcutaneously in mice, these cells displayed impaired tumor-forming ability [12▪]. However, when mice were injected with SKOV3 ovarian carcinoma cells with suppressed BCAT1 expression, tumor burden was not alleviated, although survival rates were significantly increased as compared with control animals [15] (Table 1). Likewise, suppression of BCAT1 in pancreatic ductal adenocarcinoma (PDAC) did not lead to a reduction in tumor growth, and patients with PDAC expressed low levels of tumor BCAT1 and displayed increased plasma BCAAs levels [12▪]. Thus, not all cancer types express high levels of BCAT1 and suppression of BCAT1 does not always correspond to a decrease in tumor size.
In several tumor types, the epigenetic dysregulation of BCAT1 expression has been elucidated. The best described epigenetic mechanism involves mutated IDH1/2, as discussed above [10]. However, another epigenetic mechanism, involving the disruptor of telomeric silencing 1-like (DOT1L) histone methyltransferase, was recently proposed [22]. In contrast to IDH1/2 mutations, DOT1L activates BCAT1 gene expression through histone H3K79 methylation of the coding region, but not the promoter, of BCAT1[22]. In leukemias, driven by genetic mutation of the mixed lineage leukemia 1 (MLL1) gene, DOT1L maintains an open chromatin state and gene transcription. DOT1L also forms part of the elongation assisting protein complex, along with positive transcription elongation factor b, among others, which is recruited by oncogenic MLL1 fusion proteins (such as MLL-AF9) to stimulate RNA Pol II gene transcription [23]. In addition, DOT1L can cooperate with c-Myc and p300 to enhance transcription [24]. BCAT1 has also been described as a downstream target of c-Myc in many cancers, including ovarian and liver cancer [5,15,17]. Thus, it is intriguing to speculate that at least one mechanism of BCAT1 upregulation in cancer may involve cooperation between DOT1L and c-Myc.
Most recently, a new posttranscriptional regulator of BCAT1 expression was identified in chronic myeloid leukemia (CML), the musashi RNA binding protein 2 (MSI2) [18▪▪]. MSI2 and BCAT1 are coexpressed in CML blast crisis, and a physical interaction between MSI2 protein and BCAT1 mRNA was identified, suggesting that BCAT1 expression in CML is MSI2-dependent. Moreover, the MSI2–BCAT1 axis was proposed as an important mechanism in driving cancer progression in CML [18▪▪] (Table 1).
Taken together, these studies point toward a role of BCAT1 as a prognostic cancer marker, although the mechanisms of BCAT1 gene dysregulation appear to differ between cancer types. In addition, these data suggest inhibition of BCAT1 activity may be a useful therapeutic strategy in the treatment of several cancers.
REPROGRAMING OF BRANCHED-CHAIN AMINO ACID METABOLISM TO ACTIVATE MAMMALIAN TARGET OF RAPAMYCIN SIGNALING IN CANCER
The BCAA leucine is a well described mammalian target of rapamycin (mTOR) agonist [13], and Sestrin2 was recently identified as a direct intracellular leucine sensor and mTOR complex 1 (mTORC1) regulator [25▪▪,26]. Many cancers rely on constitutive mTOR activity to maintain cellular growth and proliferation [27]. Recent reports have linked BCAT1 expression to mTOR activity in several cancers, although different mechanisms have been proposed [16,18▪▪]. Hattori et al.[18▪▪] demonstrated that BCAT1 was overexpressed in CML blast crisis. In this context, rather than deaminating BCAAs to BCKAs, BCAT1 overexpression resulted in increased intracellular concentrations of BCAAs through BCKA amination. Reduced BCAT1 expression (or activity through pharmacological inhibition) resulted in reduced mTORC1 activity, presumably through reduced intracellular BCAA concentrations. Importantly from a therapeutic standpoint, knockdown of BCAT1 in a mouse model of CML improved survival, while use of the BCAT1 inhibitor gabapentin suppressed colony formation of human patient CML [18▪▪].
Zhang and Han [16] also found BCAT1 expression promoted mTOR activity, but in the context of breast cancer cells. The authors found that BCAAs were increased in patients with breast cancer (compared with healthy controls), in both peripheral blood serum and cancer tissue, and BCAT1 was also overexpressed. BCAT1 expression also contributed to the growth of breast cancer cell lines and appeared to act through mTORC1 activity (Table 1). However, downstream BCAA catabolic enzymes were also overexpressed in breast cancer cells [16], suggesting catabolism of BCKAs into the TCA cycle may also play a role in this context. Significantly, recent metabolic analysis of BRCA1-mutant breast epithelial cells also identified increased BCAA concentrations, suggesting increased BCAA concentrations via reprogramed metabolism may be an early event in BRCA1-cancer development [28].
