

Abstract
Intoduction
Derived from heavy chain only camelid antibodies, ~15-kDa single-domain antibody fragments (sdAbs) are an attractive platform for developing molecularly specific imaging probes and targeted radiotherapeutics. The rapid tumor accumulation and normal tissue clearance of sdAbs might be ideal for use with 211At, a 7.2-h half-life α-emitter, if appropriate labeling chemistry can be devised to trap 211At in cancer cells after sdAb binding. This study evaluated two reagents, [211At]SAGMB and iso-[211At]SAGMB, for this purpose.
Methods
[211At]SAGMB and iso-[211At]SAGMB, and their radioiodinated analogues [131I]SGMIB and iso-[131I]SGMIB, were synthesized by halodestannylation and reacted with the anti-HER2 sdAb 5F7. Radiochemical purity, immunoreactivity and binding affinity were determined. Paired-label internalization assays on HER2-expressing BT474M1 breast carcinoma cells directly compared [131I]SGMIB-5F7/[211At]SAGMB-5F7 and iso-[131I]SGMIB-5F7/iso-[211At]SAGMB-5F7 tandems. The biodistribution of the two tandems was evaluated in SCID mice with subcutaneous BT474M1 xenografts.
Results
Radiochemical yields for Boc2-iso-[211At]SAGMB and Boc2-[211At]SAGMB synthesis, and efficiencies for coupling of iso-[211At]SAGMB and [211At]SAGMB to 5F7 were similar, with radiochemical purities of [211At]SAGMB-5F7 and iso-[211At]SAGMB-5F7 >98%. iso-[211At]SAGMB-5F7 and [211At]SAGMB-5F7 had immunoreactive fractions >80% and HER2 binding affinities of less than 5 nM. Internalization assays demonstrated high intracellular trapping of radioactivity, with little difference observed between corresponding 211At- and 131I-labeled 5F7 conjugates. Higher BT474M1 intracellular retention was observed from 1–6 h for the iso-conjugates (iso-[211At]SAGMB-5F7, 74.3 ± 2.8%, vs. [211At]SAGMB-5F7, 63.7 ± 0.4% at 2 h) with the opposite behavior observed at 24 h. Peak tumor uptake for iso-[211At]SAGMB-5F7 was 23.4 ± 2.2% ID/g at 4 h, slightly lower than its radioiodinated counterpart, but significantly higher than observed with [211At]SAGMB-5F7. Except in kidneys and lungs, tumor-to-normal organ ratios for iso-[211At]SAGMB-5F7 were greater than 10:1 by 2 h, and significantly higher than those for [211At]SAGMB-5F7.
Conclusion
These 211At-labeled sdAb conjugates, particularly iso-[211At]SAGMB-5F7, warrant further evaluation for targeted α-particle radiotherapy of HER2-expressing cancers.
Keywords: Astatine-211, single domain antibody fragment, HER2, α-particle, VHH, nanobody
1. Introduction
Human epidermal growth factor receptor 2 (HER2) is overexpressed in a subset of patients with multiple types of cancers including breast, non-small cell lung, gastric, colon and ovarian [1–3]. Up to 20–30% of breast cancers overexpress HER2 [4, 5] and HER2 expression has been shown to confer a more aggressive phenotype, including a greater propensity to metastasize to the central nervous system (CNS) [6–8]. Moreover, a higher incidence of brain metastases and leptomeningial carcinomatosis have been reported in patients treated with the anti-HER2 mAb trastuzumab [9, 10]. Trastuzumab frequently prolongs survival by controlling systemic disease in many patients; however, this increases the opportunity for CNS lesions, against which trastuzumab is ineffective because of poor delivery due to the blood brain barrier impermeability of this large protein [11].
Patients with HER2-positive CNS disease have a grim prognosis; thus, there is a dire need for treatments that can be more effective without compromising neurologic function, which can be an unfortunate side effect of nonspecific treatments including conventional radiation therapy [12]. An attractive approach for increasing the specificity of cancer treatment is targeted radiotherapy, in which a monoclonal antibody (mAb) or other vector is used to selectively deliver a cytotoxic radionuclide to cancer cells. In the context of disease within the CNS, α-particles, a radiation with a tissue range of only few cell diameters (50–80 μm), could be advantageous because it could minimize cross fire irradiation of normal tissue. Moreover, α-particles have a high relative biological effectiveness, requiring only a few traversals per cell to achieve its destruction [13].
As an initial investigation of the therapeutic potential of α-particles for the treatment of HER2-positive cancers, we labeled trastuzumab with the 7.2-h half-life α-emitter 211At and its cytotoxicity for 3 HER2-expressing human breast carcinoma lines was evaluated in vitro [14]. The relative biological effectiveness of 211At-labeled trastuzumab was about 10 times higher than that of conventional external beam therapy, with significant reduction in survival achieved with only a few 211At atoms bound per cell. A subsequent study was performed in a HER2-positive breast carcinomatous meningitis model to evaluate the therapeutic efficacy of a single intrathecal injection of 211At-labeled trastuzumab [15]. Significant prolongation in median survival with some long-term survivors was observed; however, even with direct injection into the intrathecal compartment, histopathological analyses revealed that regions of the neuroaxis had escaped treatment in some animals. Intact mAbs are not ideal for use in combination with short lived α-emitters such as 211At because their large size hinders homogeneous delivery and for intravenous applications, results in slow normal tissue clearance [16].
To overcome these limitations, a variety of smaller HER2-targeted proteins have been developed including recombinant fragments such as diabodies and minibodies, and smaller scaffolds such as affibodies [17]. Another attractive platform for targeted radiotherapy, derived from Camelidae heavy-chain only antibodies and known as single domain antibody fragments (sdAbs), variable domain of heavy-chain only antibodies (VHH) or nanobodies has a molecular weight of 12–15 kDa [18]. These sdAbs can be generated relatively inexpensively with nM to pM affinity, high thermal and chemical stability, and low immunogenicity [19–20]. Moreover, because of their small size, they clear rapidly from blood and normal tissues and efficiently penetrate tumors, properties that are particularly advantageous for use with short-lived α-emitters like 211At. Finally, several sdAbs with high affinity for HER2 have been generated and reported to target HER2-positive cancers in animal models and in a recent clinical imaging trial [21–23]. Particularly encouraging results in terms of both the magnitude of tumor uptake and retention as well as the rate of normal tissue clearance were obtained when anti-HER2 5F7 sdAb was labeled with N-succinimidyl 4-guanidinomethyl-3-[131I]iodobenzoate ([131I]SGMIB) [24], a residualizing prosthetic group designed to trap radioactivity in tumor cells after receptor internalization [25]. The importance of using a residualizing prosthetic group for labeling 5F7 sdAb has been described in previous publications that demonstrated increased intracellular retention and in vivo tumor uptake compared with 5F7 sdAb labeled by direct iodination [23,24] or uncharged halobenzoyl reagents [26].
In the present study, we evaluated the potential utility of the analogous reagent, N-succinimidyl 3-[211At]astato-4-guanidinomethyl benzoate ([211At]SAGMB) [27], as well as a novel residualizing agent, N-succinimidyl 3-[211At]astato-5-guanidinomethyl benzoate (iso-[211At]SAGMB), for labeling 5F7 sdAb with 211At. Motivation for the latter is based on previous work showing that N-succinimidyl 3-guanidinomethyl-5-[131I]iodobenzoate (iso-[131I]SGMIB) provided higher sdAb labeling yields than [131I]SGMIB [28], an advantage that we hypothesize could be more pronounced with the larger astatine atom. Tumor targeting properties of the four residualizing agents were evaluated in HER2-expressing breast carcinoma cells and xenografts.
