

Abstract
Lipid A is one of the core structures of bacterial lipopolysaccharides (LPSs), and it is mainly responsible for the strong immunostimulatory activities of LPS through interactions with the toll-like receptors and other molecules in the human immune system. To obtain structurally homogeneous and well-defined lipid As and their derivatives in quantities meaningful for various biological studies and applications, their chemical synthesis has become a focal point. This review has provided a survey of significant progresses made in the synthesis of lipid A and its derivatives that carry diverse saturated and unsaturated lipids, have the phosphate group at its reducing end replaced with a more stable phosphate or carboxyl group, or lack the reducing end phosphate or both phosphate groups, as well as progresses in the synthesis of LPS analogs and other lipid A conjugates. These synthetic molecules have facilitated the elucidation of the structure-activity relationships of lipid A useful for the design and development of lipid A-based therapeutics, such as those utilized to treat sepsis, and other medical applications, such as the use of monophosphoryl lipid A as a carrier molecule for the study of fully synthetic self-adjuvanting conjugate vaccines. These topics are also briefly covered in the current review.
Keywords: lipid A, lipopolysaccharide, synthesis, immunostimulant, adjuvant, immunotherapy
This review covers the chemical synthesis of natural lipid As, their unnatural analogs and derivatives, and lipid A conjugates, as well as biological evaluations, structure-activity relationship studies and medical applications of the synthetic molecules.
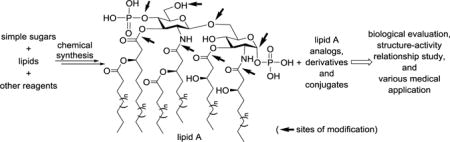
1. Introduction
Lipopolysaccharide (LPS) is one of the most abundant constituents of the outer membrane and one of the major virulent factors for Gram-negative bacteria.1–3 Structurally, LPS is composed of three components, that is, a repetitive polysaccharide O-antigen, a core polysaccharide including an inner core and an outer core, and a glycolipid portion referred to as lipid A (Figure 1). Lipid A is the hydrophobic domain of LPS that anchors LPS to the bacterial cell membrane and is also the main LPS epitope that is recognized by the human innate immune system.4–6 Consequently, lipid A is the most immunologically active component of LPS.
Figure 1.
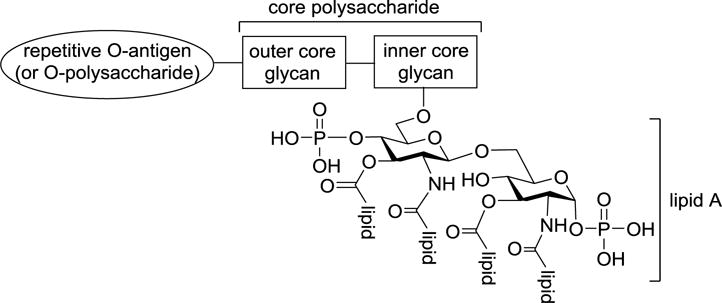
Schematic representation of the general structure of LPSs of Gram-negative bacteria.
Immunologically, lipid A functions mainly through interacting with toll-like receptor 4 (TLR-4) and the myeloid differentiation factor 2 (MD-2) complex in association with toll or interleukin-1 receptor (TIR)-domain-containing adaptor-inducing interferon-β (TRIF) and MyD88, to activate downstream signaling that stimulates cytokine and chemokine releases and upregulates immune cell expression.6–9 Thus, on lipid A activation of TLR-4/MD2, there are two consecutive intracellular signaling cascades that mediate gene expression and proinflammatory cytokine secretion. One is dependent upon MyD88, which results in the up-regulation of cytokines and chemokines, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6, monocyte chemoattractant protein 1 (MCP-1), etc. The other is TRIF-dependent, which leads to the generation of interferon-β (IFN-β). Subsequently, IFN-β stimulates the signal transducer and activator of transcription 1(STAT-1) pathway to provoke the production of interferon-inducible protein-10 (IP-10) and nitric oxide.10,11 However, lipid A is highly pro-inflammatory and highly septic to cause fatal complications, such as endotoxic shock, and thus, represents a severe clinical problem.12–15
Lipid As from different Gram-negative bacterial strains share a highly conserved construct, i.e., a β-D-glucosamine-(1→6)-α-D-glucosamine (GlcN) disaccharide with 1,4′-di-O-phosphorylation and 2,2′-N-/3,3′-O-acylation. As exemplified in Figure 2, the structures of lipid As isolated from different bacteria, and even from one bacterial strain, can vary in the number, length, saturation, and distribution of fatty acid chains and in the modification of the terminal phosphate moiety.16–21 This diversity can have a significant impact on the bioactivity of lipid A in that it alters recognition by the TLR4/MD2 complex.22–25 For example, the lipid A from Escherichia coli, which carries six lipid chains made of 12–14 carbons in length, exhibits the full spectrum of immunostimulant activities. Changing the number and length of the pendent fatty acid chains could decrease the magnitude of stimulated signals in human cells.26–28
Figure 2.
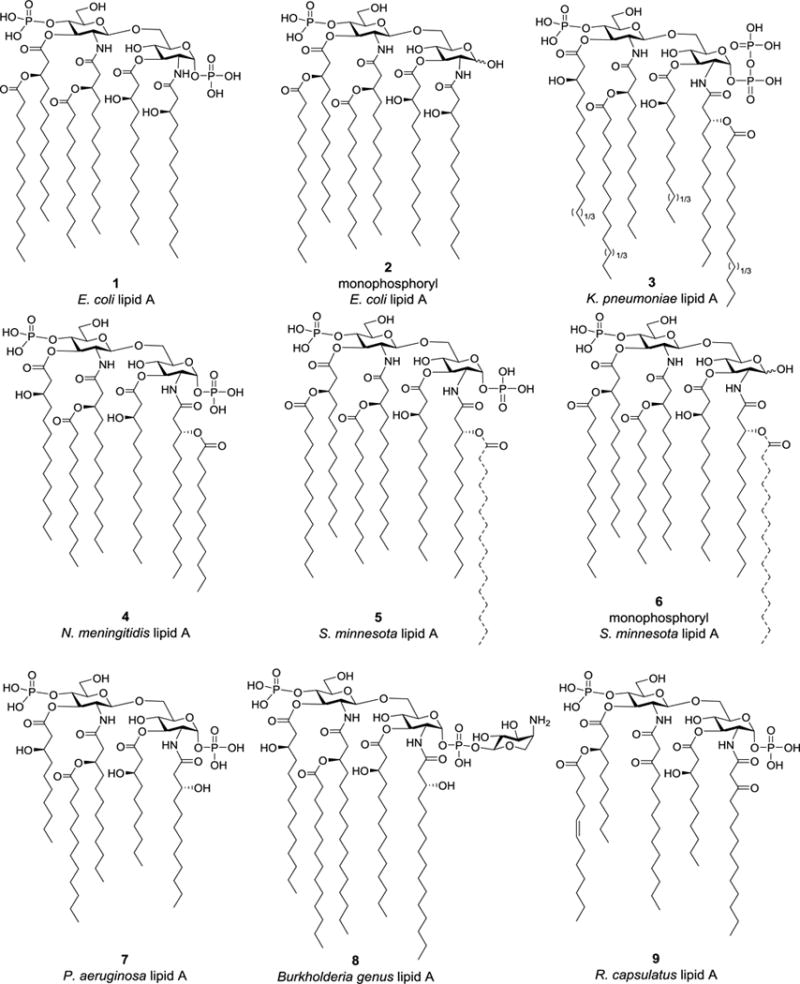
The structure of selected lipid As derived from different Gram-negative bacteria. Dashed lines represent partial structures that result from incomplete biosynthesis, and the numbers in parentheses indicate the potential variation of chain lengths.
However, extensive and detailed investigations of the structure-activity relationships (SARs) and the functional mechanisms of lipid As and LPSs at the molecular level are restricted by the difficulty to obtain structurally homogeneous lipid As and LPSs in quantities and in varied forms from biological sources. Moreover, lipid A and LPS of natural origins may be contaminated with other components from the bacterial cell wall, which can further complicate the investigations. Therefore, chemical synthesis as an important tool to resolve the accessibility problem of lipid A and related derivatives has become an attractive topic.
In the literature, there are a number of excellent reviews about the structures, biosynthesis, and bioactivities of lipid As and LPSs derived from natural sources.29–33 However, no review has been mainly focused on the chemical synthesis of lipid A and derivatives. The present article reviews the recent progress made in the synthesis of lipid A and its derivatives, as well as LPS analogs such as 3-deoxy-D-manno-octulosonic acid (Kdo)-lipid A conjugates. Their biological evaluations including SAR studies and their medical applications are also briefly discussed.
2. Chemical synthesis of lipid A and its derivatives
In 1984, Shiba et al. reported the first chemical synthesis of a lipid A derivative, i.e., lipid IVA – a biosynthetic precursor of lipid A found in a temperature sensitive E. coli mutant.34 Thereafter, from 1984 to early 1990’s, several lipid As and their analogs have been chemically achieved.35–39 Although the efficiency of these earlier syntheses was not remarkably high, these studies have unequivocally verified that lipid A is the primary endotoxin of LPS and established some general principles for lipid A synthesis, such as: (1) using the benzyl group for permanent hydroxyl group protection; (2) installing the phosphate groups at a late stage, e.g., just before the final deprotection; and (3) using N-Troc-protected glycosyl donors for the construction of the β-linked disaccharide backbone. In recent years, many more efficient syntheses have been developed, which facilitated various biological studies and SAR analysis of lipid A.40–45
Due to the presence of different acyl chains at various positions of the disaccharide backbone of lipid A, two general strategies have been adopted for lipid A and related derivative synthesis.46 One is to construct a versatile disaccharide and then install individual lipid chains at the specific positions. In this case, orthogonal protection of various disaccharide positions was a prerequisite, especially when lipid chains in the synthetic target had an asymmetrical distribution.41–44,47–49 The other strategy is to synthesize the lipidated monosaccharides first and then stitch them together by a glycosylation reaction, which can be challenging sometimes due to the increased steric hindrance of the glycosyl donors and acceptors.37,40,45,50–54 Both strategies will be discussed in great detail with individual examples presented below.
2.1 Synthesis of natural lipid As bearing saturated lipids and both phosphate groups
In 2007, Boons group reported the total synthesis of two natural lipid As derived from E. coli (1) and Salmonella typhimurium (10), as well as their derivatives 11–13 having short lipid chains or only one phosphate group (Figure 3).43 Innate immune response studies on E. coli LPS and the synthetic lipid As and derivatives 1, 10–13 revealed the impact of various structural modifications on the potency of lipid A to induce the production of cytokines, such as TNF-α, IFN-β, IL-6, IP-10, IL-1β, RANTES, pro- IL-1β, and NF-κB. It was found that the EC50 values of these compounds to activate immune cells could differ by as much as 100 folds and no bias between MyD88- and TRIF-dependent immune responses were noticed. For example, the potencies and the efficacies of these compounds to induce TNF-α and IFN-β secretion differed slightly; in contrast, they had great differences in the potency to elicit TNF-α and IL-1β production. In addition, E. coli LPS (EC50: 0.004–1.74 nM) showed much higher activity for all the eight cytokines tested than the synthetic lipid A’s 1 (EC50: 21–674 nM) and 11 (EC50: 3.6–43 nM), suggesting the biological role of the Kdo residues in E. coli LPS. Moreover, S. typhimurium lipid A 10 with seven acyl chains exhibited much lower potency than E. coli lipid A 1 with six acyl chains, whereas shortening of the lipid chains (see 11 and 12) resulted in higher potencies. For E. coli lipid As 1 and 11, the difference in their EC50 values was relatively small, whereas the difference of S. typhimurium lipid As 10 and 12 was approximately 3 orders of magnitude. These results indicated the biological importance of the number and length of the fatty acid chains.
Figure 3.
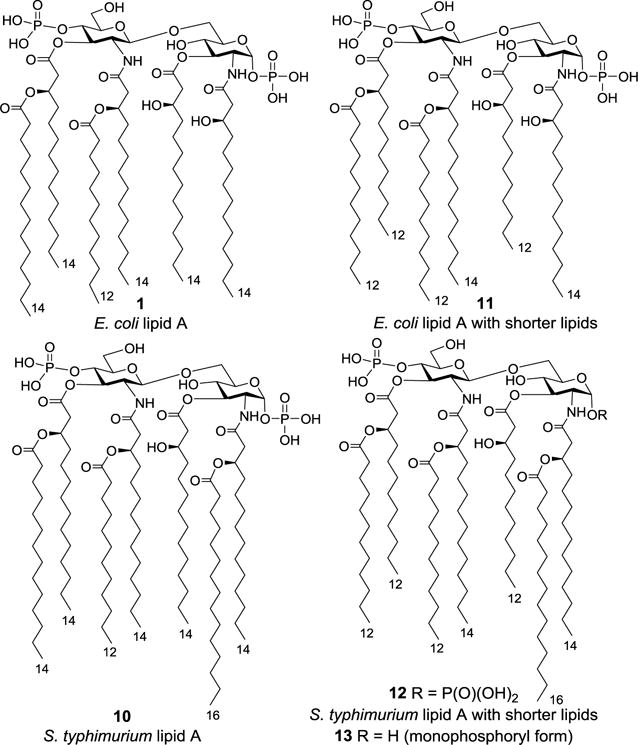
Structures of E. coli and S. typhimurium lipid As 1 and 10 and their derivatives 11–13.
The syntheses of 1 and 10–13 employed orthogonally protected disaccharide 16 as the key and common intermediate, which was obtained upon regio- and stereoselective glycosylation reaction of glycosyl acceptor 15 with N-Fmoc-protected glycosyl donor 14 in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf) (Scheme 1). This reaction gave β-1,6-linked disaccharide 16 selectively as a result of the neighboring Fmoc group participation and the higher reactivity of the primary C-6 hydroxyl group as compared to that of the secondary C-3 hydroxyl group in 15. It is worth noting that the 4′-O-phosphate group was introduced at an early stage of the synthesis, that is, in glycosyl donor 14. The phosphate group was protected as a benzyl type of xylidene diester, which was stable to subsequent reactions but readily removable in the final deprotection step. In contrast, phosphoric acid dibenzyl ester, another commonly used strategy for phosphate protection, is less stable and can be sometimes partially deprotected.29 Orthogonally protected disaccharide 16 was then utilized to prepare both 1 and 10, as well as their derivatives.
Scheme 1.
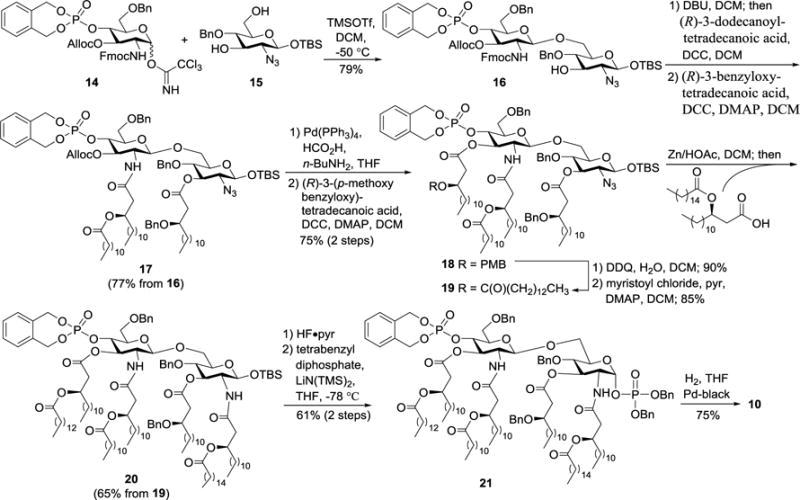
Synthesis of S. typhimurium lipid A 10.
In the synthesis of 10 (Scheme 1), 16 was first treated with 1,8-diazabicycloundec-7-ene (DBU) to remove the 2′-N-Fmoc group, which was followed by acylation of the resultant free amino group with (R)-3-dodecanoyl-tetradecanoic acid in the presence of 1,3-dicyclohexylcarbodiimide (DCC). Thereafter, the 3-O-position was acylated using (R)-3-benzyloxy-tetradecanoic acid with DCC and 4-dimethylaminopyridine (DMAP) as condensation reagents to provide 17. The allyloxycarbonyl (Alloc) group in 17 was removed with tetrakis(triphenylphosphine)palladium [Pd(PPh3)4], which was followed by acylation with (R)-3-(para-methoxybenzyloxy)-tetradecanoic acid. Cleavage of the p-methoxybenzyl (PMB) ether in 18 with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and then acylation of the exposed β-hydroxyl group on the lipid with myristoyl chloride gave 19. The azido group in 19 was reduced with Zn and HOAc, and the resultant amino group was finally acylated with (R)-3-hexadecanoyl-tetradecanoic acid in the presence of DCC to produce 20. A phosphate group was introduced to the anomeric position in two steps, including HF-pyridine mediated removal of the tert-butyldimethylsilyl (TBS) group in 20 and anomeric phosphorylation employing tetrabenzyl diphosphate in the presence of lithium bis(trimethyl)silylamide [LiN(TMS)2]. Eventually, the resultant 21 was globally deprotected through Pd-black catalyzed hydrogenolysis to obtain the synthetic target 10.
Compounds 11–13 were synthesized by a similar strategy with minor modifications, i.e., using orthogonal Alloc and azido groups to protect the 3′-O- and 2-N-positions (Scheme 2). Specifically, after stepwise acylation of 16 to get 22, the azido group was reduced with Zn/HOAc, under which condition Alloc was unaffected. Acylation of the free amine with (R)-3-hexadecanoyl-tetradecanoic acid in the presence of DCC afforded 23. Thereafter, the Alloc group was removed with Pd(PPh3)4 to generate 24, which was converted into the target molecule 12 through a series of transformations including acylation, phosphorylation, and hydrogenolysis as mentioned above.
Scheme 2.
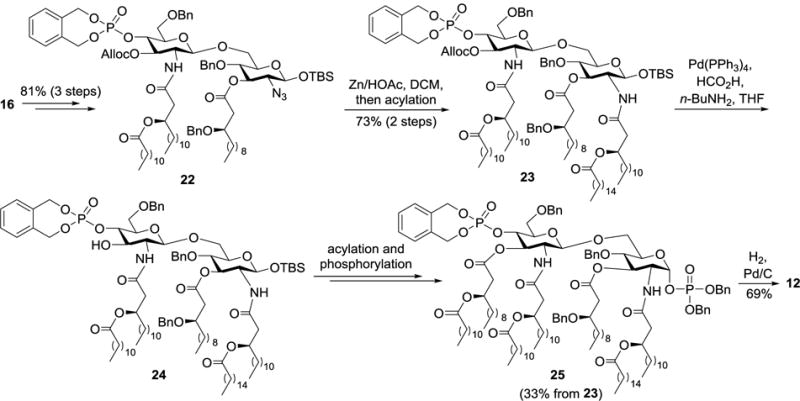
Synthesis of lipid A 12.
The above syntheses used functionalized and orthogonally protected disaccharide 16 carrying Alloc, Fmoc, TBS, and azido protecting groups as the key intermediate, which gave easy access to regioselective modifications of various positions in the disaccharide. The strategy should be useful for parallel synthesis of different lipid As and related analogs. In addition, the revealed SAR based on these structurally defined lipid A derivatives should be very helpful for further analysis and understanding of the immune modulation mechanisms of lipid A and LPS.
The natural lipid As derived from Rubrivivax gelatinosus (26) and Chlamydia trachomatis (27 and 28), as depicted in Figure 4, were synthesized by a strategy different from that for 1 and 10–13.45,54 In this case, lipidated monosaccharides were prepared and used as building blocks for glycosylation reactions to result in the disaccharide backbone of 26–28.
Figure 4.
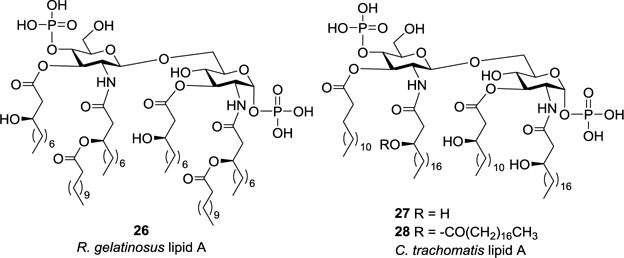
Structures of R. gelatinosus and C. trachomatis lipid As 26–28.
As represented by the synthesis of 26 (Scheme 3), in the presence of TMSOTf, regioselective coupling of the N-Troc protected imidate donor 29 and the diol acceptor 30 afforded the desired disaccharide 31 in a 72% yield. It is worth noting that the hydroxyl groups in the 3-O-linked lipid chains of 29 and 30 were protected with p-trifluoromethylbenzyl group. An unsubstituted benzyl group at this position was prone to oxidation in air and gradual conversion into the corresponding benzoyl group, which was also observed by Kusumoto et al.53 Cleavage of the 2′-N-Troc group in 31 with Zn/HOAc was followed by acylation of the amino group with (R)-3-dodecanoyl-decanoic acid utilizing 1-hydroxybenzotriazole (HOBt) and 1-ethyl-3-(dimethylaminopropyl) carbodiimide hydrochloride (WSCD•HCl) as condensation reagents to give the fully lipidated intermediate 32. Next, removal of the anomeric allyl (All) group in 32 with Ir-complex and phosphorylation of the freed anomeric hydroxyl group with tetrabenzyl pyrophosphate under the influence of LiN(TMS)2 gave 33. Finally, all of the benzyl type of protecting groups in 33 were removed by Pd-catalyzed hydrogenolysis to afford R. gelatinosus lipid A 26.45
Scheme 3.
