

Abstract
Background:
MicroRNAs are a class of small RNA molecules that inhibit protein expression through either degradation of messenger RNA or interference with protein translation. Our previous work suggested an involvement of miR-30e in myocardial fibrosis; however, the exact role of miR-30e in the pathogenesis of cardiac fibrosis and the underlying mechanisms are not known.
Methods:
Male Sprague Dawley rats were treated with isoproterenol (ISO) to induce cardiac remodeling and fibrosis and treated with either miR-30e agomir (AG) or antagomir and respective controls. The expression of miR-30e was evaluated by reverse transcription and quantitative polymerase chain reaction. Myocardial fibrosis was assessed by Masson's trichrome staining, and the level of oxidative stress and the expression of Snai1 and transforming growth factor-beta (TGF-β) were detected using Western blots.
Results:
A significant downregulation of miR-30e was found in the hearts of ISO-treated rats with cardiac fibrosis compared with nontreated controls. In vivo administration of miR-30e AG increased the survival of ISO-treated rats compared with AG-negative control administration, which was associated with reduced oxidative stress. We further identified Snai1 as a novel miR-30e target. Snai1 expression was significantly increased in hearts from ISO-treated rats, which coincided with decreased miR-30e expression and increased TGF-β expression. An miR-30e putative target sequence was identified in the 3′-untranslated region (UTR) Snai1. In a reporter assay, miR-30e greatly suppressed the activity of wild-type 3′-UTR–fused luciferase reporter, but showed no significant effect with the mutated 3′-UTR–fused reporter.
Conclusion:
MiR-30e attenuated ISO-induced cardiac dysfunction and cardiac fibrosis in a rat cardiac remodeling model. Mechanistically, miR-30e suppressed Snai1/TGF-β pathway which was involved in ISO-induced cardiac remodeling.
Key Words: miR-30e, cardiac fibrosis, Snai1
INTRODUCTION
Cardiomyopathy will eventually progress to heart failure if no effective treatment is rendered. Heart transplantation remains the most effective treatment for patients with end-stage heart failure. However, it is challenging to have a cardiac transplantation because of complex technical requirements and shortage of viable heart donors. Structural changes caused by cardiomyopathy are referred to as cardiac remodeling.1–3 At the cellular level, cardiac remodeling is associated with increased oxidative stress, apoptosis, and myocardial fibrosis. Fibrosis-increased stiffness of the heart, disrupted electrical coupling, and tissue hypoxia together contribute to cardiac contractile dysfunction and development of heart failure.4,5 Multifactorial mechanisms are implicated in the pathogenesis of cardiac fibrosis, and one of them is activated Snai1/transforming growth factor-beta (TGF-β) signaling. Snai1, a zinc finger transcription factor, was shown to induce fibrosis in various organs, including the kidneys, liver, and lungs through activating epithelial–mesenchymal transition (EMT),6–9 which plays an important role in organ fibrosis.10 In addition, it has been well documented that Snai1 activation after organ injury is associated with increased activity of the TGF-β cascade, which also plays an important role in EMT.11
MicroRNAs (miRNAs) are a class of small RNAs that negatively regulate gene expression through repression of target messenger RNAs (mRNAs).12,13 MiRNAs are often coded in clusters and transcribed as single transcriptional units, each forming a primary miRNA, which is then processed and gives rise to multiple mature miRNAs.14,15 Groups of miRNAs share the same “seed region,” which is the sequence necessary for target mRNA recognition and is usually located between nucleotides 2 and 8.16 Therefore, miRNAs play influential roles in the posttranscriptional “fine tuning” of gene expression. Previous studies have shown that a number of miRNAs are associated with cardiovascular diseases, including myocardial fibrosis, atherosclerosis, and heart failure,17 indicating that miRNAs play important roles in the pathogenesis and progression of cardiovascular disease.
MiR-30e is highly expressed in mouse hearts18 and shown to be involved in cardiovascular disease. For instance, miR-30e was mainly expressed in atherosclerotic plaques but not in healthy vessels.19 Also, doxorubicin was shown to reduce the expression of myocardial miR-30 and promoted apoptosis,20 suggesting that miR-30 might play a role in cardiac fibrosis. However, the exact role of miR-30 in cardiac fibrosis and remodeling, including its underlying mechanisms, are largely unknown.