Further mechanistic insight into the role of BCAT1 overexpression in breast cancer was recently identified by Thewes et al.[29]. In ERα-negative breast cancer, BCAT1 indirectly regulated the cell cycle regulator retinoblastoma protein through the cell cycle inhibitor p27Kip1. Here, BCAT1 controlled cell cycle progression, sustaining breast cancer proliferation. By contrast, Takegoshi et al.[30] suggested that BCAAs prevented development of hepatocellular carcinoma in mice models of nonalcoholic steatohepatitis. As in the studies described above, BCAAs appeared to act via mTORC1. These data suggest BCAAs have cancer-specific functions and that in certain contexts, BCAAs may even suppress cancer development.
Combined, these recent findings highlight the importance of BCAT1 and BCAA metabolism in reprograming cancer metabolism via mTORC1, with profound consequences on cell cycle and cancer progression.
BRANCHED-CHAIN AMINO ACIDS SUPPORT THE CANCER ENERGETIC AND BIOSYNTHETIC DEMANDS
BCAAs play an important role in energy homeostasis and nutrient signaling as well as nitrogen balance [31,32]. Several recent studies have found BCAA metabolism to be an important ‘module’ within cancer metabolism, but appear to drive cancer progression by diverse mechanisms [9,11,12▪]. For example, transamination of BCAAs leads to formation of glutamate, which can be used for biosynthesis of other nonessential amino acids such as glutamine, or recycled to α-ketoglutarate via other aminotransferases [33,34]. In NSCLC tumors, high glutamate and glutamine concentrations correlated with an increased expression of BCAT1 and higher rates of BCAA uptake [12▪]. Similarly, knockdown of BCAT1 expression (or pharmacological BCAT1 inhibition) in glioblastoma cells reduced the formation of glutamate [10]. However, not all cancers produced high levels of glutamate in response to overexpression of BCAT1. As described above, in CML blast crisis, high expression of BCAT1 correlated with lower intracellular BCKAs and glutamate concentrations [18▪▪].
Mayers et al.[12▪] provided an elegant demonstration that the same oncogenic event can result in very different BCAA metabolism and suggested that metabolic activity depended on the tissue-of-origin rather than oncogenic mutation. This study focused on mouse models of PDAC and NSCLC, both driven by Kras mutation combined with p53 deletion. Lung-derived tumors actively took up and catabolized BCAAs to BCKAs, whereas pancreas-derived tumors did not. During oncogenic transformation, PDAC cells even appeared to shut down catabolic flux of BCAAs through downregulating expression of several enzymes in the BCAA catabolic pathway, including BCAT2. In the NSCLC, nitrogen derived from BCAA deamination was used to support biosynthesis of nonessential amino acids and nucleotides [12▪] (Table 1).
However, other reports provided evidence that the genetic mutations can also influence how BCAA metabolism impacts cancer progression. Although in the above example of PDAC, BCAA metabolism was suppressed, BCAA catabolism via BCAT2 was recently found to play an important role in PDAC, driven by the chr18q21 chromosomal deletion [11]. Chromosomal region 18q21 is commonly deleted in solid tumors and can impact the expression of many housekeeping genes, including the mitochondrial malic enzyme 2 (ME2). In this study, knockdown of BCAT2 in ME2-deficient PDAC cell lines inhibited colony formation, which could be rescued by nucleotide supplementation, suggesting BCAAs to be an important nitrogen source for nucleotide biosynthesis in this cancer type (Table 1). By contrast, carbon from BCAAs could not be detected in TCA cycle metabolites by metabolic flux analysis, suggesting BCAAs were not a major source of energy in this cell type [11]. Combined, these data suggest that cancer BCAA requirements are dependent on the tissue-of-origin as well as the genetic mutation.
Thus, BCAA metabolism directly influences cancer growth, but different metabolic states are expected based on the cancer type, the genetic mutation, and/or tumor microenvironment in a complex relationship that needs to be addressed in future studies.