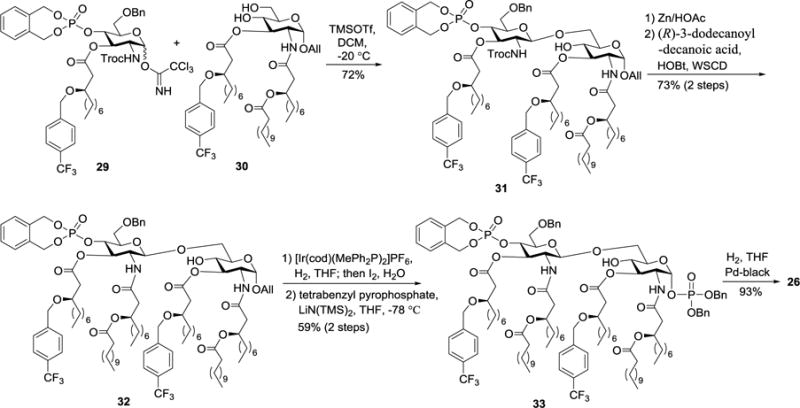
Synthesis of lipid A 26.
The above syntheses employed lipidated monosaccharides as building blocks to accomplish the disaccharide backbone. This strategy should provide an efficient approach to access lipid As and lipid A analogs bearing asymmetrically distributed lipid chains, and the products should be useful for precise and detailed analysis of the immunological activities and SARs of lipid A.
2.2 Synthesis of lipid As bearing unsaturated lipids
Qureshi et al. discovered a unique type of lipid A in Rhodobacter sphaeroides, which carried unsaturated lipid chains.55,56 It was a mixture composed of three inseparable components, and the major products were suggested to be 34a and 34b, named RsDPLA (Figure 5). Interestingly, this type of lipid A was a potent LPS antagonist, which could suppress chemokine and cytokine release induced by LPS, demonstrating the significant impact of the unsaturated lipid chain in lipid A on its biological activity.
Figure 5.
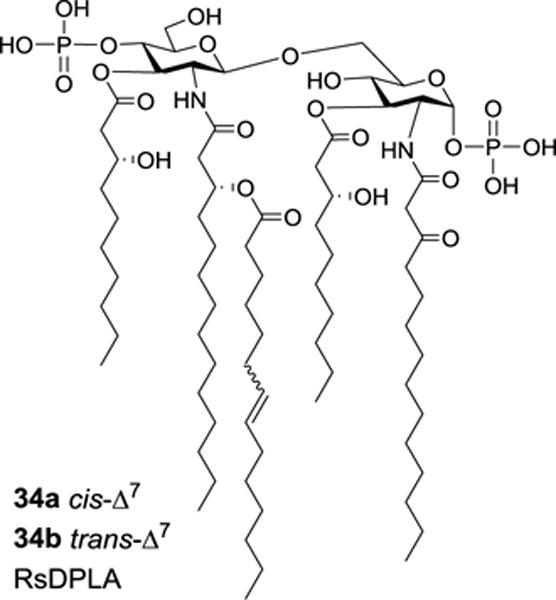
Structures of the proposed R. sphaeroides lipid As 34a and 34b bearing an unsaturated lipid.
For the chemical synthesis of lipid As carrying unsaturated lipids, the common strategy using the benzyl group for permanent hydroxyl group protection is unsuitable, as unsaturated lipids are incompatible with Pd-catalyzed hydrogenolysis employed to remove the benzyl protecting groups. Christ et al. established a synthetic strategy for these lipid As based on allylic protecting groups (Scheme 4).37 In both the glycosyl donor 35 and acceptor 36, Alloc and All groups were used to protect the hydroxyl and phosphate groups, respectively. Glycosylation of 36 with 35 under the promotion of AgOTf produced the desired β-disaccharide 37 and its anomer (β:α 2:1). Reduction of the azido group in 37 with tin(II) tris(benzenethiolate)-triethylamine complex and acylation of the resulting amino group with unsaturated lipids generated 38a and 38b. Thereafter, the anomeric TBS group was selectively removed with HF, which was followed by installation of the α-linked diallyl phosphate group to yield 39a and 39b. Finally, all allylic protecting groups were removed smoothly with Pd(Ph3P)4 to afford cis- and trans-lipid A 34a and 34b, respectively. Unfortunately, neither 34a nor 34b was a perfect match to the natural lipid A. Nevertheless, both 34a and 34b exhibited strong in vitro antagonistic activity to suppress TNF-α generation induced by LPS in human monocytes (average IC50 ≈ 1 nM) and were devoid of LPS agonistic properties even at a 100 μM concentration. Moreover, the above synthetic method may be generally applicable to lipid As and related derivatives that contain unsaturated lipid chains.
Scheme 4.
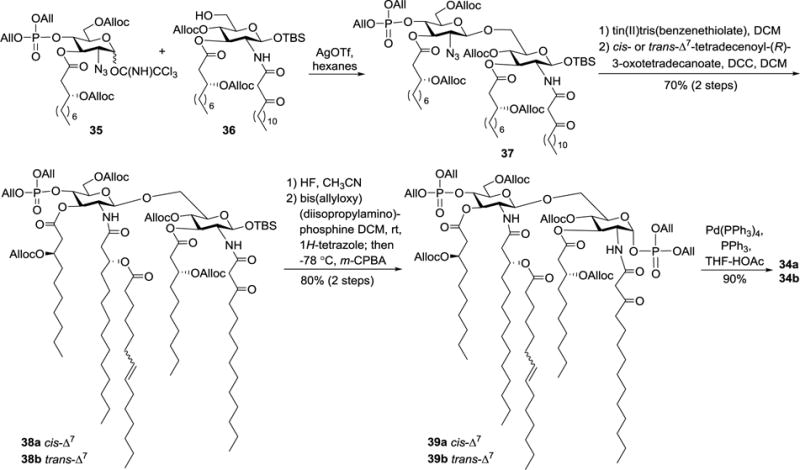
Synthesis of 34a and 34b.
Although 34a and 34b exhibited strong LPS antagonistic activity, the acyl groups at their O-3- or O-3′- or both positions can be easily hydrolyzed to result in agonistic byproducts. To deal with this issue, Christ et al. designed and prepared an artificial analog of R. sphaeroides lipid A, E5531 (40, Figure 6), which had the O-3- and O-3′-acyl groups replaced with hydrolytically stable ether linkages.57 In addition, the C-6′-hydroxyl group was blocked as methyl ether to avoid potential contaminants generated from side reactions at this position. The synthetic E5531 was shown to possess the desired chemical properties including purity, stability, and solubility. Biological study of E5531 using human monocytes revealed that it strongly antagonized the release of a variety of cytokines induced by a number of Gram-negative bacterial LPSs. More importantly, E5531 was completely devoid of LPS agonistic activity, even at the concentration 10,000-fold higher than that required for the antagonistic activity. In addition, in vivo evaluation demonstrated that E5531 could protect BCG-primed mice from LPS-induced lethality and viable E. coli infection-caused death. Structurally stable E5531 has thus exhibited great therapeutic potential for the treatment of Gram-negative sepsis.57–59
Figure 6.
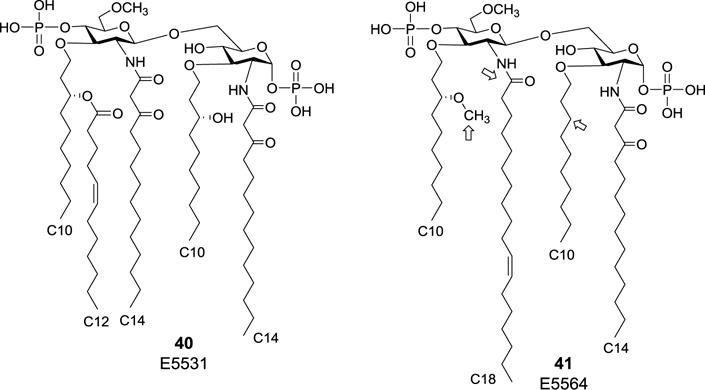
Structures of R. sphaeroides lipid A analogs E5531 and E5564 as potent LPS antagonists.
E5564 (41, Figure 6), the second generation of synthetic analogs of RsDPLA, which carried an unsaturated lipid as well, was proved to be a highly active antagonist for endotoxin-mediated immune cell activation. Compared to E5531, structural variations of E5564 were mainly focused on 3,3′-O- and 2′-N-lipid chains as marked in Figure 6, and these alterations further improved the antagonist activity.60 Currently, E5564 is utilized in clinic in the trade name of Eritoran for the treatment of severe sepsis.61,62 Due to the strong anti-sepsis activity and broad clinical prospects of E5564, many of its glucose-containing derivatives were synthesized, also based on the allylic protection strategy.63–65
2.3 Synthesis of unnatural lipid A derivatives bearing two phosphate groups
It was reported that the lipid chains of lipid A could affect its binding with polymyxin B and polymyxin B nonapeptide.66–68 To study in detail the interactions between lipid A and polymyxin B or polymyxin B nonapeptide and obtain more accurate information about the SARs that would be difficult to obtain by using heterogeneous biological lipid As, Savage group40 designed and synthesized lipid A derivatives 42–44 bearing incrementally shortened lipid chains (Figure 7). The shortening of lipid chains could also help improve the solubility in water.
Figure 7.
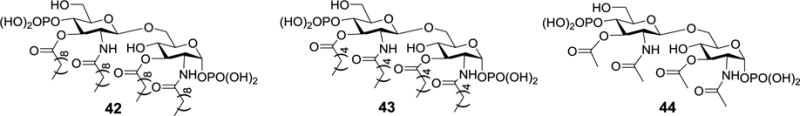
Structures of artificial diphosphorylated lipid A derivatives 42–44.
Lipid A analogs 42–44 were obtained by a convergent strategy with lipidated monosaccharides as building blocks, as shown in the synthesis of 43 (Scheme 5). First, lipidated glycosyl acceptor 48 and partially lipidated glycosyl donor 51 were prepared from 45 and 49 through a series of transformations including regioselective protecting group manipulation, lipidation, phosphorylation, and so on. Glycosylation of 48 with 51 gave disaccharide 52, of which the 2′-N-Troc group was replaced with another lipid chain to afford 53. Finally, the TBS group in 53 was removed for the introduction of anomeric phosphate group, which was followed by global deprotection to give the target molecule 43. This strategy can be useful for other similar lipid A analogs.
Scheme 5.
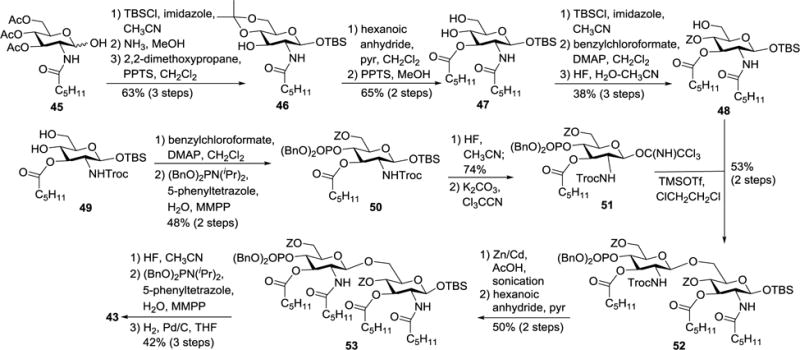
Synthesis of lipid A derivative 43.
In the structure of natural lipid A, the reducing end phosphate group is directly linked to the anomeric hydroxyl group to form glycosyl phosphate, which is rather unstable. To overcome this problem and improve the stability of lipid A, Kusama group designed and synthesized three E. coli lipid A analogs 54–56 (Figure 8) as phosphonooxyethyl glycosides to replace the labile α-glycosyl phosphate.69 It was anticipated that these lipid A analogs might have improved bioactivities, such as antitumor activity,70–73 owing to their enhanced chemical stability.
Figure 8.
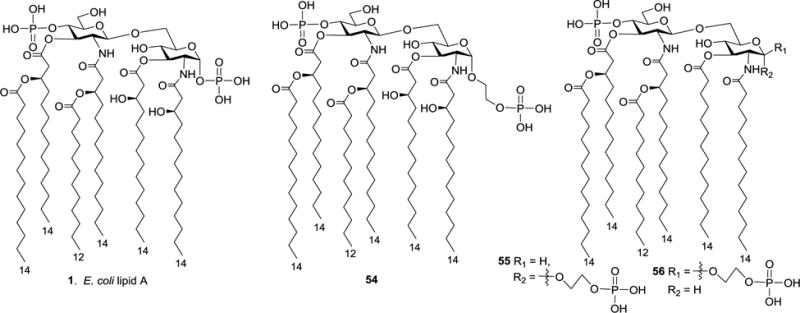
Structures of E. coli lipid A analogs 54–56.
In the synthesis of 54–56 (Scheme 6), the phosphonooxyethyl group was introduced at an early stage. Immediately after the conversion of 57 into 58 upon protection of the 4,6-O-positions with an isopropylidene group, the free primary hydroxyl group at the reducing end of 58 was selectively phosphorylated. Therefore, the phosphate group was introduced at the stage of monosaccharide. Acylation of the remaining free hydroxyl group in 58 and removal of the isopropylidene group afforded diol 59. Glycosylation of 59 with 60 under the promotion of mercuric cyanide provided the desired β-1,6-disaccharide 61, which was transformed into the synthetic target 54 following deprotection of the 2′-N- and 6′-O-Troc groups, regioselective acylation of the resulting amine, and two-step global deprotection.
Scheme 6.
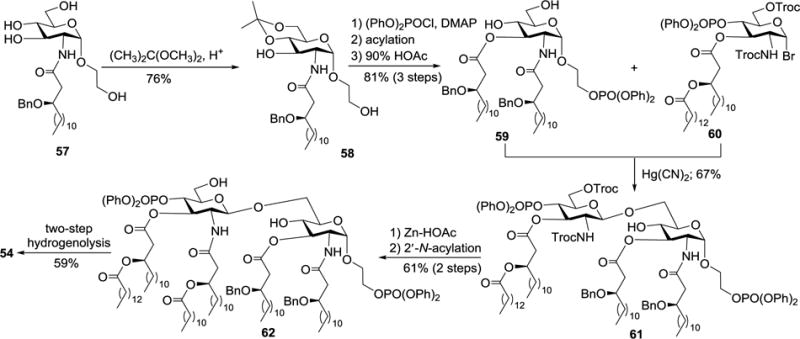
Synthesis of lipid A derivative 54.
According to the literature,70–73 E. coli lipid A and its derivatives had antitumor activities, thus the cytotoxicity of natural lipid A 1 and its synthetic analogs 54–56 to methylcholanthrene-induced murine fibrosarcoma (Meth A fibrosarcoma) was studied in vivo.69 Compounds were administered to the mice at a dose of 100 μg/mL through the tail veins three times (7, 12, and 17 days after tumor inoculation). The tumor weight ratio of tested group/control group (T/C) was measured on the 21th day, and it was revealed that phosphonooxyethyl α-glycosides 54 and 55 had potent antitumor activities (T/C: 14% for 54; 12% for 55), which were similar to natural E. coli lipid A 1 (T/C 10%) However, the β-glycoside 56 (T/C 41%) was significantly less active. It was thus suggested that the glycosyl phosphate moiety may not be critical for the antitumor activity of lipid A and can be substituted with a more stable α-linked phosphonooxyethyl moiety, but the reducing end anomeric α-conformation was important. Moreover, replacing the (R)-3-hydroxytetradecanoyl groups with tetradecanoyl groups did not exhibit a major impact on its antitumor activity. Unfortunately, these phosphonooxyethyl analogs of lipid A were still quite toxic.
To further improve the antitumor activity of the above compounds and reduce their side effects, Kusama and coworkers synthesized 64 and 65 (Figure 9), which were derivatives of the less toxic biosynthetic precursor of E. coli lipid A 63, by the same synthetic strategy for 54.74 The antitumor activities of compounds 1 and 63–65 were evaluated by the same protocol described for 54–56. It was shown that 64 (T/C 32%) and 65 (T/C 24%) had comparable antitumor activity as that of the parent lipid A analog 63 (T/C 41%), but were slightly less active than the natural E. coli lipid A 1 (T/C 10%). However, like the precursor 63, compounds 64 and 65 were weakly toxic. Particularly, 64 had no detectable toxicity at a dose (50 μg/kg) that showed significant antitumor activity. It was thus concluded that replacing the glycosidic phosphate with an α-linked phosphonooxyethyl moiety could decrease the toxicity of lipid A while maintaining its antitumor activity. Moreover, it was found that the presence or absence of a hydroxyl group on the fatty acid chains had very little influence on the antitumor activity.
Figure 9.
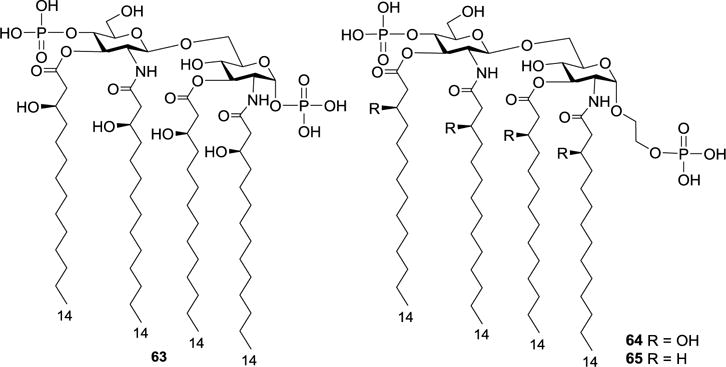
Structures of the biosynthetic precursor 63 of E. coli lipid A 1 and its artificial analogs 64 and 65.
Above investigations helped establish the first generation of potent antitumor lipid A analogs with enhanced stability and decreased toxicity. They should contain four linear fatty acid chains on the disaccharide backbone and an α-linked phosphonooxyethyloxyl group, instead of a phosphate, at the reducing end anomeric position. This structural scaffold should be useful for the design and development of novel lipid A-based antitumor agents.
Tritium-labeled lipid A derivatives, such as compounds 66 and 67 which had the tritium label on the phosphonooxyethyl group (Scheme 7),75 were also prepared. These molecules with high chemical purity and specific radioactivity were utilized to analyze the interactions between lipid A and their receptors.76 Several lipid A binding proteins were clearly detected on macrophages of both LPS-responder C3H/HeN and LPS-hyporesponder C3H/HeJ mice. For example, the p80 (a 80 kDa protein), p66, p65, p60, and p60′ proteins were found as specific lipid A-binding proteins, and no difference in the pattern of 66-binding proteins was observed between C3H/HeN and C3H/HeJ macrophages. However, these proteins had different structural requirements for the binding with lipid A and LPS. The p80, p66, and p65 proteins preferentially bind to lipid A, while the p80 protein, but not p66 or p65, was also able to bind to LPS. The p60 protein preferentially bind to LPS. In these syntheses, tritium was regiospecifically introduced via reducing aldehyde 69 with 3H-borohydride. Phosphorylation of 70 by the phosphoramidite method, followed by global deprotection through Pd-catalyzed hydrogenolysis, afforded the synthetic target 66. Compound 67 was prepared by the same method.
Scheme 7.
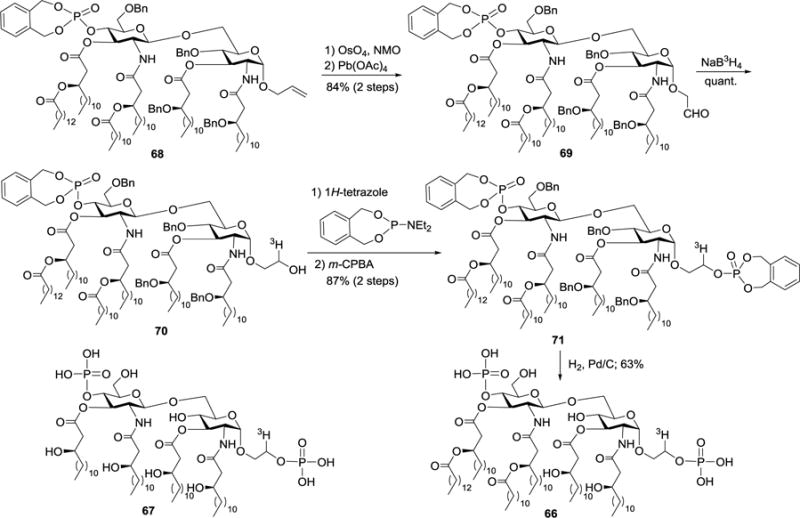
Structures of tritium-labeled lipid A analogs 66 and 67, as well as the synthesis of 66.
2.4 Synthesis of lipid A analogs bearing carboxylic acid at the reducing end
Another direction to discover biologically functional lipid A analogs with improved stabilities is replacing the labile glycosyl phosphate with an anomeric carboxylic acid moiety, which should help improve stability but can still form a negative charge. In the study of GLA-60 (72, Figure 10),77 a lipid A-related monosaccharide analog with LPS-agonistic activity, it was found that the carboxylic acid analog 73 had LPS-antagonistic property toward human monoblastic U937 cell.78 Therefore, Mochizuki et al.79,80 designed and synthesized a series of artificial carboxylic acid derivatives of lipid A, 74a-c and 75a-c (Figure 10), as potential LPS antagonists.
Figure 10.
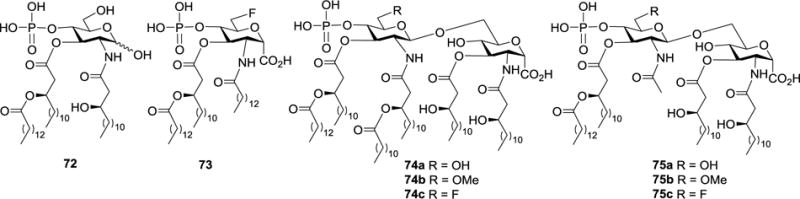
Structures of GLA-60 72, its monosaccharide pyranosyl carboxylic acid derivative 73, and pyranosyl carboxylic acid derivatives of lipid A 74a-c and 75a-c.
