

Abstract
Activation of the PI3K-AKT signaling cascade is a common critical event during malignant transformation. In this study, we used thyroid gland epithelial cells and a series of genetically engineered mouse strains as model systems to demonstrate that, while necessary, AKT activation is not sufficient for PI3K-driven transformation. Instead, transformation requires the activity of the PDK1-regulated AGC family of protein kinases. In particular, SGK1 was found to be essential for proliferation and survival of thyroid cancer cells harboring PI3K-activating mutations. Notably, co-targeting SGK1 and AKT resulted in significantly higher growth suppression than inhibiting either PI3K or AKT alone. Overall, these findings underscore the clinical relevance of AKT-independent pathways in tumors driven by genetic lesions targeting the PI3K cascade.
Introduction
Activation of PI3K signaling through a variety of mechanisms, including PIK3CA activating mutations or amplification, and PTEN mutation, deletion or silencing, is found in about 40% of all human cancers (1,2). Some specific tumor types, such as breast, prostate, colorectal, and endometrial cancer, as well as glioblastoma and anaplastic thyroid carcinoma, show even higher mutation rates for members of this pathway (2,3). PI3K-dependent generation of phosphatidylinositol-3,4,5-triphosphate (PIP3) triggers membrane recruitment of proteins containing a PH-domain, such as the three AKT isoforms and the PDK1 kinase. Furthermore, it leads to the activation of the mTORC2 complex by relieving an autoinhibitory mechanism involving its component, SIN1 (4). mTORC2 and PDK1 act then in concert to phosphorylate and activate AKT (5,6).
While, traditionally, AKT is considered the primary effector of PI3K transforming activity, there is accumulating evidence that additional PI3K-dependent pathways may play a critical role during neoplastic transformation (7) or in driving adaptive resistance to PI3K inhibition (8,9).
In fact, mTORC1 and mTORC2, two key PI3K effectors, activate, in cooperation with PDK1, several members of the AGC family of kinases, including S6K, PKC, and SGK (10). These targets, in turn, are known to control pathways with direct bearing on the transformation process (11).
In view of these notions, it is noteworthy that a formal, systematic in vivo genetic approach to address the questions of i) whether AKT activation is sufficient for tumor development and maintenance, and ii) what is the role in these processes, if any, of the other PI3K/PDK1-dependent pathways, has never been reported.
Since PI3K activation is a common feature of advanced and aggressive thyroid cancer (3), we have used the thyroid gland as a relevant model system to address this issue, and have generated a series of mouse models in which different critical components of the PI3K signaling cascade have been selectively manipulated in the thyroid epithelial cells.
We have found that while AKT activation is essential for PI3K-dependent neoplastic transformation, it is clearly not sufficient. In fact, we show that activation of PDK1-dependent AGC kinases is essential for tumor development. Furthermore, we provide evidence that the AGC kinase family member SGK1 plays a critical role downstream of PI3K activation for tumor maintenance, and that it represents a critical therapeutic target.
Methods
Animals
The Ptenthyr-/-, [Ptenthyr-/-,KrasG12D], and [Pten,Tp53]thyr-/- strains have been previously described (12-14). Pdk1L155E mice (15) were kindly provided by Dr. Dario Alessi (University of Dundee). All strains were backcrossed in the 129S6/SvEv background for at least 10 generations. All experimental procedures involving mice were conducted in accordance with IACUC-approved protocols.
Tissue analysis
Formalin fixed samples were paraffin-embedded and processed for routine H&E staining. 6 μm sections were subjected to antigen retrieval, incubated with antibodies against Ki67 (Dako), and counterstained with hematoxylin.
Alternatively, mice were injected intraperitoneally with BrdU (10 mg/Kg, Sigma) 2 hours before sacrifice. Anti-Ki67 and anti-BrdU-stained sections were analyzed using the ImageJ software.
Western blot and antibodies
Cells, thyroids, and tumor tissue were homogenized on ice in RIPA buffer (ThermoFisher Scientific) supplemented with Halt Protease and Phosphatase Inhibitor cocktail (ThermoFisher Scientific). Protein concentration was determined using the Pierce BCA protein Kit (ThermoFisher Scientific). Western blot analysis was carried out using 30–50 μg proteins run on Bis-Tris SDS precast gels (Genescript).
All the primary antibodies used were purchased from Cell Signal Technology and used at a dilution of 1:1000 in 5% BSA in TBS-T. Signals were detected with HRP-conjugated secondary antibodies (ThermoFisher Scientific) and the chemiluminescence substrate Luminata Crescendo (EMD Millipore). Equivalent loading was confirmed with anti-β-actin (Sigma-Aldrich).
Cell lines, drug treatments and cell proliferation
Cell lines were established from tumors developed in Ptenthyr−/−, [Pten, Tp53]thyr−/− and [Ptenthyr−/−,KrasG12D,Pdk1L155E] mice as previously described (16,17). All mouse cell lines, as well as the human cell lines 8505c, ACT1, and CAL62 were grown in DMEM (HyClone) supplemented with 10% FBS (HyClone) and Mycozap Plus-CL (Lonza Biologics). The human cell lines TAD2, N-Thy-Ori, T235, T238, THJ21T, SW1736, C643, Ocut2, T243, THJ16T were grown in RPMI (HyClone) supplemented with 10% FBS (HyClone) and Mycozap Plus-CL. All cell lines were maintained at 37°C with 5% CO2. Cell identity was validated by STS profiling as well as by amplifying and sequencing genomic fragments encompassing their known mutations.
Chemical inhibitors GSK650394, EMD638683, MK2206, GDC-0941 were purchased from Selleck Chemicals and dissolved in DMSO.
For drug sensitivity experiments on parental cell lines, all inhibitors were added 24 hrs after plating. After 72 hrs of treatment, cells were detached and counted using a Z2 Coulter counter (Beckman Coulter). Final DMSO concentration in media was always below 0.1%. EC50 value calculation and statistical analysis were done using GraphPad Prism (GraphPad Software).
In experiments on lentivirally-transduced cell lines, cells were pretreated with 250 ng/ml (THJ16T) or 500 ng/ml (T4888M) doxycycline hyclate (Sigma-Aldrich) for 48 hrs prior to treatment with GDC-0941. Drugs were added immediately after cell seeding.
For long term doubling rate experiments, 5 × 104 cells were plated in presence of chemical inhibitors or doxycycline hyclate. Every 3 days, cells were harvested, counted, and 5 × 104 cells were reseeded.
Statistical analysis of drug synergy was evaluated from the results of the Wst-1 assays and calculated using the Chou-Talaly method (18) and the Calcusyn Software (Biosoft). To determine synergy between two drugs, the software uses a median-effect method that determines if the drug combination produces greater effects together than expected from the summation of their individual effects. The combination index (CI) values are calculated for the different dose-effect plots (for each of the serial dilutions) based on the parameters derived from the median-effect plots of the individual drugs or drug combinations at the fixed ratios. The CI was calculated based on the assumption of mutually nonexclusive drug interactions. CI values significantly > 1 are antagonistic, not significantly different than 1 are additive, and values < 1 are synergistic.
3D spheroid assays
5 × 103 cells were plated in 96-well round bottom, ultra-low attachment plates (Corning). Spheroid volume was measured after 3 and 10 days using ImageJ.
shRNAs and doxycycline-inducible cell lines
Cells were transduced with lentiviruses encoding shRNAs against SGK1 (from the TRC Genome-Wide shRNA Collection). After preliminary experiments with a battery of previously fully validated clones (9), shRNA TRCN0000040175 was chosen for subsequent experiments due to its superior targeting efficiency.
ShRNA TRCN0000040175 was cloned into the Tet-pLKO-puro (Addgene plasmid #21915). Lentiviral constructs were packaged in HEK293 cells transfected using Lipofectamine 2000 (Invitrogen). Viral supernatant was collected 48-72 hours after transfection, combined, and filtered using 0.45 μm Nalgene SFCA syringe filters (Thermo Scientific). Target cells were plated 24 hours before infection and then exposed to viral supernatant supplemented with 20 μg/ml polybrene (Santa Cruz). Mouse cell lines were then selected for at least 72 hours with puromycin (Corning) 5 μg/ml, whereas a concentration of 2 μg/ml was used for human cell lines.