TARGETING BRANCHED-CHAIN AMINO ACID METABOLISM IN CANCER CLINICAL TRIALS
Recent clinical studies
Likely due to the ease of administration, numerous studies have investigated the consequences of BCAA supplementation on disease progression in clinical trials [35–40]. Although clinical trials investigating BCAAs in different cancers are currently ongoing, most recently published clinical studies involving BCAA in cancer treatments have focused on BCAA supplementation in liver disease and its progression to liver carcinoma [35–38]. Nojiri et al.[35] investigated the consequences of BCAA supplementation following radiative ablation of hepatocellular carcinoma in a study involving 51 patients. Several statistically significant differences were observed between the BCAA supplement patients and control group. Importantly, event-free survival was increased, whereas complications were reduced, suggesting BCAA supplementation may be efficacious in this patent population. In a second clinical study, involving BCAA supplementation in hepatocellular carcinoma, Shiozawa et al.[39] found that in a study involving 77 patients, BCAA supplementation could also improve patient outcomes. These findings were also corroborated with a third recent clinical observational study, involving 307 patients, which also found that BCAA supplementation benefited patients with advanced liver disease [38].
In addition to clinical trials involving hepatocellular carcinoma, the value of BCAT1 as a diagnostic marker was recently tested in patients with colorectal cancer, alongside the ikaros family zinc finger 1 (IKZF1) gene [41,42]. Cell-free circulating methylated DNAs of BCAT1 and IKZF1 were monitored in patient's blood of nearly 3500 patients scheduled for colonoscopy. The BCAT1/IKZF1 blood test was found to be 75% positive for recurrences, which points toward its utility in patients with remission [41]. However, further clinical studies are necessary to determine the broader diagnostic value of BCAT1 status in different cancers.
Future prospects for therapeutic targeting of branched-chain amino acid metabolism
As described above, several recent studies have found BCAT1 overexpression to be associated with cancer growth and the activity of BCAT1 to be oncogenic [18▪▪,29]. These studies suggest the prospect of using BCAT1 to develop targeted cancer therapies. Indeed, the fact that Bcat1-knockout mice are viable [13,43] suggests there may be a good therapeutic window for targeting BCAT1-dependent cancers.
An alternative approach was recently suggested by Taya et al.[44], based on their findings that hematopoietic stem cells (HSCs) required the BCAA valine. HSCs are important for homeostasis of the adult hematopoietic system and are used clinically in HSC transplantation, a curative treatment for a range of hematological diseases including leukemias. For donor HSCs to engraft, recipients must normally undergo irradiation or chemotherapy. Taya et al.[44] found that dietary depletion of valine could be used to condition the bone marrow and afford donor HSC engraftment. These findings open up the prospect of metabolic condition regimens based on BCAA modulation.
CONCLUSION
The past few years of in-depth research on BCAA metabolism in cancer has provided strong evidence for the essential role of BCAAs in tumor progression and has clearly established BCAT1 as an important prognostic cancer marker. Moreover, BCAA supplementation and BCAT1 status were tested in clinical trials for hepatocellular carcinoma and colorectal cancer. However, the recent research also revealed a complex addiction of cancer cells to BCAA metabolites, which appear dependent on both the tissue-of-origin and the cancer genetics. This heterogeneous reliance of cancer cells on BCAAs needs to be addressed with future studies so that therapeutic approaches aiming to target BCAA metabolism in cancer can be successfully developed.
Acknowledgements
None
Financial support and sponsorship
A.C.W. is supported by Bloodwise (15050) and the National Institutes of Health National Center for Advancing Translational Science Clinical and Translational Science Award (UL1 TR001085). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Reina-Campos M, Moscat J, Diaz-Meco M. Metabolism shapes the tumor microenvironment. Curr Opin Cell Biol 2017; 48:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2▪.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv 2016; 2:e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]; The recent review by DeBerardinis and Chandel provides a useful overview of the role of metabolism in cancer.