Biological evaluations of 74a-c, which carried six fatty acid chains with the acyl substitution pattern similar to that of E. coli lipid A 1, revealed that they induced the production of TNF-α in human monoblastic U937 cell, indicating LPS-agonistic activity. Clearly, replacing the phosphate group in E. coli lipid A 1 with a carboxyl group did not significantly affect its biological activity. This provided the feasibility to design more stable lipid A analogs, in which the unstably anomeric phosphate group is substituted. In contrast, 75a-c containing four fatty acid chains inhibited TNF-α production induced by LPS in a dose-dependent manner (IC50: 7, 10, and 9 nM respectively), indicating their LPS-antagonistic activity. Thus, the number of lipid chains in a lipid A analog is of great importance for its LPS-agonistic or antagonistic activity. Additionally, there is no significant difference among 75a-c in their LPS-antagonistic activity. In the meantime, 74a-c exhibited the similar level of LPS-agonistic activity. Consequently, the substituents on the 6′-C-position of these molecules appeared to be not very important for their biological activities.
In the synthesis of 74a-c and 75a-c, all of the glycosyl donors and donor precursors 76–78 were prepared from 7939 (Scheme 8). For example, 76 was readily obtained from 79 through a series of transformations including regioselective primary hydroxyl group protection, 4-O-phosphorylation, and Ir-complex mediated removal of the anomeric All group. On the other hand, 79 was converted into 80 after regioselective 6-O-silylation, 4-O-phosphorylation, and 6-O-desilylation. Compound 80 was then methylated or fluorinated to produce 81 and 82, respectively. Similarly, Ir-complex mediated removal of the anomeric All group in 81 and 82 afforded 77 and 78.
Scheme 8.
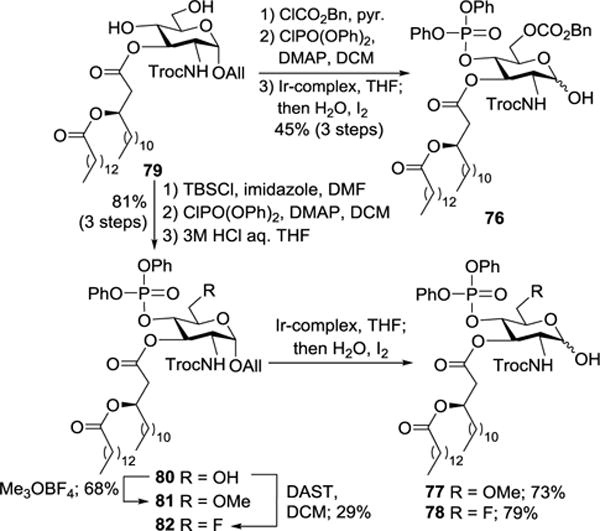
Synthesis of glycosyl donor precursors 76–78.
In the meantime, glycosyl acceptor 83 was synthesized from intermediate 84 upon protection of the 4,6-O-positions, 3-O-esterification, and removal of the 4,6-O-protection, whereas hemiacetals 76–78 were converted into glycosyl donors 86–88 on DBU and CCl3CN treatments (Scheme 9). Regioselective glycosylation of the less hindered and more reactive 6-O-position of diol 83 with imidates 86–88 in the presence of TMSOTf at −50 °C gave disaccharides 89a–c (Scheme 9). The 2′-N-Troc protection in 89a–c was removed with Zn and HOAc, and the resultant free amino group was then N-acylated with EDCI•HCl and (R)-3-(dodecanoyloxy)tetradecaonic acid or acetic acid to afford fully protected intermediates 90a–c and 91a–c that were globally deprotected via catalytic hydrogenolysis in two steps to yield the synthetic targets 74a–c and 75a–c, respectively.
Scheme 9.
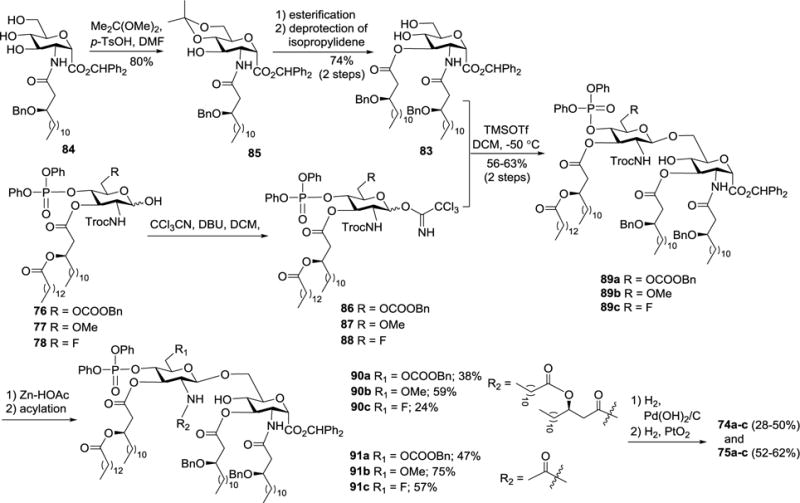
Synthesis of carboxylic acid analogs of lipid A 74a-c and 75a-c.
To obtain lipid A analogs with additionally increased structural stabilities, Shiozaki designed and synthesized another series of carboxylic acid derivatives 92a–c, 93a–c, and 94a–c (Figure 11), which contained unnatural ether linkages at the 3,3′-O-positions.81 These compounds were also evaluated for their bioactivities to inhibit or promote LPS-induced TNF-α production in human monoblastic U937 cells. Compounds 92a–c and 94a–c, which contained four lipid chains, inhibited TNF-α production with IC50 values of 0.86, 0.61, 1.2, 11, 6.4, and 10 nM, respectively, indicating their strong LPS antagonist activity. On the other hand, 93b carrying six lipid chains was almost inactive, while 93a and 93c were moderate LPS agonists to promote TNF-α production. Therefore, the total number of lipid chains in the structure of lipid A analogs was again proved to be of great importance for their LPS-antagonistic or agonistic activity, whereas the C-6′ substitution pattern had only a small impact on their bioactivity. These observations were in accordance with previous findings.79,80 Additionally, the 3-tetradecanoyloxytetradecyl or 3-dodecyloxytetradecyl group on the 3′-O-position was not critical for the inhibitory activity to human monoblastic U937 cell.
Figure 11.
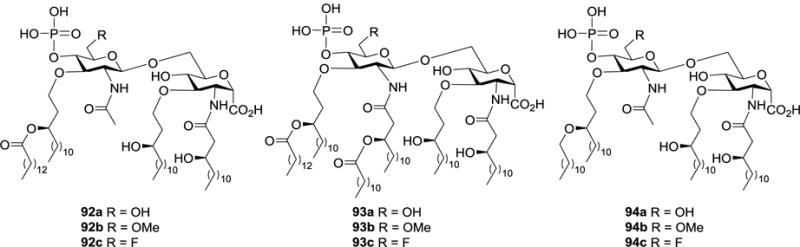
Structures of pyranosyl carboxylic acid analogs of lipid A 92a–c, 93a–c, and 94a–c bearing unnatural ether chains.
As outlined in Scheme 10, the synthesis of 94c commenced with 95, which had the amino group protected by a trifluoroacetyl group. First, 95 was converted into 98 via a series of conventional transformations including 3-O-alkylation, replacing the N-trifluoroacetyl group with a Troc group, regioselective 6-O-silylation, 4-O-phosphorylation, and selective 6-O-desilylation under acidic conditions. The 6-OH group in 98 was then substituted with a fluorine atom upon treatment with diethylaminosulfur trifluoride (DAST), which was followed by anomeric deallylation to generate hemiacetal 99. Activation of 99 was achieved by reacting with DBU and CCl3CN, and the resultant trichloroacetimidate was used to glycosylate 10082 with TMSOTf as the promoter. The reaction was regioselective for the more reactive primary hydroxyl group to afford disaccharide 101, which was readily converted into the synthetic target 94c after swapping the 2′-N-Troc group with an acetyl group and then global deprotection by a two-step hydrogenolysis procedure. The same synthetic method was also employed to achieve 92 and 93.
Scheme 10.
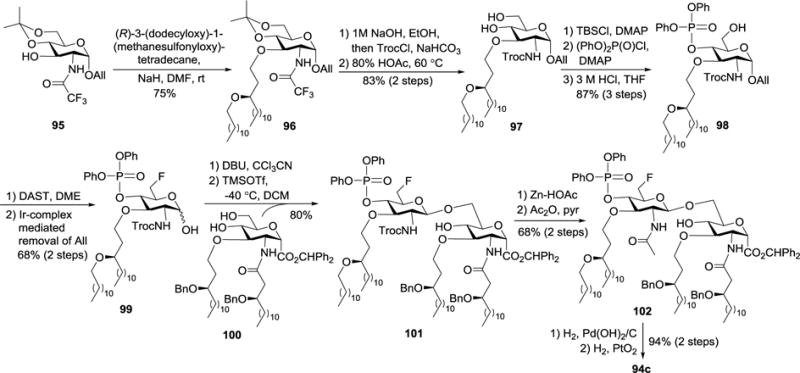
Synthesis of glycosyl carboxylic acid derivative of lipid A 94c bearing ether linked lipids.
Based on the findings that the carboxymethyl α-glycoside analog 10383 of GLA-60 and lipid A analog 92b bearing an anomeric carboxylic group and 3,3′-O-ether linkages81 exhibited potent LPS-antagonistic activities (IC50: 5.0 and 0.61 nM, respectively), Shiozaki group designed and synthesized 104a-c (Figure 12) that contained the structural components of both 103 and 92b.51 Biological evaluations showed that 104a–c were LPS-antagonists, inhibiting TNF-α production by human monoblastic U937 cell with IC50 values of 6.5, 6.1, and 12.4 nM, respectively. On the other hand, previously studies revealed that the IC50 values of corresponding carboxylic acid analogs 94a–c were 11.0, 6.4, and 10.0 nM, respectively.81 Therefore, substituting the anomeric carboxylic acid group with a carboxymethyl group did not have a significant impact on their activity to inhibit TNF-α production. However, the LPS-antagonistic activities of 94a–c and 104a–c were much lower than that of 92b, indicating that the ester linkage at the 3′-O-position played an important role in the antagonistic property.
Figure 12.
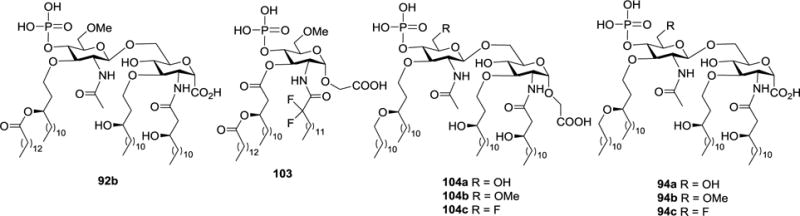
Structures of GLA-60 analog 103 and lipid A analogs 92b, 104a–c and 94a-c.
The synthesis of 104a-c (Scheme 11) started with the preparation of diphenylmethyl protected carboxymethyl glycoside 107 as a glycosyl acceptor. First, 105 was prepared and converted into 107 on dihydroxylation of its double bond with osmium tetroxide (OsO4) and 4-methylmorpholine N-oxide (NMO), oxidative cleavage of the resultant vicinal diol with lead acetate, oxidation of the aldehyde with NaClO2, esterification of the carboxylic acid with diphenyldiazomethane (Ph2CN2), and then removal of the acetonide group with 80% aq. HOAc. Glycosylation of 107 with glycosyl imidate donors 108a-c in the presence of TMSOTf gave 109a-c, respectively. Finally, swapping the 2′-N-Troc group with an acetyl group was followed by global deprotection in two steps of Pd- and Pt-catalyzed hydrogenolysis generated the synthetic targets 104a-c.
Scheme 11.
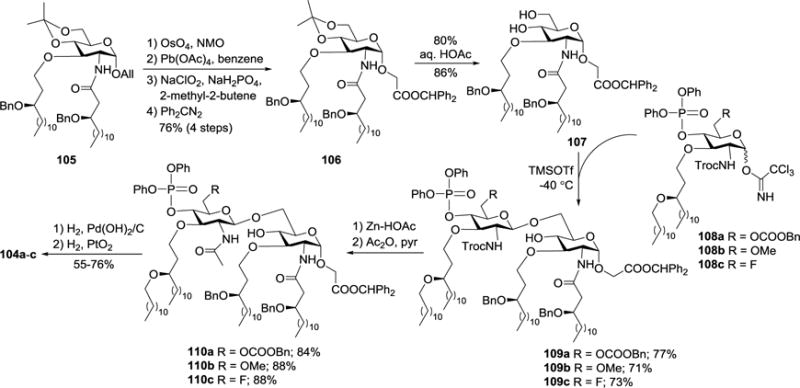
Synthesis of carboxymethyl glycoside derivatives of lipid A 104a–c.
Fukase group synthetized and investigated a series of monosaccharide analogs 111a,b–116a,b (Figure 13) of lipid A, which contained aspartic acid or phosphoserine residue as replacement of the non-reducing end phosphorylated glucosamine residue.84 These compounds were employed to elucidate the structural and conformational requirements of lipid A derivatives for their endotoxic and antagonistic activities. Biological evaluations revealed that most of the analogs did not induce IL-6 production, but 115a did exhibit a concentration-dependent induction at concentrations over 100 ng/mL. Compound 115b also showed some agonistic activity, but it was about 100-fold weaker than that of 115a. In addition, the phosphoserine-containing derivatives exhibited stronger antagonistic activity than corresponding aspartic acid-containing analogs with the same acylation pattern. As for the aspartic acid analogs, the L-form showed a stronger inhibition than the corresponding D-form, whereas D- and L-phosphoserine derivatives did not exhibit obvious differences. Furthermore, 112a,b exhibited the antagonistic activity, whereas 115a,b showed the immunostimulating activity. Thus, the endotoxic and antagonistic activities were switched by changing the phosphoryl group and the carboxylic group.
Figure 13.
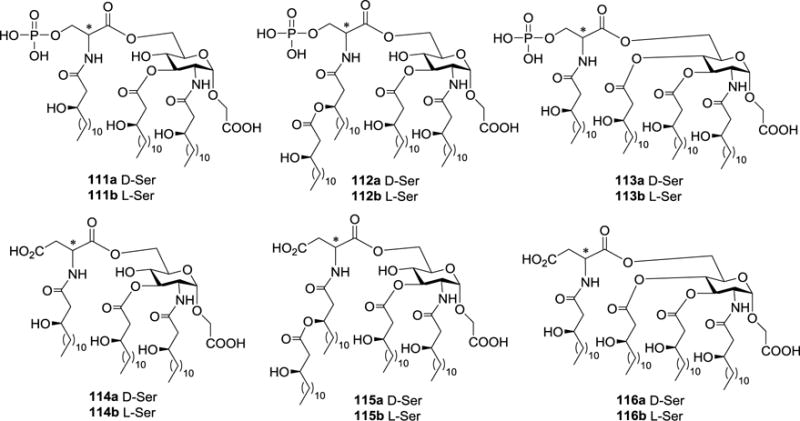
Structures of monosaccharide analogs of lipid A 111a,b–116a,b containing acidic N-acyl-amino acids as replacement of the non-reducing end phosphorylated glucosamine residue.
2.5 Synthesis of monophosphoryl lipid A and related derivatives
Although lipid A possesses strong immunostimulatory activities, its clinical applications have been limited as it can cause fatal septic shock.13 Fortunately, it was discovered that the toxicity of lipid A could be significantly reduced after its anomeric phosphate group was removed to generate 4′-O-monophosphorylated lipid A, usually known as monophosphoryl lipid A (MPLA), whereas its beneficial immunostimulatory activities were retained.2 For example, the MPLA derivative of Salmonnella minnesota lipid A was at least 99.92% less toxic than the parent compound.85,86 In fact, a MPLA preparation (MPL™) obtained by selective hydrolysis of the anomeric phosphate group of S. minnesota R595 lipid A has been approved for clinical use as a vaccine adjuvant.87 However, MPLAs derived from bacterial sources are heterogeneous and thus lack consistency in structure, property, and biological performance. To overcome this problem and to find new MPLA adjuvants with improved immunostimulatory properties, various MPLA derivatives with defined structures have been designed, synthesized, and immunologically evaluated.50,52,87–92
As mentioned above, S. minnesota R595 lipid A 117 (Figure 14) is a potent immunostimulant and endotoxin. Fortunately, its endotoxic activity could be alleviated after selective removal of its 1-phosphono and 3-O-(R)-3-hydroxytetradecanoyl groups through acidic hydrolysis.86,93 The resultant MPLA was shown to enhance both antibody responses and T cell-mediated immunity, and is an effective adjuvant for prophylactic and therapeutic vaccines.73 However, because of the inherent heterogeneity of cognate LPS and the incomplete and nonspecific reactions of hydrolysis, the resultant MPLA is highly heterogeneous, containing differently acylated products in addition to the desired hexaacyl components such as 118 (Figure 14).
Figure 14.
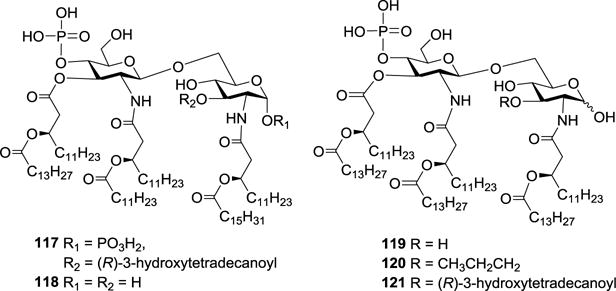
Structures of MPLAs 117–121.
To explore the SARs of MPLA and discover novel MPLA analogs with favorable properties, Jiang et al. prepared MPLAs 119–121 bearing varied 3-O-substitution and tested their adjuvant activity.50 As shown in Scheme 12, the synthesis of 119–121 employed N-Troc protected glycosyl donor 122 and lipidated glycosyl acceptor 123 as building blocks for constructing the lipid A disaccharide backbone due to the asymmetric distribution of lipid chains in the synthetic targets. Additionally, the anomeric position of the acceptor was blocked with a benzyl ether group, which could be easily deprotected at the final stage without affecting other functionalities. Glycosylation of 123 with imidate 122 in the presence of BF3•OEt2 generated disaccharide 124 in an 88% yield. Removal of the 3-O-All group in 124, acylation of the resulting hydroxyl group, and subsequent cleavage of the 2′-N-Troc group were followed by N-acetylation to afford 125. Treatment of 125 with NaBH3CN and HCl•Et2O exposed regioselectively the 4′-O-hydroxyl group, which was then phosphorylated by the phosphoramidite method to generate 126. Finally, 126 was deprotected by Pd/C catalyzed hydrogenolysis to produce the target MPLA 121.
Scheme 12.
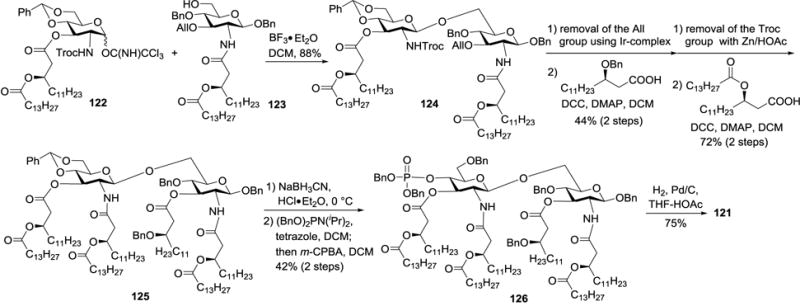
Synthesis of MPLA 121.
The immune adjuvant activities of the synthetic MPLAs 119–121 and the natural S. minnesota R595 MPLA were assessed using a synthetic BLP25 liposomal vaccine system. All three synthetic MPLAs 119–121 exhibited strong adjuvant activities, similar to that of natural S. minnesota R595 MPLA, including promoting antigen specific T cell-mediated immune responses and interferon-γ (IFN-γ) production. Compound 121 bearing a 3-O-(R)-3-hydroxytetradecanoyl group was proved to be the most active, whereas 120 containing a propyl group had only slightly lower activity. Thus, the substituent at the 3-O-position of the reducing-end sugar unit had only a small impact on the adjuvant activity of these MPLA analogs. However, it is worth noting that the preliminary lethal toxicity investigation showed that 121 containing seven lipid chains was not much more toxic than MPLA derived from natural R595 lipid A, of which the main constituent is 3-O-deacylated MPLA 118 carrying six lipid chains. This result was in contrast to the previous finding that cleavage of the 3-O-acyl group could further reduce the toxicity of MPLA.93 Consequently, the synthetic lipid As with well-defined structures were helpful for gaining insights into precise SARs.
In another study, 127a-f (Scheme 13), analogs of 118 with the side chain lipids replaced with different fatty acids containing 4 to 14 carbon atoms, were designed and synthesized for systematic analysis of the biological functions of fatty acids.52 In their synthesis, Koenigs-Knorr method was used for glycosylation of 129a-f with 128a-f as glycosyl donors and AgOTf as promoter to afford disaccharides 130a-f in 70–80% yields. Then, the Troc group was removed with Zn and HOAc, which was followed by regioselective 2′-N-acylation with 131a-f and subsequent deprotection in two steps to yield the synthetic targets 127a-f.
Scheme 13.