Transduced cell lines were pretreated with 250-500 ng/ml doxycycline hyclate for 48 hours prior to plating for growth curve experiments. After seeding, cells were counted every 24 hours and cell number was determined as previously described. In all experiments performed on transduced cell lines, doxycycline hyclate was replaced every 24 hrs.
PIP3 determination
Mass spectrometry was used to measure inositol lipid levels essentially as previously described (19), using a QTRAP 4000 (AB Sciex) mass spectrometer and employing the lipid extraction and derivitization method described for whole tissue, with the modification that frozen tissue samples were ground under liquid N2 with a mortar and pestle prior to analysis (instead of Dounce homogenizer), and that 10 ng C17:0/C16:0 PtdIns(3,4,5)P3 ISD (internal standard) and 10ng C17:0/C16:0 PtdIns ISD was added to primary extracts. Final samples were dried in a speedvac concentrator rather than under N2. Measurements were conducted in triplicate per experiment, 0.5mg per sample.
RNA extraction and Real-time PCR
Total RNA was extracted with TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. RNA yields were assessed using a NanoDrop 1000 (ThermoFisher Scientific). 1 μg of RNA was reverse transcribed using the Maxima first strand cDNA synthesis kit (ThermoFisher Scientific) according to the manufacturer's instructions. Quantitative real-time PCR (qRT-PCR) was conducted on a StepOne Plus apparatus using the Absolute Blue qPCR Rox Mix (ThermoFisher Scientific) and TaqMan expression assays (Applied Biosystem) following the manufacturer's instructions. The TaqMan expression assays used were Mm00441380_m1 (Sgk1), Mm00449845_m1 (Sgk2), Mm00453797_m1 (Sgk3), Mm02026778_g1 (Akt2) and Mm00446973_m1 (Tbp). Each sample was run in triplicate and Akt2 and Tbp were used to normalize the input RNA. Relative quantities were calculated using the 2(x0005E)(-ΔΔCt) method.
Cell cycle DNA analysis
Transduced cell lines were seeded in 10 cm plates, cultured overnight at 37°C, and incubated for 72 hr with doxocycline hyclate 250 ng/ml. Cells were harvested by trypsin treatment and fixed in 70% ethanol in ice for 30 min. After treatment with RNase (Genentech/Roche) for 5 min at RT, cells were stained with propidium iodide (Genentech/Roche) overnight, and DNA content was measured using a BD FACSCanto™ II system (BD Biosciences).
Annexin V staining
Cell lines were seeded in 100 mm plates, cultured overnight at 37°C, and incubated for 72-96-120 hrs with doxocycline hyclate. Cells were harvested by trypsinization and supernatant was also collected. Cells were stained with Annexin V FITC and PI (BD Biosciences) for 15 minutes at room temperature in the dark. Samples were analyzed by flow cytometry within 1 hr using a BD FACSCanto™ II system (BD Biosciences). Flow cytometry analysis was performed on the FloJo platform.
In vivo experiments
8 week-old NSG mice, from the Einstein Shared Facility in Stem Cell Research, were injected subcutaneously with 5 × 106 T4888M or THJ16T cells previously transduced with an inducible shRNA targeting SGK1. When tumors reached a size between 100 and 150 mm3, mice were randomized to control and doxycycline hyclate treatment groups. Doxycycline hyclate was dissolved in water and administered via oral gavage (100 mg/kg) daily, and tumor volume was calculated from two-dimensional measurements using the equation: tumor volume = (length × width2) × 0.5. Tumor weight was measured at the end of the experiment.
Results
AKT activation is indispensable for PI3K-dependent transformation
Ptenthyr-/- mice are born with mildly hyperplastic thyroid glands (12), and slowly develop solid, nodular lesions that progress to locally invasive follicular carcinomas after one year of age (20) (Fig. 1A,B). In order to test to what extent unrestrained PI3K relies on AKT activation to induce hyperproliferation and transformation of mouse thyrocytes, we generated a mouse model in which Akt1 and Akt2 are simultaneously deleted in the thyroid epithelial cells, alone or in combination with the loss-of-function Pten allele. Akt3 was excluded from this analysis, since its expression is undetectable in the mouse thyroid (Suppl. Fig. 1).
Figure 1.
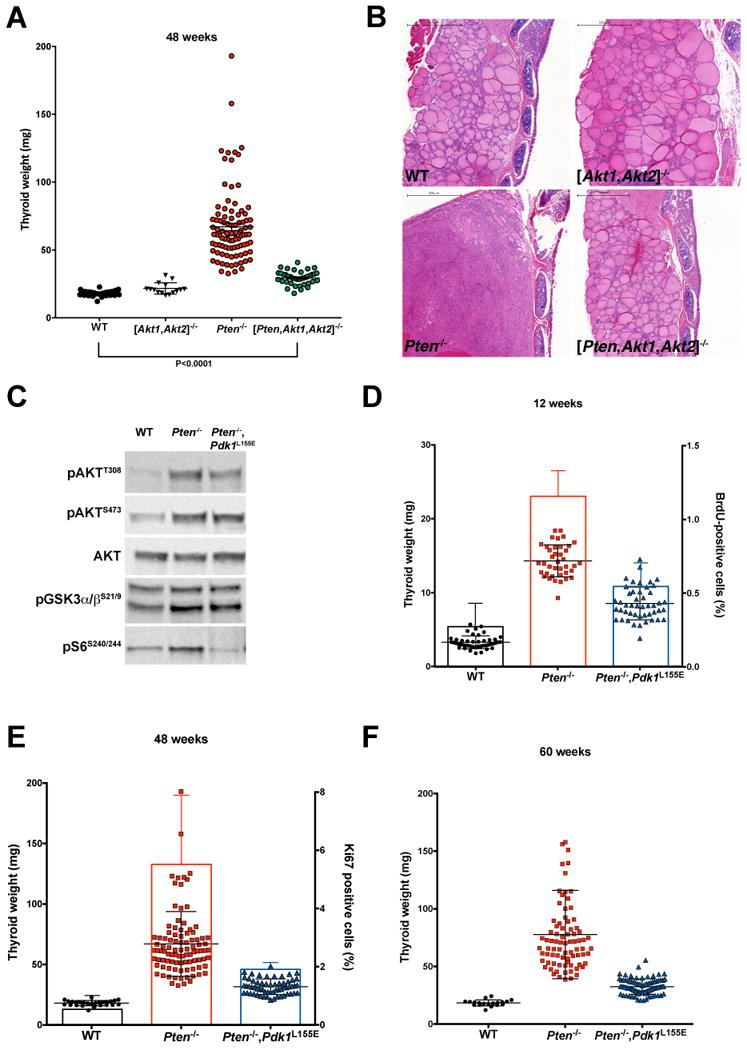
Genetic analysis of the role of AKT and PDK1-dependent AGC kinases in the development of thyroid hyperplasia. A, thyroid weight at 48 weeks of age for mice of the indicated genotypes. B, H&E staining of representative sections from thyroid glands dissected from 48-week old mice of the indicated genotypes. Bar: 500µm. C, Western blot analysis showing that the Pdk1 L155E allele does not affect AKT phosphorylation and activity, but completely impairs the activity of the PIF-pocket dependent AGC kinase S6K. D-F, thyroid weight at 12, 48, and 60 weeks of age, showing the effect of the Pdk1 L155E allele on the hyperplasia induced by Pten deletion. In addition, the thyroid proliferative index was measured by BrdU incorporation (D), or by Ki67 staining (E).
While co-deletion of Akt1 and Akt2 did not cause any overt phenotype, and did not alter thyroid size or histology at any time point analyzed, it drastically inhibited the development of both follicular hyperplasia and nodular lesions in Ptenthyr-/- mice (no triple mutant mice with nodules at 48 weeks, vs. 66.7% of Ptenthyr-/- mice) (Fig. 1A,B).
Thus, based on in vivo genetic data, AKT activation appears to be an absolute requirement for the oncogenic activity of unrestrained PI3K. It remains to be seen, however, whether it is sufficient to drive thyrocyte transformation.
Notably, we found that the size of the thyroid in the triple mutants was still slightly but significantly larger than that of wild type mice (Fig. 1A), suggesting that AKT is not the only player downstream of active PI3K to participate in the thyroid hyperplastic response.
