

Abstract
Purpose
To determine the immunologic effects of neoadjuvant chemotherapy plus ipilimumab in early stage non-small cell lung cancer (NSCLC) patients.
Experimental Design
This is a single-arm chemotherapy plus phased ipilimumab Phase II study of 24 treatment-naïve patients with Stage IB–IIIA NSCLC. Patients received neoadjuvant therapy consisting of 3 cycles of paclitaxel with either cisplatin or carboplatin and ipilimumab included in the last 2 cycles.
Results
Chemotherapy alone had little effect on immune parameters in PBMCs. Profound CD28 dependent activation of both CD4 and CD8 cells was observed following ipilimumab. Significant increases in the frequencies of CD4+ cells expressing activation markers ICOS, HLA-DR, CTLA-4, and PD-1 were apparent. Likewise, increased frequencies of CD8+ cells expressing the same activation markers, with the exception of PD-1, were observed. We also examined 7 resected tumors and found higher frequencies of activated TILs than those observed in PBMCs. Surprisingly, we found 4 cases of pre-existing tumor-associated antigens (TAA) responses against survivin, PRAME, or MAGE-A3 present in PBMC at baseline, but neither increased frequencies nor the appearance of newly detectable responses following ipilimumab therapy. Ipilimumab had little effect on the frequencies of circulating Tregs and MDSCs.
Conclusions
This study did not meet the primary endpoint of detecting an increase in blood based tumor associated antigen T cell responses after ipilimumab. Collectively, these results highlight the immune activating properties of ipilimumab in early stage NSCLC. The immune profiling data for ipilimumab alone can contribute to the interpretation of immunological data from combined immune checkpoint blockade immunotherapies.
Keywords: immunotherapy, checkpoint blockade therapy, CTLA-4 blockade, non-small cell lung cancer, T cell activation
Introduction
Lung cancer is the leading cause of cancer-related deaths in the United States (1). Early stage non-small cell lung cancer (NSCLC) is potentially curable, and there appears to be a benefit to neoadjuvant platinum-based chemotherapy in patients with locally advanced non-small cell lung cancer (2). The consensus of data suggests that preoperative chemotherapy in stage II and IIIA NSCLC provides a survival benefit compared to surgery alone (3–5). A significant number of patients who receive neoadjuvant chemotherapy followed by surgery for stage II/IIIA NSCLC experience relapse of their lung cancer and, therefore, novel therapeutic approaches are needed.
Ipilimumab is a human monoclonal antibody that binds CTLA-4 on effector T cells. CTLA-4 is upregulated following T cell activation as a regulatory mechanism to prevent over-activation and immunopathogenesis. CTLA-4 binds B7-1 and B7-2, the same ligands for CD28; however, the enrichment of CTLA-4 on the surface of T cells outcompetes CD28, resulting in the inhibition of T cell activation (6). The binding of ipilimumab to CTLA-4 enhances the co-stimulation of T-cells by allowing CD28 binding to members of the B7 family on the antigen-presenting cell. Ipilimumab (Yervoy®) monotherapy is an effective, FDA-approved therapy in metastatic melanoma (7, 8).
Immune therapy has transformed the treatment of advanced stage NSCLC, with PD-1 checkpoint inhibitors producing durable responses lasting for years (9–12). Clinical trials combining CTLA-4 and PD-1 checkpoint inhibitors have shown that the addition of ipilimumab to nivolumab exhibited promising activity in a large cohort of patients with untreated advanced stage NSCLC (13). The combination of ipilimumab and nivolumab alone and with chemotherapy is being studied in large clinical trials with advanced stage NSCLC and as well as preoperative therapy in early stage NSCLC (13). Understanding the biologic effects of ipilimumab as a single immune therapy in NSCLC will contribute important biomarker data associated with sensitivity and resistance in ipilimumab-containing combination immune therapies in NSCLC.
We conducted a single arm phase 2 study of neoadjuvant carboplatin and paclitaxel plus phased-in ipilimumab for patients with clinical stage IB, II or III NSCLC (TOP1201; NCT01820754). At the time that this study was initiated ipilimumab added to carboplatin and paclitaxel improved progression-free survival, overall response, and overall survival in phase 2 trials for advanced stage NSCLC (14, 15). The study included the collection of blood for immune profiling analysis before treatment, after chemotherapy alone, and after chemotherapy plus ipilimumab. Tumor infiltrating immune cells were disaggregated from the post-treatment resection specimen when excess tumor was available. The primary objective of the trial was to assess whether the appearance of T cells activated against select tumor-associated antigens (TAAs) increased from baseline following treatment with ipilimumab.
Materials and Methods
Patients and Treatment
Eligible patients were 18 years of age or older with histologically proven clinical stage IB, IIA, IIB or IIIA lung cancer. Eligibility criteria included histologic diagnosis of NSCLC, adequate organ function, no autoimmune disease, PET/CT scan, and brain imaging. Each participant signed an IRB-approved, protocol-specific informed consent in accordance with federal and institutional guidelines.
Patients received neoadjuvant therapy consisting of: cycle 1 paclitaxel (175 mg/m2) with either cisplatin (75 mg/m2) or carboplatin (AUC = 6, capped at 900 mg) without ipilimumab, and cycles 2 and 3 combination chemotherapy (as in cycle 1) plus ipilimumab (10 mg/kg). Patients were assessed by the thoracic surgery team before and after neoadjuvant therapy to help determine trial eligibility and evaluate for surgery after chemotherapy plus ipilimumab.
Patients who were deemed not to be surgical candidates were removed from trial treatment and were not eligible to receive adjuvant ipilimumab. At the time of surgery, if subjects were found to have bulky residual adenopathy to such a degree that the operation was aborted, they were removed from the protocol. Patients who received adjuvant radiation as standard of care were removed from the trial and were not eligible to receive adjuvant ipilimumab since, at the time of the trial, there was no safety data available for giving ipilimumab following thoracic radiation therapy.
To assess immunologic parameters, blood specimens were collected from each patient prior to the beginning of therapy, after chemotherapy alone and after the completion of chemotherapy plus ipilimumab. In addition, the surgical pathology team at the operating room pathology suite examined the resected tumor. If excess tumor not needed for standard pathology tests was available, tumor was provided to the Duke Immune Profiling Core (DIPC) for disaggregation, viable cryopreservation, and subsequent immune profiling.