- 3.Vazquez A, Kamphorst JJ, Markert EK, et al. Cancer metabolism at a glance. J Cell Sci 2016; 129:3367–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lyssiotis CA, Kimmelman AC. Metabolic interactions in the tumor microenvironment. Trends Cell Biol 2017; doi: 10.1016/j.tcb.2017.06.003. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ananieva E. Targeting amino acid metabolism in cancer growth and antitumor immune response. World J Biol Chem 2015; 6:281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allison KE, Coomber BL, Bridle BW. Metabolic reprogramming in the tumour microenvironment: a hallmark shared by cancer cells and T lymphocytes. Immunology 2017; 152:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selvan SR, Dowling JP, Kelly WK, et al. Indoleamine 2,3-dioxygenase (IDO): biology and target in cancer immunotherapies. Curr Cancer Drug Targets 2016; 16:755–764. [DOI] [PubMed] [Google Scholar]
- 8.Morris CR, Hamilton-Reeves J, Martindale RG, et al. Acquired amino acid deficiencies: a focus on arginine and glutamine. Nutr Clin Pract 2017; 32:30S–47S. [DOI] [PubMed] [Google Scholar]
- 9.Mayers JR, Vander Heiden MG. Nature and nurture: what determines tumor metabolic phenotypes? Cancer Res 2017; 77:3131–3134. [DOI] [PubMed] [Google Scholar]
- 10.Tonjes M, Barbus S, Park YJ, et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med 2013; 19:901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dey P, Baddour J, Muller F, et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 2017; 542:119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12▪.Mayers JR, Torrence ME, Danai LV, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016; 353:1161–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]; The recent article from Mayers et al. provided important evidence that tissue-of-origin plays an important role in the activity of the branched-chain amino acid (BCAA) metabolic pathway in cancer.
- 13.Ananieva EA, Powell JD, Hutson SM. Leucine metabolism in T cell activation: mTOR signaling and beyond. Adv Nutr 2016; 7:798S–805S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panosyan EH, Lin HJ, Koster J, et al. In search of druggable targets for GBM amino acid metabolism. BMC Cancer 2017; 17:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang ZQ, Faddaoui A, Bachvarova M, et al. BCAT1 expression associates with ovarian cancer progression: possible implications in altered disease metabolism. Oncotarget 2015; 6:31522–31543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L, Han J. Branched-chain amino acid transaminase 1 (BCAT1) promotes the growth of breast cancer cells through improving mTOR-mediated mitochondrial biogenesis and function. Biochem Biophys Res Commun 2017; 486:224–231. [DOI] [PubMed] [Google Scholar]
- 17.Zheng YH, Hu WJ, Chen BC, et al. BCAT1, a key prognostic predictor of hepatocellular carcinoma, promotes cell proliferation and induces chemoresistance to cisplatin. Liver Int 2016; 36:1836–1847. [DOI] [PubMed] [Google Scholar]
- 18▪▪.Hattori A, Tsunoda M, Konuma T, et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 2017; 545:500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]; Until this recent article by Hattori et al., convention held that overall flux of the BCAA metabolic pathway was catabolic. However, Hattori et al. demonstrated that branched-chain aminotransferase 1 (BCAT1) activity was reversed in chronic myeloid leukemia (CML), generating high intracellular BCAA concentrations that stimulated mammalian target of rapamycin (mTOR) activity and cancer progression. This article further identified MSI2 as a posttranscriptional regulator of BCAT1 expression, suggesting a regulatory mechanism for BCAT1 overexpression in CML.
- 19.Papathanassiu AE, Ko JH, Imprialou M, et al. BCAT1 controls metabolic reprogramming in activated human macrophages and is associated with inflammatory diseases. Nat Commun 2017; 8:16040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saygin C, Carraway HE. Emerging therapies for acute myeloid leukemia. J Hematol Oncol 2017; 10:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujii T, Khawaja MR, DiNardo CD, et al. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med 2016; 21:373–380. [PubMed] [Google Scholar]
- 22.Oktyabri D, Ishimura A, Tange S, et al. DOT1L histone methyltransferase regulates the expression of BCAT1 and is involved in sphere formation and cell migration of breast cancer cell lines. Biochimie 2016; 123:20–31. [DOI] [PubMed] [Google Scholar]
- 23.Wong M, Polly P, Liu T. The histone methyltransferase DOT1L: regulatory functions and a cancer therapy target. Am J Cancer Res 2015; 5:2823–2837. [PMC free article] [PubMed] [Google Scholar]
- 24.Cho MH, Park JH, Choi HJ, et al. DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat Commun 2015; 6:7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25▪▪.Wolfson RL, Chantranupong L, Saxton RA, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016; 351:43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]; Although leucine is a well described mTOR complex 1 (mTORC1) agonist, it was only here that a direct sensor of leucine was identified, Sestrin 2. This article therefore filled in an important part of the leucine-mTORC1 jigsaw puzzle.