2. Materials and methods
2.1. General
All reagents were purchased from Sigma-Aldrich except where noted. Sodium [131I]iodide (44.4 TBq/mmol) in 0.1 N NaOH was obtained from Perkin-Elmer Life and Analytical Sciences (Boston, MA, USA). Astatine-211 was produced on the Duke University CS-30 cyclotron via the 209Bi(α, 2n)211At reaction by bombarding natural bismuth metal targets with 28 MeV α-particles [29]. Astatine-211 was isolated from the target by dry distillation, trapped in PEEK or PTFE tubing and finally extracted with a solution of N-chlorosuccinimide (NCS) in methanol (0.2 mg/mL) as described previously [29, 30]. Succinimidyl 4/5-((1, 2-bis(tert-butoxycarbonyl)guanidino)methyl)-3-iodobenzoate (Boc2-SGMIB/iso-SGMIB) and their corresponding tin precursors (Boc2-SGMTB/iso-SGMTB) were synthesized as reported before [25, 28]. High-performance liquid chromatography (HPLC) was performed using a Beckman Gold HPLC system equipped with a Model 126 programmable solvent module, a Model 166 NM variable wavelength detector, and a ScanRam RadioTLC scanner/HPLC detector combination (LabLogic; Brandon, FL, USA). HPLC data were acquired and processed using the Laura software (LabLogic). Normal-phase HPLC was performed using a 4.6 × 250 mm Partisil silica column (10 μm; Alltech, Deerfield, IL, USA), eluted in isocratic mode with a mixture of 0.2 % acetic acid in 75:25 hexanes:ethyl acetate (v/v) at a flow rate of 1 mL/min. Disposable PD 10 desalting columns for gel filtration were purchased from GE Healthcare (Piscataway, NJ, USA). Instant thin layer chromatography (ITLC) was performed using silica gel impregnated glass fiber sheets (Pall Corporation, East Hills, NY, USA) with PBS, pH 7.4 as the mobile phase. Developed sheets were analyzed for radioactivity either using the TLC scanner described above or by cutting the sheet into small strips and counting them in an automated gamma counter. Radioactivity levels in various samples were assessed using either an LKB 1282 (Wallac, Finland) or a Perkin Elmer Wizard II (Shelton, CT, USA) automated gamma counter.
2.2. Single-domain antibody fragment 5F7
Anti-HER2 sdAb 5F7, obtained as a gift from Ablynx NV (Ghent, Belgium), was selected from phage libraries derived from llamas that had been immunized with SKBR3 human breast carcinoma cells. Its production, purification and characterization were as described previously [23] except that the glycine-glycine-cysteine (GGC) C-terminus tail was omitted, resulting in a purely monomeric preparation.
2.3. Cells and cell culture conditions
Cell culture reagents were purchased from Invitrogen (Grand Island, NY, USA). BT474M1 human breast carcinoma cells [30] were grown in DMEM/F12 medium containing 10% fetal calf serum (FCS), streptomycin (100 μg/mL), and penicillin (100 IU/mL) (Sigma-Aldrich, MO, USA). Cells were cultured at 37°C in a 5% CO2 humidified incubator.
2.4. Synthesis of [131I]SGMIB and iso-[131I]SGMIB
In most experiments, [131I]SGMIB and iso-[131I]SGMIB were synthesized as reported previously by the radioiododestannylation of the corresponding tin precursor using tert-butyl hydroperoxide (TBHP) as the oxidant and chloroform as the solvent [25, 28]. In more recent runs, NCS was used as the oxidant and the reaction was performed in methanol. For this, a solution of NCS in methanol (0.2 mg/mL; 100 μL), acetic acid (1 μL) and [131I]iodide (1–2 μL; 37–74 MBq) were added in that order to a half-dram glass vial containing 50 μg of the required tin precursor, and the reaction was allowed to proceed at 20°C for 15 min with occasional swirling of the vial. Most of the solvent was evaporated with a stream of argon, and the residue partitioned between 200 μL each of ethyl acetate and water. The ethyl acetate layer was separated, dried with anhydrous sodium sulfate and the ethyl acetate was evaporated. The residual radioactivity was reconstituted in the HPLC mobile phase (200 μL) and injected onto a normal phase column. Procedures for isolation and deprotection were as described below for [211At]SAGMB and iso-[211At]SAGMB.
2.5. Synthesis of [211At]SAGMB and iso-[211At]SAGMB
Astatine-211 in NCS/methanol (30–56 MBq) was added to a vial containing 200 μg of the required tin precursor followed by 10 μL acetic acid. The reaction mixture was incubated at 20°C for 30 min and methanol was evaporated with a gentle stream of argon. The residual mixture was re-dissolved in 20 μL of (75:25) hexanes/ethyl acetate and injected onto the normal phase HPLC column. The HPLC fractions containing Boc2-iso-[211At]SAGMB or Boc2-[211At]SAGMB (tR = ~25 min) were isolated, and the solvents from these were evaporated under a stream of argon for 20 min. Boc protecting groups were removed by treatment with 100 μL of trifluoroacetic acid (TFA) at 20°C for 10 min. To insure complete removal of TFA, the process of ethyl acetate addition (50 μL) and evaporation was performed three times. The residual radioactivity was then used as such for 5F7 sdAb labeling.
2.6. Radiolabeling of 5F7 sdAb
Iodine-131 labeling of sdAb 5F7 with [131I]SGMIB or iso-[131I]SGMIB was performed as reported previously [28]. For 211At-labeling, a solution of sdAb 5F7 in 0.1 M borate buffer, pH 8.5 (50 μL, 2 mg/mL) was added to the vial containing the [211At]SAGMB or iso-[211At]SAGMB activity and the mixture was incubated at 20°C for 20 min. The labeled 5F7 sdAb was purified by gel filtration over a PD-10 column eluted with phosphate buffered saline (PBS). Before use, the PD-10 column was preconditioned with human serum albumin to minimize nonspecific binding.
2.7. Quality control procedures
Each 131I- and 211At-labeled 5F7 preparation was evaluated by ITLC and SDS-PAGE to determine protein associated radioactivity, and the presence of aggregates and multimeric species, respectively. For ITLC, PBS, pH 7.4, was used as the mobile phase; with this system, intact protein remained at the origin (Rf = 0) and lower molecular weight radioactive species moved with an Rf value of 0.7–0.8. SDS-PAGE under non-reducing conditions and phosphor imaging were performed as previously described [23]. The immunoreactive fractions of the labeled 5F7 conjugates were determined by the Lindmo method [32] using magnetic beads coated with HER2 extracellular domain, or as a negative control, bovine serum albumin (BSA) [23, 24]. Briefly, aliquots of labeled 5F7 (~5 ng) were incubated with doubling concentrations of both HER2- and BSA-coated beads, and the immunoreactive fraction was calculated as the specific binding extrapolated to infinite HER2 excess.
2.8. Binding affinity of radiolabeled 5F7 conjugates
BT474M1 breast carcinoma cells were plated in 24-well plates at a density of 8 × 104 cells/well and incubated at 37°C for 24 h. The cells were then allowed to acclimatize at 4°C for 30 min prior to the addition of increasing concentrations of radiolabeled 5F7 conjugates (0.1–100 nM). Cells were then incubated at 4°C for 2 h, the medium containing unbound radioactivity was removed, and the cells were washed twice with cold PBS. Finally, the cells were solubilized by treatment with 1N NaOH (0.5 mL) at 37°C for 10 min. Cell-associated radioactivity was counted using an automated gamma counter. To determine non-specific binding, a parallel assay was performed as above except that a 100-fold excess of trastuzumab also was added to the incubation medium. The data were fit using GraphPad Prism software to determine the Kd values.
2.9. Internalization assays
Internalization and cell processing assays were performed in paired-label format using BT474M1 breast carcinoma cells. Cells at density of 8 × 105 cells per well in 3 mL medium were plated in 6-well plates and after overnight incubation at 37°C, were brought to 4°C and incubated for 30 min. Medium was removed and replenished with fresh medium containing 5 nmol each of either [211At]SAGMB-5F7 plus [131I]SGMIB-5F7, or iso-[211At]SAGMB-5F7 plus iso-[131I]SGMIB-5F7, and the cells were further incubated at 4°C for 1 h. Cell culture supernatants containing unbound radioactivity were removed and fresh medium at 37°C was added. The fraction of initial cell-bound radioactivity that was internalized, on the cell membrane, or released into the cell culture supernatant after incubation at 37°C for 1, 2, 4, 6, and 24 h was determined as described previously [23, 24]. To determine nonspecific uptake, parallel experiments were performed as above except that a 100-fold molar excess of trastuzumab also was added to the wells.
2.10. Paired-label biodistribution experiments
Animal experiments were performed following the guidelines established by the Duke University Institutional Animal Care and Use Committee. Subcutaneous BT474M1 tumor xenografts were established in SCID mice as described previously [24] and two paired-label biodistribution studies were performed when tumors reached a volume of about 350–500 mm3. Groups of 5 mice received tail vein injections of ~185 kBq each of the labeled molecules. In the first experiment, [211At]SAGMB-5F7 (178 MBq/mg) and [131I]SGMIB-5F7 (174 MBq/mg) were administered, and in the second, iso-[211At]SAGMB-5F7 (85 MBq/mg) and iso-[131I]SGMIB-5F7 (89 MBq/mg) were injected. In this way, we could directly compare the effect of 211At-for-131I substitution on tumor targeting and in vivo stability for each of the two isomer configurations. Biodistribution was evaluated at 1 h, 2 h, 4 h, and 21 h after injection; an additional time point of 14 h was included in the second study. Blood and urine were collected, and mice were killed by an overdose of isofluorane. Tumor and normal tissues were isolated, blot-dried, and weighed along with blood and urine. All tissue samples together with 5% injection standards were counted for 131I and 211At activity using an automated gamma counter, and the percentage of injected dose (%ID) per organ and per gram of tissue were calculated.