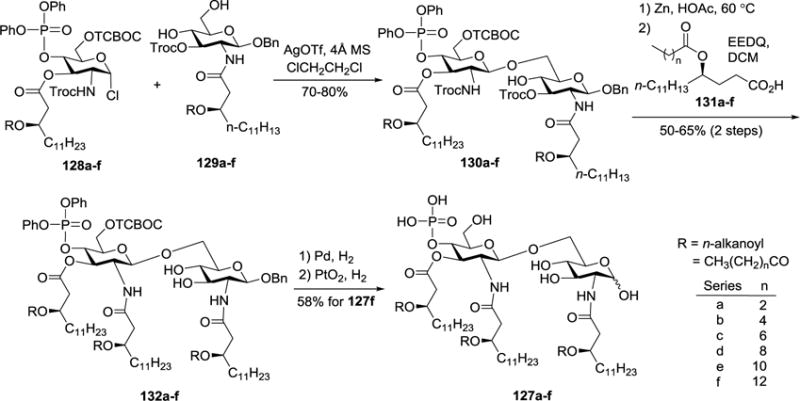
Synthesis of MPLAs 127a-f.
Biological assays of 127a-f revealed that the fatty acid chain length was a critical determinant of iNOS gene expression in activated mouse macrophages and proinflammatory cytokine (TNF-α and IL-1β) induction in human peripheral monocytes. For example, the ED50 of 127c-f to induce iNOS were 0.3, 0.05, 1, and 5 ng/mL, whereas the ED50 of 127a,b was more than 10 mg/mL, and the potencies of 127a-f to induce proinflammatory cytokines reached the maximum when n = 8 (127d). Thus, in contrast to the relatively small biological significance of the 3-O-substituent in MPLAs 119–121 carrying three large side chain lipids, the length of these side chains in 3-O-deacylated S. minnesota R595 MPLA 118 and related analogs 127a-f was of great importance for their immunostimulatory activities.52
Corsaro et al developed a semisynthetic method to prepare novel lipid A derivatives 133–140 (Figure 15).94 Based on the lipid A 2 purified from the fed-batch fermentation of E. coli K4, a series of selective chemical reactions including functionalization of the C-6′ hydroxyl group, modification of the lipid pattern, and methylation of the phosphate group gave novel lipid A derivatives 133–140. This facile methodology represented an alternative to lengthy total synthesis or extraction from bacterial sources. Preliminary in vitro immunological test showed that some of them, especially 136, were the promising, novel immunoadjuvant candidates. For example, 136 (20 μg/mL) exhibited a stronger potency in inducing TNF-α than that of the clinical S. minnesota R595 MPL with the TNF-α concentration were about 700 pg/mL and 40 pg/mL, respectively.
Figure 15.
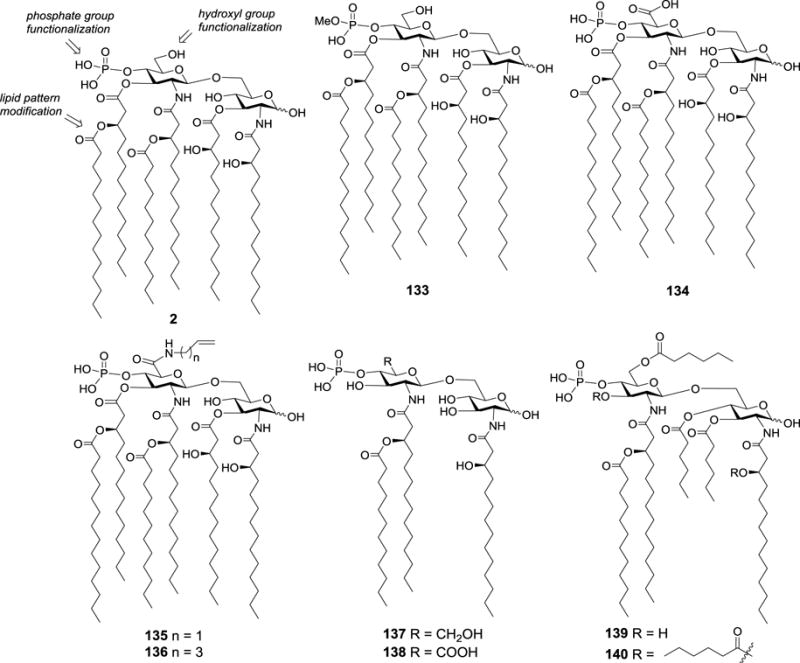
Structures of E. coli MPLA 2 and its semisynthetic derivatives 133–140.
Since MPLA has strong immunostimulatory and adjuvant activities but is almost nontoxic, it can be a useful molecular carrier for the development of fully synthetic vaccines that may also have self-adjuvanting properties. In this context, Guo and co-workers designed and synthesized a series of MPLA derivatives carrying functionalities to facilitate their further elaboration, such as coupling with antigens or other biomolecules, and applied them to biological studies. For example, they prepared a unique monophosphoryl derivative 146 of Neisseria meningitidis lipid A, which carried an amino group at the reducing end.95 This amino group could be selectively derivatized for coupling 146 or its deprotected form with other molecules. In the synthesis of 146 (Scheme 14), monosaccharide 142 with a 2-azidoethyl group attached to its reducing-end was used as a glycosyl acceptor, and the azido group could be selectively reduced later on to form the desirable free amino group. Glycosylation of 142 with 141 in the presence of NIS and AgOTf afforded the β-linked disaccharide backbone 143. Thereafter, the phthalyl and acetyl groups in 143 were simultaneously removed with hydrazine to pave the way for regioselective acylation of the amino groups and then acylation of the remaining 3-OH and 3′-OH groups to obtain 144. Regioselective opening of the benzylidene ring in 144 using NaBH3CN and HCl•Et2O was followed by phosphorylation of the resulting free hydroxyl group to give fully protected MPLA derivative 145. Selective reduction of its azido group was achieved with Zn and HOAc to yield 146, which was eventually coupled with tumor-associated carbohydrate antigens (TACAs) for the development of fully synthetic carbohydrate-based cancer vaccines.
Scheme 14.
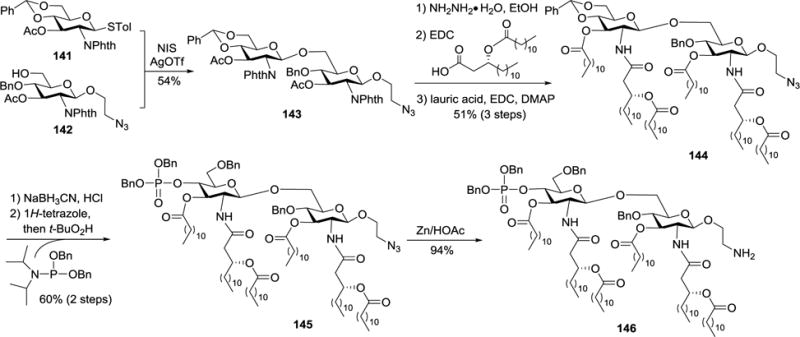
Synthesis of N. meningitidis MPLA derivative 146.
The above synthetic strategy was also used to prepare other N. meningitidis MPLA derivatives 147–150 (Figure 16) carrying symmetric 2-N/3-O- and 2′-N/3′-O-lipids of varied chain length and structure. These MPLA derivatives were employed to systematically study the SARs of MPLA, such as how the lipid chain length and structure may affect the immunostimulatory and adjuvant activity of MPLA, to discover optimized vaccine carriers and adjuvants.96
Figure 16.
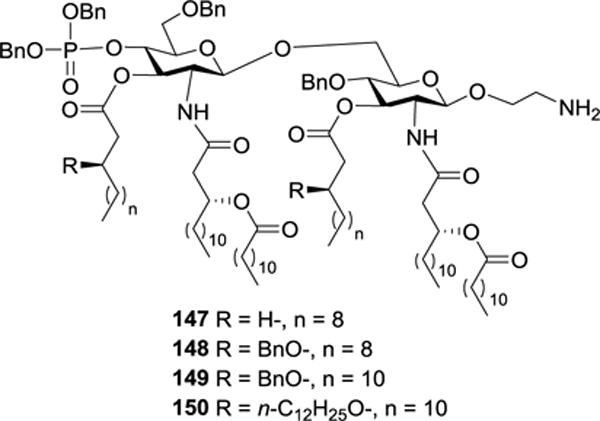
Structures of N. meningitidis MPLA derivatives 147–150.
For the synthesis of 2-N/3-O- and 2′-N/3′-O-asymmetrically acylated MPLA derivatives, such as that of E. coli lipid A 156, partially and fully lipidated monosaccharides 151 and 152 were used as building blocks (Scheme 15).46 The 2-N-position in 151 was protected with a Troc group to minimize steric hindrance of the donor and to assistant β-glycosylation through neighboring group participation as well. Moreover, the Troc group could be readily removed later on to facilitate the introduction of the desired lipid moiety. In the presence of NIS and AgOTf, glycosylation of 152 with 151 afforded the desired β-anomer 153 in a 75% yield. Subsequently, the benzylidene group in 153 was regioselectively opened with Et3SiH and BF3•Et2O to expose the 4′-OH group, which was phosphorylated upon reaction with dibenzyl phosphoramidite in the presence of 1H-tetrozole and then in situ oxidation with t-BuO2H to generate 154. The azide group in 154 was reduced with 1,3-dithiopropane, which was followed by the introduction of an alkyne group via reaction with 4-pentynoic acid under the influence of EDC. Finally, treatment of 155 with Zn/AcOH to remove the 2′-N-Troc group and reaction of the resulting amine with (R)-3-dodecanoyloxy-tetradecanoic acid in the presence of HATU and DIPEA afforded the target MPLA analog 156, which was eventually coupled with TACA via a click reaction for biological studies.
Scheme 15.
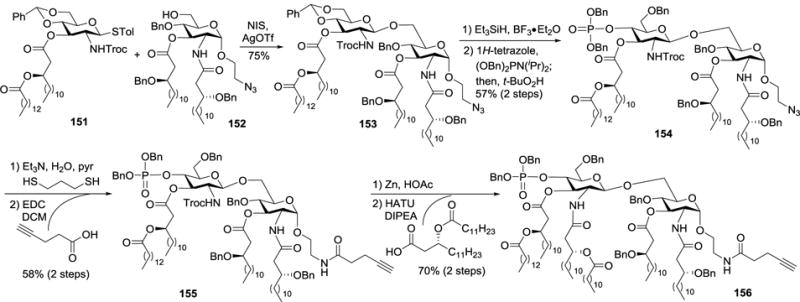
Synthesis of E. coli MPLA derivative 156.
In contrast, the lipid A unit of the Porphyromonas gingivalis LPS possessed antagonist activity of human TLR4.97,98 The absence of ester-linked phosphate at the 4′-O position and the presence of unusual branched fatty acids such as R-(3)-hydroxy-13-methyltetradecanic acid and R-(3)-hydroxy-15-methyltetradecanic acid make P. gingivalis lipid As unique. To explore their accurate SAR and explore MPLA derivatives with promising antagonistic activity, Boons and Ogawa groups synthesized P. gingivalis lipid As 157–158 and their tetra-acylated analogs 159–160 (Figure 17).99,100 Biological evaluation showed that both 159 and 160 were able to antagonize TNF-α production induced by E. coli LPS in human monocytic cells and that 159 was a significantly more potent antagonist than 160 (IC50 for 159 and 160 were 160 nM and 3.2 μM, respectively). Thus, the acylation pattern of 159 was of great importance for optimal activity.
Figure 17.
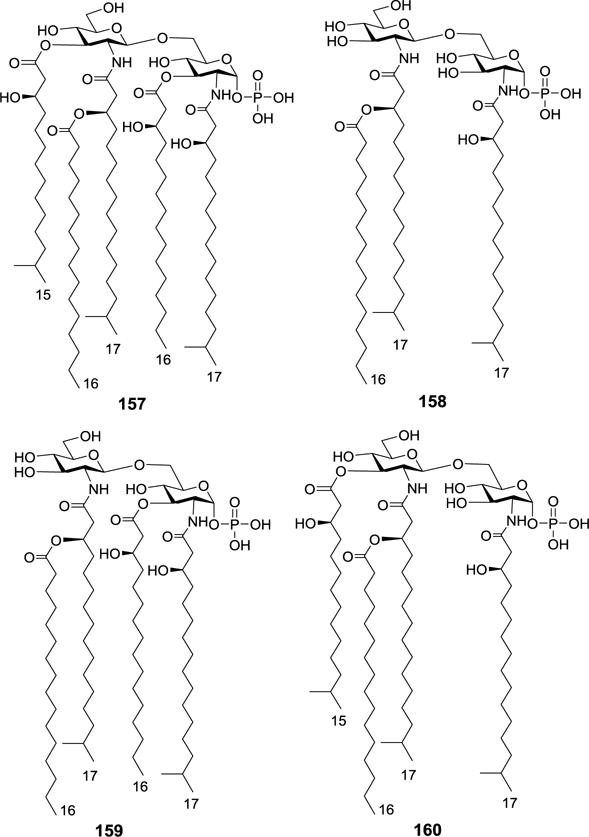
Structures of P. gingivalis MPLA 157, 158 and tetra-acylated derivatives 159, 160.
2.6 Synthesis of non-phosphorylated lipid As and related derivatives
Vandenplas et al. isolated a LPS from the symbiotic and nitrogen-fixing bacterial species Rhizobium sin-1, which could significantly inhibit the TNF-α release by human monocytes.101,102 The R. sin-1 LPS contained structurally unusual lipid As 161 and 162 (Figure 18), which are different from other lipid As in that they are devoid of both phosphate moieties and the reducing end glucosamine is replaced with a 2-aminogluconate residue. Interestingly, under mildly acidic conditions, 2-aminogluconate in 161 and 162 can easily convert into 2-aminogluconolactone (→ 163 and 164, Figure 18) with elimination of the fatty acid chain at the 3-O-position. In addition, the R. sin-1 lipid As contain a long lipid chain, 27-hydroxyoctacosanoic acid, which in turn can be esterified by β-hydroxybutyrate.
Figure 18.
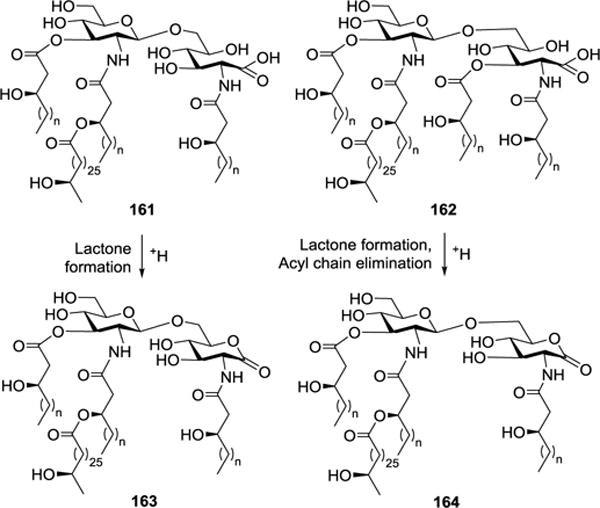
Structures of R. sin-1 lipid As 161–164.
To explore the intriguing structure and bioactivity of R. sin-1 lipid A, Boons and co-workers carried out extensive studies to establish synthetic strategies for these molecules and their analogs and investigate the SAR.41,42,44,49,103 For example, they employed an advanced intermediate 169 derived from selective glycosylation of 168 with 167 to obtain R. sin-1 lipid A derivatives 165 and 166 (Scheme 16).41 Compound 169 was useful to access various lipid A analogs, as its anomeric center, 2- and 2′-amino groups, and 3- and 3′-hydroxyl groups were orthogonally protected and could be regioselectively functionalized. Thus, treatment of 169 with ethylenediamine in refluxing n-butanol to deprotect the phthalimido and acetyl ester groups and regioselective 2′-N-acylation with (R)-3-octacosanoyloxy-hexadecanoic acid using DCC generated 170. Reduction of the azido group in 170 with 1,3-dithiopropane and then synchronous acylation of the free amino group and two hydroxyl groups of the resultant intermediate with (R)-3-benzyloxy-hexadecanoic acid, DCC, and DMAP afforded fully protected 171. Finally, NIS/TfOH-mediated hydrolysis of the thioglycoside in 171 and removal of the benzyl ethers and benzylidene acetal via Pd/C-catalyzed hydrogenolysis gave the target lipid A analog 165. En route to 166, PCC-mediated oxidation of the anomeric position after NIS/TfOH-mediated anomeric hydrolysis of 171 produced lactone 172, whereas oxidation reactions under the Moffatt (DMSO/Ac2O), Swern (DMSO/oxallyl chloride) or NMO/TPAP conditions led mainly to 1,2-oxazoline and elimination byproducts. Pd/C-catalyzed hydrogenolysis of 172 finally generated 166.
Scheme 16.
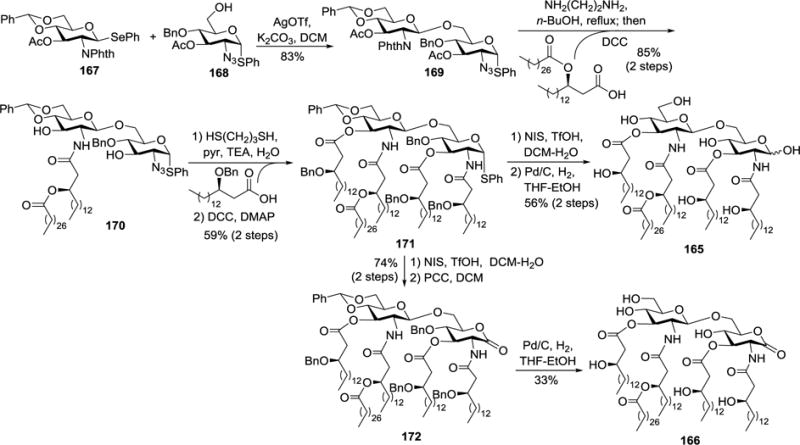
Synthesis of R. sin-1 lipid A derivatives 165 and 166.
Compounds 165 and 166 were structurally similar, but their reducing end anomeric carbon atoms had different oxidation states. Biological studies showed that, in contrast to E. coli LPS, both 165 and 166 lacked proinflammatory activities as indicated by their absence of abilities to induce TNF-α secretion or TNF-α mRNA expression. In addition, 166 was able to antagonize enteric E. coli LPS, but 165 was not, indicating that the gluconolactone moiety in R. sin-1 lipid A was important for its antagonistic property.
Based on the above SAR findings, Boons and co-workers synthesized two other putative R. sin-1 lipid A analogs 173 and 174 (Figure 19), which were devoid of 3-O-acylation, to further examine the biological effect of the 3-O-acyl group.42 It was shown that 173 and 174 not only lacked the proinflammatory activity but also antagonized E. coli LPS. However, its antagonistic activity was significantly less potent than that of 166.41 Thus, the β-hydroxyl fatty acyl group at the 3-O-position was important for the antagonistic activity of R. sin-1 lipid A. Moreover, R. sin-1 LPS, which consists of two Kdo units, an O-polysaccharide and a core oligosaccharide, exhibited 60 times stronger activity than that of synthetic R. sin-1 lipid A analog 166, indicating that the Kdo units contributed to the antagonistic effects of R. sin-1 LPS as well.
Figure 19.
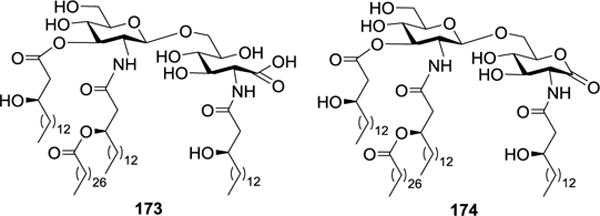
Structures of R. sin-1 lipid A analogs 173 and 174.
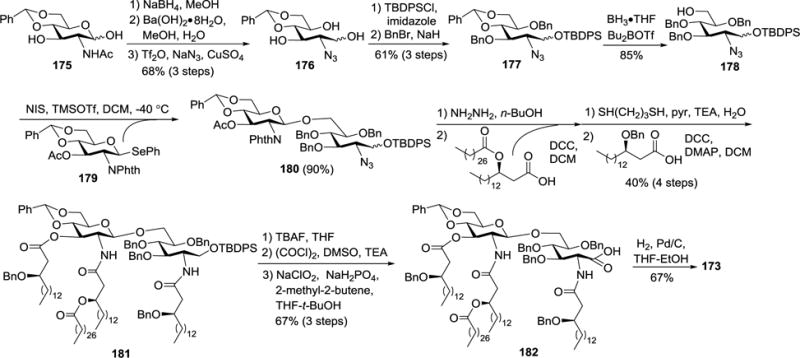
The synthesis of 173 (Scheme 17) was commenced with the reduction of the hemiacetal group in 175 with NaBH4, which was followed by Ba(OH)2-mediated hydrolysis of the acetamido group and swap of the amine for azide to give 176. Subsequently, the primary hydroxyl group in 176 was regioselectively silylated, and the remaining hydroxyl groups were benzylated to afford 177. Since the synthetic target 173 did not contain the 3-O-acyl group, this position was protected with a benzyl group. Regioselective ring-opening of the benzylidene group in 177 using BH3•THF and Bu2BOTf converted it to glycosyl acceptor 178. Glycosylation of 178 with 179 in the presence of NIS and TMSOTf gave β-linked disaccharide 180 stereoselectively as a result of the neighboring phthalimido group participation. Compound 180 was then consecutively deprotected and acylated. The phthalimido and 3′-O-acetyl groups were removed upon treatment with hydrazine hydrate in n-BuOH, which was followed by selective 2′-N-acylation. Thereafter, the azido group was reduced, and the resulting amine and the free 3′-OH group were concomitantly acylated in the presence of DCC and DMAP to yield 181. Compound 181 was transformed into carboxylic acid 182 through TBAF treatment to remove the TBDPS group and then a two-step oxidation procedure including Swern oxidation and in situ oxidation of the resulting aldehyde with NaClO2/NaH2PO4. Finally, global deprotection of 182 via Pd/C-catalyzed hydrogenolysis gave the target lipid A 173.
Scheme 17.