- 26.Saxton RA, Knockenhauer KE, Wolfson RL, et al. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 2016; 351:53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell 2017; 169:361–371. [DOI] [PubMed] [Google Scholar]
- 28.Cuyas E, Fernandez-Arroyo S, Alarcon T, et al. Germline BRCA1 mutation reprograms breast epithelial cell metabolism towards mitochondrial-dependent biosynthesis: evidence for metformin-based ‘starvation’ strategies in BRCA1 carriers. Oncotarget 2016; 7:52974–52992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thewes V, Simon R, Hlevnjak M, et al. The branched-chain amino acid transaminase 1 sustains growth of antiestrogen-resistant and ERalpha-negative breast cancer. Oncogene 2017; 36:4124–4134. [DOI] [PubMed] [Google Scholar]
- 30.Takegoshi K, Honda M, Okada H, et al. Branched-chain amino acids prevent hepatic fibrosis and development of hepatocellular carcinoma in a nonalcoholic steatohepatitis mouse model. Oncotarget 2017; 8:18191–18205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conway ME, Hutson SM. BCAA metabolism and NH3 homeostasis. Adv Neurobiol 2016; 13:99–132. [DOI] [PubMed] [Google Scholar]
- 32.Yoon MS. The emerging role of branched-chain amino acids in insulin resistance and metabolism. Nutrients 2016; 8:E405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cluntun AA, Lukey MJ, Cerione RA, et al. Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer 2017; 3:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 2016; 16:749. [DOI] [PubMed] [Google Scholar]
- 35.Nojiri S, Fujiwara K, Shinkai N, et al. Effects of branched-chain amino acid supplementation after radiofrequency ablation for hepatocellular carcinoma: a randomized trial. Nutrition 2017; 33:20–27. [DOI] [PubMed] [Google Scholar]
- 36.Iwasa M, Sugimoto R, Ishihara T, et al. Usefulness of Levocarnitine and/or branched-chain amino acids during invasive treatment for hepatocellular carcinoma. J Nutr Sci Vitaminol (Tokyo) 2015; 61:433–440. [DOI] [PubMed] [Google Scholar]
- 37.Higashi T, Hayashi H, Kaida T, et al. Prognostic impact of visceral fat amount and branched-chain amino acids (BCAA) in hepatocellular carcinoma. Ann Surg Oncol 2015; 22 Suppl 3:S1041–S1047. [DOI] [PubMed] [Google Scholar]
- 38.Park JG, Tak WY, Park SY, et al. Effects of branched-chain amino acids (BCAAs) on the progression of advanced liver disease: a Korean nationwide, multicenter, retrospective, observational, cohort study. Medicine (Baltimore) 2017; 96:e6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shiozawa S, Usui T, Kuhara K, et al. Impact of branched-chain amino acid-enriched nutrient on liver cirrhosis with hepatocellular carcinoma undergoing transcatheter arterial chemoembolization in Barcelona Clinic Liver Cancer Stage B: a prospective study. J Nippon Med Sch 2016; 83:248–256. [DOI] [PubMed] [Google Scholar]
- 40.Budhathoki S, Iwasaki M, Yamaji T, et al. Association of plasma concentrations of branched-chain amino acids with risk of colorectal adenoma in a large Japanese population. Ann Oncol 2017; 28:818–823. [DOI] [PubMed] [Google Scholar]
- 41.Symonds EL, Pedersen SK, Baker RT, et al. A blood test for methylated BCAT1 and IKZF1 vs. a fecal immunochemical test for detection of colorectal neoplasia. Clin Transl Gastroenterol 2016; 7:e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Young GP, Pedersen SK, Mansfield S, et al. A cross-sectional study comparing a blood test for methylated BCAT1 and IKZF1 tumor-derived DNA with CEA for detection of recurrent colorectal cancer. Cancer Med 2016; 5:2763–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ananieva EA, Patel CH, Drake CH, et al. Cytosolic branched chain aminotransferase (BCATc) regulates mTORC1 signaling and glycolytic metabolism in CD4+ T cells. J Biol Chem 2014; 289:18793–18804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taya Y, Ota Y, Wilkinson AC, et al. Depleting dietary valine permits nonmyeloablative mouse hematopoietic stem cell transplantation. Science 2016; 354:1152–1155. [DOI] [PubMed] [Google Scholar]