2.11. Statistical analyses
Data are presented as mean ± standard deviation. Differences in the behavior of co-incubated (in vitro) or co-administered (in vivo) labeled conjugates were analyzed for statistical significance with a paired two-tailed Student t-test using the Microsoft Office excel program, while differences in the behavior of labeled conjugates that were not co-incubated or co-administered were tested with an unpaired Student t-test. Differences with a P value < 0.05 were considered statistically significant.
3. Results
3.1. Radiolabeling
The scheme for synthesis of the four radiohalogenated 5F7 conjugates is illustrated in Fig. 1. The radiochemical yield for the synthesis of iso-[211At]SAGMB-Boc2 was 66.8 ± 2.4% (n=7) compared with 62.6 ± 2.3% (n=6) for [211At]SAGMB-Boc2 under identical conditions. Although the difference in the two yields was small, it was statistically significant (P < 0.05). The radiochemical yield for the synthesis of [211At]SAGMB-Boc2 was similar to that reported previously [27] when TBHP was used as the oxidant and chloroform as the solvent. In most experiments reported herein, [131I]SGMIB and iso-[131I]SGMIB were synthesized using TBHP as the oxidant; however, in a few studies, [131I]SGMIB and iso-[131I]SGMIB were synthesized using NCS as the oxidant and methanol as the solvent, which resulted in radiochemical yields of 69.2 ± 4.2% (n=4) and 84.0 ± 4.5% (n=2), respectively, considerably higher than those obtained using TBHP and chloroform [28].
Fig. 1.

Scheme for the synthesis of 5F7 single domain antibody labeled with 211At using [211At]SAGMB/iso-[211At]SAGMB or with radioiodine using [*I]SGMIB/iso-[*I]SGMIB
Labeling sdAb 5F7 with 211At was accomplished by reaction with [211At]SAGMB and iso-[211At]SAGMB, which were obtained by treatment of Boc2-[211At]SAGMB and Boc2-iso-[211At]SAGMB with TFA. When performed under identical conditions, the conjugation efficiency of iso-[211At]SAGMB (39.5 ± 6.8%; n=5) and [211At]SAGMB (38.4 ± 15.6%; n=6) to 5F7 was similar (P > 0.05). Conjugation efficiencies for labeling 5F7 with [131I]SGMIB and iso-[131I]SGMIB were 28.9 ± 13.0 % (n = 6) and 33.1 ± 7.1 % (n = 6), respectively. The radiochemical purity obtained by ITLC analysis was 98.9%, 97.8%, 98.6%, and 98.4% for iso-[211At]SAGMB-5F7, [211At]SAGMB-5F7, iso-[131I]SGMIB-5F7 and [131I]SGMIB-5F7, respectively. As shown in Fig. 2, SDS-PAGE performed under non-reducing conditions demonstrated that more than 98% of the radioactivity for the 4 radiohalogenated 5F7 conjugates was present in a single band with a molecular weight of about 15 kDa, corresponding to the molecular weight of an sdAb monomer.
Fig. 2.
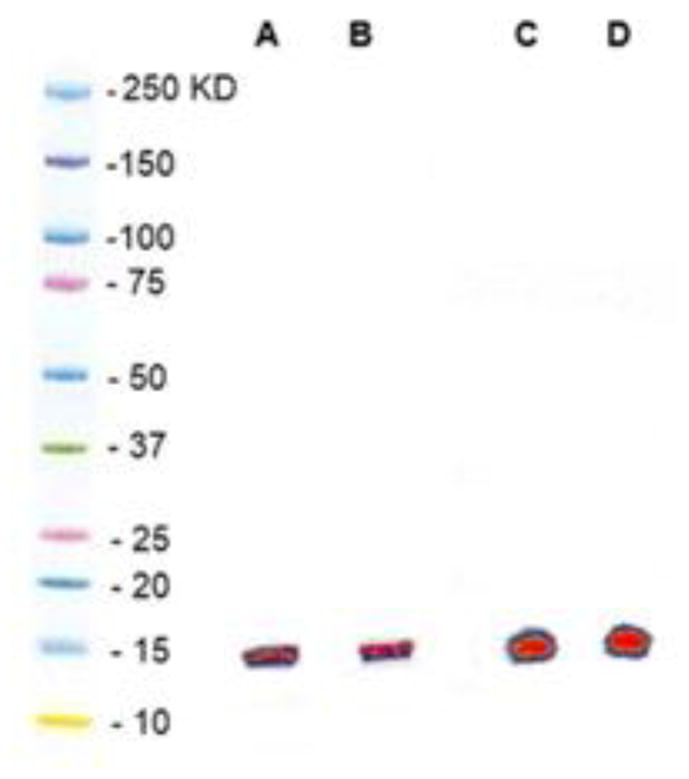
Non-reducing SDS-PAGE/phosphor imaging profiles of (A) [211At]SAGMB-F7, (B) [131I]SGMIB-5F7, (C) iso-[211At]SAGMB-5F7, and (D) iso-[131I]SGMIB-5F7. Molecular weight standards in left lane for comparison.
3.2. Immunoreactive fraction and binding affinity
To determine whether labeling 5F7 sdAb compromised HER2 binding, immunoreactive fractions were determined in paired-label format using the extracellular domain of HER2 as the molecular target. The immunoreactive fractions were determined to be 81.3 ± 0.9%, 83.5 ± 1.1%, 81.8 ± 1.4% and 84.5 ± 0.8% for iso-[211At]SAGMB-5F7, [211At]SAGMB-5F7, iso-[131I]SGMIB-5F7 and [131I]SGMIB-5F7, respectively, suggesting that 5F7 retained immunoreactivity to a similar degree irrespective of the prosthetic agent used. The dissociation constant (Kd) values obtained from saturation binding assays performed on HER2-expressing BT474M1 human breast carcinoma cells were <5 nM for the four labeled conjugates (Fig. 3). However, significantly higher affinity binding (P < 0.05) was observed for iso-[211At]SAGMB-5F7 (3.0 ± 0.1 nM) compared with [211At]SAGMB-5F7 (4.5 ± 0.4 nM). The Kd values for iso-[131I]SGMIB-5F7 and [131I]SGMIB-5F7 were 1.3 ± 0.2 nM and 2.4 ± 0.2 nM, respectively. Thus, the 131I-labeled conjugates had significantly higher binding affinity than their corresponding 211At-labeled 5F7 counterparts (P < 0.05).
Fig. 3.

Saturation binding assays performed on HER2-expressing BT474M1 breast carcinoma cells with 5F7 labeled using [131I]SGMIB, iso-[131I]SGMIB, [211At]SAGMB and iso-[211At]SAGMB. Cells (8 × 104) were incubated with increasing concentrations of the labeled sdAbs and specific cell-associated radioactivity determined as described in text. Plots were generated and Kd values calculated using GraphPad Prism software.
3.3. Internalization assays
Paired-label internalization assays were performed using HER2-expressing BT474M1 cells to determine the extent of intracellular trapping of radioactivity in vitro with [211At]SAGMB-5F7 and iso-[211At]SAGMB-5F7, both in absolute terms and in comparison to co-incubated [131I]SGMIB-5F7 and iso-[131I]SGMIB-5F7, respectively. As shown in Fig. 4, the percentage of initially bound radioactivity that was cell associated (membrane bound + internalized) and internalized for [211At]SAGMB-5F7 remained nearly constant for 24 h, when values of 77.4 ± 0.8% and 67.2 ± 1.1%, respectively, were observed. In general, changing the nature of the prosthetic agent did not affect residualization of radioactivity in HER2-positive cancer cells. For example, at 6 h, 69.5 ± 1.2% and 73.2 ± 1.7% of initially bound radioactivity remained in the intracellular compartment for iso-[211At]SAGMB-5F7 and iso-[131I]SGMIB-5F7, respectively (Fig. 5). However, unlike the behavior of [131I]SGMIB-5F7 and [211At]SAGMB-5F7, intracellular radioactivity levels from iso-[131I]SGMIB-5F7 (49.0 ± 3.6%) and iso-[211At]SAGMB-5F7 (48.4 ± 5.5%) at 24 h was significantly lower (P < 0.05) than those observed from 1–6 h.
Fig. 4.