Also AGC kinase activation is essential for PI3K-dependent transformation
In addition to AKT, PI3K controls, via mTORC1 and mTORC2, the activation of several members of the AGC family of protein kinases, including S6K, SGK, and a number of PKC isoforms (10). Similar to AKT, activation of these kinases requires two phosphorylation events, one in the activation loop, mediated by PDK1, and the other in the hydrophobic motif, mediated by mTORC1 or mTORC2. Other AGC kinases such as RSK, instead, require ERK1/2 activity for the hydrophobic motif phosphorylation. In stark contrast with AKT, phosphorylation of the AGC kinases activation loop is absolutely dependent on prior phosphorylation of the hydrophobic motif, which creates a docking site for the PIF-pocket of PDK1 (21). Consequently, a point mutation in the PIF-pocket, L155E, completely abolishes PDK1 ability to activate all the AGC kinases, without impacting its ability to phosphorylate AKT (22).
We have taken advantage of this differential activation mechanism to explore the role and relevance of AGC kinases activation downstream of PI3K.
To this end, Ptenthyr-/- mice were crossed to mice in which the Pdk1 L155E mutant allele is expressed from its endogenous locus in a Cre-dependent manner (15). The resulting compound mutant progeny was then compared to sex- and age-matched wild type and single mutant mice. Western blot analysis of thyroid extracts confirmed that AKT was still fully phosphorylated in the double mutants, and was able to phosphorylate its targets, GSK3α/β, while the PIF-pocket dependent S6K was, as predicted, unable to phosphorylate ribosomal protein S6 (Fig. 1C).
Analysis of control, Ptenthyr-/-, and compound mutant mice at 12, 48, and 60 weeks of age revealed that impairment of AGC kinases activation significantly mitigates the hyperplastic phenotype observed in the thyroid of Ptenthyr-/- mice. Both thyroid weight and thyrocyte proliferative index were reduced by at least 50% at each time point analyzed, strongly suggesting that AKT activation is only partially responsible for the hyperproliferative phenotype, and that AGC kinase-driven pathways are critical for thyroid proliferation (Fig. 1D-F).
Histological analysis of these thyroids revealed striking differences: at 48 weeks of age, when over 60% of Ptenthyr-/- female mice display nodular lesions, and a few of them (3%) already have progressed to invasive carcinomas, 97% of Ptenthyr-/-,Pdk1L155E compound mutant mice displayed only mild hyperplasia (Fig. 2A, B). At 60 weeks, when over 46% of Ptenthyr-/- female mice display invasive carcinomas, and 43% have nodular lesions, 94% of Ptenthyr-/-,Pdk1L155E compound mutant mice still displayed simple hyperplasia (Fig. 2A, B). We have previously reported (20) that male Ptenthyr-/- mutants show a delay in the development of thyroid neoplastic lesions. At 60 weeks of age, 7% of Ptenthyr-/- male mice display invasive carcinomas, and 64% have nodular lesions; on the other hand, 94% of Ptenthyr-/-,Pdk1L155E compound mutant males displayed simple hyperplasia (Suppl. Fig. 2). These data strongly suggest that, despite constitutive AKT activity, the simple thyroid hyperplasia developed by Ptenthyr-/-,Pdk1L155E compound mutants does not progress to neoplastic lesions.
Figure 2.
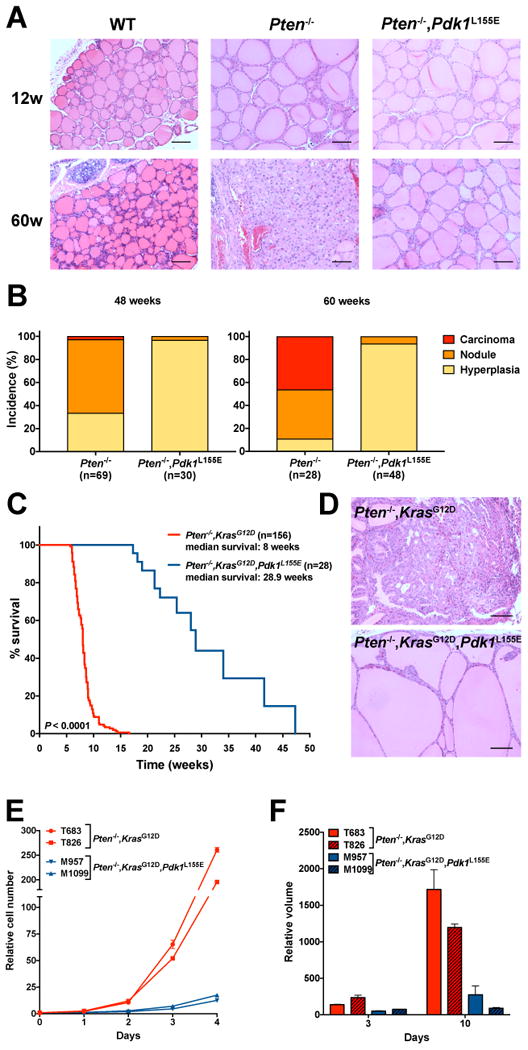
The Pdk1 L155E allele prevents PI3K-dependent tumor development. A, H&E staining of representative sections from thyroid glands dissected from 12- and 60-week old mice of the indicated genotypes. Bar: 100µm. B, Effect of the Pdk1 L155E allele on the incidence of hyperplasia, solid nodules, and carcinomas in Ptenthyr-/- mice at 48 and 60 weeks of age. C, Kaplan-Meier analysis of the effect of the Pdk1 L155E allele on the survival of Ptenthyr-/-,KrasG12D mice. D, H&E staining of representative sections from thyroid glands dissected from mice of the indicated genotypes, showing that the Pdk1 L155E allele prevents the development of solid, invasive tumors. E, analysis of the effect of the Pdk1 L155E allele on the proliferation of cell lines established from thyroid glands dissected from mice of the indicated genotypes. F, 3D growth analysis of spheroids derived from the same cell lines as in E. Size is expressed as final volume at day 7 divided by the initial volume measured at 24 hours after plating.
AGC kinase inactivation drastically impairs tumor progression in aggressive models of thyroid cancer
The thyroid lesions developed by Ptenthyr-/- mice progress very slowly to invasive tumors. This prompted us to analyze the effect of ablating AGC kinase activation in more aggressive models of thyroid cancer. We have reported that simultaneous thyroid-specific activation of Kras and PI3K leads to the rapid development of aggressive carcinomas (14). When we introduced the Pdk1 L155E mutant allele into this strain, we found that the median survival of the triple compound mutants was increased from 8 to 29 weeks (Fig. 2C). Furthermore, histological analysis revealed that the lesions developed by the triple mutants were not solid carcinomas, as in the Ptenthyr-/-,KrasG12D mice, but rather massive hyperplasias ultimately leading to mouse death via airway compression (Fig. 2D).
We have generated stable cell lines from thyroid lesions developed by Ptenthyr-/-,KrasG12D (20) as well as by Ptenthyr-/-,KrasG12D,Pdk1L155E mice. In agreement with the in vivo data, cell lines carrying the Pdk1 L155E mutant allele proliferated dramatically slower than their Pdk1 wild type counterpart, both in standard culture conditions and as spheroids in the more physiological 3D growth conditions (Fig. 2E,F).
Finally, we tested the effect of the Pdk1 L155E mutant allele on the development of anaplastic thyroid carcinoma, the most aggressive and undifferentiated subtype of thyroid cancer, using the [Pten, Tp53]thyr-/- model (16). [Pten, Tp53]thyr-/- mice develop follicular carcinomas early on, which progress to anaplastic carcinomas in over 70% of cases by 48 weeks of age.
Impairment of AGC kinase activation reduced the weight of the compound mutant glands by 75%, to the same level observed in single mutant Ptenthyr-/- mice (Fig. 3A). Histological analysis of these lesions showed that only 5% of cases had nests of undifferentiated cells, while 65% of cases showed only well-differentiated follicular carcinomas, and 30% nodular hyperplasia (Fig. 3B,C).
Figure 3.