Isolation and storage of peripheral blood mononuclear cells (PBMCs) and tumor infiltrating lymphocytes (TILs)
Peripheral blood was obtained by venipuncture and collected in Vacutainer tubes containing acid-citrate-dextrose anticoagulant (BD Vacutainer). Peripheral blood mononuclear cells (PBMCs) and plasma were separated by Ficoll density gradient centrifugation, and PBMCs were resuspended in a 90% FBS (Gemini, Sacramento, CA) and 10% DMSO (Sigma-Aldrich, St. Louis, MO) solution. PBMCs were viably cryopreserved and stored in vapor phase liquid nitrogen, while plasma was frozen and stored at −80°C for future use.
Tumor cells and TILs were isolated from freshly resected tumor tissue using Miltenyi’s tumor dissociation kit and GentleMACS mechanical dissociator in accordance with manufacturer’s recommendations for lung tumor. Following disaggregation, the tumor cells and TILs were viably cryopreserved in a manner identical to PBMCs described above.
Flow cytometry analysis
Immune profiling of PBMCs and TILs was performed using two polychromatic flow cytometry panels: an immune cell subsetting panel (Supplementary Table 1A) and a T cell function panel (Supplementary Table 1B). Due to the complexity of the subsetting panel, CD8+ frequencies were ascertained as CD4 negative cells within the CD3+ gate. For staining with the immune cell subsetting panel, two million PBMCs or TILs were first incubated in a 12x75 staining tube, with Fc Block (BD Biosciences, San Jose, CA), followed by a surface stain with ICOS, CD14, CD16, CD19, CD20, CD56, PD-1, CD28, HLA-DR, Viability dye, CD3, CD25, CD4, CD33, and CD11b for 25 minutes at 4°C. Following cell surface staining, cells were treated with the FOXP3 Fix/Perm solution (eBioscience, San Diego, CA) for 45 minutes at 4°C. Intranuclear staining was then performed for 30 minutes at 4°C using fluorescent antibodies against FOXP3 and CTLA-4. Matched isotype controls were used for ICOS, CD14, HLA-DR, CTLA-4, and FOXP3. Cells were fixed with 1% paraformaldehyde (Sigma-Adrich) and acquired on a BD LSRII flow cytometer.
For staining with the T cell function panel, two million PBMCs or TILs were plated in a 96-well plate in RPMI + 10% FBS. Cells were left untreated or stimulated with overlapping peptide pools (15-mers with 11 aa overlaps) representing MAGE-A3, PRAME, and survivin (JPT, Berlin Germany), along with anti-CD3 and anti-CD28 as a positive control and cells in media alone as a negative control. All conditions also included anti-CD107a for the duration of the stimulation and cells were incubated for 18 hours at 37°C in 5% CO2 humidified incubator. At 18 hours, Brefeldin A and Monensin (BD Biosciences) were added to all tubes and incubated for an additional 6 hours. After the stimulation period, cells were surface stained with 50μL of a cocktail mix consisting of fluorescent conjugates for CD45RA, CCR7, CD14, and viability dye for 25 minutes at 4°C. Following cell surface staining, cells were treated with cytofix/cytoperm (BD Biosciences) in accordance with manufacturer’s recommendations. Intracellular staining was then performed for 25 minutes at 4°C using fluorescent antibodies against TNF-α, IFN-γ, IL-2, CD4, CD3, CD8, CD154, and CD69. Isotype controls were used for TNF-α, IFN-γ, CD45RA, CD107a, CCR7, and IL-2. Cells were fixed with 1% paraformaldehyde and acquired on a LSRII flow cytometer (BD Biosciences).
Statistical analysis
Student’s t-tests were used to assess differences in cell frequencies between visits. Overall survival was defined as the time between the initiation of protocol therapy and date of death or last follow-up. Kaplan-Meier methods were used to describe overall survival. Analyses were not adjusted for multiple testing. Flow cytometric analysis was performed using Flowjo software (Tree Star, Ashland, OR). Statistical analysis was performed using Prism software (Graph Pad, LaJolla, CA) and SAS (version 9.4, Cary, NC).
Results
Patients and Treatment
Twenty-four eligible patients with NSCLC were enrolled between March 2013 and December 2015 and received neoadjuvant therapy on TOP1201: 12 (50%) female, 23 (96%) current or former smokers, 15 (62%) adenocarcinoma, 9 (37%) squamous carcinoma, and median age of 65 years (Supplementary Table 2). Three patients (13%) were lung cancer stage 2A, 2 (8%) were stage 2B, and 19 (79%) were stage 3A with 18 (75%) patients having pretreatment pathologically-positive N2 lymph node metastases. Surgical resection post neoadjuvant therapy was not attempted on eleven patients for the following reasons: persistent N2 cancer (n=5), inadequate pulmonary function (n=2), cancer progression (n=2), location of tumor (n=1), and preoperative complications leading to the patient not reporting for surgery (n=1). The preoperative complications preventing surgery for one patient were a pulmonary embolism followed by renal failure from the contrast dye given for the CT scan to diagnose the embolism, and these complications were deemed not likely secondary to ipilimumab. Twenty-two (92%) received carboplatin and two (8%) cisplatin. Two patients had a delay in surgery of four and five weeks due to ipilimumab-related diarrhea. Thirteen patients were treated with chemotherapy and ipilimumab, followed by surgical resection less than twelve weeks after neoadjuvant therapy.
Adverse events
Fifty-four percent of patients experienced a treatment-related grade 1 or 2 adverse event, while 46% had a grade 3 or 4 event. There were no treatment-related deaths. Most of the adverse events were those expected for carboplatin and paclitaxel chemotherapy, although toxicities such as rash, fatigue, peripheral neuropathy, and others could be attributed to chemotherapy or ipilimumab. Immune-related adverse events felt to be related to ipilimumab were: grade 2 pneumonitis (n=1, 4%), grade 3 adrenal insufficiency (n=4, 17%), and diarrhea/colitis 8 (grade 1 or 2: n=6, 25%; grade 3: n=3, 13%).