Paired label internalization of [211At]SAGMB-5F7 (red) and [131I]SGMIB-5F7 (blue) by BT474M1 breast carcinoma cells in vitro. Data presented as percentage of initial specifically bound radioactivity that was (A) cell associated (membrane bound + internalized) and (B) internalized at selected time points. All measurements performed in triplicate.
Fig. 5.

Paired label internalization of iso-[211At]SAGMB-5F7 (brown) and iso-[131I]SGMIB-5F7 (green) by BT474M1 breast carcinoma cells in vitro. Data presented as percentage of initial specifically bound radioactivity that was (A) cell associated (membrane bound + internalized) and (B) internalized at selected time points. All measurements performed in triplicate.
3.4. Biodistribution studies
Two-paired label experiments were performed in SCID mice with subcutaneous BT474M1 breast carcinoma xenografts to directly compare the tissue distribution of [211At]SAGMB-5F7 and iso-[211At]SAGMB-5F7 to their 131I-labeled counterparts. The results obtained over a 21 h period, corresponding to approximately three half-lives of 211At decay, are summarized in Table 1 and Table 2, respectively. Tumor uptake of [211At]SAGMB-5F7 remained at 15–16% ID/g from 1–4 h post injection and then declined to 9.49 ± 1.22% ID/g at 21 h (Fig. 6). Similar tumor uptake values were observed for co-administered [131I]SGMIB-5F7 except at 21 h when values for the radioiodinated conjugate were about 20% higher (11.8 ± 1.5% ID/g; P < 0.05). In the second experiment, similar trends were observed with regard to tumor uptake of iso-[211At]SAGMB-5F7 in comparison to its radioiodinated counterpart. However, tumor accumulation of iso-[211At]SAGMB-5F7 was almost 50% higher than that of [211At]SAGMB-5F7 at all time points, peaking at 23.4 ± 2.2% ID/g at 4 h (difference significant, P < 0.05, except at 21 h by unpaired t test). Likewise, tumor uptake of iso-[131I]SGMIB-5F7 was significantly higher than that of [131I]SGMIB-5F7 at all time points. With the exception of the kidneys, normal tissue uptake of the four 5F7 radioconjugates was low, particularly for iso-[211At]SAGMB-5F7 and iso-[131I]SGMIB-5F7. In kidneys, activity levels for the iso-conjugates were significantly lower than those for the corresponding non-iso-conjugate (P < 0.05 by unpaired t test), with the difference less pronounced for the 211At-labeled conjugates. Because of the lower carbon-halogen bond strength expected for 211At-labeled compounds, comparison of activity levels in the thyroid and the stomach, tissues known to sequester free astatide and iodide, can shed light on the relative in vivo stability of these conjugates. The uptake of 211At and 131I activity in thyroid and stomach after injection of the four 5F7 sdAb conjugates is summarized in Fig. 7. Thyroid and stomach accumulation for both 211At-labeled 5F7 conjugates was significantly higher than seen with their 131I-labeled co-administered counterparts. However, thyroid and stomach activity levels were about twofold lower for iso-[211At]SAGMB-5F7 compared with [211At]SAGMB-5F7, suggesting a lower degree of deastatination in vivo for iso-[211At]SAGMB-5F7.
Table 1.
Paired label biodistribution of [211At]SAGMB-5F7 and [131I]SGMIB-5F7 in SCID mice with subcutaneous B474M1 human breast carcinoma xenografts.
| Tissue | Percent injected dose per grama | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 h | 2 h | 4 h | 21 h | |||||
| 211At | 131I | 211At | 131I | 211At | 131I | 211At | 131I | |
| Liver | 3.0 ± 0.3 | 3.7 ± 0.4 | 2.4 ± 0.2 | 3.0 ± 0.3 | 2.1 ± 0.2 | 2.4 ± 0.4b | 0.7 ± 0.1 | 0.5 ± 0.1 |
| Spleen | 2.5 ± 0.4 | 2.1 ± 0.1b | 2.3 ± 0.2 | 1.8 ± 0.2 | 2.2 ± 0.3 | 1.8 ± 0.5b | 1.0 ± 0.3 | 0.5 ± 0.0 |
| Lungs | 11.7 ± 0.7 | 14.3 ± 2.0 | 7.9 ± 0.8 | 9.7 ± 1.6 | 5.4 ± 1.3 | 5.7 ± 1.7b | 1.5 ± 0.3 | 0.4 ± 0.2 |
| Heart | 1.8 ± 0.3 | 0.7 ± 0.1 | 1.3 ± 0.2 | 0.4 ± 0.0 | 1.1 ± 0.2 | 0.2 ± 0.1 | 0.5 ± 0.2 | 0.1 ± 0.0 |
| Kidneys | 85.3 ± 12.2 | 96.0 ± 13.6 | 46.2 ± 4.2 | 56.0 ± 3.9 | 23.0 ± 2.8 | 29.5 ± 3.4 | 6.1 ± 1.1 | 8.0 ± 1.3 |
| Sm. Int. | 1.4 ± 0.3 | 1.0 ± 0.2 | 1.1 ± 0.1 | 0.6 ± 0.1 | 0.9 ± 0.2 | 0.4 ± 0.1 | 0.6 ± 0.2 | 0.1 ± 0.0 |
| Lg. Int. | 0.9 ± 0.2 | 0.5 ± 0.1 | 1.1 ± 0.2 | 1.3 ± 0.2 | 0.9 ± 0.2 | 1.2 ± 0.2 | 1.1 ± 0.4 | 0.8 ± 0.2 |
| Muscle | 0.9 ± 0.2 | 0.6 ± 0.3 | 0.5 ± 0.2 | 0.3 ± 0.2 | 0.6 ± 0.2 | 0.4 ± 0.2 | 0.1 ± 0.0 | 0.0 ± 0.0 |
| Blood | 1.3 ± 0.2 | 1.0 ± 0.3 | 0.7 ± 0.1 | 0.3 ± 0.1 | 0.5 ± 0.1 | 0.1 ± 0.0 | 0.2 ± 0.1 | 0.0 ± 0.0 |
| Bone | 1.1 ± 0.2 | 0.6 ± 0.1 | 0.8 ± 0.2 | 0.4 ± 0.2 | 0.6 ± 0.2 | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.1 ± 0.0 |
| Brain | 0.2 ± 0.0 | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 |
| Tumor | 15.7 ± 1.4 | 15.1 ± 1.3 | 15.7 ± 1.7 | 16.3 ± 1.9 | 15.5 ± 3.1 | 15.4 ± 3.1b | 9.5 ± 1.2 | 11.8 ± 1.5 |
Mean ± SD (n =5).
Difference in uptake between the two agents statistically NOT significant.
Table 2.
Paired label biodistribution of iso-[211At]SAGMB-5F7 and iso-[131I]SGMIB-5F7 in SCID mice with subcutaneous B474M1 human breast carcinoma xenografts.
| Tissue | Percent injected dose per grama | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 h | 2 h | 4 h | 21 h | |||||
| 211At | 131I | 211At | 131I | 211At | 131I | 211At | 131I | |
| Liver | 2.7 ± 0.4 | 2.2 ± 0.4 | 2.2 ± 0.3 | 1.7 ± 0.3 | 1.4 ± 0.3 | 0.9 ± 0.2 | 0.6 ± 0.5 | 0.2 ± 0.1 |
| Spleen | 1.9 ± 0.7 | 1.4 ± 0.7 | 1.4 ± 0.3 | 0.9 ± 0.3 | 1.1 ± 0.1 | 0.7 ± 0.1 | 1.4 ± 2.2 | 0.1 ± 0.1b |
| Lungs | 4.6 ± 0.5 | 5.2 ± 0.9b | 3.7 ± 0.4 | 3.2 ± 0.7b | 2.6 ± 0.2 | 2.5 ± 0.9b | 2.1 ± 2.3 | 0.2 ± 0.1b |
| Heart | 1.4 ± 0.2 | 0.7 ± 0.2 | 1.0 ± 0.1 | 0.4 ± 0.1 | 0.7 ± 0.1 | 0.2 ± 0.0 | 0.7 ± 1.1 | 0.0 ± 0.0b |
| Kidneys | 65.5 ± 14.5 | 58.9 ± 15.1 | 37.8 ± 9.7 | 32.2 ± 7.0 | 15.8 ± 1.4 | 13.0 ± 1.1 | 3.1 ± 1.5 | 1.6 ± 0.4 |
| Sm. Int. | 3.0 ± 2.3 | 2.9 ± 2.7b | 1.1 ± 0.3 | 0.7 ± 0.2 | 0.7 ± 0.1 | 0.3 ± 0.1 | 0.9 ± 1.3 | 0.3 ± 0.5b |
| Lg. Int. | 1.9 ± 1.3 | 1.8 ± 1.4b | 0.8 ± 0.1 | 0.6 ± 0.1 | 2.0 ± 0.4 | 1.8 ± 0.4b | 2.2 ± 3.7 | 2.8 ± 5.3b |
| Muscle | 0.8 ± 0.1 | 0.7 ± 0.2 | 0.4 ± 0.1 | 0.3 ± 0.1 | 0.2 ± 0.0 | 0.1 ± 0.0 | 0.2 ± 0.2 | 0.0 ± 0.0b |
| Blood | 1.1 ± 0.2 | 0.8 ± 0.2 | 0.6 ± 0.1 | 0.3 ± 0.0 | 0.4 ± 0.1 | 0.2 ± 0.1 | 0.2 ± 0.2 | 0.0 ± 0.0 |
| Bone | 0.9 ± 0.1 | 0.7 ± 0.1 | 0.7 ± 0.1 | 0.5 ± 0.2 | 0.4 ± 0.1 | 0.2 ± 0.2 | 0.3 ± 0.3 | 0.0 ± 0.0b |
| Brain | 0.2 ± 0.0 | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.1 ± 0.1 | 0.0 ± 0.0b |
| Tumor | 21.2 ± 0.7 | 22.8 ± 0.9 | 22.5 ± 5.3 | 25.0 ± 6.1 | 23.4 ± 2.2 | 25.2 ± 2.4 | 14.6 ± 6.1 | 17.4 ± 6.2 |
Mean ± SD (n =5).