Therefore, our hypothesis is that miR-30e may protect the heart from agonist-induced cardiac fibrosis and heart failure. In this study, we use a rat cardiac remodeling model induced by isoproterenol (ISO), a synthetic catecholamine and β-adrenergic receptor agonist, to explore the effects of miR-30e on cardiac function during heart failure and to investigate the molecular mechanisms involved in rat cardiac remodeling after miR-30e treatment.
MATERIALS AND METHODS
Animals and Experimental Protocols
Male rats (Sprague Dawley rats, 8–10 weeks old) were obtained from Jilin University of Medicine Laboratory and were maintained in a pathogen-free facility. Both animal husbandry and experimental procedures complied with the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health, DHEW publication No. NIH 85-23, 1996. All procedures were reviewed and approved by the Institutional Animal Care and Use Committee of Jilin University. Rats were randomly divided into 2 groups: rats were intraperitoneally injected with ISO (2 mg/kg) once a day for 10 days or injected with the same volume of 0.9% normal saline. ISO-treated rats were then further randomly assigned into 4 subgroups (n = 6/group); each subgroup was injected with miR-30e agomir (AG, 10 nmol), antagomir (AN, 10 nmol), or their respective negative controls (NCs) AG-NC (10 nmol) and AN-NC (10 nmol).
Reagents
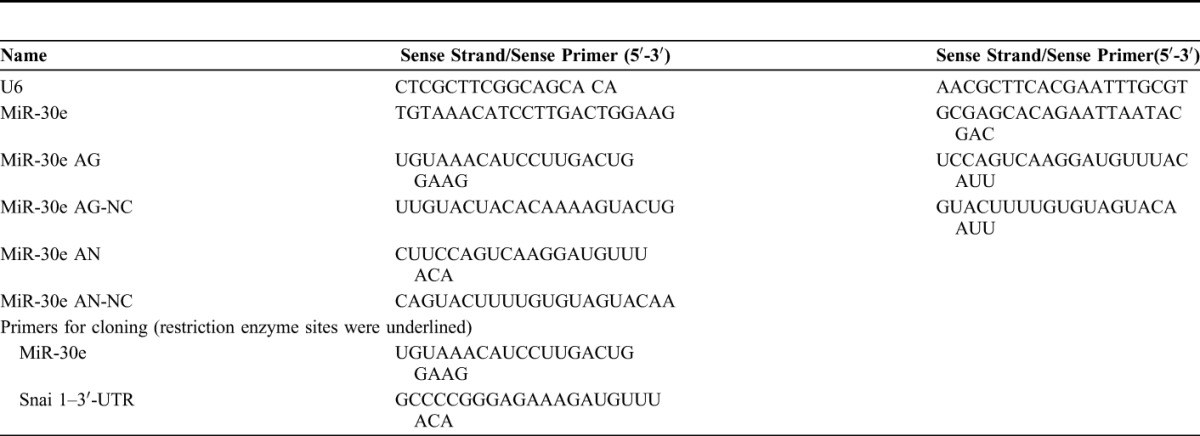
Masson's trichrome staining kit was purchased from Jiancheng Biological Engineering Institute (Nanjing, Jiangsu, China). Antibody against nitrotyrosine was purchased from Invitrogen (Carlsbad, CA). Anti-TGF-β and anti-β-actin antibodies were purchased from Cell Signaling Technology (Beverly, MA). Anti-Snai1 antibody and horseradish peroxidase–conjugated secondary antibody were obtained from Santa Cruz Biotechnology Inc (Dallas, TX). MiR-30e AG/AN and their respective NCs were purchased from RiboBio (Guangzhou, China). All miR-30e–related sequences are shown in Table 1.
TABLE 1.
Sequences of RNA and DNA Oligonucleotides
Histology
Cardiac fibrosis was evaluated with Masson's trichrome staining. Briefly, the hearts were collected from rats of control and ISO-treated groups after 10 days of ISO injection, followed by fixation in 10% formalin for 48 hours. Specimens were then embedding in paraffin, cut into 4 µm thick sections, and stained according to the manufacturer's instructions.