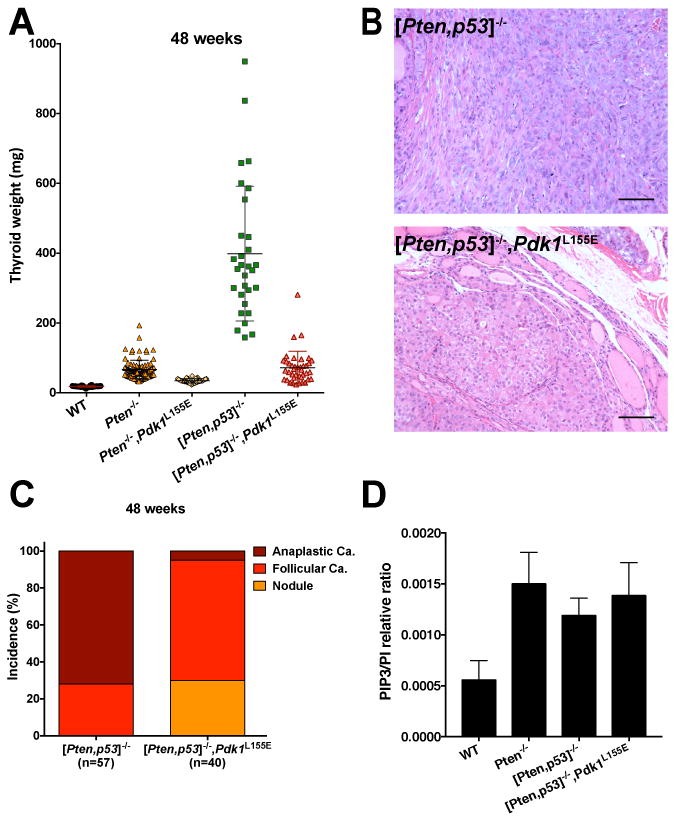
The Pdk1 L155E allele prevents the development of anaplastic carcinomas in [Pten, Tp53]thyr-/- mice. A, thyroid weight at 48 weeks of age for mice of the indicated genotypes. B, H&E staining of representative sections from thyroid glands dissected from mice of the indicated genotypes, showing that the Pdk1 L155E allele prevents the development of undifferentiated tumors. C, Effect of the Pdk1 L155E allele on the incidence of solid nodules, follicular, and anaplastic carcinomas in [Pten, Tp53]thyr-/- mice at 48 weeks of age. D, Mass spectrometry measurement of the PIP3/PI ratio in extracts from thyroids of the indicated genotypes.
Thus, despite constitutive PI3K and AKT activation, neoplastic lesions developed by [Pten, Tp53]thyr-/-,Pdk1L155E thyroids are unable to progress to aggressive, undifferentiated anaplastic carcinomas.
Genetic manipulation of PI3K downstream effectors might lead to unexpected feedbacks that could impact PI3K catalytic activity. In order to assess the activation status of PI3K in our genetic models, we measured the levels of phosphatidylinositol-3,4,5-triphosphate (PIP3) in wild type, single, double, and triple mutant thyroids, using a well-established mass spectrometry approach, and found that PIP3 levels were increased, as expected, in Ptenthyr-/- mice, but did not change upon introduction of additional mutant alleles (Fig. 3D).
Thus, despite a fully active PI3K/AKT axis, Ptenthyr-/- thyrocytes absolutely need AGC kinase activity to reach their full transformation potential, which is dictated by the additional genetic alterations introduced (Kras, Tp53, etc.).
Activation of SGK family members is required for PI3K-active cancer cell proliferation
To identify the critical AGC kinase that is required for full PI3K oncogenic activity, we used a large panel of mouse and human thyroid cancer cell lines encompassing the spectrum of mutations most commonly found in advanced thyroid tumors, and performed a sensitivity screen using small molecule inhibitors of PI3K and mTORC1 (as controls), and of the PI3K-regulated AGC kinase families, S6K, PKC, and SGK.
Strikingly, we found that PKC and S6K inhibitors had little to no effect on the proliferation of thyroid cancer cells at concentrations commonly used in cell culture assays. On the other hand, GSK650394, an SGK inhibitor, had a dramatic growth suppressive effect, especially in cells carrying driver mutations that activate PI3K signaling (Fig. 4A).
Figure 4.
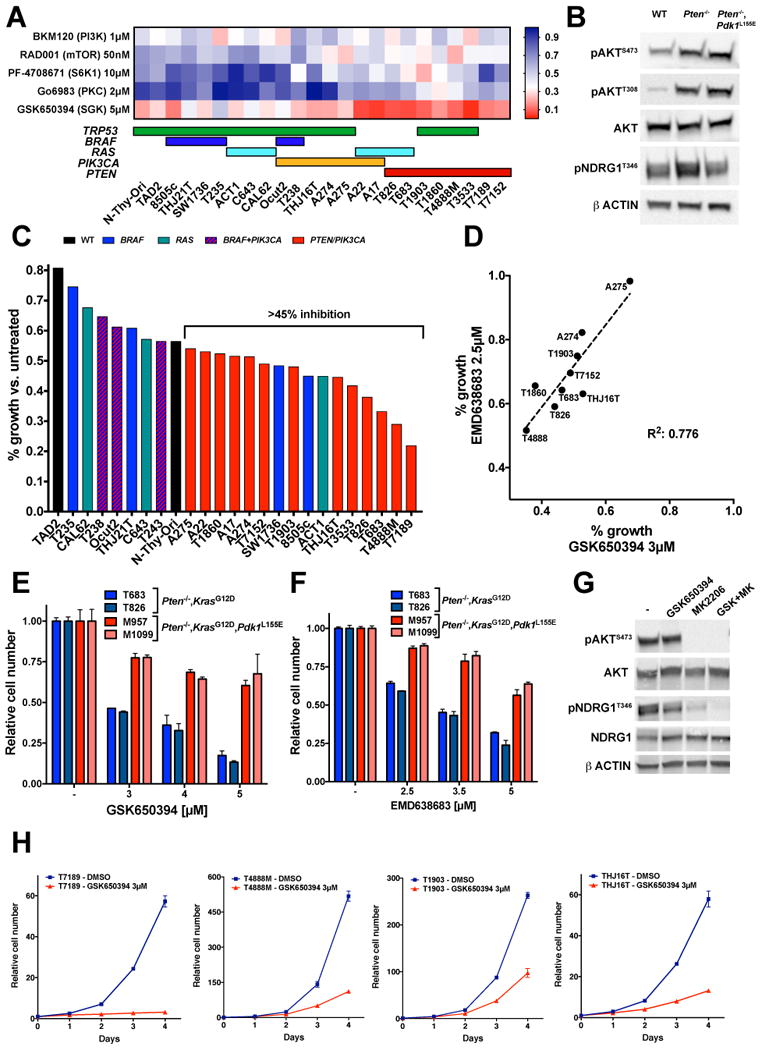
SGK activity is critical for the proliferation of thyroid cancer cells carrying PI3K activating mutations. A, heat map illustrating the effect of inhibitors of different PI3K-dependent AGC kinases on the proliferation of mouse and human thyroid cancer cell lines. Colored horizontal bars below the heat map indicate the known driver mutation for each cell line. B, Western blot analysis of the phosphorylation of the SGK target, NDRG1, in the thyroids from mice of the indicated genotypes. C, Effect of a reduced concentration (3µM) of the SGK inhibitor GSK650394 on the proliferation of mouse and human thyroid cancer cell lines. D, Correlation between the responses of representative thyroid cancer cell lines to low concentrations of the SGK inhibitors GSK650394 and EMD638683. E-F, Differential response to two different SGK inhibitors of mouse thyroid cancer cell lines proficient or impaired in AGC kinase activation. G, Western blot analysis of the phosphorylation of the SGK1 target, NDRG1, in the T4888M cells upon 6 hours treatment with the indicated inhibitors. H, Analysis of the proliferation of different PI3K-active thyroid cancer cell lines treated with the SGK inhibitor GSK650394.
Given the lack of antibodies specifically recognizing active SGK, we tested the phosphorylation level of NDRG1, a bona fide SGK target, in the thyroids of wild type, Pten mutant, and double mutant mice, and found that, indeed, SGK activation is increased in Ptenthyr-/- thyroids and returns at baseline levels in the glands of Ptenthyr-/-,Pdk1L155E compound mutant mice (Fig. 4B).
These data suggest that SGK is the critical AGC kinase that is required for PI3K full oncogenic activity.