Synthesis of putative R. sin-1 lipid A 173.
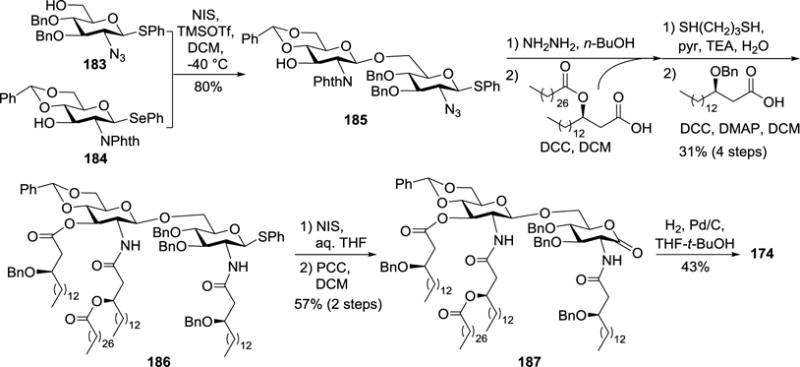
Compound 174 was synthesized by a similar strategy (Scheme 18). After selenoglycoside 184 was selectively activated with NIS/TMSOTf for the glycosylation of 183, the resultant 185 was consecutively deprotected and acylated at the 2′-N- and 2-N-/3′-O-positions, as discussed above, to afford 186. It is worth noting that NIS/TfOH-mediated hydrolysis of thioglycoside 186 afforded the synthetic target together with a 1,2-oxazoline byproduct, while the NIS/aq. THF system in the absence of TfOH gave a high yield of the target compound. In addition, NMR and MS analyses revealed that 173 and 174 were in equilibrium even under neutral conditions.
Scheme 18.
Synthesis of putative R. sin-1 lipid A 174.
The relationship between the unusual long lipid chain, i.e., the 27-hydroxyoctacosanoic group, at the 2′-N-position of R. sin-1 lipid A and its antagonistic property was also studied by comparing the natural product 189 and its derivatives 166 and 188 bearing different 2′-N-lipid chains (Figure 20).44 All of these compounds, as well as the natural R. sin-1 lipid A and LPS, were evaluated for their ability to antagonize TNF-α secretion by monocytic cells in the presence of E. coli LPS (10 ng/mL). Compounds 189 and 166 exhibited the similar and strong inhibition on TNF-α production with IC50 values of 9.1 and 7.5 μM, respectively. However, 188 only showed very weak inhibitory activity. Thus, while the hydroxyl group on the 27-hydroxyoctacosanoic residue did not account for its antagonistic properties, the length of the lipid chain played an important role in the activity. In addition, the R. sin-1 LPS was found to possess the strongest antagonized potency (IC50 = 6.5 nM), indicating that the KDO moiety of R. sin-1 LPS was also important for the biological activity, which was in accordance with the previous report.42
Figure 20.
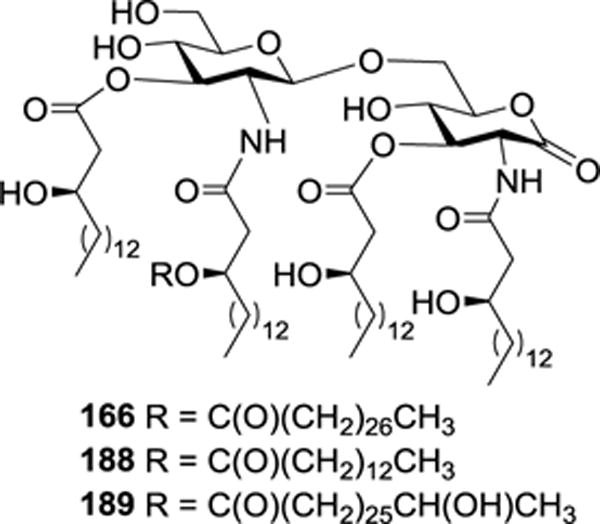
Structures of R. sin-1 lipid A 189 and its derivatives 166 and 188.
Synthesis of 188 was commenced with the glycosylation of the β-isomer of 168 with 184 (Scheme 19). At a low temperature, NIS and TfOH could selectively activate selenoglycoside 184 in the presence of thioglycoside acceptor, and the reaction gave a higher yield (84%) of disaccharide 190 when compared to that with AgOTf/K2CO3 as the promoters.104 Treatment of 190 with ethylenediamine to remove the acetyl and phthalimido groups was followed by regioselective acylation of the 2′-N-position with (R)-3-tetradecanoyloxy-hexadecanoic acid under the influence of DCC. Thereafter, the azido moiety was reduced to free amine, which was acylated together with the 3-OH and 3′-OH groups to afford 191. NIS/TfOH-promoted hydrolysis of the thiophenyl moiety in 191 generated 193, and the reaction also formed a byproduct, that is, 1,2-oxazoline 192, which could be converted into 193 after treatment with dibenzylphosphate. Finally, the anomeric hydroxyl group in 193 was oxidized with PCC, which was followed by global deprotection via Pd/C-catalyzed hydrogenolysis to yield the synthetic target 188.
Scheme 19.
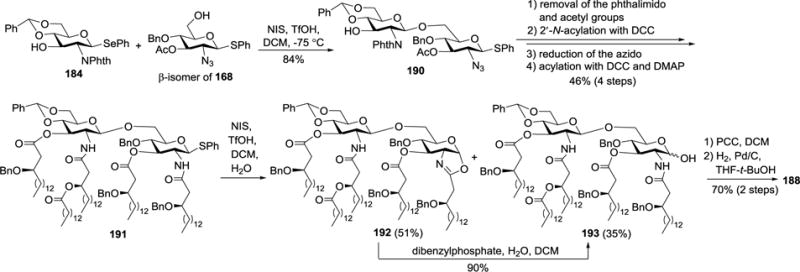
Synthesis of R. sin-1 lipid A derivative 188.
In the synthesis of 166 and 189 (Scheme 20), α-thioglycoside 194, instead of its β-isomer 190, was used as a key intermediate. By the similar deprotection and acylation procedures as shown in Scheme 19, 194 was readily converted into 195. One of the side chain hydroxyl groups of its lipids was selectively deprotected and acylated to give 196, which was subjected to NIS/TfOH-promoted hydrolysis of the thiophenyl moiety, oxidation of the resultant hemiacetal with PCC, and global deprotection via Pd/C-catalyzed hydrogenolysis to get the synthetic target 189. It was worth noting that hydrolysis of α-thioglycoside did not give the 1,2-oxazoline byproduct, possibly because the C-2 amide could not directly displace the anomeric leaving group, resulting in effective trapping of the generated oxa-carbenium ion with water to give the desired lactol.
Scheme 20.
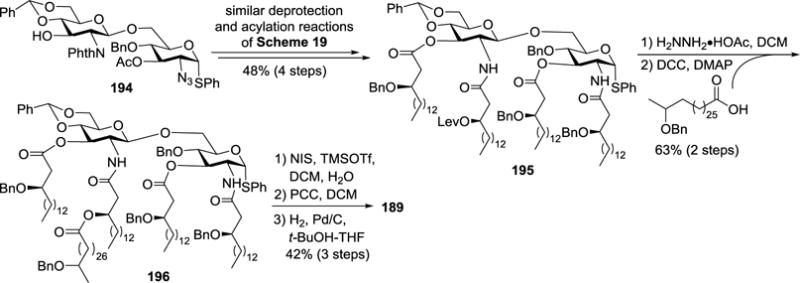
Synthesis of R. sin-1 lipid A 189.
In summary, the above synthetic strategies provided easy access to different R. sin-1 lipid A derivatives with well-defined structures, which are useful for understanding their SARs and action mechanisms at the molecular level. Importantly, the antagonistic properties of R. sin-1 lipid A derivatives and R. sin-1 LPS make them leading compounds for the design and development of therapeutic antagonists for endotoxin-related diseases.37,57
2.7 Synthesis of miscellaneous lipid A analogs
To obtain structurally simplified lipid A mimics, peptides were used to replace the disaccharide moiety of lipid A. For example, mono- and bis-O-phosphorylated Nδ-L-homoserinyl-D-ornithinol pseudodipeptides 197 and 198 with N-acylation were designed and synthesized to mimic E. coli lipid A OM-174105 (Scheme 21).106 Compounds 197 and 198 were demonstrated to have partial immunostimulatory or immunomodulating activities of OM-174. Importantly, they exhibited very low endotoxicity and pyrogenicity.
Scheme 21.
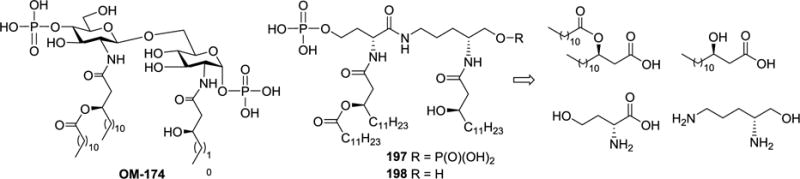
Structure of E. coli lipid A OM-174 and retrosynthesis of its analogs 197 and 198.
3. Synthesis of LPS and Kdo-lipid A derivatives
Lipid A is the artificial product produced by acidic hydrolysis of LPS, thus it is not exposed freely on the living bacterial cell surface. As depicted in Figure 1, the 6′-O-position of lipid A is covalently linked to a Kdo residue, which is in turn linked to the outer core oligosaccharide and O-antigen. Kdo is important for the LPS molecule as it not only serves as the linkage between lipid A and O-antigen but also plays a critical role in the bioactivity of LPS.41,42,107,108 As mentioned above,43 E. coli LPS that contains two Kdo units exhibited more potent cytokine-inducing activity than E. coli lipid A and its analogs. Furthermore, Meningococcal lipid A expressed by a bacterial strain that is defect of Kdo biosynthesis possessed considerably reduced activity compared to Kdo bearing Meningococcal lipooligosaccharide.109 However, these findings are still difficult to be more vigorously verified due to the difficulty to purify LPSs from bacteria, which results in heterogeneous and even contaminated products that may affect the observed immunostimulatory activity. Therefore, totally chemical synthesis of homogeneous and structurally well-defined LPS or Kdo-containing lipid A derivatives would be of great value for elucidating their precise SARs and for other biological studies.
3.1 Total synthesis of Re LPS
Re LPS 199 (Figure 21) was originally isolated from an E. coli mutant.110 Consisting of lipid A and two Kdo residues, it is known as the simplest LPS identified so far. In 2001, Kusumoto et al. reported the first total synthesis of Re LPS 199 in 23 steps and a 0.9% overall yield.53
Figure 21.
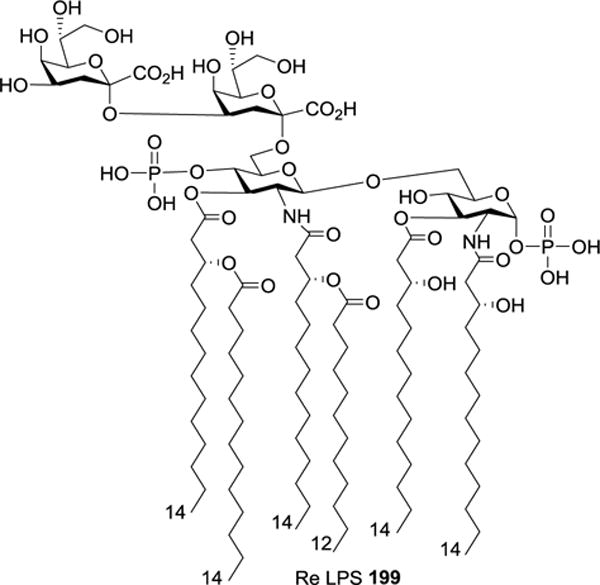
Structure of Re LPS 199.
As shown in Scheme 22, regioselective glycosylation of 201 with imidate 200 in the presence of polymer-supported Nafion-TMS111 afforded disaccharide 202 in a 73% yield. Acylation of the remaining free hydroxyl group in 202 was followed by successive cleavage of the 2′-N-Troc group, 2′-N-acylation, and removal of the benzylidene group with TFA in DCM-H2O to give tetraacylated 203. It is worth noting that the side chain hydroxyl groups on the lipids at the 2,3-N,O-positions in 203 were protected with the p-trifluoromethylbenzyl group, which was found to be resistant to oxidation but readily removable by hydrogenolysis.112
Scheme 22.
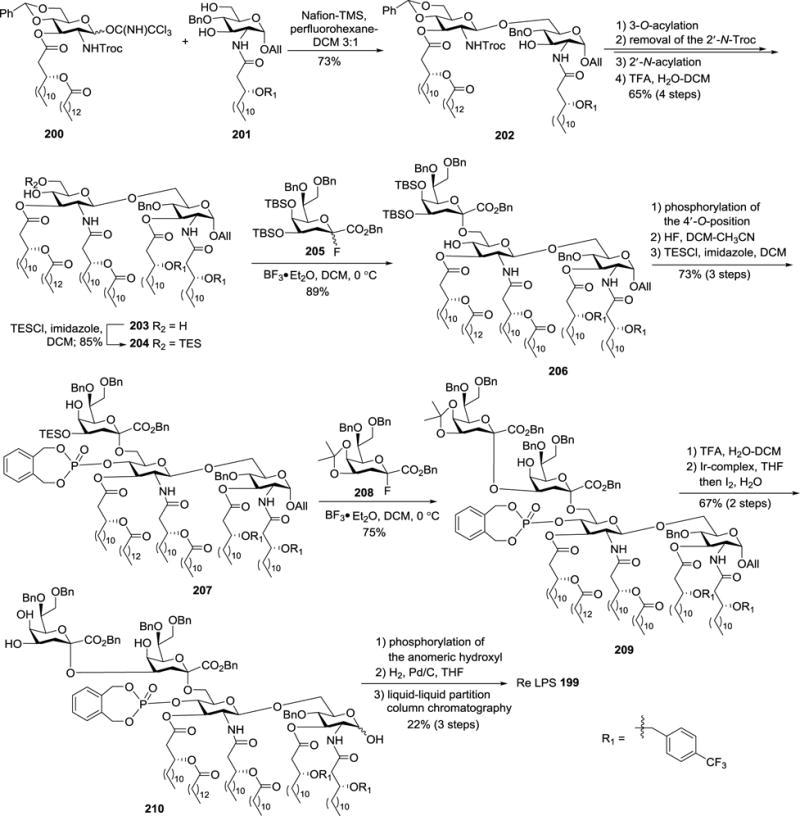
Total synthesis of Re LPS 199.
Model reactions showed that TBS-protected Kdo fluoride 205 and triethylsilyl (TES)-protected glycosyl acceptors are the best coupling partners to provide Kdo glycosides in high yields and stereoselectivity. Consequently, 203 was converted to 6′-O-TES ether 204, which was glycosylated with TBS-protected Kdo donor 205 under the promotion of BF3•Et2O to generate an 89% yield of the desired α-2,6-linked disaccharide 206 with high regio- and stereoselectivity. Phosphorylation of the 4′-hydroxyl group in 206 was followed by desilylation and regioselective triethylsilylation to give trisaccharide 207, which was ready for introducing the second Kdo unit. Glycosylation of 207 was achieved with the less bulky isopropylidene-protected Kdo fluoride donor α-208 to afford tetrasaccharide 209 in a 75% yield. It is worth noting that glycosylation of 207 with 205 resulted in a low yield (26%) of the desired product, probably because of the steric hindrance. Removal of the isopropylidene group in 209 with TFA/DCM-H2O was followed by Ir-complex mediated cleavage of the anomeric All group to afford the hemiacetal intermediate 210. Finally, regioselective phosphorylation of the anomeric hydroxyl group, catalytic hydrogenolysis to remove all of the benzyl-type of protecting groups, and purification of the product using liquid-liquid partition chromatography on a Sephadex LH-20 column produced the target molecule 199. Consecutive cytokine-inducing tests showed that natural Re LPS was slightly more potent than synthetic 199. It was speculated that natural Re LPS contained some potent contaminants, most probably molecules bearing longer saccharide residues.
It was the first chemical synthesis of a LPS molecule. This work established the strategy for α-glycosylation using Kdo fluorides bearing bulky protecting groups, such as isopropylidene and TBS groups, at the 4,5-O-positions, which prevented the undesired β-side attack. Nevertheless, stepwise installation of Kdo units was not especially efficient, which restricted parallel preparation of various LPS analogs and further investigation of their SAR.
3.2 Synthesis of H. pylori Kdo-lipid A conjugates
Chemical synthesis of Kdo-lipid A conjugates bearing one Kdo unit, especially those related to Helicobacter pylori LPS, was also accomplished.107,113–117 Compared to enterobacterial LPS, H. pylori LPS has significantly lower endotoxicity.118,119 In addition, the lipid A component of H. pylori LPS is greatly different from the enterobacterial lipid A in that: (1) it has fewer and longer fatty acid chains; (2) it is devoid of the 4′-O-phosphate group; and (3) its 1-O-phosphate group is occasionally modified with an ethanolamine group.120,121
In 2007, Fukase et al. described the synthesis of H. pylori lipid A 211 and its Kdo conjugate 212 (Figure 22).107 As shown in Scheme 23, acylated 214 was glycosylated with N-Troc-protected imidate 213 at −78 °C, under the promotion of TMSOTf, to give disaccharide 215 in an 80% yield. The 2′-N-Troc group in 215 was removed upon treatment with Zn-Cu/HOAc, which was followed by acylation of the resulting amine group to generate 216. Thereafter, the 6′-O-Alloc group was swapped for a TES group after sequential removal of the Alloc group with Pd(PPh3)4 and then reaction with TESCl in the presence of imidazole to produce 217.
Figure 22.
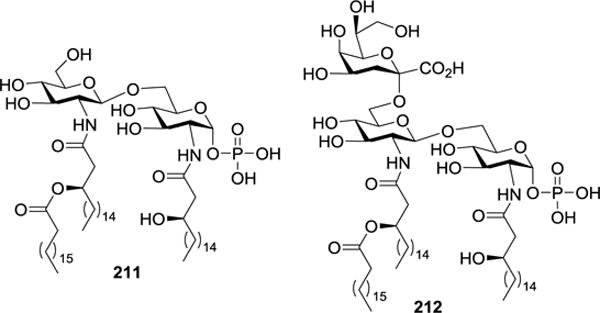
Structures of H. pylori lipid A 211 and its Kdo conjugate 212.
Scheme 23.
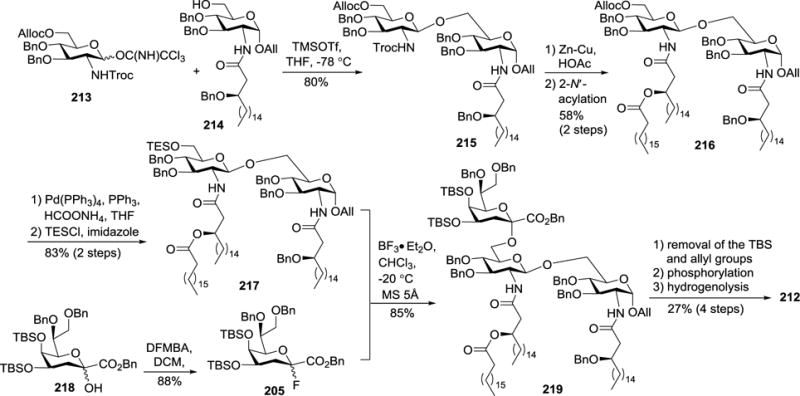
Synthesis of H. pylori Kdo-lipid A conjugate 212.
Consecutively, 217 was glycosylated with 20553 using BF3•Et2O as the promoter to afford 219 in an 85% yield. Compound 205 was obtained from hemiketal 218 in a high yield on fluorination with N,N-diethyl-α,α-difluoro-(m-methylbenzyl)amine (DFMBA) at a low temperature,122 under which condition the formation of glycal byproduct was suppressed. For the glycosylation reaction between 217 and 205, the MS 5Å was proved to be more effective than MS 4Å, probably because the calcium ions in MS 5Å could trap fluoride anions generated from the reaction, thus promoting the glycosylation. Subsequently, the TBS and All groups in 219 were selectively removed with HF and Ir-complex, respectively, which was followed by phosphorylation of the anomeric hydroxyl group with tetrabenzyl pyrophosphate and, finally, debenzylation via catalytic hydrogenolysis to produce the target Kdo-lipid A conjugate 212.
The immunostimulatory activity of 211 and 212 was tested by measuring their influence on IL-6 secretion from human whole blood cells (HWBC).123 It was demonstrated that neither 211 nor 212 induced IL-6 secretion; instead, both competitively inhibited IL-6 secretion induced by E. coli LPS, verifying that 211 and 212 were TLR-4 antagonists. Moreover, 212 showed a 10-fold increase in inhibitory activity than 211, demonstrating the biological significance of the Kdo unit. In combination with the previous finding that the ethanolamine-containing H. pylori lipid A 220 (Figure 23) showed weak immunostimulatory activity,112,124 it was concluded that the number of anionic charges were of importance for the bioactivity of lipid A.
Figure 23.
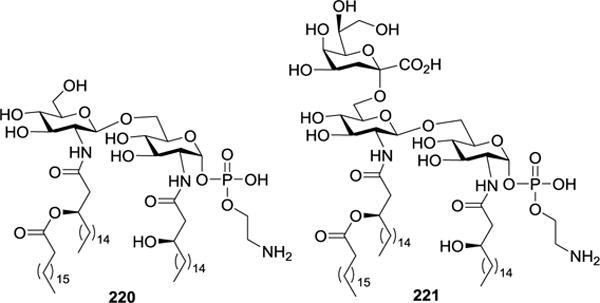
Structures of the ethanolamine-containing H. pylori lipid A 220 and its Kdo conjugate 221.