Western Blot
Total proteins from myocardial tissue were extracted using RIPA lysis buffer containing protein phosphatase inhibitors. Forty µg protein lysate of each sample was stacking on 4% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, then separated in 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene fluoride membrane. The membrane was blocked for 1 hour at room temperature with 5% milk or 5% bovine serum albumin, followed by incubation with anti-nitrotyrosine antibody (1:2000), anti-TGF-β antibody (1:3000), and anti-Snai1 antibody (1:2000) at 4°C overnight. Membranes were then washed 3 times with TBS-T and incubated with a horseradish peroxidase–conjugated secondary antibody (1:5000) for 1 hour at room temperature. Membranes were also immunoblotted with an anti-β-actin antibody (1:4000) as an internal control. Protein bands were visualized by luminol chemiluminescence (Bio-Rad; Hercules, CA). The intensity of each band was quantified using ImageJ (NIH).
Reverse Transcription and Quantitative Polymerase Chain Reaction
Total RNA was extracted from the hearts of control and ISO groups using Trizol reagent (Invitrogen, NY) according to the manufacturer's instruction. Methods to detect miR-30e expression were as follows: Reverse transcription was performed with the TaqMan miRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA), followed by quantitative polymerase chain reaction using SYBR Green Master Mix (Applied Biosystems) with the following thermal cycling profile: 95°C for 20 seconds, 40 cycles of amplification (95°C for 10 seconds, 60°C for 20 seconds, and 72°C for 10 seconds), and melt curve analysis. The relative expression levels of miR-30e were calculated using the 2−ΔΔCt method, and standardization was performed using U6 primers. The primer sequences are shown in Table 1.
Dual-luciferase Reporter Assays
Two target prediction applications: TargetScan and miRWALK, were used to predict Snai1 as the potential target of miR-30e. Wild-type (WT) or mutant 3′-untranslated regions (UTRs) of Snai1 were subcloned into the psiCHECKTM-2 reporter vector (Promega, Madison, WI), and the sequences of the synthesized oligos are shown in Table 1. WT or mutant reporter vector (50 ng/well) was cotransfected with miR-30e mimics (50 nM) or scrambled oligonucleotides (Ambion miRNA Precursors; Life Technologies, Carlsbad, CA) into 293T cells (1 × 104 cells/well) for 48 hours with Lipofectamine 2000 in 96-well plates. The luciferase activity was measured 48 hours after transfection using a Dual-Luciferase Reporter Assay System (Promega). Each sample was assayed in triplicate and the dual-luciferase reporter assay was repeated 3 times.
Echocardiography
Echocardiography was performed 10 days after ISO injection using a VIVID 7 dimension system (General Electric Vingmed Ultrasound; Horton, Norway). Images were captured using a 10S transducer (5.5–12.0 MHz) with a high temporal and spatial resolution. In brief, rats were anesthetized with an intraperitoneal injection of 25 mg/kg pentobarbital sodium. The transducer was placed directly on the shaved chest wall. A complete 2-dimensional, M-mode, color, and tissue Doppler echocardiogram was performed as previously described, according to the standards of the American Society of Echocardiography.21 Data of 3 consecutive heart cycles were digitally recorded and analyzed (Echo Pac; GE Medical Systems, Chicago,IL).
Statistical Analysis
All the data were expressed as mean ± SEM. Comparisons between 2 observations were assessed by Student's paired t test. Two-way analysis of variance was used, followed by the Bonferroni test to evaluate the difference among multiple groups. A P value of < 0.05 was considered statistically significant.
RESULTS
MiR-30e Is Downregulated in ISO-treated Rat Hearts
We first evaluated the expression level of miR-30e in the rat hearts of control and ISO-treated groups by reverse transcription quantitative polymerase chain reaction. The miR-30e expression level in the ISO-treated rats was significantly lower than that of control rats (P < 0.05) (Fig. 1).
FIGURE 1.