Difference in uptake between the two agents statistically NOT significant.
Fig. 6.

Paired-label uptake of 211At and 131I activity after injection of radiolabeled 5F7 sdAb conjugates in SCID mice with subcutaneous BT474M1 breast carcinoma xenografts. (A) [211At]SAGMB-5F7 (red) and [131I]SGMIB-5F7 (blue). (B) iso-[211At]SAGMB-5F7 (brown) and iso-[131I]SGMIB-5F7 (green).
Fig. 7.

Percentage of the injected dose of 211At and 131I activity present after injection of [211At]SAGMB-5F7 (red) and [131I]SGMIB-5F7 (blue) in (A) thyroid and (C) stomach and iso-[211At]SAGMB-5F7 (brown) and iso-[131I]SGMIB-5F7 (green) in (B) thyroid and (D) stomach, tissues known to accumulate free [211At]astatine and [131I]iodide.
As shown in Fig. 8, tumor-to-normal tissue ratios for iso-[211At]SAGMB-5F7 were significantly higher than those for [211At]SAGMB-5F7 in all tissues. For example, tumor-to-liver, tumor-to-blood, tumor-to-spleen and tumor-to-kidney ratios were 18 ± 4, 63 ± 13, 21 ± 3, and 1.50 ± 0.25, respectively, for iso-[211At]SAGMB-5F7 at 4 h, compared with 7.31 ± 1.26, 32 ± 4, 7.11 ± 1.47, and 0.67 ± 0.08 for [211At]SAGMB-5F7. Likewise, tumor-to-normal tissue ratios for iso-[131I]SGMIB-5F7 were significantly higher than those for [131I]SGMIB-5F7 in all tissues (Fig. 9). Finally, tumor-to-normal tissue ratios for the radioiodinated 5F7 conjugates were considerably higher than those for the corresponding 211At-labeled 5F7 conjugates.
Fig. 8.

Tumor-to-normal tissue ratios after injection of [211At]SAGMB-5F7 (red) and iso-[211At]SAGMB-5F7 (brown) in SCID mice bearing subcutaneous breast carcinoma BT474M1 xenografts.
Fig. 9.

Tumor-to-normal tissue ratios after injection of [131I]SGMIB-5F7 (blue) and iso-[131I]SGMIB-5F7 (green) in SCID mice bearing subcutaneous breast carcinoma BT474M1 xenografts.
4. Discussion
In the present study, the anti-HER2 sdAb 5F7 was successfully labeled with the α-particle emitting radiohalogen 211At using two related prosthetic agents, [211At]SAGMB and iso-[211At]SAGMB, designed to trap the radionuclide in HER2-expressing cancer cells after receptor-mediated internalization through the generation of positively charged, labeled catabolites [25, 27]. The high cytotoxicity of 211At α-particles for HER2 expressing breast carcinoma cells has been demonstrated with 211At-labeled trastuzumab both in vitro [14] and in vivo in compartmental settings [15, 33]. Although 211At has many potential advantages for targeted radiotherapy [16], the combination of the short tissue range of its α-particles and it’s 7.2-h half-life necessitates the development of strategies for rapidly achieving homogeneous and prolonged delivery to cancer cells with rapid clearance from normal tissues. Most approaches for achieving this goal utilize a small molecule such as a mAb fragment; however, unlike the case with whole mAbs, 211At-labeled mAb fragments exhibit high uptake in thyroid and stomach, indicating release of free 211At in vivo [34, 35]. Within the HER2 targeting space, this behavior has been observed with an affibody (7 kDa) labeled using N-succinimidyl 3-[211At]astatobenzoate (SAB), which exhibited 25–55 times higher stomach and thyroid levels than the corresponding 125I-labeled construct [36]. An anti-HER2 diabody also has been labeled with 211At using N-succinimidyl N-(4-[211At]astatophenethylsuccinamate (SAPS) and although some encouraging therapeutic responses were obtained, biodistribution results for the 211At-labeled diabody were not reported [37].
In attempting to develop optimal 211At-labeled anti-HER2 constructs, it is important to not only consider the in vivo stability issue noted above but also how to maximize the extent and duration of radioactivity entrapment in cancer cells after binding and internalization of the labeled molecule. In addition, one must select a protein format that offers rapid tumor targeting at therapeutically relevant levels without prolonged residence times in normal tissues. The excellent results obtained with anti-HER2 sdAb SGMIB conjugates provided motivation for the current study evaluating the potential utility of guanidinomethyl substituted prosthetic groups for labeling 5F7 with 211At. Single domain antibody fragment 5F7 with [24] and without [26] a GGC tail has been evaluated after SGMIB labeling in SCID mice with BT474M1 xenografts and with both constructs, tumor uptake peaked 2 h after injection, suggesting that this sdAb had localization kinetics compatible with the 7.2-h half-life of 211At. Because the version without the GGC tail exists as a pure monomer vs. a mixture of monomer and dimer with 5F7-GGC [24] and exhibited significantly higher tumor localization, the tailless 5F7 construct was selected for use in these experiments.
Because of the larger size of the astatine atom compared with the iodine atom, steric hindrance could be an even more important factor for 211At labeling. Based on the significantly higher radioiodination and protein conjugation yields observed for iso-[131I]SGMIB compared with [131I]SGMIB [27], both iso-[211At]SAGMB (1,3,5-isomer) and [211At]SAGMB (1,3,4-isomer) were evaluated for labeling 5F7 sdAb. Although radiolabeling and sdAb conjugation yields for iso-[211At]SAGMB were higher than those for [211At]SAGMB, these differences were not significant. Conjugation of these prosthetic groups, as well as their radioiodinated counterparts, resulted in monomeric products with excellent immunoreactivity and affinity (<5 nM) for binding to HER2-overexpressing BT474M1 breast carcinoma cells. The results obtained for [131I]SGMIB-5F7 were in good agreement with those reported previously for the [131I]SGMIB-5F7-GGC construct [24]. With both isomers, the affinity for the 211At-labeled 5F7 conjugate was about half that of the corresponding 131I-labeled 5F7 conjugate. We speculate that the larger size of the astatine atom and/or radiolytic effects of 211At α-particles could have reduced binding affinity. Nevertheless, the binding affinities for iso-[211At]SAGMB-5F7 (3.0 ± 0.1 nM) and [211At]SAGMB-5F7 (4.5 ± 0.4 nM) should be compatible with their use as targeted radiotherapeutics.