Fukase et al. also synthesized H. pylori lipid A-Kdo 221 (Figure 23) bearing an aminoethanol phosphate through a divergent strategy.114 As shown in Scheme 24, the 4′,6′-O-benzylidene group in 222 was regioselectively opened with Me2NH•BH3 and BF3•Et2O to obtain 223. Previous study showed that the acid-labile TBS or isopropylidene group in the Kdo donor could be occasionally cleaved when a large excess of Lewis acid was employed to activate Kdo fluoride donors, which decreased the glycosylation reaction stereoselectivity.53 Based on this finding, they probed a new synthetic method using Kdo N-phenyltrifluoroacetimidate 224 as a donor for the coupling reaction with disaccharide 223.113,115 Although in α-promoting solvent cyclopentyl methyl ether (CPME) the reaction under the promotion of TBSOTf afforded the desired trisaccharide 225 in a good yield (70%) and stereoselectivity, a large excess of donor 224 (5 equiv.) had to be utilized because the reaction produced a significant amount of glycal byproduct 226.
Scheme 24.
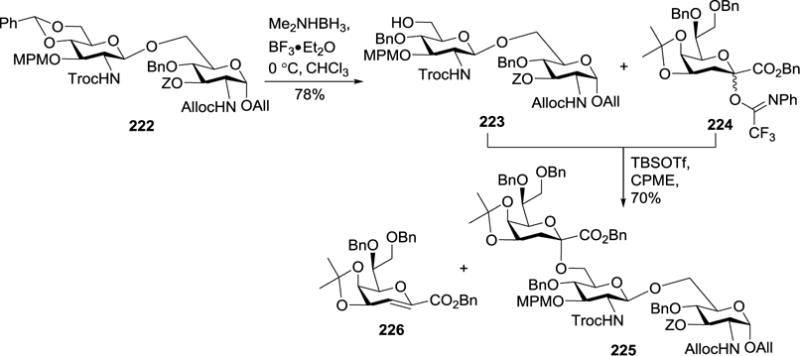
Glycosylation of 223 and 224 in the presence of Lewis acid.
The low efficiency of the above glycosylation reaction might be ascribed to the difficulties in temperature control since imidate 224 was highly reactive and the reaction produced substantial heat. To address the issue, a microfluidic glycosylation protocol was developed,125 in which the CPME solution of donor 224 (1.5 equiv.) and acceptor 223 (1 equiv.) was mixed with the TBSOTf (1 equiv.) solution in a microreactor at a flow rate of 0.5 mL/min (Scheme 25). Then, the desired trisaccharide 225 was formed in a 72% yield, with the glycal byproduct suppressed. In addition, the protecting groups were intact when relatively high concentration of Lewis acid was used. This represented some significant improvement over the previous glycosylation methods.53 After the trisaccharide backbone 225 was obtained, its 2′-N-Troc group was removed with Zn-Cu/HOAc, followed by acylation of the resultant amino group with acyloxycarboxylic acid in the presence of 2-methyl-6-nitrobenzoic anhydride (MNBA) and N,N-diisopropylethylamine (DIPEA). Thereafter, the 2-N-position was deprotected with Pd(PPh3)4 and acylated with benzyloxycarboxylic acid in the presence of HATU and Et3N to produce 227. Its isopropylidene group was then swapped with a benzylidene group, as the isopropylidene group was relatively unstable under the condition of 1-O-phosphorylation. Ir-complex mediated removal of 1-O-All group in 228 and phosphorylation of the exposed anomeric hydroxyl group using phosphoramidate were followed by catalytic hydrogenolysis to afford the target molecule 221.
Scheme 25.
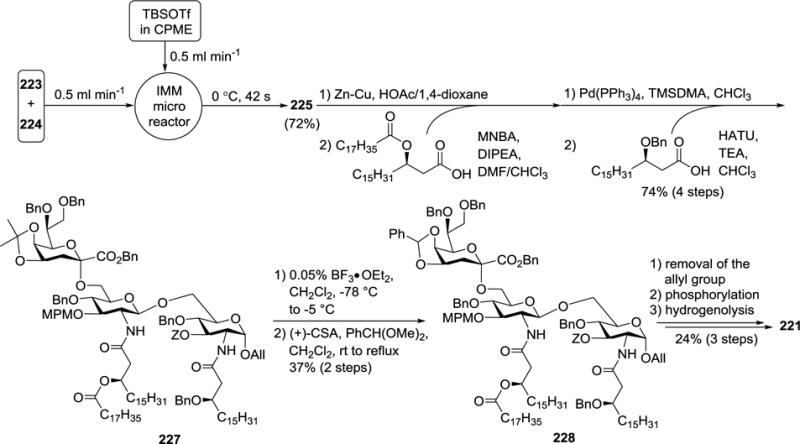
Synthesis of H. pylori lipid A-Kdo conjugate 221.
The immunomodulatory activities of 211, 212, 220 and 221 were evaluated via the analysis of their impacts on cytokine production, using E. coli strain O111:B4 LPS as a positive control. The results showed that 220 exhibited weak stimulating effects on IL-1β, IL-6, and IL-8 release. On the other hand, 211, 212, and 221 possessed inhibitory activities toward cytokine induction by E. coli LPS. Thus, elongation of 220 with a Kdo unit reversed its bioactivity, complementing previous findings that the Kdo residue enhanced the activities of other lipid A derivatives.108 More detailed SARs of these compounds were summarized in a review.116
In summary, to address the challenge of efficient and stereoselective glycosylation with Kdo in the synthesis of LPS or Kdo-lipid A conjugates, glycosylation strategies using Kdo fluoride and imidate donors were exploited in combination with microfluidic reaction conditions. Studies on the synthetic compounds contributed to understanding the SARs of LPS or Kdo-lipid A conjugates at molecular levels. Nevertheless, Kdo installation to lipid A remains a great synthetic challenge, especially in the synthesis of complex LPS analogs containing more Kdo units and O-antigens. As a result, more efficient glycosylation and synthetic strategies for LPS are in high demand.
In recent years, great advancements have been made towards understanding the biosynthetic processes of LPSs,30,126 such as the characterization of various glycosyltransferases, polymerases, and other enzymes involved in and controlling O-antigen and LPS assembly.127–129 These studies have significantly enhanced the potential of using enzymatic procedures to achieve some key steps of LPS synthesis in vitro. It is therefore anticipated that chemoenzymatic synthesis of structurally defined LPSs and their analogs will be an attractive and promising research topic.
4. Application of MPLA to vaccine development
TACAs are useful targets for the development of therapeutic cancer vaccines, but TACAs are typically poorly immunogenic and T cell independent antigens. To make them more immunogenic and T cell dependent antigens, they have to be covalently linked to immunologically active carrier molecules to form conjugate vaccine. In recent years, fully synthetic TACA-based conjugate cancer vaccines have become a hot topic, because they possess a number of advantages, such as defined structures, reproducible physical, chemical and biological properties, and promising immunologic activity.130–133 For the development of fully synthetic cancer vaccine, MPLA can be a particularly useful carrier molecule. First, MPLA is an extremely strong immunostimulator. Second, owing to its bacterial origin, MPLA can function as a “danger signal” to the immune system to enhance the immune responses to TACAs covalently linked to it.134 Third, as mentioned, the endotoxicity of lipid A could be dramatically reduced after removal of its anomeric phosphate group, whereas its beneficial immunostimulatory activity was unaffected.2 In reality, MPLA has been approved for clinical use as a vaccine adjuvant and immunostimulator.87
To probe the potential of MPLA as a carrier molecule for the development of conjugate cancer vaccines, Guo et al. designed and synthesized a monophosphoryl derivative 146 (Scheme 14) of N. meningitidis lipid A, which had lipid chains symmetrically distributed on the two sugar units. In 146, the reducing end phosphate of lipid A was replaced with an aminoalkyl group, to which TACAs could be linked.95 For example, as depicted in Scheme 26, after acylation of the amino group in 146 with succinic anhydride and then esterification, 146 was converted into active ester 229 to enable the coupling of MPLA with a modified form of GM3, NPhAcGM3 230. Finally, the resultant conjugate 231 was deprotected to afford conjugate vaccine 232.
Scheme 26.
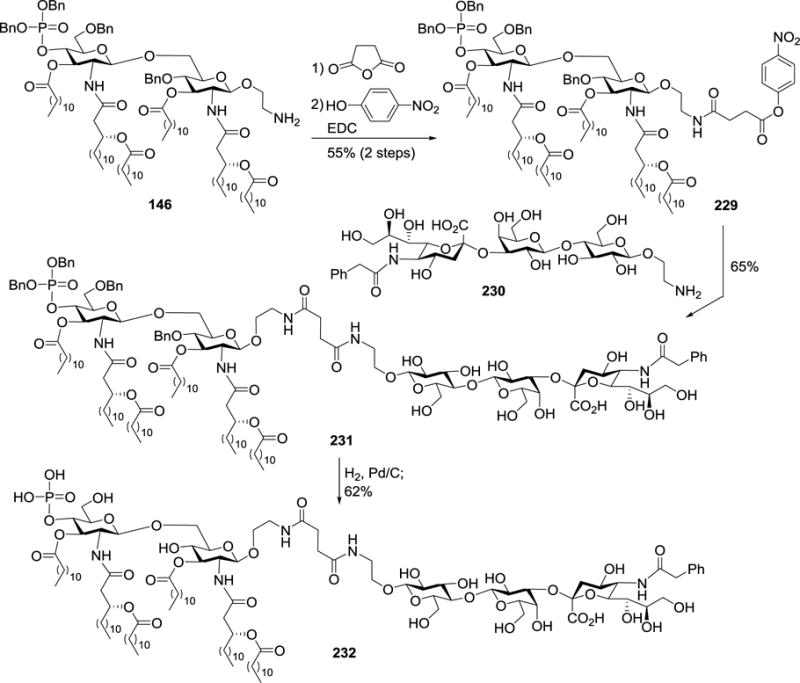
Synthesis of N. meningitidis MPLA–NPhAcGM3 conjugate 232.
Immunological studies of the MPLA–NPhAcGM3 conjugate 232 in mice revealed that it alone, namely without the use of an external adjuvant, elicited robust IgG antibody responses. The results have demonstrated that MPLA could act not only as a carrier molecule to significantly improve the immunogenicity of TACAs but also as a built-in adjuvant to form self-adjuvanting vaccines.135,136 Therefore, MPLA was identified as a highly promising carrier for the design and development of fully synthetic glycoconjugate cancer vaccines.
Guo and coworkers have also developed a convergent synthetic strategy, as shown in Scheme 15, for a monophosphorylated E. coli lipid A derivative 156, which carried lipids asymmetrically on the two sugar units.46 Compound 156 was smoothly coupled with an NPhAcGM3 derivative 233 that carried an azido group through click reaction in the presence of CuI/DIPEA to create the desired MPLA-GM3NPhAc conjugate 234 (Scheme 27).
Scheme 27.
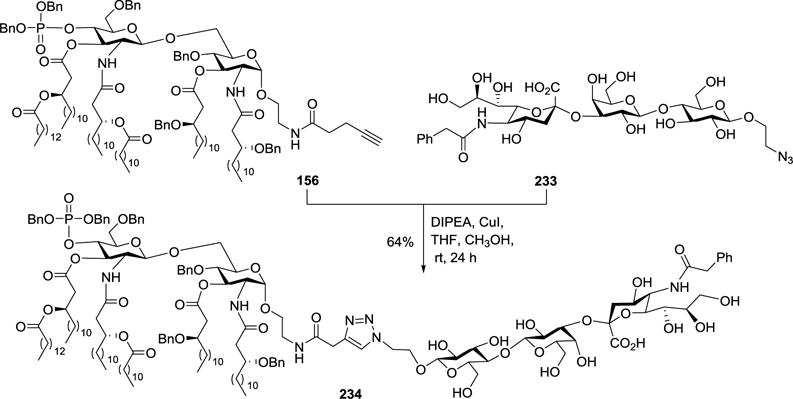
Synthesis of E. coli MPLA–NPhAcGM3 conjugate 234.
To optimize MPLA as a vaccine carrier, Guo and coworkers prepared several N. meningitidis MPLA derivatives 147–150 (Figure 16), which contained different lipids, and studied their SAR in the form of NPhAcsTn conjugates 240–243.96 Following the above mentioned synthetic strategy for 229, 147–150 were converted into active esters 235–238, which were coupled with NPhAcsTn 239 (Scheme 28). The resultant conjugates were deprotected via Pd/C-catalyzed hydrogenolysis to afford the desired MPLA-NPhAcsTn conjugates 240–243.
Scheme 28.
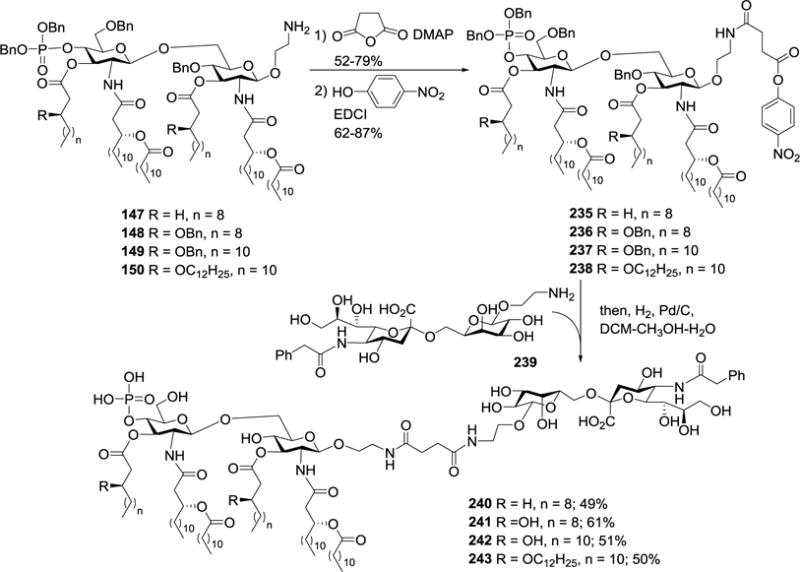
Synthesis of N. meningitidis MPLA–NPhAcsTn conjugates 240–243.
Immunological studies of 240–243 revealed that all of them provoked robust antigen-specific immune responses in mice without the use of an external adjuvant. Moreover, they stimulated high titers of IgG3 and IgG1 antibodies, indicating T cell dependent immune responses.137,138 Despite that 240–243 elicited similar patterns of immune responses, they did show significant difference in the immunological activity. For example, 241 provoked much higher and consistent titers of both total and IgG3 antibodies than 240, 242, and 243. Structurally, 241 contained the monophosphoryl form of natural N. meningitidis (H44/76 strain) lipid A.139 In 240, the two free hydroxyl groups on the lipid chains were removed; 242 and 243 were different from 241 in the lipid chain length and number. Thus, the lipid chain length and number and the free hydroxyl groups on the lipid chains in MPLA exhibited a significant impact on its immunological property.
After an optimized MPLA as vaccine carrier was established, it was employed to couple with globo H, a TACA expressed by several tumors, to formulate MPLA and globo H-based anticancer vaccines.140 As shown in Scheme 29, coupling of the active ester 236 of MPLA with a globo H derivative 244141 followed by Pd/C-catalyzed hydrogenolysis generated the target MPLA-globo H conjugate 245. It was evaluated in mouse and compared with the keyhole limpet hemocyanin (KLH) conjugate of globo H 246, the positive control, which is at Phase III clinical trial for the treatment of colon cancer. It was discovered that MPLA-globo H conjugate 245 elicited much faster and stronger immune responses than that of the KLH conjugate 246, in terms of both total and IgG antibodies. It was demonstrated that both 245 and 246 induced the similar patterns of immune responses. These results suggested the great promise of conjugate 245 as a therapeutic cancer vaccine.
Scheme 29.
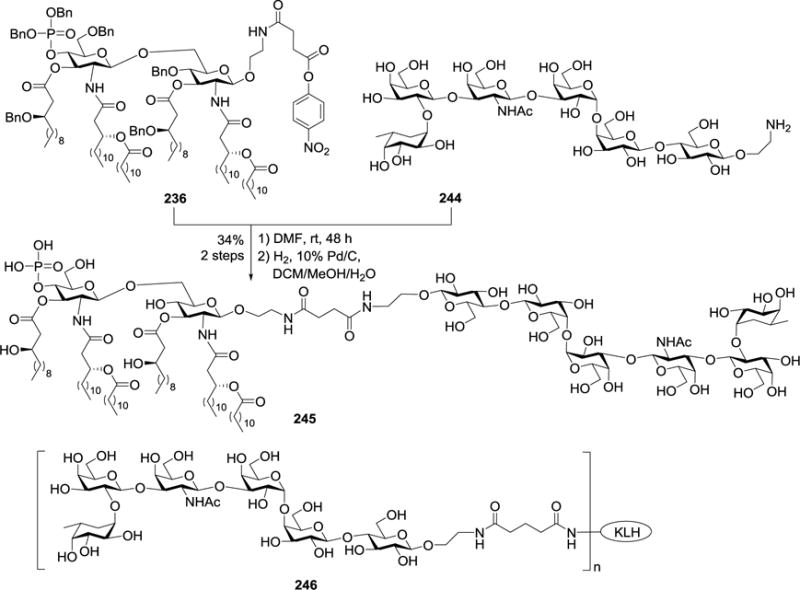
Synthesis of N. meningitidis MPLA-globo H conjugate 245 and the structure of KLH-globo H conjugate 246.
Recently, MPLA was also employed as a carrier molecule in the development of fully synthetic antibacterial glycoconjugate vaccines.142 For this purpose, a series of α-2,9-linked oligosialic acids, which were short analogs of α-2,9-polysialic acid – a unique and important capsular polysaccharide antigen of serotype C N. meningitidis, were synthesized143 and coupled with MPLA by the same strategy described above. Immunological studies of the resultant conjugates 247–251 (Figure 24) in mouse provided detailed SARs about the antigens and the carrier molecule. It was revealed that all of the conjugates alone elicited robust T cell-dependent immunities, suggesting their self-adjuvanting properties. It was also shown that their antisera had strong and specific binding to α-2,9-polysialic acid and meningococcal cells, indicating the ability of the elicited antibodies to selectively target the bacteria. It was eventually demonstrated that the antisera could mediate strong bactericidal activities. It was thus concluded that the MPLA conjugates of α-2,9-oligosialic acids, especially tri- and tetrasialic acids, are promising self-adjuvanting anti-group C meningitis vaccines worthy further investigation.
Figure 24.
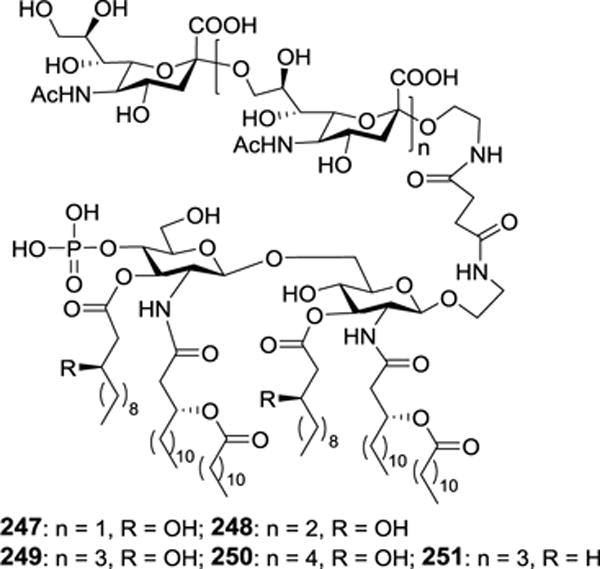
Structures of MPLA-oligosialic acid conjugates 247–251 explored as fully synthetic anti-group C meningitis vaccines.
Introduction of unique functional groups, such as an azido or alkyne group, to the reducing end of MPLA can provide the flexibility for its regioselective coupling with other molecules, such as carbohydrates, peptides, and proteins to quickly generate complex multicomponent conjugates in a convergent manner. This should be very helpful for the broad applications of MPLA. For example, current results have proved that MPLA can be a useful carrier molecule in the development of fully synthetic self-adjuvanting anticancer and antibacterial glycoconjugate vaccines.
5. Conclusion
Since the first chemical synthesis of a lipid A derivative reported by Shiba et al. in 1984, great progresses have been made in the synthesis and biological studies of lipid A and related molecules. As a result, a series of convergent and efficient strategies are currently available for the preparation of both natural lipid As and their artificial analogs, such as various lipid A mimics and derivatives that contain different lipids, truncated carbohydrate chains and no or only one phosphate group or have one or both of the two phosphate groups replaced with more stable functionalities (Table 1). Chemical syntheses of simple LPS analogs were also accomplished. These synthetic studies have facilitated access to structurally defined lipid As and their rationally designed derivatives or analogs, thus enabling thorough and systematic examination of the structures, functions, and SARs of lipid As at molecular levels. This has led to not only in-depth understanding of the functional mechanisms of lipid A and LPS but also the discovery of new, lipid A-based therapeutics. For example, several lipid A analogs have found clinical applications as endotoxin antagonists for the treatment of severe sepsis, and recently MPLA has been approved for human use as a vaccine adjuvant. MPLA has also been extensively inspected as a promising carrier molecule for the development of fully synthetic self-adjuvanting glycoconjugate vaccines.
Table 1.