Commercially available SGK inhibitors are notoriously suboptimal in terms of target selectivity. To verify that SGK is indeed the critical target affecting thyroid cancer cell proliferation, we first reduced the dose of GSK650394 by 40% and found that a 3µM concentration was still able to profoundly inhibit proliferation of PI3K-active cell lines (Fig. 4C). Then we compared the response of nine thyroid cancer cell lines to both GSK650394 (3µM) and to another SGK inhibitor, EMD638686, again used at a lower concentration (2.5µM) than usually found in published studies (10µM). We found a strong correlation between the sensitivity of these cells to the two inhibitors, further suggesting that they are indeed targeting SGK (Fig. 4D).
Next, we used the four Pten-/-,KrasG12D cell lines described above, two of which also carry the Pdk1L155E mutation, to demonstrate that these two inhibitors were able to decrease cell proliferation in a dose-dependent manner in the AGC kinase-proficient cells, but not in the AGC kinase-defective lines (Fig. 4E,F). Furthermore, we tested whether GSK650394 could affect the ability of SGK to phosphorylate NDRG1 in T4888M cells, and found that it significantly reduced NDRG1 phosphorylation. Interestingly, concomitant AKT inhibition was required for full ablation of phosphorylation, suggesting that, at least in specific cellular contexts, NDRG1 is a common target of SGK and AKT (Fig. 4G).
Finally, we performed a time course analysis of the effect of 3µM GSK650394 on the proliferation of PI3K-active thyroid cancer cells, and found that SGK inhibition profoundly affected cell growth, with inhibition rates ranging between 63% and 95% after four days (Fig. 4H), thus formally validating SGK as a critical component of the PI3K-dependent oncogenic machinery.
SGK1 depletion reduces proliferation and viability of PI3K-active cancer cells
SGK is a family of three closely related kinases. In order to assess their relative contribution to the transformation process, we analyzed the RNA level of the three isoforms in the thyroid of control, Pten mutant, and double mutant mice, and found that Sgk1 is the most highly expressed of the three genes, while Sgk2 expression levels are several orders of magnitude lower than those of Sgk1 and Sgk3 (Fig. 5A). These results were also confirmed at the protein level, in thyroid extracts from wild type and Pten mutant mice (Fig. 5B). More variability, instead, was observed in mouse thyroid cancer cell lines, where, although Sgk1 is still the most expressed gene, the relative expression of Sgk2 and Sgk3 is different in each line (Fig. 5C). The almost undetectable expression of Sgk2 suggests that this isoform is unlikely to play a critical role in the control of thyroid cell proliferation. On the other hand, Sgk3 is unique among the SGK genes, since it has a N-terminal PX-domain targeting the protein to the endosomes, where it is activated in response to the class III PI3K Vps34 (23). Based on these notions, as well as on preliminary shRNA-based experiments showing very limited effects of Sgk3 depletion on cell proliferation (Suppl. Fig. 3), we focused on Sgk1 as the primary conduit of this AKT-independent, PI3K-driven pathway.
Figure 5.
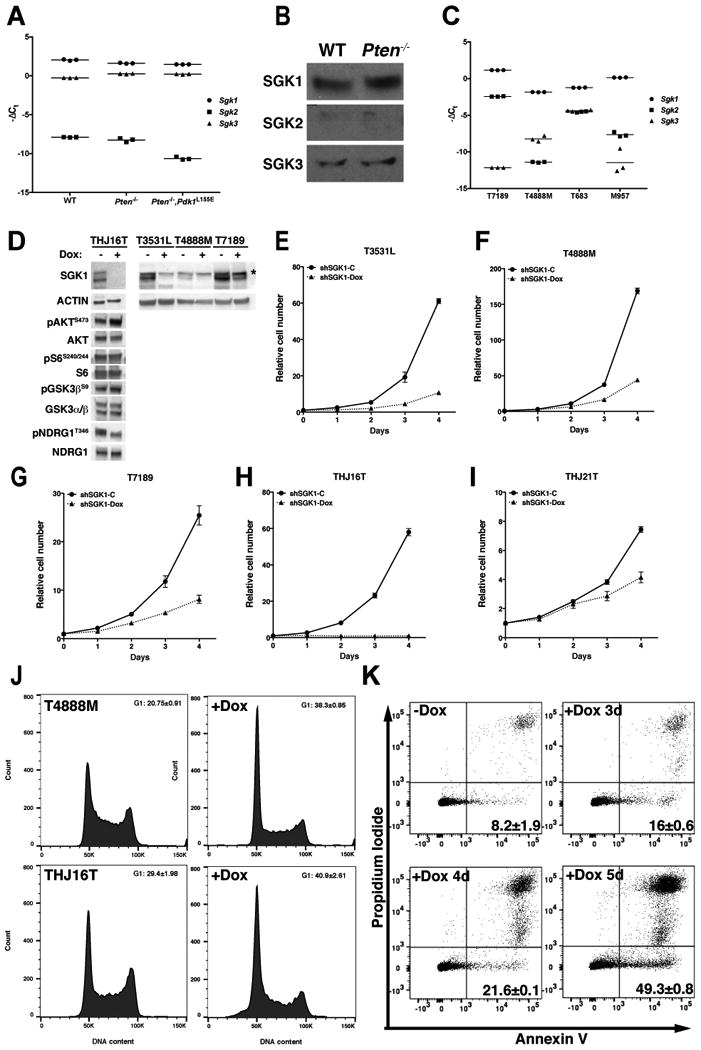
SGK1 depletion impairs PI3K-dependent thyroid cancer cell proliferation. A, qPCR analysis of the expression of Sgk1, -2, and -3 in the thyroids of mice of the indicated genotypes, showing predominant expression of Sgk1. B, Western blot validation of the qPCR results using thyroid extracts from the indicated genotypes. C, qPCR analysis of the expression of Sgk1, -2, and -3 in mouse thyroid cancer cell lines, showing predominant expression of Sgk1. D, Western blot analysis of shRNA-mediated SGK1 depletion upon 96 hours doxycycline treatment of the indicated cell lines. Asterisk marks a non-specific band only observed in mouse cells. E-H, Analysis of the effect of doxycycline-regulated Sgk1 depletion on the proliferation of PI3K-active mouse and human thyroid cancer cells. I, Representative example of the minimal effect of SGK1 depletion on the proliferation of a human BRAFV600E-driven thyroid cancer cell line. J, shRNA-mediated Sgk1 depletion induces G1 arrest 48 hours after doxycycline treatment. K, shRNA-mediated Sgk1 depletion induces a time-dependent increase in apoptosis upon doxycycline treatment.
Preliminary experiments revealed that constitutive and sustained shRNA-mediated Sgk1 depletion is not tolerated by PI3K-active thyroid cancer cells, and that no stably transduced pools could be maintained for more than a few passages. Thus we generated doxycycline-inducible mouse and human thyroid cancer cell lines, using a previously validated shRNA (9) that targets both mouse and human SGK1 (Fig. 5D). Four days of SGK1 depletion resulted in reduced NDRG1 phosphorylation and in a slight increase in AKT activation, accompanied by a minor increase in GSK3β phosphorylation (Fig. 5D). However, no changes in S6 phosphorylation were observed.
Analysis of cell proliferation upon shSgk1 induction revealed dramatic growth suppression in both mouse (Fig. 5E-G) and human (Fig. 5H) thyroid cancer cells harboring driver mutations that activate PI3K signaling, with inhibition rates ranging between 68% and 98% after six days of doxycycline treatment. The growth suppressive effect, conversely, was significantly lower in human thyroid cancer cell lines without mutations activating PI3K (44%, Fig. 5I and Suppl. Fig. 4).
Flow cytometric analysis of control and shSgk1-induced thyroid cancer cells showed that SGK depletion caused arrest in G1 at early time points (Fig. 5J), followed, 24 to 48 hours later, by induction of a significant degree of apoptosis (Fig. 5K).
Taken together, these data show that PI3K-active thyroid cancer cells absolutely need SGK1 activity to support sustained proliferation and survival.
In order to test the long-term effect of SGK1 depletion on the growth of PI3K-active thyroid cancer cells, we performed a three-week long analysis of cell proliferation using the THJ16T cell line. Induction of shSGK1 drastically reduced cell proliferation, which reached its lowest level by day 6 and then remained rather constant for the remainder of the follow up (Fig. 6A). SGK1-depleted cells, however, did not undergo irreversible growth inhibition or senescence, since doxycycline removal after 12 days promptly restored the normal proliferation rate for these cells (Fig. 6A, green line).
Figure 6.