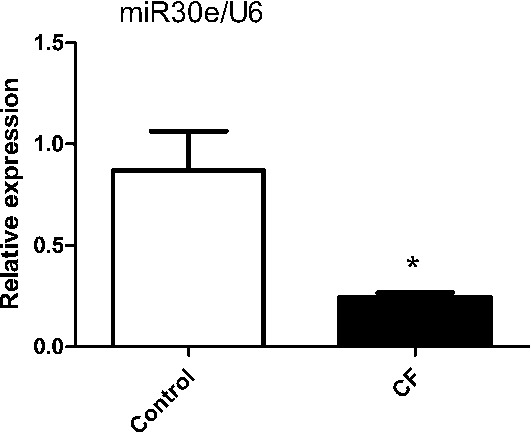
MiR-30e is downregulated in ISO-treated rat hearts. MiR-30e expression levels were evaluated using quantitative reverse transcription polymerase chain reaction. Data were compiled from at least 3 independent experiments each performed in triplicate. *P < 0.05; CF, cardiac fibrosis.
MiR-30e Improves the Survival Rate and Attenuates Cardiac Fibrosis of ISO-treated Rats
To explore the effects of miR-30e on the phenotype of ISO-treated mice, rats were injected with miR-30e AG or AN through the tail vein. MiR-30e AG administration increased the survival rate of ISO-treated rats, but miR-30e AN significantly decreased the survival rate (Fig. 2A). Consistent with the above observation, Masson's trichrome staining revealed that miR-30e AG greatly diminished the left ventricular fibrosis in ISO-injected rats. However, miR-30e AG aggravated cardiac fibrosis (Fig. 2B). Thus, we conclude that miR-30e possesses the capability to provoke ISO-induced cardiac fibrosis.
FIGURE 2.

MiR-30e improves the survival rate and attenuates cardiac fibrosis of ISO-treated rats. A, Survival rates of each group. B, Myocardial collagen fiber content was evaluated by Masson staining. *P < 0.05, n = 6 in each group.
MiR-30e Improves Cardiac Function of ISO-treated Rats
Next, we asked whether miR-30e could improve cardiac function of ISO-treated rats. Echocardiography was performed on rats from 4 groups: AG-NC, AG, AN-NC, and AN. As shown in Figure 3, although rats in AG-NC, AN-NC, and AN groups showed no difference in left ventricular wall thickness, diameters, fractional shortening (FS), and ejection fraction (EF), the AG group showed significantly improved EF%, FS%, left ventricular end-diastolic posterior wall thickness, and left ventricular internal dimension in diastole (*P < 0.05 vs. AG-NC). Consistent with the above observations, miR-30e substantially attenuates ISO-induced cardiac dysfunction.
FIGURE 3.
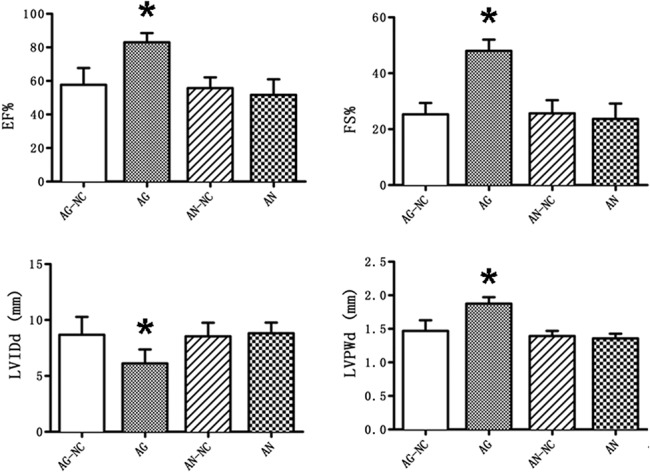
MiR-30e AG improves cardiac function impaired by ISO infusion. LVID, LVPW, EF% and FS% were detected by Doppler ultrasound. *P < 0.05, n = 6 in each group.
Snai1 Is a Putative Target of MiR-30e
miRNA regulates gene expression through repression of the mRNA level of target genes. We predicted Snai1 as a potential target of miR-30e using TargetScan and miRWALK. Prediction algorithms showed that miR-30e was one of the potential miRNAs binding on the 3′-UTR of Snai1. We next fused WT Snai1 3′-UTR sequence into a luciferase reporter vector and generated a mutation from UGUUUAC to ACAAAUGA on 630 to 637 on the miR-30e–binding sequence (Fig. 4). The WT or mutant reporter was cotransfected with miR-30e mimics into 293T cells, followed by luciferase activity measurement after 48-hour transfection. Although miR-30e greatly reduced the WT 3′-UTR–fused reporter activity, it did not have any significant effect on the mutated reporter activity. Thus, we conclude that Snai1 is a direct target of miR-30e.