Natural lipid As and various analogs and derivatives of lipid As achieved by chemical synthesis
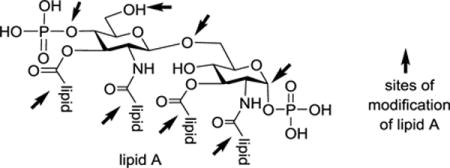
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Compound | Bacterial origin | Group that realized the synthesis | Year | Reference | Key structural and synthetic features | Biological outcomes |
| Natural lipid As and lipid A derivatives bearing saturated lipids and both phosphate groups | ||||||
| 1 | E. coli | Boons | 2007 | 43 | Natural lipid A with six acyl chains; synthesis based on an orthogonally protected disaccharide | Lower potency to induce cytokine production than E. coli LPS, suggesting the role of Kdo residues in LPS |
| 10 | S. typhimurium | Boons | 2007 | 43 | Natural lipid A with seven acyl chains. | Significantly lower potency to induce cytokine production than 1 |
| 11 | E. coli | Boons | 2007 | 43 | A lipid A derivative with shortened lipids | Higher potency to induce cytokine production than 1 |
| 12 | S. typhimurium | Boons | 2007 | 43 | A lipid A derivative with shortened lipids | Higher potency to induce cytokine production than 10 |
| 13 | S. typhimurium | Boons | 2007 | 43 | A lipid A derivative devoid of the anomeric phosphate group | Lower potency to induce cytokine production than 12 |
|
|
||||||
| 1 and 10–13 were synthesized by a similar strategy; their difference in biological activities indicated the importance of the number and length of the fatty acid lipid chains in lipid A | ||||||
|
|
||||||
| 26 | Ru. gelatinosus | Fukase | 2008 | 45 | Natural lipid A with six acyl chains; synthesis by using lapidated monosaccharide blocks | Potent ability to antagonize LPS endotoxic activity. |
| 27, 28 | C. trachomatis | Kosma | 2004 | 54 | C. trachomatis lipid A with tetra- or pentaacyl lipid chains. | |
|
| ||||||
| Natural lipid As and lipid A derivatives bearing unsaturated lipids | ||||||
| 34a,b | R. sphaeroides | Christ | 1994 | 37, 55, 56 | A derivative of natural lipid A; use of Alloc and All groups to protect the hydroxyl and phosphate groups | Devoid of LPS agonistic properties; strong in vitro antagonistic activity; suppress TNF-α production induced by LPS |
|
40 (E5531) |
R. sphaeroides | Yamatsu | 1995 | 57–59 | A derivative of natural lipid A with 3,3′-O-acyl groups substituted for hydrolytically stable ether linkages and C-6′-hydroxyl group blocked as methyl ether | Strong antagonist of cytokine release of induced by LPSs; completely devoid of LPS agonistic activity; protecting BCG-primed mice from LPS-induced lethality and E. coli infection-caused death. |
|
41 (E5564) |
R. sphaeroides | Christ | 2002 | 60–62 | Second generation of the above lipid derivatives | Excellent antagonist activity; clinical use in the trade name of Eritoran for the treatment of severe sepsis |
|
| ||||||
| Unnatural lipid A derivatives bearing two phosphate groups | ||||||
| 42–44 | artificial | Savage | 2003 | 40 | Containing four lipid chains with incrementally shortened length | Different solubility in water; used to study the interaction of lipid A with polymyxin B |
| 54–56 | E. coli | Kusama | 1990 | 69 | Lipid A derivatives containing anomeric α/β-phosphonooxyethyl group instead of labile α-glycosylphosphate. | α-glycosides had comparable antitumor activity as natural lipid A; β-glycoside was less active |
| 64, 65 | E. coli | Kusama | 1991 | 74 | Derivatives of lipid A biosynthetic precursor; containing anomeric α-phosphonooxyethyl | The first generation of potent antitumor lipid A derivatives with enhanced stability and reduced toxicity. |
| 66, 67 | E. coli | Fukase | 2001 | 75, 76 | Lipid A derivatives with tritium-labeled α-phosphonooxyethyl | Useful for the study of the interactions between lipid A and their acceptors |
|
| ||||||
| Lipid A analogs carrying an anomeric carboxylic acid | ||||||
| 74a–c, 75a–c | E. coli | Shiozaki | 2000 | 79, 80 | Lipid A derivatives with a carboxylic acid moiety in place of the glycosyl phosphate; 74a–c and 75a–c had six and four lipid chains; different 6′-O-substituents | 74a–c was a LPS agonist but 75a–c was a LPS antagonist, indicating the importance of the number of lipid chains; the carboxyl group and the 6′-O-substituent did not have a significant impact |
| 92a–c, 93a–c, 94a–c | artificial | Shiozaki | 2001 | 81 | Anomeric carboxylic acid; unnatural 3,3′-O-ether linkage; different 6′-C-substituents; 92 and 94 had four lipid chains; 93 had six | Improved stability; 92 and 94 were strong LPS antagonists; 93b was inactive; 93a,c were moderate LPS agonists; ether linkage and anomeric carboxylic acid did not have a major impact |
| 104a–c | artificial | Shiozaki | 2003 | 51 | Anomeric carboxymethyl group and 3,3′-O-ether linkages. | Improved stability; LPS antagonists; ether linkage and anomeric carboxylic acid did not have a major impact |
| 111a,b–116a,b | artificial | Fukase | 2006 | 84 | Monosaccharide analogs of lipid A with aspartic acid or phosphoserine in place of non-reducing end phosphorylated glucosamine unit | Most compounds except 115a,b showed antagonistic activity |
|
| ||||||
| Monophosphoryl lipid A and related derivatives | ||||||
| 119–121 | S. minnesota | Jiang | 2007 | 50 | S. minnesota R595 MPLA derivatives; different substituents at the 3-O-position | All were strong adjuvants; 121 was not more toxic than natural R595 MPLA 118 that devoid of 3-O-acyl chain |
| 127a-f | S. minnesota | Johnson | 1999 | 52 | S. minnesota R595 MPLA derivatives with variant fatty acids | Fatty acid chain length was important for inducing proinflammatory cytokines |
| 133–140 | E. coli K4 | Corsaro | 2016 | 94 | Semi-synthetic lipid A analogs derived from selective modification of the C-6′-OH, lipid pattern and phosphate | Some of them, especially 136, showed promising immunoadjuvant activity |
| 146 | N. meningitidis | Guo | 2009 | 95 | MPLA derivative with an amino group at the reducing end, readied for coupling with other carbohydrate antigens | Useful as a vaccine carrier for the development of self-adjuvant conjugate vaccines |
| 147–150 | N. meningitidis | Guo | 2014 | 96 | MPLA derivative with varied acyl chains with an amino group at the reducing end. | Used to discover new and optimized vaccine carriers and adjuvants. |
| 156 | E. coli | Guo | 2010 | 46 | MPLA derivative with an alkyne group at the reducing end for click reaction | Useful as a vaccine carrier for the development of self-adjuvant conjugate vaccines |
| 157–160 | P. gingivalis | Ogawa, Boons | 2007, 2008 | 99, 100 | Tetra-acylated MPLAs with branched lipid chains and devoid of 4′-O-phosphate | 159 and 160 showed potent antagonist activity, which was determined by their acylation pattern. |
|
| ||||||
| Non-phosphorylated lipid As and derivatives | ||||||
| 161–164 | R. sin-1 | Carlson, Vandenplas | 2002 | 101, 102 | Lack of both phosphate groups; aminogluconate in place of the reducing end glucosamine unit | Inhibited TNF-α release |
| 165, 166 | R. sin-1 | Boons | 2003 | 41 | Reducing end anomeric carbon had different oxidation states | The gluconolactone unit in 166 was important for its antagonistic activity. |
| 173, 174 | R. sin-1 | Boons | 2004 | 42 | Devoid of 3-O-acylation | Antagonists of LPS but less potent than 166, indicating the biological importance of 3-O-acyl chain |
| 188, 189 | R. sin-1 | Boons | 2007 | 44 | 189 contained the 27-hydroxyoctacosanoic group at its 2′-N-position | Lipid chain length, not the OH group on it, was critical for its antagonistic activity |
|
| ||||||
| LPS analogs and Kdo-lipid A conjugates | ||||||
|
199 (ReLPS) |
E. coli mutant | Kusumoto | 2001 | 53, 110 | First chemical synthesis of a short LPS with two Kdo units linked to lipid A; Kdo fluorides with bulky 4,5-O-protecting groups were used for α-glycosylation | 199 showed slightly less potent cytokine-inducing activity than that of the natural ReLPS |
| 212 | H. pylori | Fukase. | 2007 | 107 | Lipid A-Kdo conjugate with fewer but longer acyl chains; devoid of 4-O′-phosphate; Kdo fluorides was used for α-glycosylation | TLR-4 antagonists; Kdo residue could enhance the inhibitory activity |
| 221 | H. pylori | Fukase, Fujimoto | 2011 | 114 | Lipid A-Kdo conjugate with an ethanolamine unit in its structure; Kdo N-phenyltrifluoroacetimidate and a microfluidic glycosylation protocol was developed and used for α-glycosylation | Inhibition of cytokine production; the presence of a Kdo unit in the structure reversed the bioactivity of its parent lipid A 220 |
It has been demonstrated that the Kdo residues and the O-antigen in LPS can have a significant influence on the activities of lipid A. To gain more insights into the mechanisms of these structural motifs to affect the functions of lipid A, it is necessary to have access to structurally defined LPSs and LPS derivatives. However, this remains a significant challenge despite the great achievements discussed above. The sheer complex structure of LPSs makes their chemical assembly particularly difficult. In addition, the efficiency of some glycosylation reactions, such as that of Kdo, involved in LPS synthesis is still far from satisfactory. Therefore, an important future direction in this research area would be development of new efficient strategies and glycosylation methods for the assembly of LPSs. In this regard, chemoenzymatic synthesis that combines enzyme-catalyzed glycosylation with chemical synthesis should be particularly attractive, as enzymatic glycosylation reactions can be especially efficient and selective. In addition, various oligosaccharide transferases, polymerases, and transglycosidases are also powerful tools that can be employed for rapid and highly convergent assembly of complex polysaccharides.
Owing to the extensive biological activities of lipid A, its derivatives can have broad medical applications. As mentioned above, some therapies have already been developed based on lipid A and utilized in clinic. Therefore, another anticipated important direction in this research area would be to develop new lipid A-based therapeutics.144 However, although in general a series of efficient synthetic strategies have been established for lipid As, their chemical synthesis is still challenging because of their complex structure. This is at least the partial reason for why the MPLA adjuvant currently used in clinic is obtained through controlled hydrolysis of LPSs derived from biological sources, which can cause a number of problems. Consequently, it is preferable that the new lipid A-based therapeutics should have simplified structures, increased stabilities, enhanced beneficial activities, and improved toxicological profiles. In this regard, the SAR analysis results of synthetic lipid As and related structures should be very useful, as they can be employed to guide the design of novel molecular entities with desirable properties.
Acknowledgments
We thank National Institutes of Health (NIH) for the generous support (R01 CA095142 and R01 GM090270) of our research work.
References
- 1.Rietschel ET, Brade H, Holst O, Brade L, Muller-Loennies S, Mamat U, Zahringer U, Beckmann F, Seydel U, Brandenburg K, Ulmer AJ, Mattern T, Heine H, Schletter J, Loppnow H, Schonbeck U, Flad HD, Hauschildt S, Schade UF, Di Padova F, Kusumoto S, Schumann RR. Bacterial endotoxin: Chemical constitution, biological recognition, host response, and immunological detoxification. Curr Top Microbiol Immunol. 1996;216:39–81. doi: 10.1007/978-3-642-80186-0_3. [DOI] [PubMed] [Google Scholar]
- 2.Erridge C, Bennett-Guerrero E, Poxton IR. Structure and function of lipopolysaccharides. Microbes Infect. 2002;4:837–851. doi: 10.1016/s1286-4579(02)01604-0. [DOI] [PubMed] [Google Scholar]
- 3.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexander C, Rietschel E. Bacterial lipopolysaccharides and innate immunity. J Endotox Res. 2001;7:167–202. [PubMed] [Google Scholar]
- 5.Alexander C, Zahringer U. Chemical structure of lipid A - The primary immunomodulatory center of bacterial lipopolysaccharides. Trends Glycosci Glycotechnol. 2002;14:69–86. [Google Scholar]
- 6.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 7.Dobrovolskaia MA, Vogel SN. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect. 2002;4:903–914. doi: 10.1016/s1286-4579(02)01613-1. [DOI] [PubMed] [Google Scholar]
- 8.Wang ZM, Liu C, Dziarski R. Chemokines are the main proinflammatory mediators in human monocytes activated by Staphylococcus aureus, peptidoglycan, and endotoxin. J Biol Chem. 2000;275:20260–20267. doi: 10.1074/jbc.M909168199. [DOI] [PubMed] [Google Scholar]
- 9.Di Lorenzo F, Kubik Ł, Oblak A, Lorè NI, Cigana C, Lanzetta R, Parrilli M, Hamad MA, De Soyza A, Silipo A, Jerala RB,A, Valvano MA, Martín-Santamaría S, Molinaro A. Activation of human toll-like receptor 4 (TLR4)-myeloid differentiation factor 2 (MD-2) by hypoacylated lipopolysaccharide from a clinical isolate of Burkholderia cenocepacia. J Biol Chem. 2015;290:21305–21319. doi: 10.1074/jbc.M115.649087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 11.Karaghiosoff M, Steinborn R, Kovarik P, Kriegshauser G, Baccarini M, Donabauer B, Reichart U, Kolbe T, Bogdan C, Leanderson T, Levy D, Decker T, Muller M. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat Immunol. 2003;4:471–477. doi: 10.1038/ni910. [DOI] [PubMed] [Google Scholar]
- 12.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 13.Van Amersfoort ES, Van Berkel TJ, Kuiper J. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clin Microbiol Rev. 2003;16:379–414. doi: 10.1128/CMR.16.3.379-414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kilbourn RG, Griffith OW, Gross SS. Pathogenetic mechanisms of septic shock. New Engl J Med. 1993;329:1427–1428. doi: 10.1056/NEJM199311043291916. [DOI] [PubMed] [Google Scholar]
- 15.Parrillo JE. Pathogenetic mechanisms of septic shock. New Engl J Med. 1993;328:1471–1477. doi: 10.1056/NEJM199305203282008. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Shi F, Li Y, Wang X. Structure and function of lipopolysaccharide lipid A in bacteria–a review. Wei Sheng Wu Xue Bao. 2008;48:844–849. [PubMed] [Google Scholar]
- 17.Trent MS. Biosynthesis, transport, and modification of lipid A. Biochem Cell Biol. 2004;82(1):71–86. doi: 10.1139/o03-070. [DOI] [PubMed] [Google Scholar]
- 18.Ernst RK, Hajjar AM, Tsai JH, Moskowitz SM, Wilson CB, Miller SI. Pseudomonas aeruginosa lipid A diversity and its recognition by Toll-like receptor 4. J Endotoxin Res. 2003;9:395–400. doi: 10.1179/096805103225002764. [DOI] [PubMed] [Google Scholar]
- 19.Aygun-Sunar S, Bilaloglu R, Goldfine H, Daldal F. Rhodobacter capsulatus OlsA is a bifunctional enyzme active in both ornithine lipid and phosphatidic acid biosynthesis. J Bacteriol. 2007;189:8564–8574. doi: 10.1128/JB.01121-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ieranò T, Silipo A, Sturiale L, Garozzo D, Brookes H, Khan CMA, Bryant C, Gould FK, Corris PA, Lanzetta R, Parrilli M, De Soyza A, Molinaro A. The structure and proinflammatory activity of the lipopolysaccharide from Burkholderia multivorans and the differences between clonal strains colonizing pre and posttransplanted lungs. Glycobiology. 2008;18:871–881. doi: 10.1093/glycob/cwn074. [DOI] [PubMed] [Google Scholar]
- 21.De Soyza A, Ellis CD, Khan CMA, Corris PA, de Hormaeche RD. Burkholderia cenocepacia lipopolysaccharide, lipid A, and proinflammatory activity. Am J Respir Crit Care Med. 2004;170:70–77. doi: 10.1164/rccm.200304-592OC. [DOI] [PubMed] [Google Scholar]
- 22.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zahringer U, Seydel U, Di Padova F, et al. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 23.Stover AG, Da Silva Correia J, Evans JT, Cluff CW, Elliott MW, Jeffery EW, Johnson DA, Lacy MJ, Baldridge JR, Probst P, Ulevitch RJ, Persing DH, Hershberg RM. Structure-activity relationship of synthetic toll-like receptor 4 agonists. J Biol Chem. 2004;279:4440–4449. doi: 10.1074/jbc.M310760200. [DOI] [PubMed] [Google Scholar]
- 24.Kanegasaki S, Tanamoto K, Yasuda T, Homma JY, Matsuura M, Nakatsuka M, Kumazawa Y, Yamamoto A, Shiba T, Kusumoto S, et al. Structure-activity relationship of lipid A: comparison of biological activities of natural and synthetic lipid A’s with different fatty acid compositions. J Biochem. 1986;99:1203–1210. doi: 10.1093/oxfordjournals.jbchem.a135583. [DOI] [PubMed] [Google Scholar]
- 25.Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol. 2005;3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 26.Schromm AB, Brandenburg K, Loppnow H, Zahringer U, Rietschel ET, Carroll SF, Koch MHJ, Kusumoto S, Seydel U. The charge of endotoxin molecules influences their conformation and IL-6-inducing capacity. J Immunol. 1998;161:5464–5471. [PubMed] [Google Scholar]
- 27.Schromm AB, Brandenburg K, Loppnow H, Moran AP, Koch MHJ, Rietschel ET, Seydel U. Biological activities of lipopolysaccharides are determined by the shape of their lipid A portion. Eur J Biochem. 2000;267:2008–2013. doi: 10.1046/j.1432-1327.2000.01204.x. [DOI] [PubMed] [Google Scholar]
- 28.Somerville JE, Cassiano L, Darveau RP. Escherichia coli msbB gene as a virulence factor and a therapeutic target. Infect Immun. 1999;67:6583–6590. doi: 10.1128/iai.67.12.6583-6590.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knirel YA, Valvano MA. Bacterial lipopolysaccharides: structure, chemical synthesis, biogenesis, and interaction with host cells. Wien: Springer; 2011. pp. 1–440. [Google Scholar]
- 30.Wang X, Quinn PJ. Lipopolysaccharide: Biosynthetic pathway and structure modification. Prog Lipid Res. 2010;49:97–107. doi: 10.1016/j.plipres.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 31.Caroff M, Karibian D. Structure of bacterial lipopolysaccharides. Carbohydr Res. 2003;338:2431–2447. doi: 10.1016/j.carres.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 32.Brandenburg K, Andra J, Muller M, Koch MH, Garidel P. Physicochemical properties of bacterial glycopolymers in relation to bioactivity. Carbohydr Res. 2003;338:2477–2489. doi: 10.1016/j.carres.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 33.Molinaro A, Holst O, Di Lorenzo F, Callaghan M, Nurisso A, D’Errico G, Zamyatina A, Peri F, Berisio R, Jerala R, Jiménez-Barbero J, Silipo A, Martín-Santamaría S. Chemistry of lipid A: At the heart of innate immunity. Chem Eur J. 2015;21:500–519. doi: 10.1002/chem.201403923. [DOI] [PubMed] [Google Scholar]
- 34.Imoto M, Yoshimura H, Yamamoto M, Shimamoto T, Kusumoto S, Shiba T. Chemical Synthesis of Phosphorylated Tetraacyl Disaccharide Corresponding to a Biosynthetic Precursor of Lipid-A. Tetrahedron Lett. 1984;25:2667–2670. [Google Scholar]
- 35.Imoto M, Yoshimura H, Sakaguchi N, Kusumoto S, Shiba T. Total Synthesis of Escherichia-Coli Lipid-A. Tetrahedron Lett. 1985;26:1545–1548. [Google Scholar]
- 36.Imoto M, Yoshimura H, Yamamoto M, Shimamoto T, Kusumoto S, Shiba T. Chemical Synthesis of a Biosynthetic Precursor of Lipid-a with a Phosphorylated Tetraacyl Disaccharide Structure. Bull Chem Soc Jap. 1987;60:2197–2204. [Google Scholar]