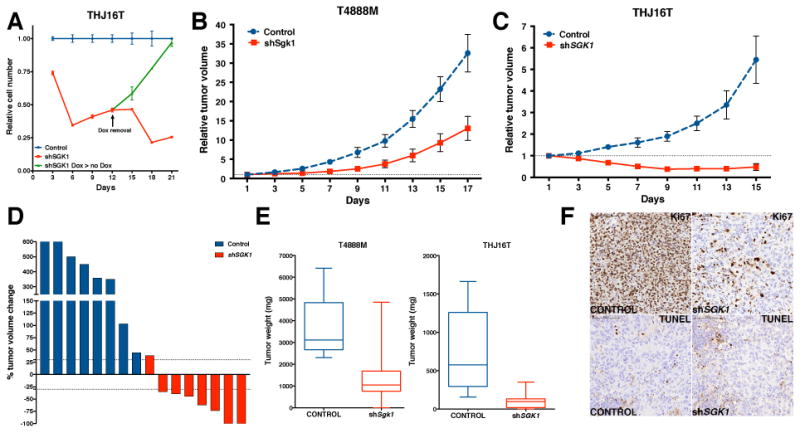
SGK1 depletion impairs thyroid cancer growth in vivo. A, long-term Sgk1 depletion induces reversible growth suppression. 50,000 THJ16T-shSGK1 cells were plated, then counted and re-plated at the same density every three days. B-C, T4888M-shSgk1 and THJ16T-shSGK1 cells were injected into the flank of NSG mice; doxycycline treatment was initiated when tumors reached approximately 100mm3, and tumor growth was monitored every two days. D, waterfall plot showing that SGK1 depletion in THJ16T cells caused a 30% or more decrease in tumor size in 7 of 8 tumors. E, endpoint measurement of excised tumor weight. The boxes extend from the 25th to the 75th percentiles; the line in the middle of the box is plotted at the median; the whiskers are drawn down to the 10th percentile and up to the 90th. F, Ki67 and TUNEL immunostaining of representative sections from control and shSGK1 THJ16T tumors.
SGK1 depletion impairs tumor growth in vivo
To test the effect of SGK1 depletion on tumor growth in vivo, we generated xenograft models using the murine T4888M cell line (Pten-/-, Tp53-/-) and the human THJ16T cell line (PIK3CAE545K, TP53-/-). Doxycycline-mediated induction of the shRNA targeting SGK1 severely impaired tumor growth in both models (Fig. 6B-C). In line with the higher knockdown level (see Fig. 5D) the effect was more dramatic in the THJ16T xenograft: SGK1 depletion resulted in tumor regression in almost 90% of cases, with complete tumor disappearance in 25% of the mice (Fig. 6D). These results were further validated by tumor weight analysis at endpoint, demonstrating that, despite continuous PI3K signaling, SGK1 depletion reduced mean tumor size by over 80% (Fig. 6E).
IHC analysis of these tumors showed no significant changes in the number of apoptotic cells. However, the fraction of proliferating cells was strikingly reduced in SGK1-depleted cells (Fig. 6F).
SGK1 inhibition increases the efficacy of AKT targeting
The notion that SGK1 activation is fully dependent on PI3K signaling leads to the prediction that simultaneous targeting of both kinases should not result in a significant advantage over PI3K inhibition alone.
We tested this postulate using the doxycycline-inducible shSGK1 T4888M and THJ16T cell lines. As expected, the pan-PI3K inhibitor GDC0941 was clearly effective in reducing thyroid cancer cell proliferation over a three-day period. When comparing each time point to the proliferation of untreated cells, simultaneous depletion of SGK1 dramatically increased the overall growth suppressive effect (Fig. 7A). However, when we normalized cell proliferation at each GDC0941 concentration to the basal proliferation in the presence or absence of SGK1, thus obtaining a measure of the “net effect” of GDC0941 treatment, it became clear that SGK1 depletion paradoxically reduced the net ability of GDC0941 to inhibit cell proliferation, in particular in the THJ16T cells (Fig. 7B). These data demonstrate that SGK1 is a key PI3K downstream effector for the induction of proliferation, and thus its absence reduces the net ability of PI3K inhibitors to impact cell proliferation, despite complete abolishment of AKT and S6 phosphorylation (Fig. 7C).
Figure 7.
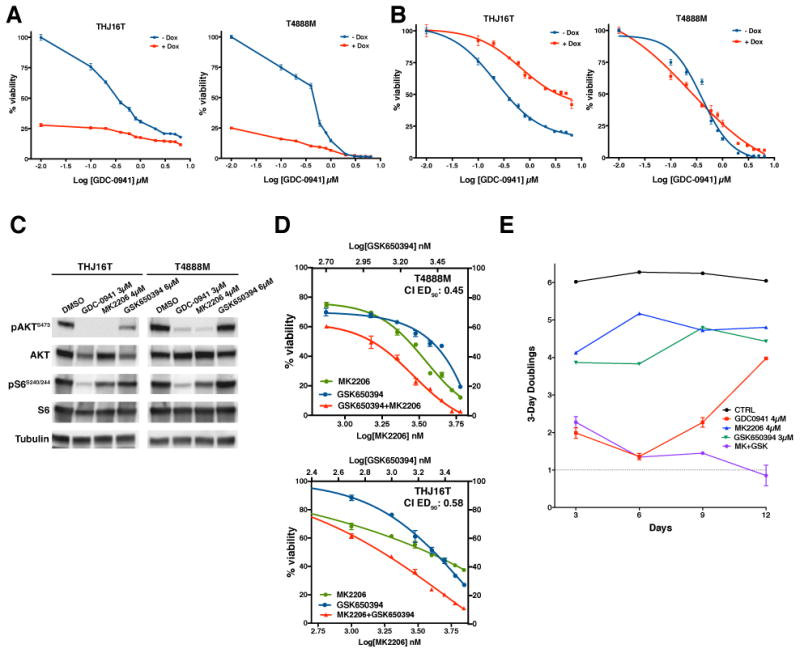
Combined targeting of AKT and SGK completely inhibits proliferation of PI3K-active cancer cells. A-B dose-response plots showing the effect of SGK1 depletion on the efficacy of PI3K targeting. Dox+ values are shown as ratio to completely untreated cells in A, and as ratio to doxycycline-treated, GDC-0941-untreated cells in B. C, Western blot analysis of the phosphorylation of AKT and S6 in the THJ16T and T4888M cells upon 6 hours treatment with the indicated inhibitors at concentrations corresponding to EC75-EC90 as single drug. D, pharmacological co-targeting of AKT and SGK at a constant inhibitors ratio, showing synergistic interaction between the two compounds. E, long-term treatment of T4888M cells with the indicated inhibitors, showing progressive resistance to PI3K inhibition, and complete growth suppression with combined AKT and SGK inhibition.
Along the same lines, simultaneous pharmacological co-targeting of PI3K and SGKs did not result in strong synergistic activity (Suppl. Fig. 5).
Our model also predicts that targeting in combination the two major PI3K effectors, AKT and SGK, should be more effective than targeting each kinase separately. In fact, we found that the AKT inhibitor MK2206 strongly synergized with SGK inhibition in both T4888M and THJ16T cells, with a Combination Index at EC90 of 0.58 and 0.45, respectively (Fig. 7C,D).
Prolonged inhibition of certain signaling pathways can elicit adaptive responses that result in the development of resistance to the inhibitor. These responses are usually not evident in the standard three-day proliferation assays. To assess the effect of long term pathway inhibition on cell proliferation, we measured the doubling rate of T4888M cells over a two-week period, and found that the growth-inhibitory effect of the PI3K inhibitor GDC0941, used at EC90, was rapidly and progressively lost, beginning after six days of continuous treatment (Figure 7E, red line). On the other hand, single agent treatment with the AKT inhibitor MK2206 or the SGK inhibitor GSK650394, at their EC80, had a more steady growth suppressive effect. Strikingly, when administered in combination, these two inhibitors dramatically synergized, leading to almost complete inhibition of cell proliferation (Fig. 7E).
Thus, combined inhibition of the two critical PI3K effectors, AKT and SGK, has a clearly increased and prolonged therapeutic potential, compared to direct PI3K inhibition.