Clinical outcomes
Based on RECIST 1.1 criteria, clinical responses evaluated after completion of neoadjuvant carboplatin, paclitaxel, and ipilimumab included 14 (58%) patients with a partial response (PR), 8 (33%) with stable disease (SD), and 2 (8%) with progression (PD). Median overall survival (OS) was 29.2 months (95% CI: 22.1–∞), while 12-month OS was 82.2% (95% CI: 59.2–92.9) and 24-month OS was 73.0% (95% CI: 44.1–88.6) for all patients initiating neoadjuvant therapy. There have been no deaths at 24 months for the patients who underwent surgical resection of their lung cancer.
Upregulation of activation markers on CD4 and CD8 T cells after ipilimumab therapy
Flow cytometry analyses were performed on PBMCs collected at baseline (V1), after chemotherapy alone (V2), and after chemotherapy plus ipilimumab (V3) to profile the effect of ipilimumab on the immune parameters of patients with NSCLC (Fig. 1A). The immune subsetting panel consisted of 17 unique immune markers that identify both innate and adaptive cell types along with specific subsets (Supplementary Table 1). Markers including ICOS, HLA-DR, CTLA-4, and PD-1 are associated with T cell activation. Similar expression patterns in samples from V1 and V2 suggest that chemotherapy alone had little effect on lymphocyte activation. In contrast, ipilimumab had a broad activating effect on both CD4 and CD8 lymphocyte populations, as evidenced by the increased marker expression at V3 compared to baseline. The mean frequencies and corresponding SDs of positive subsets of CD4 T cells at V1 vs V3 for ICOS were 8.99; 3.25 vs 23.59; 7.51, for HLA-DR positive were 7.06; 3.89 vs 14.80; 5.56, for CTLA-4 positive were 17.20; 6.48 vs 29.39; 10.52, and for PD-1 positive were 3.51; 3.12 vs 6.30; 4.34 (Fig. 1). For CD8 T cells, the frequency and corresponding SDs of ICOS positive (4.91; 1.72 vs 11.36; 5.30), HLA-DR positive (14.35; 8.61 vs 35.85; 12.60), and CTLA-4 positive (14.35; 8.61 vs 35.85; 12.60) cells significantly increased after ipilimumab therapy, but PD-1 frequency and corresponding SD on CD8 T cells remained unchanged from V1 to V3 (5.51;5.50 vs 5.50; 5.32). Stratification of lymphocyte activation by overall response after neo-adjuvant cycle 3 did not reveal differences over time between the PD, SD, and PR patients (Supplementary Fig. 1). An extended comparison of immune subsets and activation phenotypes of CD4 and CD8 T cells is listed in Supplementary Table 3. Overall, ipilimumab had a profound effect on CD4 and CD8 T cell activation regardless of the clinical response.
Figure 1.
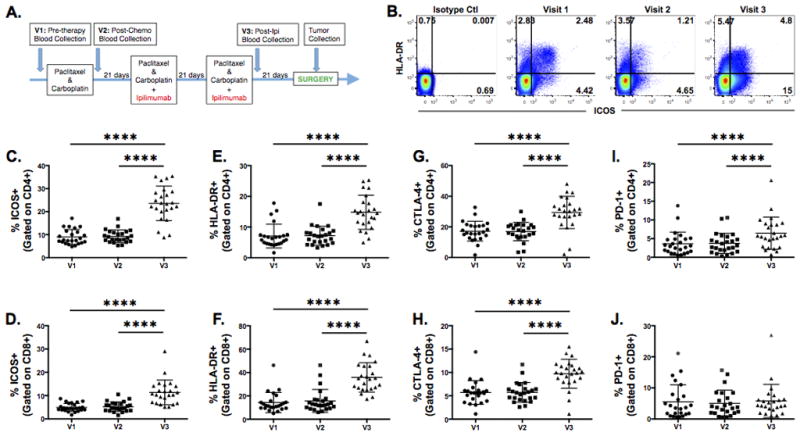
Ipilimumab enhances the expression of activation markers on peripheral CD4 and CD8 T cells. A, Study design of treatment and sample collection. B, Immune snapshot of HLA-DR and ICOS expression on CD4 T cells in a representative patient. C and D, Changes in the expression of ICOS, (E and F) HLA-DR, (G and H) CTLA-4, and (I and J) PD-1 on CD4 (C, E, G, and I) and CD8 T cells (D, F, H, and J) were examined by flow cytometry analysis. Frequency of gated CD4 or CD8 T cells are shown for 24 patients at pre-treatment (V1), post-chemotherapy only (V2), and post-chemotherapy and ipilimumab (V3) timepoints. Statistical significance is represented by **** p ≤ 0.0001. Mean and SD are shown.
TILs in NSCLC are highly activated
Based on the impact of ipilimumab on the expression pattern of activation markers on PBMCs, we performed a parallel analysis of ICOS, CTLA-4, HLA-DR, and PD-1 expression on TILs isolated from tumors of seven patients who underwent resection and for which there was adequate excess tumor to perform disaggregation of TILs, approximately 1 gram. Relative to the expression of these markers on peripheral T cells at visit 3, tumor-derived CD4 T cells expressed higher frequencies of ICOS (TIL mean with SD vs PBMC mean with SD); 60.44; 6.84 vs 23.59;7.51), CTLA-4 (60.30; 25.81 vs 29.39; 10.52), HLA-DR (82.31; 22.14 vs 14.80; 5.56), and PD-1 (36.61; 11.33 vs 6.30; 4.34) (Fig. 2A). The frequencies of activated CD8 T cells were also higher in TILs based on the expression of ICOS (TIL vs PBMC; 32.84; 9.45 vs 11.36; 5.30), CTLA-4 (43.43; 19.87 vs 35.85; 12.60), HLA-DR (87.10; 15.70 vs 35.85; 12.60), and PD-1 (35.50; 16.94 vs 5.50; 5.32) (Fig. 2B). Although we did not have access to pre-treatment tumor samples for comparison, the highly-activated phenotype of the CD4 and CD8 TILs is indicative of an immunogenic tumor microenvironment.
Figure 2.