FIGURE 4.
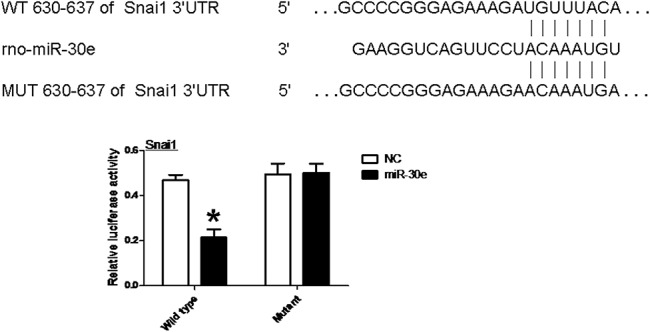
Snai1 is a putative target of miR-30e. MiR-30e and its putative-binding sequence on the 3′-UTR of Snai1 gene are shown. WT and mutated 3′-UTR were cotransfected with empty vector or miR-30e expression vector as indicated. Luciferase reporter activity was measured 48 hours after transfection. *P < 0.05 versus empty vector of WT.
MiR-30e Attenuates ISO-induced Cardiac Fibrosis Through Snai1/TGF-β Pathway
Previous studies have shown that the expression of Snai1 in epithelial cells has contributed to the generation of EMT, which promotes the progression of organ fibrosis.22–24 Indeed, Snai1 plays an essential role in myocardial fibrosis by activating cardiac fibroblasts, and subsequently promoting the generation of collagen synthesis. Because the putative miR-30e–binding sites in the 3′-UTR of Snai1 was verified, we assumed that miR-30e directly inhibits the Snai1-TGF-β signaling cascade and thus attenuated myocardial fibrosis. Western blots were performed on the protein lysates purified from the hearts from the 4 groups treated with ISO. MiR-30e AG significantly reduced the expression of Snai1, TGF-β, and nitrotyrosine, a bio marker of oxidative stress, whereas miR-30e AN increased their expression (Fig. 5). These data suggest that miR-30e may attenuate oxidative stress level and myocardial fibrosis at least in part through suppressing the Snai1/TGF-β signaling cascade.
FIGURE 5.
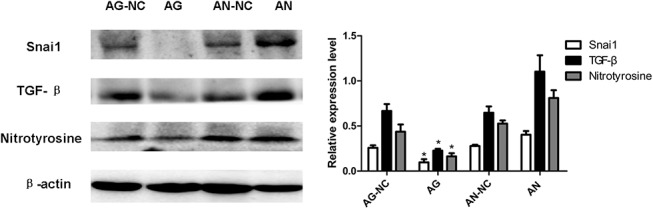
MiR-30e AG inhibits the expression of Snai1, TGF-β, and nitrotyrosine. Western blots were performed on heart lysates from rats of 4 groups as indicated to assess the expression of Snai1, TGF-β, and nitrotyrosine. β-actin was used as an internal control. *P < 0.05, n = 6 in each group.
DISCUSSION
Myocardial fibrosis is one of the major pathological changes in the failing hearts and contributes to the cardiac stiffness, arrhythmia, and decreased cardiac function. Therefore, reducing myocardial fibrosis represents a valid therapeutic avenue and is an active research area. The major findings from this study are as follows: (1) miR-30e improves cardiac function and reduces myocardial fibrosis in ISO-treated mice, and (2) miR-30e significantly reduces Snai1/TGF-β signaling, which plays an important role in the pathogenesis of cardiac fibrosis.