Maximizing radionuclide trapping in cancer cells after binding and cellular processing of radiolabeled receptor-targeted proteins should increase effectiveness for targeted radiotherapy. Internalization assays performed with both trastuzumab and 5F7 sdAb demonstrated that labeling these HER2-targeted proteins with either [*I]SGMIB or iso-[*I]SGMIB resulted in a similar degree of cellular trapping of radioiodine up to 6 h; however, at 24 h, total cell associated and internalized activities were significantly lower for the iso-[*I]SGMIB conjugates [28]. Although these results suggest that the residualizing capability of iso-[*I]SGMIB is not as prolonged as that of [*I]SGMIB, this might not be a significant disadvantage with 211At because of its 7.2-h half-life. Paired label experiments on BT474M1 breast carcinoma cells permitted direct comparison of cell associated and intracellular activity for both iso-[211At]SAGMB-5F7 and [211At]SAGMB-5F7 to their radioiodinated counterparts. Our results indicated that astatine-for-iodine substitution had no effect on residualizing capacity with both the 1,3,4- and 1,3,5-isomers; however, for the latter, a significant decrease in intracellular trapping was observed with both iso-[211At]SAGMB-5F7 and iso-[131I]SGMIB-5F7 at 24 h. Although the mechanism responsible for this behavior is not known, it seems likely that a higher rate of catabolism and/or egress of labeled catabolites for the 1,3,5-isomers could play a role. Nonetheless, even with iso-[211At]SAGMB-5F7, 48.4 ± 5.5% of initially bound radioactivity remained internalized at 24 h, which is encouraging because more than 90% of 211At atoms would have decayed by this time.
The primary focus of this study was the evaluation of the 211At-labeled 5F7 conjugates which to the best of our knowledge, represents the first attempt to evaluate this promising α-emitter for labeling sdAbs. One of these agents, [211At]SAGMB, has been used successfully for labeling the internalizing intact mAb L8A4 that reacts with a mutant form of the epidermal growth factor receptor [27]. However, extrapolation of results from one type of protein construct to another must be done with caution. For example, Nε-(3-[131I]iodobenzoyl)-Lys5-Nα-maleimido-Gly1-GEEEK (131I-IB-Mal-D-GEEEK) was shown to be an excellent reagent for labeling intact mAb L8A4 but offered no advantages in terms of tumor uptake, and a distinct disadvantage in terms of kidney uptake, for labeling 5F7 sdAb [23, 38]. Importantly, the high and prolonged retention of radioactivity in HER2-expressing BT474M1 cancer cells observed in the internalization assays with [211At]SAGMB-5F7 and iso-[211At]SAGMB-5F7 was replicated in the paired-label biodistribution studies performed in SCID mice with xenografts derived from the same BT474M1 cell line. The magnitude of tumor accumulation observed with these 211At-labeled 5F7 conjugates was two- to threefold higher than reported for another HER2-targeted sdAb, 2Rs15d, labeled with 99mTc [21], 177Lu [39], 68Ga [40] and 18F [41] as well as HER2-specific affibodies labeled with a variety of radionuclides including 211At [36, 42].
Regarding the possibility of isomer substitution pattern affecting tumor activity levels, iso-[131I]SGMIB-5F7 and iso-[211At]SAGMB-5F7 exhibited a significant ~1.5-fold tumor delivery advantage compared with [131I]SGMIB-5F7 and [211At]SAGMB-5F7 at all time points. However, this does not appear to reflect differences in residualization capacity because similar degrees of intracellular trapping were observed for both isomers in the in vitro internalization assays until the last time point. With regard to differences in the in vivo behavior of the 211At- and 131I-labeled conjugates, the localization of [211At]SAGMB-5F7 and iso-[211At]SAGMB-5F7 in HER2-positive BT474M1 xenografts was comparable to that of their co-administered 131I-labeled analogues at early time points but about 20% lower at 21 h. This likely reflects halogen-dependent differences in in vivo stability, with a higher rate of dehalogenation for astatine the most probable cause, consistent with the lower C-X bond strength for astatine [43]. This is supported by the observation of higher levels of 211At compared with 131I in thyroid and stomach, tissues known to sequester free radiohalides, with both isomers. However, activity levels in the thyroid and stomach after injection of [211At]SAGMB-5F7 were 0.4–0.6% and 1.0–2.3% ID, respectively, while those for iso-[211At]SAGMB-5F7 were 0.2–0.3% and 0.6–1.7% ID, respectively, suggesting a lower degree of deastatination for the iso-[211At]SAGMB conjugate. Likewise, stomach and thyroid radioactivity levels after injection of iso-[131I]SGMIB-5F7 were lower than those for [131I]SGMIB-5F7, suggesting isomer-dependent differences in the in vivo stability of these radiohalogenated sdAb conjugates. Nonetheless, the degree of 211At uptake in thyroid and stomach for both [211At]SAGMB-5F7 and iso-[211At]SAGMB-5F7 were lower than those reported for a variety of lower molecular weight proteins labeled using several different methods [34–36]. Even though the loss of 211At[astatide] from [211At]SAGMB-5F7 and iso-[211At]SAGMB-5F7 was relatively low, it could increase normal tissue toxicity, which can be reduced significantly through the use of blocking agents [44] as we have done in clinical studies with 211At-labeled antibodies [45].
Tumor-to-normal tissue ratios were generally higher for the radioiodinated conjugates compared with the astatinated versions, presumably reflecting the higher in vivo stability of the iodo versions. Unexpectedly, tumor-to-normal tissue ratios were significantly higher with both radionuclides when 5F7 was labeled using the iso- prosthetic agents. As summarized in Tables 1 and 2, this reflects not only small advantages in tumor uptake but also considerably lower activity levels in normal tissues, particularly with the 131I-labeled conjugates. A possible explanation for this behavior is a mass effect wherein a certain mass of sdAb is needed to block nonspecific uptake of the labeled sdAb in normal organs such as the liver spleen and lungs [40]. This could be relevant here because the [211At]SAGMB-5F7 plus [131I]SGMIB-5F7 biodistribution experiment was performed at a total 5F7 dose of 2.1 μg while in the iso-[211At]SAGMB-5F7 plus iso-[131I]SGMIB-5F7 study, a total 5F7 dose of 4.3 μg was administered. However, this is likely not a factor because the biodistribution observed for [131I]SGMIB-5F7 in the current study at a total sdAb dose of 2.1 μg was quite similar to those reported previously for [131I]SGMIB-5F7 at total 5F7 doses of 4.3. and 6.8 μg [26]. Moreover, significant mass dependent localization differences were observed for the anti-HER2 sdAb 2Rs15d after labeling with 68Ga between 0.1 and 1 μg doses but not between doses of 1 and 10 μg [40], which encompasses the doses used in the current study.
The differences observed in the biological behavior with the two isomer versions with the same radiohalogen were unexpected, particularly given the similarity in tissue distribution observed previously when iso-[125I]SGMIB-trastuzumab and [131I]SGMIB-trastuzumab were compared in the same animal model [28]. However, sdAb are about 10 times smaller than intact mAbs, which may lead to more rapid degradation to species that are small enough to allow easy access to deiodinases and other enzymes such as cytochrome P450 that can lead to dehalogenation [46]. The greater metabolic stability of iso-[125I]SGMIB-5F7 compared with [131I]SGMIB-5F7 could be explained by differences in the catabolism of the two conjugates and the susceptibility of the labeled catabolites towards in vivo deiodination. As summarized in a recent review [46], subtle differences in the design of radioiodinated compounds can lead to increased rates of deiodination. Consistent with our observations, Wieland et al [47] reported that the deiodination of meta-iodobenzylguanidine (structural element of iso-SGMIB) was less than that of ortho-iodobenzylguanidine (structural element of SGMIB). Studies are planned to evaluate the chemical nature of the labeled catabolites generated from iso-SGMIB-sdAb and SGMIB-sdAb conjugates to better understand the differences observed in their in vivo behavior.
A potential problem with using sdAb as a platform for targeted radiotherapeutics is the high accumulation and prolonged retention of radioactivity in the kidney, which could result in dose limiting renal toxicity. This behavior has been observed with radiometals such as 177Lu [39,48] as well as with some residualizing radiohalogenation agents such as 131I-IB-Mal-D-GEEEK [23]. For example, when 5F7-GGC was labeled using 131I-IB-Mal-D-GEEEK, kidney levels were greater than 150% ID/g from 1–8 h after injection and about 100% ID/g at 24 h [24]. In contrast, with all four radiohalogenated 5F7 conjugates evaluated in the current study, initial kidney radioactivity levels were high (60–100% ID/g) but decreased rapidly with renal clearance half-lives of about 1–2 h. Renal radioactivity levels for both the 131I- and 211At-labeled iso- conjugates were significantly lower than those observed for their corresponding 1,3,4-isomer conjugates at all time points with the difference in kidney retention increasing with time. For example, the renal radioactivity level observed 21 h after injection of iso-[131I]SGMIB-5F7 was more than 4 times lower than that for [131I]SGMIB-5F7. Radionuclide-dependent differences in kidney activity levels also were observed although to a lesser extent than those between the two isomeric versions for a given radionuclide, Paradoxically, kidney radioactivity levels after injection of iso-[211At]SAGMB-5F7 were higher than those for co-administered iso-[131I]SGMIB-5F7 while renal radioactivity levels after injection of [211At]SAGMB-5F7 were lower than those for co-administered [131I]SGMIB-5F7. The differences in renal uptake and retention of the four 5F7 sdAb radioconjugates cannot be explained at this time and were unexpected considering the similarity of the acylation agents in physical properties that might influence kidney retention such as polarity and hydrophilicity. Moreover, previous studies showed no significant differences between kidney uptake values for intact mAb L8A4 labeled with [131I]SGMIB and [211At]SAGMB [26], and trastuzumab labeled using [131I]SGMIB and iso-[125I]SGMIB [27]. Although the mechanism(s) responsible for their lower kidney radioactivity levels are not clear, the iso-[211At]SAGMB and iso-[131I]SGMIB conjugates are the reagents of choice for minimizing radiation absorbed dose to the kidneys with 5F7 and potentially other sdAb. If further reduction in renal radiation dose is needed, D’Huyvetter et al [48] have shown that this can be accomplished, at least with a 177Lu-labeled sdAb by co-infusion with the plasma expander Gelofusin.