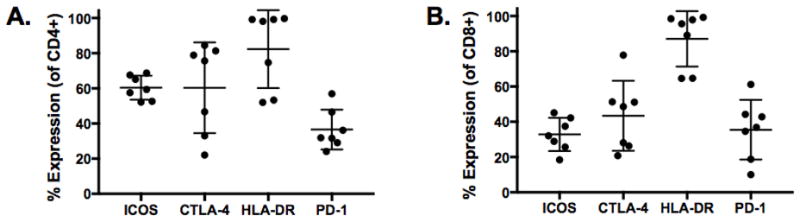
Enhanced expression of activation markers on TILs. A and B, Expression pattern of ICOS, CTLA-4, HLA-DR, and PD-1 on CD4 and CD8 TILs from 7 patients undergoing reductive surgery following ipilimumab treatment. Mean and SD are shown.
Magnitude of T cell activation is dependent on CD28 expression
A recent report by Kamphorst et al highlighted the finding that proliferating CD8 T cells in the peripheral circulation of lung cancer patients following PD-1 therapy predominantly expressed CD28 (16). Although a proliferation marker was not included among our profiling panels, we sought to determine whether the post-ipilimumab activated CD8 T cells also co-expressed CD28. CD28 expression did not change in response to chemotherapy or ipilimumab (Fig. 3A). However, dissection of the CD8 T cells based on CD28 expression revealed that the ipilimumab-induced activation of CD8 T cells was CD28 dependent. The mean frequencies and corresponding SDs of ICOS (9.41; 3.47 vs 24.71; 7.85) or CTLA-4 (4.28; 1.91 vs 9.12; 3.41) in CD28+ T cells were significantly higher at V3 than V1 or V2 (Fig. 3B and 3C). The singular exception was the frequency of V3 CD8 T cells expressing HLA-DR, where the frequencies and corresponding SDs among CD28+ and CD28− (35.01; 13.24 vs 36.58; 17.86) populations were similar (Fig. 3D). Overall, our results provide support that ipilimumab-induced activation of CD8 T cells is CD28 dependent.
Figure 3.
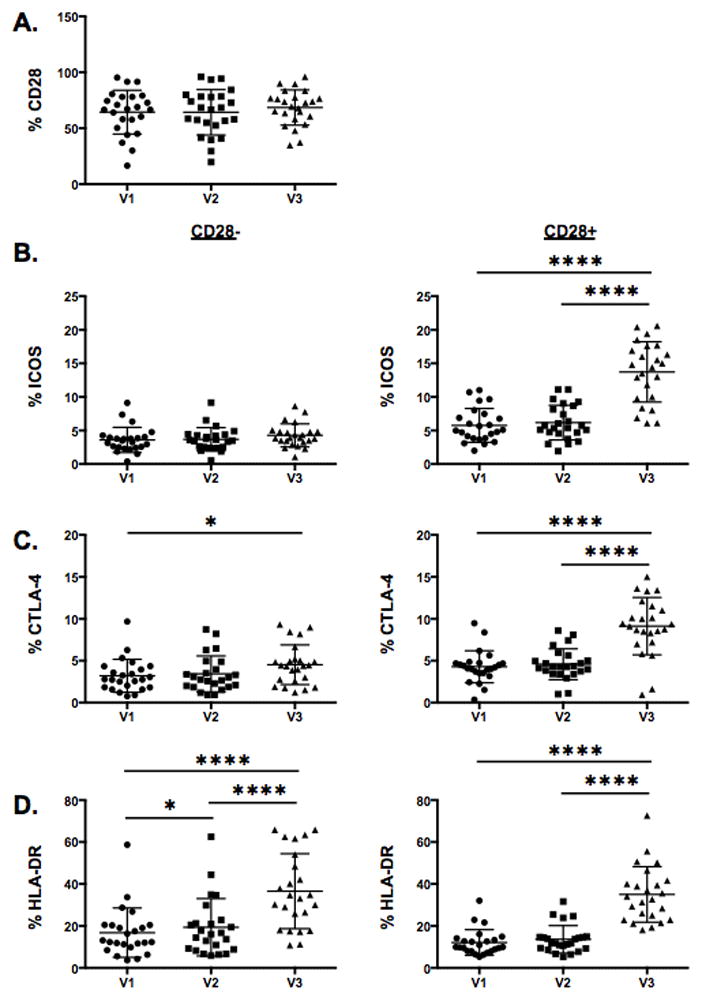
Increase in CD8 T cell activation following ipilimumab treatment is CD28 dependent. A, Composite data from 24 patients showing the fraction of CD8 T cells expressing CD28 at pre-treatment (V1), post-chemotherapy only (V2), and post-chemotherapy and ipilimumab (V3) timepoints. B–D, Comparison of ICOS, CTLA-4, and HLA-DR expression on CD28− and CD28+ CD8 T cells. Statistical significance is represented by * p ≤ 0.05, **** p ≤ 0.0001. Mean and SD are shown.
Tumor-Associated Antigen Specific T Cell Reactivities
After observing a significant increase in the expression of activation markers after ipilimumab therapy, we next examined whether this increase was associated with increases in functional TAA-specific CD4 or CD8 T cell responses. Functional T cell responses, reflected by intracellular accumulation of IFN-γ, TNF-α, and IL-2 as well as surface expression of the degranulation marker CD107a, were examined following PBMC stimulation with overlapping peptide pools representing 3 of the most prevalent antigens found in NSCLC (17–19), namely MAGE-A3, survivin, and PRAME. Among 24 NSCLC patients in this study, CD4 or CD8 T cell responses to MAGE-A3, survivin, or PRAME were detectable in four patients (Fig. 4). The responses of each patient varied in the antigen-specificity that induced the highest frequency of IFN-γ positivity and T cell subset that was responsive to antigen stimulation. Among the patients with IFN-γ+ responses, the majority of T cell responses were polyfunctional, as we also observed intracellular production of TNF-α, and expression of CD107a (data not shown). Collectively, in patients with detectable T cell responses to MAGE-A3, survivin, or PRAME, these responses were present at baseline prior to treatment, and ipilimumab therapy had little or no effect on their relative frequencies, although several appeared to decline following ipilimumab treatment. No new anti-TAA reactivities were observed in conjunction with ipilimumab therapy, nor were any TAA reactivities detected within the tumor microenvironment of the seven resected tumors available for study (data not shown). Unfortunately, none of the four patients who had pre-existing TAA reactivities at baseline had tumor resections and, therefore, we did not have an opportunity to profile the functional status of their TILs.
Figure 4.