It has been well documented that several miRNAs are involved in the development and/or progression of cardiovascular diseases and might serve as potential therapeutic targets.25 For example, miR-133 and miR-24 are 2 of the major antifibrotic miRNAs that reduced cardiac fibrosis in heart failure,26–28 whereas some miRNAs including miR-21 promoted cardiac fibrosis.29 In this study, we used an ISO-treated rat model to explore the changes of miR-30e on cardiac function in hypertrophic, failing hearts. We found that miR-30e was downregulated in ISO-induced cardiac remodeling and heart failure. Also, miR-30e AG significantly improved the survival rate of ISO-treated rats, attenuated ISO-triggered cardiac dysfunction, and diminished cardiac fibrosis. On the contrary, miR-30e AN heightened the cardiac hypertrophy and dysfunction induced by ISO. These findings together suggest that miR-30e plays a protective role in cardiomyopathy and heart failure by reducing cardiac fibrosis. Our findings are in agreement with several previous reports, in which downregulation of miR-30e expression was observed in dilated cardiomyopathy,30 and in doxorubicin-treated mouse hearts, doxorubicin-treated cultured cardiomyocytes, and infarcted myocardium.20 Moreover, decreased expression of miR-30e was believed to contribute to increased activity of β-adrenergic signaling and heart failure.20 More interestingly, miR-30e-5p, an isoform of miR-30e, was significantly reduced in circulating blood of patients with coronary artery diseases.31 Taken together, we believe that miR-30e could be a potential therapeutic target for treatment of cardiomyopathy and heart failure. It will be of great interest to explore whether and how the miR-30e level is altered in the myocardium of patients with different stages of heart failure.
As mentioned above, the molecular mechanisms leading to cardiac fibrosis are complex, and several molecules and signaling pathways are involved. For example, it has been well recognized that TGF-β signaling plays an important role in the pathogenesis of cardiac fibrosis, as evidenced by findings showing that myocardial TGF-β was upregulated in hypertrophic and infarcted hearts, in patients with cardiac muscle disorders, and in a mouse model that overexpressed TGF-β.32 Also, reactive oxygen species (ROS), which are activated in most diseased hearts,33 activate TGF-β signaling and thus are implicated in cardiac fibrosis.34,35 In addition, Snai1, a zinc finger-containing transcription factor, is well known for its role in promoting EMT,8 which plays a role in fibrosis of a number of organs including the heart.36 Thus, Snai1 is believed to act as a cardiac fibrosis promoter in diseased hearts. Indeed, a recent study revealed Snai1 as a potential target in ischemia–reperfusion–induced cardiac fibrosis.11 Moreover, Snai1 and TGF-β signaling were also functionally correlated. Consistent with the above reports, in this study, we found that miR-30e AN significantly increased the myocardial expression of Snai1 and TGF-β, suggesting that Snai1 and TGF-β contribute to the pathogenesis of ISO-induced cardiac remodeling and fibrosis. In addition, miR-30e AG was found to substantially reduce the expression of Snai1 and TGF-β in ISO-treated rat hearts, which coincided with the improved cardiac function of ISO-treated rats, indicating that the increased level of miR-30e benefited ISO-treated rats by decreasing Snai1 and TGF-β expression. In addition, miR-30e AG suppressed the production of ROS, as evidenced by the decreased level of nitrotyrosine, an indicator of the level of oxidative stress and the activity of ROS.37 Taken together, these findings suggest that miR-30e ameliorated cardiac fibrosis at least in part through suppressing the activation of Snai1/TGF-β signaling and oxidative stress in the hearts linked to ISO treatment. Previous findings suggested that downregulation of miR-30–mediated apoptosis through upregulated p38 and DRP138 and that miR-30e mimic downregulated Beclin-1 and protected primary cardiomyocytes against apoptosis in vitro.39 Given that p38, Drp1, and Beclin-1 were all involved in ISO-induced cardiac dysfunction,40–42 we speculate that miR-30e protects the heart against ISO-induced cardiac phenotypes through multiple mechanisms. Further studies are needed to corroborate our premise.
In this study, we further identified Snai1 as a novel miR-30e target through mutagenesis and reporter analysis on the 3′-UTR of Snail gene. We found that miR-30 directly mediated Snai1 expression through binding to its putative binding element localized in the 3′-UTR of Snail gene. Although miR-30e has been previously studied for its potential implication in human disease and in animal models, studies attempting to identify its direct targets have been limited. A previous study uncovered Bmi1 as a direct putative target of miR-30e.43 In addition, a recent study suggested that Snai1 was a direct target of miR-30e-3p in clear cell renal cell carcinoma.44 Taken together, these findings support the notion that miR-30e may be involved in a number of pathological processes and human diseases through mediating the expression of Snai1.