Discussion
Three decades of research have formally established the critical role of the PI3K/AKT/mTOR pathway in tumor development and progression (24). At the same time, the development of genetic tools for accurate pathway dissection and of small molecule inhibitors of most members of this signaling cascade have uncovered the existence of an intricate network of signals and feedbacks, which strongly underlines the need for a deeper understanding of this pathway, if we want to effectively target it for therapeutic purposes (25).
One of the tenets that have been recently challenged by several studies is the universal role of AKT activation as the critical conduit of PI3K transforming signals (26). As an example, activation of SGK3 has been shown to be essential for the growth of a subset of PIK3CA-mutant breast tumors exhibiting only minimal AKT activation (7).
Our data address for the first time, in a clinically relevant in vivo model, the question of AKT sufficiency in a tumor type that instead relies on AKT activity as an effector of PI3K activation. In fact, we show that simultaneous genetic ablation of Akt1 and Akt2 completely suppresses thyroid cancer development in mice with thyroid-specific loss of the Pten tumor suppressor gene, thus formally proving that AKT activity is essential for tumorigenesis downstream of PI3K in this tissue. At the same time, the residual hyperplasia observed in [Pten, Akt1, Akt2]thyr-/- mice suggests that additional PI3K-dependent, AKT-independent pathways contribute to thyrocyte hyperproliferation.
The Pdk1 L155E genetic model that uncouples AKT-dependent and AKT-independent pathways downstream of PI3K has been previously utilized to show that PI3K-dependent, AKT-independent pathways are required for T cell progenitors proliferation (27), leucine-induced mTORC1/S6K activation in the cardiac muscle (28), and proper brain patterning (29). Our data further extend these concepts by providing clear genetic evidence that activation of AGC kinases controlled by PDK1 via its PIF pocket is critical not only for the proliferative response to PI3K activation, but also for the ability of PI3K to drive neoplastic transformation and tumor progression in three well-characterized mouse models of follicular, poorly differentiated, and anaplastic thyroid cancer, respectively.
The AGC kinases co-controlled by PI3K and PDK1 are grouped in three major families, S6K, PKC, and SGK, each with multiple members. This renders impractical any in vivo genetic approach to pinpoint the critical kinases that act downstream of PI3K to facilitate tumor initiation.
We elected instead to use a small scale inhibitor-based screen, combined with shRNA approaches in established thyroid cancer cell lines, to identify which AGC kinase is required for the maintenance of the transformed phenotype in cells that depend on PI3K activity. This approach has resulted in the identification of SGK1 as the primary candidate for this role.
SGK1 was originally discovered as an immediate early gene, induced by glucocorticoids and serum (30), and has historically been associated with the control of ion transport, in particular in the kidney (31). Notably, AKT and SGK1 display a high level of target promiscuity, which has often complicated the attribution of specific functions to either of them (32). Accumulating data in recent years support the notion that SGK1 might play a distinct, AKT-independent role in cellular transformation as an effector of activated PI3K signaling. As an example, Sgk1-/- mice develop less colon tumors than wild type mice when subjected to chemical carcinogenesis (33), and ApcMin/+/Sgk1-/- mice display a 75% reduction in the number of tumors per mouse, compared to ApcMin/+ mice (34).
However, the best characterized mechanism through which SGK1 has been described to contribute to transformation is via opportunistic compensation upon PI3K or AKT inhibition. In fact, in breast cancer cells expressing high levels of SGK1 and treated with a PI3K inhibitor, SGK1 was shown to stimulate mTORC1 activity by directly phosphorylating TSC2, thus inducing resistance to PI3K inhibition (8). Along the same lines, high levels of SGK1 have been found to predict resistance to AKT inhibitors in breast cancer cells (9).
Here, we have demonstrated that loss of SGK1 profoundly impacts thyroid cancer cell proliferation and survival, both in vitro and in vivo, despite intact PI3K and AKT activity. As such, our results support a new model in which SGK1 plays an essential role as an integral part of PI3K transforming machinery.
Further in depth work is of course warranted to identify the specific pathways controlled by SGK1 in its oncogenic role. This function might be attained by impinging upon classical AKT targets and contributing to their phosphorylation, or by controlling known SGK targets that impact cell proliferation, such as the Ca2+ release-activated Ca2+ channel (ICRAC) (35), potassium channels such as Kv1.3 (36), proton exchangers such as NHE3 (37), or the epithelial sodium channel, ENAC (38). Alternatively, SGK1 could phosphorylate still unknown targets, which may contribute to neoplastic transformation either directly, or by controlling additional downstream players.
The notion that SGK1 is a critical component of the PI3K oncogenic pathway has obvious clinical consequences. Our data clearly show that, upon short-term exposure, SGK1 genetic depletion does not significantly increase the efficacy of PI3K inhibition, since SGK1 activation appears to be completely PI3K-dependent. At the same time, however, we show that prolonged exposure to a pan-PI3K inhibitor elicits a rapid adaptive response that dramatically dampens the efficacy of the inhibitor. This effect correlates with and might be mechanistically linked to the limited and short-lived responses to different PI3K inhibitors observed in clinical trials (39-41).
Pharmacological SGK inhibition, instead, appears to strongly cooperate with AKT inhibition, does not instigate the adaptive resistance response, and leads to sustained and powerful growth inhibition, a result of a magnitude rarely observed in anaplastic thyroid cancer models, and which might apply to additional PI3K-dependent aggressive tumor types. Of note, the absence of any major phenotype in mice lacking both Sgk1 and Sgk2 (42), or both Sgk1 and Sgk3 (43) suggests that SGK inhibition should cause limited toxicity in patients.
In conclusion, our results uncover an essential role of SGK1 in the PI3K-mediated neoplastic transformation process, and warrant an increased effort in the development of small molecule SGK inhibitors for clinical use, with the promise of more effective and long-lasting therapeutic effects.
Supplementary Material
Acknowledgments
We acknowledge the Animal Housing, Histology, shRNA, Analytical Imaging, and Flow Cytometry Core Facilities of Albert Einstein College of Medicine, which are partially supported by the NIH Cancer Center Support Grant to the Albert Einstein Cancer Center (P30CA013330). This work utilized a High-Speed/Resolution Whole Slide Scanner that was purchased with funding from a National Institutes of Health SIG grant 1S10OD019961-01. The Einstein Shared Facility in Stem Cell Research is supported in part by the ESSCF, NYS-DOH, Contract #C029154A.
Research reported in this publication was supported by NIH grants CA172012, CA128943 and CA167839 to A. Di Cristofano.