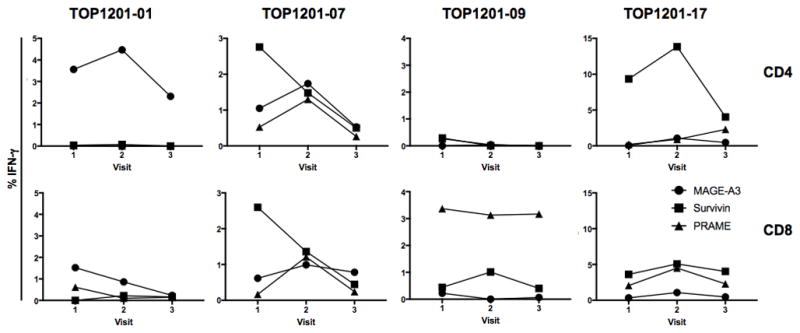
Detection of TAA-specific CD4 or CD8 T cell responses in a subset of ipilimumab treated patients. CD4 and CD8 T cell function, as measured by IFN-γ production, was evaluated pre-treatment (V1), post-chemotherapy only (V2), and post-chemotherapy and ipilimumab (V3). The graphs represent the four patients with detectable TAA reactivity against MAGE-A3, survivin, or PRAME. The values are the frequency of IFN-γ+ cells gated on total CD4 (top row) or CD8 (bottom row) T cells.
Effect of ipilimumab on regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSC)
To determine whether the ipilimumab induced T cell activation was accompanied by a compensatory decrease in putative suppressive cell populations, we examined the changes in frequencies Tregs and MDSCs from baseline (V1) to post neoadjuvant chemotherapy plus ipilimumab (V3). From chemotherapy only to post-ipilimumab therapy, the median frequency of CD4+CD25+FOXP3+ Tregs increased slightly by 1.05% (p=0.012; Fig. 5A). Further examination of CTLA-4+ Tregs revealed no changes in this subset after ipilimumab therapy. Earlier clinical studies of ipilimumab therapy in patients with advanced malignant melanomas suggested that the presence of high levels of MDSC (i.e. >15%) in the peripheral blood at baseline predicted non-responsiveness to ipilimumab therapy (20). Collectively, the frequency of MDSCs, identified as CD14+ cells gated off the lineage negative, HLA-DRlow/negative population, remained unchanged after ipilimumab therapy (Fig. 5B). Outlying MDSC frequencies (>15%) by three patients were not predictive of clinical outcomes, as 2 of the 3 patients had a PR response and the 3rd patient had a response of SD. Overall, ipilimumab had little to no effect on the frequency of circulating Tregs and MDSCs.
Figure 5.
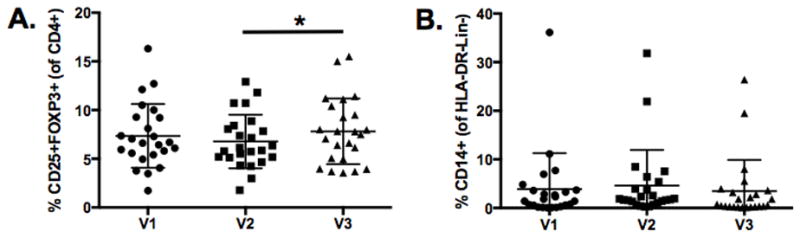
Maintenance of Tregs and MDSCs after ipilimumab therapy. A, Frequency of CD4+CD25+FOXP3+ Tregs at pre-treatment (V1), post-chemotherapy only (V2), and post-chemotherapy and ipilimumab (V3). B, Frequency of MDSCs based on CD14 expression of HLA-DR-Lin- cells. 24 patients were analyzed at each visit and statistical significance is represented by * p ≤ 0.05. Mean and SD are shown.
Discussion
Immune checkpoint therapy has transformed the care of advanced stage NSCLC (9–12). Combination immune therapy, including CTLA-4 checkpoint blockade, has shown promise compared to PD-1 checkpoint blockade alone (13). Understanding how immune therapy combinations work by themselves and together to overcome immune resistance mechanisms will require an understanding of the activation and inhibitory factors associated with each individual immune therapy component. Randomized phase 3 trials studying chemotherapy with or without ipilimumab in advanced squamous non-small cell lung cancer showed no survival benefit for the addition of ipilimumab to chemotherapy (NCT01285609, data available in clinicaltrials.gov). However, the combination of ipilimumab and the anti-PD1 antibody nivolumab in untreated advanced NSCLC showed a response rate of approximately 40% (13). Ipilimumab is being studied in randomized trials in combination with PD1 checkpoint immune therapy for untreated early and advanced stage NSCLC (NCT02998528, NCT02477826). Therefore, while single agent CTLA-4 blockade is no longer being studied in NSCLC, ipilimumab is being actively studied in combination immune therapy in multiple large randomized trials in lung cancer, and the immune profiling data reported here is of particular interest in helping to understand how CTLA-4 inhibitors may be best utilized in immune therapy combinations. To our knowledge, this is the first report of a broad immunological study in early stage NSCLC for the activation of blood T cells, MDSCs, and Tregs at baseline, after chemotherapy, and after ipilimumab. We also report some of the first data supporting ipilimumab immune activation in TILs from early stage NSCLC.
The patients referred by the surgical service for this trial had either a large primary tumor and/or mediastinal lymph node metastasis that needed to respond to neoadjvant therapy in order to make the cancer appropriate for surgical resection by our institutional standard of care. Thus, only 13 of 24 patients underwent resection mostly because of tumor stage after neoadjuvant therapy, rather than some side effect from neoadjuvant treatment. The adverse events on this study were modestly increased compared to what would be expected for chemotherapy alone despite using an ipilimumab dose of 10mg/kg that was of interest at the time the study was designed. There were cases of colitis and endocrinopathies reported, as expected with ipilimumab therapy. Hypothyroidism, adrenal insufficiency, colitis, and other immune related side effects may require chronic therapy effecting quality of life in early stage lung cancer patients who receive immune therapy as part of their treatment regimen. No patient was unable to go to surgery because of adverse effects of neoadjuvant therapy, although two patients had surgery delayed due to diarrhea thought to be caused by ipilimumab. The major objective response rate of 58% after neoadjuvant platinum, paclitaxel, and ipilimumab compares favorably to the 41% objective response rate reported for the Southwest Oncology Group trial that also studied neoadjuvant carboplatin and paclitaxel in early stage lung cancer (21). However, the sample size in our study is small, so it cannot be concluded that our results indicate that ipilimumab plus chemotherapy is more active than chemotherapy alone in early stage NSCLC. It was not possible to compare the immune effects of ipilimumab in patients responding to versus resistant to neoadjuvant therapy because only two tumors progressed after three cycles of neoadjuvant therapy.