In conclusion, we demonstrated that miR-30e improves the survival rate of ISO-treated rats, which results from miR-30e–linked amelioration of cardiac dysfunction and reduction in cardiac fibrosis. Mechanistically, miR-30e significantly diminishes the ISO-triggered activation of Snai1/TGF-β signaling and oxidative stress. We further demonstrate that Snai1 is a direct putative target of miR-30e. Therefore, we propose that miR-30e may serve as a potential target for treatment of cardiomyopathy and heart failure.
Footnotes
The authors report no conflicts of interest.
REFERENCES
- 1.Boström P, Mann N, Wu J, et al. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collier P, Watson CJ, van Es MH, et al. Getting to the heart of cardiac remodeling; how collagen subtypes may contribute to phenotype. J Mol Cell Cardiol. 2012;52:148–153. [DOI] [PubMed] [Google Scholar]
- 3.Giam B, Chu PY, Kuruppu S, et al. N-acetylcysteine attenuates the development of cardiac fibrosis and remodeling in a mouse model of heart failure. Physiol Rep. 2016;4:e12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lei L, Mason S, Liu D, et al. Hypoxia-inducible factor-dependent degeneration, failure, and malignant transformation of the heart in the absence of the von Hippel-Lindau protein. Mol Cell Biol. 2008;28:3790–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheikh AY, Chun HJ, Glassford AJ, et al. In vivo genetic profiling and cellular localization of apelin reveals a hypoxia-sensitive, endothelial-centered pathway activated in ischemic heart failure. Am J Physiol Heart Circ Physiol. 2008;294:H88–H98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rowe RG, Lin Y, Shimizu-Hirota R, et al. Hepatocyte-derived Snai1 propagates liver fibrosis progression. Mol Cell Biol. 2011;31:2392–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boutet A, Esteban MA, Maxwell PH, et al. Reactivation of Snail genes in renal fibrosis and carcinomas: a process of reversed embryogenesis? Cell Cycle. 2007;6:638–642. [DOI] [PubMed] [Google Scholar]
- 8.Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–3161. [DOI] [PubMed] [Google Scholar]
- 9.Medici D, Potenta S, Kalluri R. Transforming growth factor-beta2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem J. 2011;437:515–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalluri R, Weinberg RAJ. The basics of epithelial-mesenchymal transition. Clin Invest. 2009;119:1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee SW, Won JY, Kim WJ, et al. Snail as a potential target molecule in cardiac fibrosis: paracrine action of endothelial cells on fibroblasts through snail and CTGF axis. Mol Ther. 2013;21:1767–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pers YM, Jorgensen C. MicroRNA in 2012: biotherapeutic potential of microRNAs in rheumatic diseases. Nat Rev Rheumatol. 2013;9:76–78. [DOI] [PubMed] [Google Scholar]
- 13.Zhou X, Yang PC. MicroRNA: a small molecule with a big biological impact. Microrna. 2012;1:1. [DOI] [PubMed] [Google Scholar]
- 14.Marco A, Ninova M, Ronshaugen M, et al. Clusters of microRNAs emerge by new hairpins in existing transcripts. Nucleic Acids Res. 2013;41:7745–7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. [DOI] [PubMed] [Google Scholar]
- 16.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. [DOI] [PubMed] [Google Scholar]
- 17.Romaine SP, Tomaszewski M, Condorelli G, et al. MicroRNAs in cardiovascular disease: an introduction for clinicians. Heart. 2015;101:921–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ro S, Park C, Young D, et al. Tissue-dependent paired expression of miRNAs. Nucleic Acids Res. 2007;35:5944–5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bidzhekov K, Gan L, Denecke B, et al. microRNA expression signatures and parallels between monocyte subsets and atherosclerotic plaque in humans. Thromb Haemost. 2012;107:619–625. [DOI] [PubMed] [Google Scholar]
- 20.Roca-Alonso L, Castellano L, Mills A, et al. Myocardial MiR-30 downregulation triggered by doxorubicin drives alterations in beta-adrenergic signaling and enhances apoptosis. Cell Death Dis. 2015;6:e1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leitmann M, Lysyanski P, Sidenko S, et al. Two-dimensional strain a novel software for real-time quantitative echocardiographic assessment of myocardial function. J Am Soc Echocardiogr. 2004;17:1021–1029. [DOI] [PubMed] [Google Scholar]