5. Conclusion
In summary, we have shown that the anti-HER2 sdAb 5F7 can be labeled with 211At in reasonable yields with excellent retention of affinity and immunoreactivity after labeling. Studies in preclinical models with [211At]SAGMB-5F7 demonstrated high and prolonged tumor targeting and rapid normal tissue clearance, with even more favorable observed with iso-[211At]SAGMB-5F7. Moreover, iso-[131I]SGMIB-5F7 was shown to offer significantly improved tumor targeting compared with [131I]SGMIB-5F7. Taken together, our results suggest that iso-[211At]SAGMB-5F7 and iso-[131I]SGMIB-5F7 warrant further evaluation as α-particle and β-particle emitting targeted radiotherapeutics for the treatment of HER2 expressing malignancies.
Acknowledgments
The authors acknowledge the National Cancer Institute for providing support for this research under Grant CA42324. The 5F7 single domain antibody fragment was generously provided by Dr. Hilde Revets from Ablynx N.V., Ghent, Belgium. We thank Xiao-Guang Zhao, Elizabetha Krol, and Phil Welsh for excellent technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Menard S, Casalini P, Campiglio M, Pupa S, Agresti R, Tagliabue E. HER2 overexpression in various tumor types, focussing on its relationship to the development of invasive breast cancer. Ann Oncol. 2001;12:S15–9. doi: 10.1093/annonc/12.suppl_1.s15. [DOI] [PubMed] [Google Scholar]
- 2.Scholl S, Beuzeboc P, Pouillart P. Targeting HER2 in other tumor types. Ann Oncol. 2001;12:S81–7. doi: 10.1093/annonc/12.suppl_1.s81. [DOI] [PubMed] [Google Scholar]
- 3.Hede K. Gastric cancer: trastuzumab trial results spur search for other targets. J Natl Cancer Inst. 2009;101:1306–7. doi: 10.1093/jnci/djp341. [DOI] [PubMed] [Google Scholar]
- 4.Luque-Cabal M, Garcia-Teijido P, Fernandez-Perez Y, Sanchez-Lorenzo L, Palacio-Vazquez I. Mechanisms behind the resistance to trastuzumab in HER2-amplified breast cancer and strategies to overcome It. Clin Med Insights Oncol. 2016;10:21–30. doi: 10.4137/CMO.S34537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zambrano J, Yeh ES. Autophagy and Apoptotic Crosstalk: Mechanism of Therapeutic Resistance in HER2-Positive Breast Cancer. Breast Cancer (Auckl) 2016;10:13–23. doi: 10.4137/BCBCR.S32791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koo T, Kim IA. Brain metastasis in human epidermal growth factor receptor 2-positive breast cancer: from biology to treatment. Radiat Oncol J. 2016;34:1–9. doi: 10.3857/roj.2016.34.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaeckle KA. Neoplastic meningitis from systemic malignancies: diagnosis, prognosis and treatment. Semin Oncol. 2006;33:312–23. doi: 10.1053/j.seminoncol.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 8.Kordbacheh T, Law WY, Smith IE. Sanctuary site leptomeningeal metastases in HER-2 positive breast cancer: A review in the era of trastuzumab. Breast. 2016;26:54–8. doi: 10.1016/j.breast.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Stemmler HJ, Schmitt M, Willems A, Bernhard H, Harbeck N, Heinemann V. Ratio of trastuzumab levels in serum and cerebrospinal fluid is altered in HER2-positive breast cancer patients with brain metastases and impairment of blood-brain barrier. Anticancer Drugs. 2007;18:23–8. doi: 10.1097/01.cad.0000236313.50833.ee. [DOI] [PubMed] [Google Scholar]
- 10.Stemmler HJ, Schmitt M, Harbeck N, Willems A, Bernhard H, Lassig D, et al. Application of intrathecal trastuzumab (Herceptintrade mark) for treatment of meningeal carcinomatosis in HER2-overexpressing metastatic breast cancer. Oncol Rep. 2006;15:1373–7. doi: 10.3892/or.15.5.1373. [DOI] [PubMed] [Google Scholar]
- 11.Tomasello G, Bedard PL, de Azambuja E, Lossignol D, Devriendt D, Piccart-Gebhart MJ. Brain metastases in HER2-positive breast cancer: the evolving role of lapatinib. Crit Rev Oncol/Hematol. 2010;75:110–121. doi: 10.1016/j.critrevonc.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Chien AJ, Rugo HS. Emerging treatment options for the management of brain metasases in patients with HER2-positive metastatic breast cancer. Breast Cancer Res Treat. 2013;137:1–12. doi: 10.1007/s10549-012-2328-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larson SM, Carrasquillo JA, Cheung NKV, Press OW. (2015) Radioimmunotherapy of human tumours. Nature Rev Cancer. 2015;15:347–360. doi: 10.1038/nrc3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akabani G, Carlin S, Welsh P, Zalutsky MR. In vitro cytotoxicity of 211At-labeled trastuzumab in human breast cancer cell lines: effect of specific activity and HER2 receptor heterogeneity on survival fraction. Nucl Med Biol. 2006;33:333–347. doi: 10.1016/j.nucmedbio.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 15.Boskovitz A, McLendon RE, Okamura T, Sampson JH, Bigner DD, Zalutsky MR. Treatment of HER2 positive breast carcinomatous meningitis with intrathecal administration of α-particle emitting 211At-labeled trastuzumab. Nucl Med Biol. 2009;36:659–669. doi: 10.1016/j.nucmedbio.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zalutsky MR, Vaidyanathan G. Astatine-211-labeled radiotherapeutics: an emerging approach to targeted alpha-particle radiotherapy. Current Pharm Design. 2000;6:1433–55. doi: 10.2174/1381612003399275. [DOI] [PubMed] [Google Scholar]
- 17.Freise AC, Wu AM. In vivo imaging with antibodies and engineered fragments. Mol Immunol. 2015;67:142–52. doi: 10.1016/j.molimm.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kijanka M, Dorresteijn B, Oliveira S, van Bergen en Henegouwen PM. Nanobody-based cancer therapy of solid tumors. Nanomedicine. 2015;10:161–74. doi: 10.2217/nnm.14.178. [DOI] [PubMed] [Google Scholar]
- 19.Dmitriev OY, Lutsenko S, Muyldermans S. Nanobodies as Probes for Protein Dynamics in Vitro and in Cells. J Biol Chem. 2016;291:3767–75. doi: 10.1074/jbc.R115.679811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82:775–97. doi: 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- 21.Vaneycken I, Devoogdt N, Van Gassen N, Vincke C, Xavier C, Wernery U, et al. Preclinical screening of anti-HER2 nanobodies for molecular imaging of breast cancer. FASEB J. 2011;25:2433–46. doi: 10.1096/fj.10-180331. [DOI] [PubMed] [Google Scholar]
- 22.Keyaerts M, Xavier C, Heemskerk J, Devoogdt N, Everaert H, Ackaert C, et al. Phase I Study of 68Ga-HER2-nanobody for PET/CT assessment of HER2 expression in breast carcinoma. J Nucl Med. 2016;57:27–33. doi: 10.2967/jnumed.115.162024. [DOI] [PubMed] [Google Scholar]
- 23.Pruszynski M, Koumarianou E, Vaidyanathan G, Revets H, Devoogdt N, Lahoutte T, et al. Targeting breast carcinoma with radioiodinated anti-HER2 Nanobody. Nucl Med Biol. 2013;40:52–9. doi: 10.1016/j.nucmedbio.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pruszynski M, Koumarianou E, Vaidyanathan G, Revets H, Devoogdt N, Lahoutte T, et al. Improved tumor targeting of anti-HER2 nanobody through N-succinimidyl 4-guanidinomethyl-3-iodobenzoate radiolabeling. J Nucl Med. 2014;55:650–6. doi: 10.2967/jnumed.113.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaidyanathan G, Zalutsky MR. Synthesis of N-succinimidyl 4-guanidinomethyl-3- [*I]iodobenzoate: a radio-iodination agent for labeling internalizing proteins and peptides. Nature Prot. 2007;2:282–6. doi: 10.1038/nprot.2007.20. [DOI] [PubMed] [Google Scholar]