Highly-standardized polychromatic flow cytometric assay platforms were utilized to profile both functional and phenotypic changes brought about by ipilimumab therapy. Given the small sample of 24 patients, our analysis is designed as a descriptive study, with the goal to validate our observations in a large cohort study. By far, the most profound early immunologic effects seen in this study following administration of ipilimumab were the significantly increased frequencies of highly activated T cells in the peripheral circulation. Both CD4+ and CD8+ cells expressing the activation markers ICOS, HLA-DR, and CTLA-4 increased dramatically following phased ipilimumab with chemotherapy. Increased expression of CD4+ cells, but not CD8+ cells, co-expressing PD-1 were also noted. Earlier studies in advanced metastatic melanoma patients treated with ipilimumab also noted increased frequencies of ICOS+ CD4 and CD8 cells, and suggested that the magnitude of the increase might be predictive of a clinical response (22, 23). The stable frequency of CD4 and CD8 T cells before and after ipilimumab suggest that the increase in activation is not due to cell expansion, but rather the activation of preexisting T cells in the periphery.
In the present study, the ICOS+ cells were nearly exclusively contained within the CD28+ subset of CD8+ T cells, whereas the HLA-DR+ cells appeared to be equally distributed among both CD28+ and CD28− subpopulations. Interestingly, a recent murine study demonstrated that the promotion of the CD8 T cell response, after PD-1 blockade, is dependent on CD28 (16). In the same study, the proliferating CD8 T cells found in the blood of NSCLC patients, following PD-1 targeted therapy, were predominantly CD28+. CD28 expression has been demonstrated to decrease with age consequently resulting in a dampened immune response (24). Interestingly, the loss of CD28 expression may be prevented or even reversed using cytokines IL-12 or IL-21 (25, 26). While we do not suggest that CD28 is a predictive biomarker for immunotherapy response due to the small sample size, CD28 expression may be a worthwhile pharmacodynamic biomarker to evaluate in a large cohort study to identify patients who will benefit from immunotherapy.
Similar immune profiling was performed on the tumor microenvironment of all seven resected tumors available for study. Once again, high frequencies of CD4+ and CD8+ T-cells expressing the activation markers ICOS, HLA-DR, CTLA-4, and PD-1 were observed. Overall, the frequencies of activated T-cells within the tumor microenvironment were notably higher than those of their counterparts in the peripheral circulation. Unfortunately, unlike the available PBMC samples, we did not have pre-treatment tumor samples available for immune profiling and, therefore, could not assess the relative contribution of ipilimumab therapy to the post-treatment activation status of the T cells populating the tumor microenvironment. Also, the tumors studied for immune phenotyping were only those tumors that could be resected and had excess tumor after neoadjvant therapy, thus representing a selected subset of early stage NSCLC.
Previous studies in advanced melanoma patients receiving ipilimumab therapy revealed that anti-CTLA-4 treament potentiated CD4+ and CD8+ T cell responses in the peripheral circulation against NY-ESO-1, MART-1, and gp100 (27). Utilizing our highly standardized intracellular cytokine staining (ICS) assay, we found baseline anti-TAA reactivities in a total of 4 patients against MAGE-A3, survivin, or PRAME, 3 of the most common TAA expressed among non-small cell lung tumors (28, 29). In contrast to the melanoma studies, we failed to demonstrate any potentiation of pre-existing TAA responses following ipilimumab therapy. Furthermore, we saw no evidence of the development of de novo anti-TAA responses during or after ipilimumab therapy. These differences could most likely be due to differences in the natural history of anti-TAA responses in melanoma versus NSCLC patients or the study of advanced stage (melanoma) versus early stage NSCLC patient populations. We also failed to detect anti-TAA reactivities in any of the seven available tumor resection samples. Unfortunately, there were no tumor samples available for any of the four patients who had detectable anti-TAA responses in their PBMCs.
Tregs and MDSCs are seen as formidable obstacles to most immunotherapeutic strategies and have been previously studied in the context of ipilimumab therapy in advanced melanoma. Tarhini et al reported a significant increase in circulating Tregs that was associated with improved progression-free survival, while Treg levels within the tumor microenvironment were significantly reduced (27). In the present study, we did not observe significant changes in the frequences of circulating Tregs following ipilimumab therapy. CTLA-4 blockade has been demonstrated to deplete intratumoral Tregs through Fc receptor mediated ADCC (30); however, the Treg levels among the seven tumors were higher and more highly variable than in PBMCs. Once again, in the absence of pre-treatment tumor samples, we are unable to adequately assess the effect of therapy on Treg frequencies within the tumor microenvironment of NSCLC patients. MDSC frequencies were the focus of a previous study of ipilimumab treatment of advanced melanoma patients (20). This group reported that advanced melanoma patients with baseline PBMC levels of MDSC (defined as lin-CD14+HLA-DRlow/neg) less than 14.9% had a significantly increased likelihood of prolonged overall survival following ipilimumab therapy. Those patients with greater MDSC levels were far less likely to benefit from ipilimumab therapy. Thus, baseline circulating MDSC levels may represent a valuable predictive biomarker to identify patients who will most benefit from ipilumab therapy. Of the 24 patients enrolled in the present NSCLC study, only 2 had MDSC levels >15%, and neither of them was classified as having progressive disease. Once again, the lower levels of MDSC in the current study could simply be a reflection of the earlier stage of disease that we are studying or differences in the biology of melanomas versus NSCLC. In a comprehensive study of 123 immune subsets following avelumab (anti-PD-L1 monoclonal Ab) in patients with solid tumors, PD-L1 blockade had no effect on any of the 123 immune subsets in the peripheral blood including Tregs and MDSCs (31). Observations in the peripheral blood between CTLA-4 and PD-L1 blockades differ because their function is site-specific. Anti-CTLA-4 blockade is mainly involved during initial T cell priming in lymphoid tissues, whereas anti-PD-1/PD-L1 impacts the interaction of PD-1 and PD-L1 in the tumor microenvironment where PD-L1 expressing tumor cells are located (32–34).