References
- 1.Zhang Y, Kwok-Shing Ng P, Kucherlapati M, Chen F, Liu Y, Tsang YH, et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell. 2017;31(6):820–32 e3. doi: 10.1016/j.ccell.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of Phosphatidylinositol-3-Kinase Pathway Alterations Across 19784 Diverse Solid Tumors. JAMA Oncol. 2016;2(12):1565–73. doi: 10.1001/jamaoncol.2016.0891. [DOI] [PubMed] [Google Scholar]
- 3.Xing M. Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer. Thyroid. 2010;20(7):697–706. doi: 10.1089/thy.2010.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu P, Gan W, Chin YR, Ogura K, Guo J, Zhang J, et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015;5(11):1194–209. doi: 10.1158/2159-8290.CD-15-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 6.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7(4):261–9. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 7.Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16(1):21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castel P, Ellis H, Bago R, Toska E, Razavi P, Carmona FJ, et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kalpha Inhibition. Cancer Cell. 2016;30(2):229–42. doi: 10.1016/j.ccell.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sommer EM, Dry H, Cross D, Guichard S, Davies BR, Alessi DR. Elevated SGK1 predicts resistance of breast cancer cells to Akt inhibitors. Biochem J. 2013;452(3):499–508. doi: 10.1042/BJ20130342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11(1):9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 11.Arencibia JM, Pastor-Flores D, Bauer AF, Schulze JO, Biondi RM. AGC protein kinases: from structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochim Biophys Acta. 2013;1834(7):1302–21. doi: 10.1016/j.bbapap.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 12.Yeager N, Klein-Szanto A, Kimura S, Di Cristofano A. Pten loss in the mouse thyroid causes goiter and follicular adenomas: insights into thyroid function and Cowden disease pathogenesis. Cancer Res. 2007;67(3):959–66. doi: 10.1158/0008-5472.CAN-06-3524. [DOI] [PubMed] [Google Scholar]
- 13.Antico Arciuch VG, Russo MA, Dima M, Kang KS, Dasrath F, Liao XH, et al. Thyrocyte-specific inactivation of p53 and Pten results in anaplastic thyroid carcinomas faithfully recapitulating human tumors. Oncotarget. 2011;2(12):1109–26. doi: 10.18632/oncotarget.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller KA, Yeager N, Baker K, Liao XH, Refetoff S, Di Cristofano A. Oncogenic Kras requires simultaneous PI3K signaling to induce ERK activation and transform thyroid epithelial cells in vivo. Cancer Res. 2009;69(8):3689–94. doi: 10.1158/0008-5472.CAN-09-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bayascas JR, Sakamoto K, Armit L, Arthur JS, Alessi DR. Evaluation of approaches to generation of tissue-specific knock-in mice. J Biol Chem. 2006;281(39):28772–81. doi: 10.1074/jbc.M606789200. [DOI] [PubMed] [Google Scholar]
- 16.Dima M, Miller KA, Antico-Arciuch VG, Di Cristofano A. Establishment and characterization of cell lines from a novel mouse model of poorly differentiated thyroid carcinoma: powerful tools for basic and preclinical research. Thyroid. 2011;21(9):1001–7. doi: 10.1089/thy.2011.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Champa D, Russo MA, Liao XH, Refetoff S, Ghossein RA, Di Cristofano A. Obatoclax overcomes resistance to cell death in aggressive thyroid carcinomas by countering Bcl2a1 and Mcl1 overexpression. Endocr Relat Cancer. 2014;21(5):755–67. doi: 10.1530/ERC-14-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 19.Clark J, Anderson KE, Juvin V, Smith TS, Karpe F, Wakelam MJ, et al. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat Methods. 2011;8(3):267–72. doi: 10.1038/nmeth.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antico-Arciuch VG, Dima M, Liao XH, Refetoff S, Di Cristofano A. Cross-talk between PI3K and estrogen in the mouse thyroid predisposes to the development of follicular carcinomas with a higher incidence in females. Oncogene. 2010;29(42):5678–86. doi: 10.1038/onc.2010.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biondi RM, Kieloch A, Currie RA, Deak M, Alessi DR. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. Embo J. 2001;20(16):4380–90. doi: 10.1093/emboj/20.16.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins BJ, Deak M, Arthur JS, Armit LJ, Alessi DR. In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. Embo J. 2003;22(16):4202–11. doi: 10.1093/emboj/cdg407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bago R, Sommer E, Castel P, Crafter C, Bailey FP, Shpiro N, et al. The hVps34-SGK3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC1 and tumour growth. EMBO J. 2016 doi: 10.15252/embj.201670010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayer IA, Arteaga CL. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu Rev Med. 2016;67:11–28. doi: 10.1146/annurev-med-062913-051343. [DOI] [PubMed] [Google Scholar]
- 25.Rozengurt E, Soares HP, Sinnet-Smith J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol Cancer Ther. 2014;13(11):2477–88. doi: 10.1158/1535-7163.MCT-14-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lien EC, Dibble CC, Toker A. PI3K signaling in cancer: beyond AKT. Current opinion in cell biology. 2017;45:62–71. doi: 10.1016/j.ceb.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly AP, Finlay DK, Hinton HJ, Clarke RG, Fiorini E, Radtke F, et al. Notch-induced T cell development requires phosphoinositide-dependent kinase 1. EMBO J. 2007;26(14):3441–50. doi: 10.1038/sj.emboj.7601761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanchez Canedo C, Demeulder B, Ginion A, Bayascas JR, Balligand JL, Alessi DR, et al. Activation of the cardiac mTOR/p70(S6K) pathway by leucine requires PDK1 and correlates with PRAS40 phosphorylation. Am J Physiol Endocrinol Metab. 2010;298(4):E761–9. doi: 10.1152/ajpendo.00421.2009. [DOI] [PubMed] [Google Scholar]
- 29.Cordon-Barris L, Pascual-Guiral S, Yang S, Gimenez-Llort L, Lope-Piedrafita S, Niemeyer C, et al. Mutation of the 3-phosphoinositide-dependent protein kinase-1 (PDK1) substrate-docking site in the developing brain causes microcephaly with abnormal brain morphogenesis independently of Akt, leading to impaired cognition and disruptive behaviors. Mol Cell Biol. 2016 doi: 10.1128/MCB.00230-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Webster MK, Goya L, Ge Y, Maiyar AC, Firestone GL. Characterization of sgk, a novel member of the serine/threonine protein kinase gene family which is transcriptionally induced by glucocorticoids and serum. Mol Cell Biol. 1993;13(4):2031–40. doi: 10.1128/mcb.13.4.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loffing J, Flores SY, Staub O. Sgk kinases and their role in epithelial transport. Annu Rev Physiol. 2006;68:461–90. doi: 10.1146/annurev.physiol.68.040104.131654. [DOI] [PubMed] [Google Scholar]
- 32.Di Cristofano A. SGK1: The Dark Side of PI3K Signaling. Curr Top Dev Biol. 2017;123:49–71. doi: 10.1016/bs.ctdb.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nasir O, Wang K, Foller M, Gu S, Bhandaru M, Ackermann TF, et al. Relative resistance of SGK1 knockout mice against chemical carcinogenesis. IUBMB Life. 2009;61(7):768–76. doi: 10.1002/iub.209. [DOI] [PubMed] [Google Scholar]
- 34.Wang K, Gu S, Nasir O, Foller M, Ackermann TF, Klingel K, et al. SGK1-dependent intestinal tumor growth in APC-deficient mice. Cell Physiol Biochem. 2010;25(2-3):271–8. doi: 10.1159/000276561. [DOI] [PubMed] [Google Scholar]
- 35.Lang F, Shumilina E. Regulation of ion channels by the serum- and glucocorticoid-inducible kinase SGK1. FASEB J. 2013;27(1):3–12. doi: 10.1096/fj.12-218230. [DOI] [PubMed] [Google Scholar]
- 36.Urrego D, Tomczak AP, Zahed F, Stühmer W, Pardo LA. Potassium channels in cell cycle and cell proliferation. Philosophical Transactions of the Royal Society B: Biological Sciences. 2014;369(1638):20130094. doi: 10.1098/rstb.2013.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steffan JJ, Williams BC, Welbourne T, Cardelli JA. HGF-induced invasion by prostate tumor cells requires anterograde lysosome trafficking and activity of Na+-H+ exchangers. Journal of cell science. 2010;123(Pt 7):1151–9. doi: 10.1242/jcs.063644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu C, Zhu LL, Xu SG, Ji HL, Li XM. ENaC/DEG in Tumor Development and Progression. J Cancer. 2016;7(13):1888–91. doi: 10.7150/jca.15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarker D, Ang JE, Baird R, Kristeleit R, Shah K, Moreno V, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21(1):77–86. doi: 10.1158/1078-0432.CCR-14-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doi T, Fuse N, Yoshino T, Kojima T, Bando H, Miyamoto H, et al. A Phase I study of intravenous PI3K inhibitor copanlisib in Japanese patients with advanced or refractory solid tumors. Cancer chemotherapy and pharmacology. 2017;79(1):89–98. doi: 10.1007/s00280-016-3198-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Munster P, Aggarwal R, Hong D, Schellens JH, van der Noll R, Specht J, et al. First-in-Human Phase I Study of GSK2126458, an Oral Pan-Class I Phosphatidylinositol-3-Kinase Inhibitor, in Patients with Advanced Solid Tumor Malignancies. Clin Cancer Res. 2016;22(8):1932–9. doi: 10.1158/1078-0432.CCR-15-1665. [DOI] [PubMed] [Google Scholar]
- 42.Schnackenberg CG, Costell MH, Bernard RE, Minuti KK, Grygielko ET, Parsons MJ, et al. Compensatory role for Sgk2 mediated sodium reabsorption during salt deprivation in Sgk1 knockout mice. The FASEB Journal. 2007;21(5):A508. [Google Scholar]
- 43.Grahammer F, Artunc F, Sandulache D, Rexhepaj R, Friedrich B, Risler T, et al. Renal function of gene-targeted mice lacking both SGK1 and SGK3. Am J Physiol Regul Integr Comp Physiol. 2006;290(4):R945–50. doi: 10.1152/ajpregu.00484.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.