- 26.Vaidyanathan G, McDougald D, Choi J, Koumarianou E, Weitzel D, Osada T, et al. Preclinical evaluation of 18F-labeled anti-HER2 nanobody conjugates for imaging HER2 receptor expression by immuno-PET. J Nucl Med. 2016;57:967–73. doi: 10.2967/jnumed.115.171306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaidyanathan G, Affleck DJ, Bigner DD, Zalutsky MR. N-succinimidyl 3-[211At]astato-4-guanidinomethylbenzoate: an acylation agent for labeling internalizing antibodies with alpha-particle emitting 211At. Nucl Med Biol. 2003;30:351–9. doi: 10.1016/s0969-8051(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 28.Choi J, Vaidyanathan G, Koumarianou E, McDougald D, Pruszynski M, Osada T, et al. N-Succinimidyl guanidinomethyl iodobenzoate protein radiohalogenation agents: influence of isomeric substitution on radiolabeling and target cell residualization. Nucl Med Biol. 2014;41:802–12. doi: 10.1016/j.nucmedbio.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zalutsky MR, Zhao X-G, Alston K, Bigner D. High-level production of α-particle-emitting 211At and preparation of 211At-labeled antibodies for clinical use. J Nucl Med. 2001;42:1508–1515. [PubMed] [Google Scholar]
- 30.Pozzi OR, Zalutsky MR. Radiopharmaceutical chemistry of targeted radiotherapeutics III: solvent and radiation dose effects on astatination chemistry at high radiation doses of astatine-211 α-particles. J Nucl Med. 2007;48:1190–1206. doi: 10.2967/jnumed.106.038505. [DOI] [PubMed] [Google Scholar]
- 31.Yu Z, Xia W, Wang HY, Wang SC, Pan Y, Kwong KY, et al. Antitumor activity of an Ets protein, PEA3, in breast cancer cell lines MDA-MB-361DYT2 and BT474M1. Molecular Carcin. 2006;45:667–75. doi: 10.1002/mc.20212. [DOI] [PubMed] [Google Scholar]
- 32.Lindmo T, Boven E, Cuttitta F, Fedorko J, Bunn PA., Jr Determination of the immunoreactive fraction of radiolabeled monoclonal antibodies by linear extrapolation to binding at infinite antigen excess. J Immunol Methods. 1984;72:77–89. doi: 10.1016/0022-1759(84)90435-6. [DOI] [PubMed] [Google Scholar]
- 33.Palm S, Bäck T, Claesson I, Danielsson A, Elgqvist J, Frost S, et al. Therapeutic efficacy of astatine-211-labeled trastuzumab on radioresistant SKOV-3 tumors in nude mice. Int J Radiat Oncol Biol Phys. 2007;69:572–579. doi: 10.1016/j.ijrobp.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 34.Hadley SW, Wilbur DS, Gray MA, Atcher RW. Astatine-211 labeling of an antimelanoma antibody and its Fab fragment uning N-succinimidyl p-astatobenzoate: comparison in vivo with the p-[125I]iodobenzoyl conjugate. Bioconjugate Chem. 1991;2:171–179. doi: 10.1021/bc00009a006. [DOI] [PubMed] [Google Scholar]
- 35.Garg PK, Harrison CL, Zalutsky MR. comparative tissue distribution in mice of the alpha-emitter 211At and 131I as labels of a monoclonal antibody and F(ab′)2 fragment. Cancer Res. 1990;50:3514–20. [PubMed] [Google Scholar]
- 36.Steffen A-C, Almqvist Y, Chyan M-K, Lundqvist H, Tolmachev V, Wilbur DS, et al. Biodistribution of 211At labeled HER2 binding affibody molecules in mice. Oncol Reports. 2007;17:1141–7. [PubMed] [Google Scholar]
- 37.Robinson MK, Shaller C, Gamestani K, Plascjak PS, Hodge KM, Yuan Q-A, et al. Effective treatment of established breast tumor xenografts in immunodeficient mice with a single dose of α-emitting radioisotope astatine-211 conjugated to anti-HER2/neu diabodies. Clin Cancer Res. 2008;14:875–882. doi: 10.1158/1078-0432.CCR-07-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vaidyanathan G, Alston KL, Bigner DD, Zalutsky MR. Nε-(3-[*I]iodobenzoyl)- Lys5-Nα -maleimido-Gly1-GEEEK ([*I]IB-Mal-D-GEEEK): a radioiodinated prosthetic group containing negatively charged D-glutamates for labeling internalizing monoclonal antibodies. Bioconjug Chem. 2006;17:1085–1092. doi: 10.1021/bc0600766. [DOI] [PubMed] [Google Scholar]
- 39.D’Huyvetter M, Aerts A, Xavier C, Vaneycken I, Devoogdt N, Gijs M, et al. Development of 177Lu-nanobodies for radioimmunotherapy of HER2-positive breast cancer: evaluation of different bifunctional chelators. Contrast Media Mol Imaging. 2012;7:254–264. doi: 10.1002/cmmi.491. [DOI] [PubMed] [Google Scholar]
- 40.Xavier C, Vaneycken I, D’Huyvetter M, Heemskerk J, Keyaerts M, Vincke C, et al. Synthesis, preclinical validation, dosimetry, and toxicity of 68Ga-NOTA-anti-HER2 nanobodies for iPET imaging of HER2 receptor expression in cancer. J Nucl Med. 2013;54:776–784. doi: 10.2967/jnumed.112.111021. [DOI] [PubMed] [Google Scholar]
- 41.Xavier C, Blykers A, Vaneycken I, D’Huyvetter M, Heemskerk J, et al. 18F-nanobody for PET imaging of HER2 overexpressing tumors. Nucl Med Biol. 2016;43:247–52. doi: 10.1016/j.nucmedbio.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 42.Glaser M, Iveson P, Hoppmann S, Indrevoll B, Wilson A, Arukwe J, et al. Three methods for 18 F labeling. of the HER2 binding affibody molecule ZHER2:2891 including preclinical assessment. J Nucl Med. 2013;54:1981–1988. doi: 10.2967/jnumed.113.122465. [DOI] [PubMed] [Google Scholar]
- 43.Teze D, Sergentu D-C, Kalichuk V, Barbet J, Deniaud D, Galland N, et al. Targeted radionuclide therapy with astatine-211: oxidative dehalogenation of astatobenzoate conjugates. Scientific Reports. 2017;7:2579. doi: 10.1038/s41598-017-02614-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larsen RH, Slade S, Zalutsky MR. Blocking [211At]astatide accumulation in normal tissues: preliminary evaluation of seven potential compounds. Nucl Med Biol. 1998;25:351–357. doi: 10.1016/s0969-8051(97)00230-8. [DOI] [PubMed] [Google Scholar]
- 45.Zalutsky MR, Reardon DA, Akabani G, Coleman RE, Friedman AH, Freidman HS, et al. Clinical experience with α-emitting astatine-211: treatment of recurrent brain tumor patients with 211At-labeled chimeric 81C6 anti-tenascin monoclonal antibody. J Nucl Med. 2008;49:30–38. doi: 10.2967/jnumed.107.046938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cavina L, van der Born D, Klaren PHM, Feiters MC, Boerman OC, Rutjes FPJT. Design of radioiodinated pharmaceuticals: structural features affecting metabolic stability towards in vivo deiodination. Eur J Org Chem. 2017;2017:3387–3414. doi: 10.1002/ejoc.201601638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wieland DM, Wu J-I, Brown LE, Manger TJ, Swanson DP, Beiewaltes WH. Radiolabeled adrenergic neuron-blacking agents: adrenomedullary imaging with [131I]iodobenzylguanidine. J Nucl Med. 1980;21:349–353. [PubMed] [Google Scholar]
- 48.D’Huyvetter M, Vincke C, Xavier C, Aerts A, Impens N, Baatout S, et al. Targeted radionuclide therapy with a 177Lu-labeled antt-HER2 nanobody. Theranostics. 2014;4:708–720. doi: 10.7150/thno.8156. [DOI] [PMC free article] [PubMed] [Google Scholar]