This study did not meet the primary endpoint of detecting an increase in circulating T cells with specificities against tumor-associated antigens from zero percent of subjects prior to therapy to 20% of subjects after neoadjuvant chemotherapy plus ipilimumab. Instead, we discovered that almost 20% of patients on the study had blood T cells with specificity against survivin, MAGE-A3, or PRAME at baseline, and ipilimumab did not increase the number of cases. Ipilimumab, but not chemotherapy alone, caused dramatic activation in blood CD4+ and CD8+ T cells, but did not have a marked effect on blood Tregs or MDSCs. These results suggest that the activation of T cells observed in this study with ipilimumab may be necessary but not sufficient by itself for a therapeutic immune effect in a subset of non-small cell lung cancer, since ipilimumab alone did not improve survival when added to chemotherapy and the addition of ipilimumab to nivolumab appears to increase the number of durable responses in non-small cell lung cancer (13). We were able to isolate variable but sufficient numbers of viable and functional immune cells from early stage lung cancers after neoadjuvant immune therapy to perform flow cytometry analysis. Finally, much of what we know about the immunologic effects of ipilimumab therapy has been gleaned from studies conducted in the context of advanced malignant melanomas. Great care must be taken in attempting to extrapolate those findings to other tumor immunotherapy settings, as exemplified by our current studies of early stage NSCLC patients. The data from this study will contribute to the understanding of how ipilimumab may be influencing therapeutic efficacy in combination immune therapy for NSCLC. Thus, an understanding of immunotherapeutic effects in the context of early treatment becomes an important objective. Our current neoadjuvant study is extending immune profiling to studying neoadjuvant PD-1 checkpoint therapy in early stage NSCLC. It is hoped that the observations reported herein will contribute to the development of a immune based rationale for future combination immunotherapies in NSCLC patients.
Supplementary Material
Translational Relevance.
This study is one of the first immune profiling efforts to characterize the immune response to ipilimumab in early stage non-small cell lung cancer (NSCLC). Our findings provide immunological evidence supporting the use of ipilimumab to induce T cell activation in NSCLC. Notably, we demonstrate that CD8 T cell activation seen after ipilimumab therapy is CD28 dependent and the expression of CD28 may improve the efficacy of ipilimumab. Furthermore, ipilimumab did not affect the frequency of Tregs or Myeloid-derived suppressor cells (MDSCs), or PD-1 expression on CD8 T cells. With the rising use of immunotherapies in the treatment of NSCLC, the optimal immunotherapy or combination of immunotherapies will require the promotion of T cell activation, migration into the tumor microenvironment, avoidance of T cell inhibition, and long-lasting cytotoxic function. Collectively, ipilimumab serves an important function in strengthening the anti-tumor response.
Acknowledgments
Financial Support: Research costs supported in part by a grant from Bristol Myers Squibb, Duke University grant number (393-8326)
We would like to thank the Duke Immune Profiling Core (DIPC), a Duke School of Medicine and Duke Cancer Center Institute Shared Resource, for providing their expertise in flow cytometry methods and data analysis. We also thank the BioRepository & Precision Pathology Center (BRPC), a shared resource of the Duke University School of Medicine and Duke Cancer Institute, for providing access to the human biospecimens used under IRB oversight in this work. The BRPC and DIPC receive support from the P30 Cancer Center Support Grant (P30 CA014236).
Footnotes
Conflict of Interest: Neal Ready, MD, PhD: scientific advisor: BMS, Celgene, MERCK, Astra Zeneca, Arriad Honoria: BMS
Jeffrey Crawford MD: scientific advisor: AstraZeneca, Merck, Pfizer
Jeffrey Clarke MD: scientific advisor: Medpacto
Statement of Author Contributions: N.R., J. Clarke., K.W., J. Crawford., F.D, J.D.C., M.B., B.T., T.D., S.M., D.S., and D.H. designed the study, obtained clinical specimens, and/or clinical data; J.Y., C.D., R.O., and K.W. designed, performed, and/or analyzed experiments; J.Y., N.R., and K.W. wrote the manuscript; P.H., F.M., and X.W. provided statistical support
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Song W-A, Zhou N-K, Wang W, Chu X-Y, Liang C-Y, Tian X-D, et al. Survival Benefit of Neoadjuvant Chemotherapy in Non-small Cell Lung Cancer: An Updated Meta-Analysis of 13 Randomized Control Trials. Journal of Thoracic Oncology. 5:510–6. doi: 10.1097/JTO.0b013e3181cd3345. [DOI] [PubMed] [Google Scholar]
- 3.Rosell R, Gomez-Codina J, Camps C, Maestre J, Padille J, Canto A, et al. A randomized trial comparing preoperative chemotherapy plus surgery with surgery alone in patients with non-small-cell lung cancer. The New England journal of medicine. 1994;330:153–8. doi: 10.1056/NEJM199401203300301. [DOI] [PubMed] [Google Scholar]
- 4.Roth JA, Fossella F, Komaki R, Ryan MB, Putnam JB, Jr, Lee JS, et al. A randomized trial comparing perioperative chemotherapy and surgery with surgery alone in resectable stage IIIA non-small-cell lung cancer. J Natl Cancer Inst. 1994;86:673–80. doi: 10.1093/jnci/86.9.673. [DOI] [PubMed] [Google Scholar]
- 5.Scagliotti GV, Pastorino U, Vansteenkiste JF, Spaggiari L, Facciolo F, Orlowski TM, et al. Randomized phase III study of surgery alone or surgery plus preoperative cisplatin and gemcitabine in stages IB to IIIA non-small-cell lung cancer. J Clin Oncol. 2012;30:172–8. doi: 10.1200/JCO.2010.33.7089. [DOI] [PubMed] [Google Scholar]