- 22.Ohnuki K, Umezono T, Abe M, et al. Expression of transcription factor Snai1 and tubulointerstitial fibrosis in progressive nephropathy. J Nephrol. 2012;25:233–239. [DOI] [PubMed] [Google Scholar]
- 23.Villar AV, Llano M, Cobo M, et al. Gender differences of echocardiographic and gene expression patterns in human pressure overload left ventricular hypertrophy. J Mol Cell Cardiol. 2009;46:526–535. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Du J, Zhang J, et al. Snai1 is involved in de novo cardiac fibrosis after myocardial infarction in mice. Acta Biochim Biophys Sin (Shanghai). 2012;44:902–910. [DOI] [PubMed] [Google Scholar]
- 25.Condorelli G, Latronico MV, Dorn GW. microRNAs in heart disease: putative novel therapeutic targets? Eur Heart J. 2010;31:649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meder B, Katus HA, Rottbauer W. Right into the heart of microRNA-133a. Genes Dev. 2008;22:3227–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matkovich SJ, Wang W, Tu Y, et al. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res. 2010;106:166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Huang W, Xu R, et al. MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. Cell Mol Med. 2012;16:2150–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang KC, Ku YC, Lovett M, et al. Combined deep microRNA and mRNA sequencing identifies protective transcriptomal signature of enhanced PI3Kα signaling in cardiac hypertrophy. J Mol Cell Cardiol. 2012;53:101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ikeda S, Kong SW, Lu J, et al. Altered microRNA expression in human heart disease. Physiol Genomics. 2007;31:367–373. [DOI] [PubMed] [Google Scholar]
- 31.Weber M, Baker MB, Patel RS, et al. MicroRNA expression profile in CAD patients and the impact of ACEI/ARB. Cardiol Res Pract. 2011;2011:532915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sugamura K, Keaney JF., Jr Reactive oxygen species in cardiovascular disease. Free Radic Biol Med. 2011;51:978–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purnomo Y, Piccart Y, Coenen T, et al. Oxidative stress and transforming growth factor-beta1-induced cardiac fibrosis. Cardiovasc Hematol Disord Drug Targets. 2013;13:165–172. [DOI] [PubMed] [Google Scholar]
- 35.Shao D, Oka S, Brady CD, et al. Redox modification of cell signaling in the cardiovascular system. J Mol Cell Cardiol. 2012;52:550–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225:631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mehlhorn U, Krahwinkel A, Geissler HJ, et al. Nitrotyrosine and 8-isoprostane formation indicate free radical-mediated injury in hearts of patients subjected to cardioplegia. Thorac Cardiovasc Surg. 2003;125:178–183. [DOI] [PubMed] [Google Scholar]
- 38.Li J, Donath S, Li Y, et al. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010;6:e1000795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lai L, Chen J, Wang N, et al. MiRNA-30e mediated cardioprotection of ACE2 in rats with doxorubicin-induced heart failure through inhibiting cardiomyocytes autophagy. Life Sci. 2017;169:69–75. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Xu J, Long Z, et al. Hydrogen (H2) inhibits isoproterenol-induced cardiac hypertrophy via antioxidative pathways. Front Pharmacol. 2016;7:392. eCollection 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okada M, Morioka S, Kanazawa H, et al. Canstatin inhibits isoproterenol-induced apoptosis through preserving mitochondrial morphology in differentiated H9c2 cardiomyoblasts. Apoptosis. 2016;21:887–895. [DOI] [PubMed] [Google Scholar]
- 42.Dong RQ, Wang ZF, Zhao C, et al. Toll-like receptor 4 knockout protects against isoproterenol-induced cardiac fibrosis: the role of autophagy. J Cardiovasc Pharmacol Ther. 2015;20:84–92. [DOI] [PubMed] [Google Scholar]
- 43.Sugihara H, Ishimoto T, Watanabe M, et al. Identification of miR-30e regulation of Bmi1 expression mediated by tumor-associated macrophages in gastrointestinal cancer. PLoS One. 2013;8:e81839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang D, Zhu C, Zhang Y, et al. MicroRNA-30e-3p inhibits cell invasion and migration in clear cell renal cell carcinoma by targeting Snai1. Oncol Lett. 2017;13:2053–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]