- 6.Lenschow DJ, TLW, Bluestone JA. CD28/B7 SYSTEM OF T CELL COSTIMULATION. Annual review of immunology. 1996;14:233–58. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 7.Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus Dacarbazine for Previously Untreated Metastatic Melanoma. New England Journal of Medicine. 2011;364:2517–26. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 8.Eggermont AMM, Chiarion-Sileni V, Grob J-J, Dummer R, Wolchok JD, Schmidt H, et al. Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. New England Journal of Medicine. 2016;375:1845–55. doi: 10.1056/NEJMoa1611299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. The New England journal of medicine. 2015;373:1627–39. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. The New England journal of medicine. 2015;373:123–35. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387:1837–46. doi: 10.1016/S0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 12.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. The New England journal of medicine. 2016;375:1823–33. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 13.Hellmann MD, Rizvi NA, Goldman JW, Gettinger SN, Borghaei H, Brahmer JR, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017;18:31–41. doi: 10.1016/S1470-2045(16)30624-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Genova C, Rijavec E, Barletta G, Sini C, Dal Bello MG, Truini M, et al. Ipilimumab (MDX-010) in the treatment of non-small cell lung cancer. Expert Opinion on Biological Therapy. 2012;12:939–48. doi: 10.1517/14712598.2012.681371. [DOI] [PubMed] [Google Scholar]
- 15.Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in Combination With Paclitaxel and Carboplatin As First-Line Treatment in Stage IIIB/IV Non–Small-Cell Lung Cancer: Results From a Randomized, Double-Blind, Multicenter Phase II Study. Journal of Clinical Oncology. 2012;30:2046–54. doi: 10.1200/JCO.2011.38.4032. [DOI] [PubMed] [Google Scholar]
- 16.Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355:1423–7. doi: 10.1126/science.aaf0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yasumoto K, Hanagiri T, Takenoyama M. Lung cancer-associated tumor antigens and the present status of immunotherapy against non-small-cell lung cancer. Gen Thorac Cardiovasc Surg. 2009;57:449–57. doi: 10.1007/s11748-008-0433-6. [DOI] [PubMed] [Google Scholar]
- 18.Huang LN, Wang DS, Chen YQ, Zhao CL, Gong BL, Jiang AB, et al. Expression of survivin and patients survival in non-small cell lung cancer: a meta-analysis of the published studies. Mol Biol Rep. 2013;40:917–24. doi: 10.1007/s11033-012-2132-8. [DOI] [PubMed] [Google Scholar]
- 19.Thongprasert S, Yang PC, Lee JS, Soo R, Gruselle O, Myo A, et al. The prevalence of expression of MAGE-A3 and PRAME tumor antigens in East and South East Asian non-small cell lung cancer patients. Lung Cancer. 2016;101:137–44. doi: 10.1016/j.lungcan.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 20.Kitano S, Postow MA, Ziegler CG, Kuk D, Panageas KS, Cortez C, et al. Computational algorithm-driven evaluation of monocytic myeloid-derived suppressor cell frequency for prediction of clinical outcomes. Cancer Immunol Res. 2014;2:812–21. doi: 10.1158/2326-6066.CIR-14-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pisters KM, Vallieres E, Crowley JJ, Franklin WA, Bunn PA, Jr, Ginsberg RJ, et al. Surgery with or without preoperative paclitaxel and carboplatin in early-stage non-small-cell lung cancer: Southwest Oncology Group Trial S9900, an intergroup, randomized, phase III trial. J Clin Oncol. 2010;28:1843–9. doi: 10.1200/JCO.2009.26.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Giacomo AM, Calabro L, Danielli R, Fonsatti E, Bertocci E, Pesce I, et al. Long-term survival and immunological parameters in metastatic melanoma patients who responded to ipilimumab 10 mg/kg within an expanded access programme. Cancer Immunol Immunother. 2013;62:1021–8. doi: 10.1007/s00262-013-1418-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng Tang D, Shen Y, Sun J, Wen S, Wolchok JD, Yuan J, et al. Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti-CTLA-4 therapy. Cancer Immunol Res. 2013;1:229–34. doi: 10.1158/2326-6066.CIR-13-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boucher N, Dufeu-Duchesne T, Vicaut E, Farge D, Effros RB, Schachter F. CD28 expression in T cell aging and human longevity. Exp Gerontol. 1998;33:267–82. doi: 10.1016/s0531-5565(97)00132-0. [DOI] [PubMed] [Google Scholar]
- 25.Warrington KJ, Vallejo AN, Weyand CM, Goronzy JJ. CD28 loss in senescent CD4+ T cells: reversal by interleukin-12 stimulation. Blood. 2003;101:3543–9. doi: 10.1182/blood-2002-08-2574. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen H, Weng NP. IL-21 preferentially enhances IL-15-mediated homeostatic proliferation of human CD28+ CD8 memory T cells throughout the adult age span. Journal of leukocyte biology. 2010;87:43–9. doi: 10.1189/jlb.0209086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tarhini AA, Edington H, Butterfield LH, Lin Y, Shuai Y, Tawbi H, et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PloS one. 2014;9:e87705. doi: 10.1371/journal.pone.0087705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karanikas V, Tsochas S, Boukas K, Kerenidi T, Nakou M, Dahabreh J, et al. Co-expression patterns of tumor-associated antigen genes by non-small cell lung carcinomas: implications for immunotherapy. Cancer Biol Ther. 2008;7:345–52. doi: 10.4161/cbt.7.3.5424. [DOI] [PubMed] [Google Scholar]
- 29.Babiak A, Steinhauser M, Gotz M, Herbst C, Dohner H, Greiner J. Frequent T cell responses against immunogenic targets in lung cancer patients for targeted immunotherapy. Oncol Rep. 2014;31:384–90. doi: 10.3892/or.2013.2804. [DOI] [PubMed] [Google Scholar]
- 30.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1:32–42. doi: 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 31.Donahue RN, Lepone LM, Grenga I, Jochems C, Fantini M, Madan RA, et al. Analyses of the peripheral immunome following multiple administrations of avelumab, a human IgG1 anti-PD-L1 monoclonal antibody. J Immunother Cancer. 2017;5:20. doi: 10.1186/s40425-017-0220-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory Pathways in Immunotherapy for Cancer. Annual review of immunology. 2016;34:539–73. doi: 10.1146/annurev-immunol-032414-112049. [DOI] [PubMed] [Google Scholar]
- 33.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–61. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nature reviews Cancer. 2016;16:275–87. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.