- 37.Christ WJ, Mcguinness PD, Asano O, Wang Y, Mullarkey MA, Perez M, Hawkins LD, Blythe TA, Dubuc GR, Robidoux AL. Total Synthesis of the Proposed Structure of Rhodobacter-Sphaeroides Lipid-a Resulting in Synthesis of New Potent Lipopolysaccharide Antagonists. J Am Chem Soc. 1994;116:3637–3638. [Google Scholar]
- 38.Ikeda K, Takahashi T, Kondo H, Achiwa K. Lipid-a and Related-Compounds .10. Total Synthesis of Proteus-Mirabilis Lipid-A. Chem Pharm Bull. 1987;35:1311–1314. [Google Scholar]
- 39.Imoto M, Yoshimura H, Shimamoto T, Sakaguchi N, Kusumoto S, Shiba T. Total Synthesis of Escherichia-Coli Lipid-a, the Endotoxically Active Principle of Cell-Surface Lipopolysaccharide. Bull Chem Soc Japan. 1987;60:2205–2214. [Google Scholar]
- 40.Yin N, Marshall RL, Matheson S, Savage PB. Synthesis of lipid A derivatives and their interactions with polymyxin B and polymyxin B nonapeptide. J Am Chem Soc. 2003;125:2426–2435. doi: 10.1021/ja0284456. [DOI] [PubMed] [Google Scholar]
- 41.Demchenko AV, Wolfert MA, Santhanam B, Moore JN, Boons GJ. Synthesis and biological evaluation of Rhizobium sin-1 lipid A derivatives. J Am Chem Soc. 2003;125:6103–6112. doi: 10.1021/ja029316s. [DOI] [PubMed] [Google Scholar]
- 42.Santhanam B, Worfent MA, Moore JN, Boons GJ. Synthesis and biological evaluation of a lipid A drivative that contains an aminogluconate moiety. Chem Eur J. 2004;10:4798–4807. doi: 10.1002/chem.200400376. [DOI] [PubMed] [Google Scholar]
- 43.Zhang YH, Gaekwad J, Wolfert MA, Boons GJ. Modulation of innate immune responses with synthetic lipid a derivatives. J Am Chem Soc. 2007;129:5200–5216. doi: 10.1021/ja068922a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang YH, Wolfert MA, Boons GJ. The influence of the long chain fatty acid on the antagonistic activities of Rhizobium sin-1 lipid A. Bioorg Med Chem. 2007;15:4800–4812. doi: 10.1016/j.bmc.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fukase Y, Fujimoto Y, Adachi Y, Suda Y, Kusumoto S, Fukase K. Synthesis of Rubrivivax gelatinosus lipid a and analogues for investigation of the structural basis for immunostimulating and inhibitory activities. Bull Chem Soc Jap. 2008;81:796–819. [Google Scholar]
- 46.Tang S, Wang Q, Guo Z. Synthesis of a monophosphoryl derivative of Escherichia coli lipid A and its efficient coupling to a tumor-associated carbohydrate antigen. Chem Eur J. 2010;16:1319–1325. doi: 10.1002/chem.200902153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rembold H, Schmidt RR. Synthesis of Kdo-alpha-glycosides of lipid A derivatives. Carbohydr Res. 1993;246:137–159. doi: 10.1016/0008-6215(93)84029-6. [DOI] [PubMed] [Google Scholar]
- 48.Fujimoto Y, Kimura E, Murata S, Kusumoto S, Fukase K. Synthesis and bioactivity of fluorescence- and biotin-labeled lipid A analogues for investigation of recognition mechanism in innate immunity. Tetrahedron Lett. 2006;47:539–543. [Google Scholar]
- 49.Santhanam B, Boons GJ. Preparation of a lipid a derivative that contains a 27-hydroxyoctacosanoic acid moiety. Org Lett. 2004;6:3333–3336. doi: 10.1021/ol048746f. [DOI] [PubMed] [Google Scholar]
- 50.Jiang ZH, Budzynski WA, Qiu DX, Yalamati D, Koganty RR. Monophosphoryl lipid A analogues with varying 3-O-substitution: synthesis and potent adjuvant activity. Carbohydr Res. 2007;342:784–796. doi: 10.1016/j.carres.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 51.Watanabe Y, Miura K, Shiozaki M, Kanai S, Kurakata S, Nishijima M. Synthesis of lipid A type carboxymethyl derivatives with ether chains instead of ester chains and their LPS-antagonistic activities. Carbohydr Res. 2003;338:47–54. doi: 10.1016/s0008-6215(02)00357-9. [DOI] [PubMed] [Google Scholar]
- 52.Johnson DA, Keegan DS, Sowell CG, Livesay MT, Johnson CL, Taubner LM, Harris A, Myers KR, Thompson JD, Gustafson GL, Rhodes MJ, Ulrich JT, Ward JR, Yorgensen YM, Cantrell JL, Brookshire VG. 3-O-desacyl monophosphoryl lipid a derivatives: Synthesis and immunostimulant activities. J Med Chem. 1999;42:4640–4649. doi: 10.1021/jm990222b. [DOI] [PubMed] [Google Scholar]
- 53.Yoshizaki H, Fukuda N, Sato K, Oikawa M, Fukase K, Suda Y, Kusumoto S. First total synthesis of the Re-type lipopolysaccharide. Angew Chem Int Ed. 2001;40:1475–1479. doi: 10.1002/1521-3773(20010417)40:8<1475::AID-ANIE1475>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 54.Zamyatina A, Sekljic H, Brade H, Kosma P. Synthesis and purity assessment of tetra- and pentaacyl lipid A of Chlamydia containing (R)-3-hydroxyicosanoic acid. Tetrahedron. 2004;60:12113–12137. [Google Scholar]
- 55.Qureshi N, Honovich JP, Hara H, Cotter RJ, Takayama K. Location of Fatty-Acids in Lipid-a Obtained from Lipopolysaccharide of Rhodopseudomonas-Sphaeroides Atcc-17023. J Biol Chem. 1988;263:5502–5504. [PubMed] [Google Scholar]
- 56.Qureshi N, Takayama K, Kurtz R. Diphosphoryl-Lipid-a Obtained from the Nontoxic Lipopolysaccharide of Rhodopseudomonas-Sphaeroides Is an Endotoxin Antagonist in Mice. Infect Immun. 1991;59:441–444. doi: 10.1128/iai.59.1.441-444.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Christ WJ, Asano O, Robidoux ALC, Perez M, Wang YA, Dubuc GR, Gavin WE, Hawkins LD, Mcguinness PD, Mullarkey MA, Lewis MD, Kishi Y, Kawata T, Bristol JR, Rose JR, Rossignol DP, Kobayashi S, Hishinuma L, Kimura A, Asakawa N, Katayama K, Yamatsu I. E5531, a Pure Endotoxin Antagonist of High Potency. Science. 1995;268:80–83. doi: 10.1126/science.7701344. [DOI] [PubMed] [Google Scholar]
- 58.Kawata T, Bristol JR, Rossignol DP, Rose JR, Kobayashi S, Yokohama H, Ishibashi A, Christ WJ, Katayama K, Yamatsu I, Kishi Y. E5531, a synthetic non-toxic lipid A derivative blocks the immunobiological activities of lipopolysaccharide. Br J Pharmacol. 1999;127:853–862. doi: 10.1038/sj.bjp.0702596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bunnell E, Lynn M, Habet K, Neumann A, Perdomo CA, Friedhoff LT, Rogers SL, Parrillo JE. A lipid A analog, E5531, blocks the endotoxin response in human volunteers with experimental endotoxemia. Crit Care Med. 2000;28:2713–2720. doi: 10.1097/00003246-200008000-00005. [DOI] [PubMed] [Google Scholar]
- 60.Rossignol DP, Lynn M. Antagonism of in vivo and ex vivo response to endotoxin by E5564, a synthetic lipid A analogue. J Endotoxin Res. 2002;8:483–488. doi: 10.1179/096805102125001127. [DOI] [PubMed] [Google Scholar]
- 61.Barochia A, Solomon S, Cui X, Natanson C, Eichacker PQ. Eritoran tetrasodium (E5564) treatment for sepsis: review of preclinical and clinical studies. Expert Opin Drug Metab Toxicol. 2011;7:479–494. doi: 10.1517/17425255.2011.558190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Czeslick E, Struppert A, Simm A, Sablotzki A. E5564 (Eritoran) inhibits lipopolysaccharide-induced cytokine production in human blood monocytes. Inflamm Res. 2006;55:511–515. doi: 10.1007/s00011-006-6057-3. [DOI] [PubMed] [Google Scholar]
- 63.Shiozaki M, Doi H, Tanaka D, Shimozato T, Kurakata S. Syntheses of glucose analogues of E5564 as a highly potent anti-sepsis drug candidate. Bioorg Med Chem. 2006;14:3011–3016. doi: 10.1016/j.bmc.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 64.Shiozaki M, Iwano Y, Doi H, Tanaka D, Shimozato T, Kurakata S. Syntheses of glucose derivatives of E5564-related compounds and their LPS-antagonistic activities. Carbohydr Res. 2006;341:811–822. doi: 10.1016/j.carres.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 65.Shiozaki M, Doi H, Tanaka D, Shimozato T, Kurakata S. Syntheses of glucose-containing E5564 analogues and their LPS-antagonistic activities. Tetrahedron. 2006;62:205–225. [Google Scholar]
- 66.Kirikae T, Schade FU, Zahringer U, Kirikae F, Brade H, Kusumoto S, Kusama T, Rietschel ET. The Significance of the Hydrophilic Backbone and the Hydrophobic Fatty-Acid Regions of Lipid-a for Macrophage Binding and Cytokine Induction. FEMS Immunol Med Microbiol. 1994;8:13–26. doi: 10.1111/j.1574-695X.1994.tb00421.x. [DOI] [PubMed] [Google Scholar]
- 67.Koch PJ, Frank J, Schuler J, Kahle C, Bradaczek H. Thermodynamics and structural studies of the interaction of Polymyxin B with deep rough mutant lipopolysaccharides. J Coll Interf Sci. 1999;213:557–564. doi: 10.1006/jcis.1999.6137. [DOI] [PubMed] [Google Scholar]
- 68.Bruch MD, Cajal Y, Koh JT, Jain MK. Higher-order structure of polymyxin B: The functional significance of topological flexibility. J Am Chem Soc. 1999;121:11993–12004. [Google Scholar]
- 69.Kusama T, Soga T, Shioya E, Nakayama K, Nakajima H, Osada Y, Ono Y, Kusumoto S, Shiba T. Synthesis and antitumor activity of lipid A analogs having a phosphonooxyethyl group with alpha- or beta-configuration at position 1. Chem Pharm Bull. 1990;38:3366–3372. doi: 10.1248/cpb.38.3366. [DOI] [PubMed] [Google Scholar]
- 70.Nakatsuka M, Kumazawa Y, Matsuura M, Homma JY, Kiso M, Hasegawa A. Enhancement of nonspecific resistance to bacterial infections and tumor regressions by treatment with synthetic lipid A-subunit analogs. Critical role of N- and 3-O-linked acyl groups in 4-O-phosphono-D-glucosamine derivatives. Int J Immunopharmacol. 1989;11:349–358. doi: 10.1016/0192-0561(89)90080-5. [DOI] [PubMed] [Google Scholar]
- 71.Matsumoto N, Aze Y, Akimoto A, Fujita T. ONO-4007, an antitumor lipid A analog, induces tumor necrosis factor-alpha production by human monocytes only under primed state: different effects of ONO-4007 and lipopolysaccharide on cytokine production. J Pharmacol Exp Ther. 1998;284:189–195. [PubMed] [Google Scholar]
- 72.Kumazawa E, Tohgo A, Soga T, Kusama T, Osada Y. Significant antitumor effect of a synthetic lipid A analogue, DT-5461, on murine syngeneic tumor models. Cancer Immunol Immunother. 1992;35:307–314. doi: 10.1007/BF01741143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cluff CW. Monophosphoryl Lipid A (MPL) as an Adjuvant for Anti-Cancer Vaccines: Clinical Results. Adv Exp Med Biol. 2009;667:111–123. doi: 10.1007/978-1-4419-1603-7_10. [DOI] [PubMed] [Google Scholar]
- 74.Kusama T, Soga T, Ono Y, Kumazawa E, Shioya E, Osada Y, Kusumoto S, Shiba T. Synthesis and biological activities of analogs of a lipid A biosynthetic precursor: 1-O-phosphonooxyethyl-4′-O-phosphono-disaccharides with (R)-3-hydroxytetradecanoyl or tetradecanoyl groups at positions 2, 3, 2′ and 3′. Chem Pharm Bull. 1991;39:1994–1999. doi: 10.1248/cpb.39.1994. [DOI] [PubMed] [Google Scholar]
- 75.Fukase K, Kirikae T, Kirikae F, Liu WC, Oikawa M, Suda Y, Kurosawa M, Fukase Y, Yoshizaki H, Kusumoto S. Synthesis of [H-3]-labeled bioactive lipid A analogs and their use for detection of lipid A-binding proteins on murine macrophages. Bull Chem Soc Jap. 2001;74:2189–2197. [Google Scholar]
- 76.Kobayashi M, Saitoh S, Tanimura N, Takahashi K, Kawasaki K, Nishijima M, Fujimoto Y, Fukase K, Akashi-Takamura S, Miyake K. Regulatory roles for MD-2 and TLR4 in ligand-induced receptor clustering. J Immunol. 2006;176:6211–6218. doi: 10.4049/jimmunol.176.10.6211. [DOI] [PubMed] [Google Scholar]
- 77.Kiso M, Ogawa Y, Fujishima Y, Fujita M, Tanaka S, Hasegawa A. Nonreducing-Sugar Subunit Analogs of Bacterial Lipid a Carrying the Ester-Bound (3r)-3-(Acyloxy)Tetradecanoyl Group. J Carbohydr Chem. 1987;6:625–638. [Google Scholar]
- 78.Shiozaki M, Kurakata S, Tatsuta T, Maeda H, Nishijima M. Syntheses of 2,6-anhydro-3-deoxy-5-O-phosphono-3-tetradecanamido-4-O-[(R)-3-(tetradecanoyloxy)tetradecanoyl]-D-glycero-D-ido-heptonic acid, its dimeric analogue, and related compounds. Tetrahedron. 1997;53:16041–16060. [Google Scholar]
- 79.Mochizuki T, Iwano Y, Shiozaki M, Kurakata S, Kanai S, Nishijima M. Synthesis and biological activities of lipid A-type pyrancarboxylic acid derivatives. Carbohydr Res. 2000;324:225–230. doi: 10.1016/s0008-6215(99)00326-2. [DOI] [PubMed] [Google Scholar]
- 80.Mochizuki T, Iwano Y, Shiozaki M, Kurakata S, Kanai S, Nishijima M. Lipid A-type pyrancarboxylic acid derivatives, their synthesis and their biological activities. Tetrahedron. 2000;56:7691–7703. doi: 10.1016/s0008-6215(99)00326-2. [DOI] [PubMed] [Google Scholar]
- 81.Watanabe Y, Mochizuki T, Shiozaki M, Kanai S, Kurakata S, Nishijima M. Synthesis of lipid A type pyran carboxylic acids with ether chains and their biological activities. Carbohydr Res. 2001;333:203–231. doi: 10.1016/s0008-6215(01)00134-3. [DOI] [PubMed] [Google Scholar]
- 82.Watanabe Y, Miura K, Shiozaki M, Kanai S, Kurakata S, Nishijima M. Synthesis of GLA-60 type pyran carboxylic acids with an alkyl chain instead of an ester chain as LPS-antagonists. Carbohydr Res. 2001;332:257–277. doi: 10.1016/s0008-6215(01)00055-6. [DOI] [PubMed] [Google Scholar]
- 83.Shiozaki M, Deguchi N, Macindoe WM, Arai M, Miyazaki H, Mochizuki T, Tatsuta T, Ogawa J, Maeda H, Kurakata S. Syntheses of 1-O-carboxyalkyl GLA-60 analogues. Carbohydr Res. 1996;283:27–51. doi: 10.1016/0008-6215(95)00402-5. [DOI] [PubMed] [Google Scholar]
- 84.Akamatsu M, Fujimoto Y, Kataoka M, Suda Y, Kusumoto S, Fukase K. Synthesis of lipid A monosaccharide analogues containing acidic amino acid: Exploring the structural basis for the endotoxic and antagonistic activities. Bioorg Med Chem. 2006;14:6759–6777. doi: 10.1016/j.bmc.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 85.Qureshi N, Takayama K, Ribi E. Purification and Structural Determination of Nontoxic Lipid-a Obtained from the Lipopolysaccharide of Salmonella-Typhimurium. J Biol Chem. 1982;257:1808–1815. [PubMed] [Google Scholar]
- 86.Qureshi N, Mascagni P, Ribi E, Takayama K. Monophosphoryl Lipid-a Obtained from Lipopolysaccharides of Salmonella-Minnesota R595 - Purification of the Dimethyl Derivative by High-Performance Liquid-Chromatography and Complete Structural Determination. J Biol Chem. 1985;260:5271–5278. [PubMed] [Google Scholar]
- 87.Casella CR, Mitchell TC. Putting endotoxin to work for us: Monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell Mol Life Sci. 2008;65:3231–3240. doi: 10.1007/s00018-008-8228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jiang ZH, Bach MV, Budzynski WA, Krantz MJ, Koganty RR, Longenecker BM. Lipid a structures containing novel lipid moieties: Synthesis and adjuvant properties. Bioorg Med Chem Lett. 2002;12:2193–2196. doi: 10.1016/s0960-894x(02)00362-1. [DOI] [PubMed] [Google Scholar]
- 89.Johnson DA, Sowell CG, Johnson CL, Livesay MT, Keegan DS, Rhodes MJ, Ulrich JT, Ward JR, Cantrell JL, Brookshire VG. Synthesis and biological evaluation of a new class of vaccine adjuvants: Aminoalkyl glucosaminide 4-phosphates (AGPs) Bioorg Med Chem Lett. 1999;9:2273–2278. doi: 10.1016/s0960-894x(99)00374-1. [DOI] [PubMed] [Google Scholar]
- 90.Johnson DA, Sowell CG, Keegan DS, Livesay MT. Chemical synthesis of the major constituents of Salmonella minnesota monophosphoryl lipid A. J Carbohydr Chem. 1998;17:1421–1426. [Google Scholar]
- 91.Akamatsu M, Fujimoto Y, Kataoka M, Suda Y, Kusumoto S, Fukase K. Synthesis of lipid A monosaccharide analogues containing acidic amino acid: exploring the structural basis for the endotoxic and antagonistic activities. Bioorg Med Chem. 2006;14:6759–6777. doi: 10.1016/j.bmc.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 92.Maiti KK, DeCastro M, El-Sayed AMA, Foote MI, Wolfert MA, Boons GJ. Chemical synthesis and proinflammatory responses of monophosphoryl lipid A adjuvant candidates. Eur J Org Chem. 2010:80–91. doi: 10.1002/ejoc.200900973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Erwin AL, Munford RS. Deacylation of Structurally Diverse Lipopolysaccharides by Human Acyloxyacyl Hydrolase. J Biol Chem. 1990;265:16444–16449. [PubMed] [Google Scholar]
- 94.D’Alonzo D, Cipolletti M, Tarantino G, Ziaco M, Pieretti G, Iadonisi A, Palumbo G, Alfano A, Giuliano M, De Rosa M, Schiraldi C, Cammarota M, Parrilli M, Bedini E, Corsaro MM. A semisynthetic approach to new immunoadjuvant candidates: Site-selective chemical manipulation of Escherichia coli monophosphoryl lipid A. Chem Eur J. 2016;22:11053–11063. doi: 10.1002/chem.201601284. [DOI] [PubMed] [Google Scholar]
- 95.Wang QL, Xue J, Guo ZW. Synthesis of a monophosphoryl lipid A derivative and its conjugation to a modified form of a tumor-associated carbohydrate antigen GM3. Chem Commun. 2009:5536–5537. doi: 10.1039/b907351e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhou ZF, Mondal M, Liao GC, Guo ZW. Synthesis and evaluation of monophosphoryl lipid A derivatives as fully synthetic self-adjuvanting glycoconjugate cancer vaccine carriers. Org Biomol Chem. 2014;12:3238–3245. doi: 10.1039/c4ob00390j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ogawa T. Chemical structure of lipid A from porphyromonas (bacteroides) gingivalis lipopolysaccharide. FEBS Lett. 1993;332:197–201. doi: 10.1016/0014-5793(93)80512-s. [DOI] [PubMed] [Google Scholar]
- 98.Kumada H, Haishima Y, Umemoto T, Tanamoto K. Structural study on the free lipid A isolated from lipopolysaccharide of Porphyromonas gingivalis. J Bacteriol. 1995;177:2098–2106. doi: 10.1128/jb.177.8.2098-2106.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang Y, Gaekwad J, Wolfert MA, Boons G-J. Synthetic tetra-acylated derivatives of lipid A from Porphyromonas gingivalis are antagonists of human TLR4. Org Biomol Chem. 2008;6:3371–3381. doi: 10.1039/b809090d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sawada N, Ogawa T, Asai Y, Makimura Y, Sugiyama A. Toll-like receptor 4-dependent recognition of structurally different forms of chemically synthesized lipid As of Porphyromonas gingivalis. Clin Exp Immunol. 2007;148:529–536. doi: 10.1111/j.1365-2249.2007.03346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jeyaretnam B, Glushka J, Kolli VSK, Carlson RW. Characterization of a novel lipid-A from Rhizobium species Sin-1. J Biol Chem. 2002;277:41802–41810. doi: 10.1074/jbc.M112140200. [DOI] [PubMed] [Google Scholar]
- 102.Vandenplas ML, Carlson RW, Jeyaretnam BS, McNeill B, Barton MH, Norton N, Murray TF, Moore JN. Rhizobium Sin-1 lipopolysaccharide (LPS) prevents enteric LPS-induced cytokine production. J Biol Chem. 2002;277:41811–41816. doi: 10.1074/jbc.M205252200. [DOI] [PubMed] [Google Scholar]
- 103.Lee HS, Wolfert MA, Zhang YH, Boons GJ. The 2-aminogluconate isomer of Rhizobium sin-1 lipid A can antagonize TNF-alpha production induced by enteric LPS. ChemBioChem. 2006;7:140–148. doi: 10.1002/cbic.200500298. [DOI] [PubMed] [Google Scholar]
- 104.Mehta S, Pinto BM. Phenylselenoglycosides as Novel, Versatile Glycosyl Donors - Selective Activation over Thioglycosides. Tetrahedron Lett. 1991;32(35):4435–4438. [Google Scholar]
- 105.Brandenburg K, Lindner B, Schromm A, Koch MHJ, Bauer J, Merkli A, Zbaeren C, Davies JG, Seydel U. Physicochemical characteristics of triacyl lipid A partial structure OM-174 in relation to biological activity. Eur J Biochem. 2000;267:3370–3377. doi: 10.1046/j.1432-1327.2000.01370.x. [DOI] [PubMed] [Google Scholar]
- 106.Martin OR, Zhou W, Wu XF, Front-Deschamps S, Moutel S, Schindl K, Jeandet P, Zbaeren C, Bauer JA. Synthesis and immunobiological activity of an original series of acyclic lipid a mimics based on a pseudodipeptide backbone. J Med Chem. 2006;49:6000–6014. doi: 10.1021/jm060482a. [DOI] [PubMed] [Google Scholar]
- 107.Fujimoto Y, Iwata M, Imakita N, Shimoyama A, Suda Y, Kusumoto S, Fukase K. Synthesis of immunoregulatory Helicobacter pylori lipopolysaccharide partial structures. Tetrahedron Lett. 2007;48:6577–6581. [Google Scholar]
- 108.Gaekwad J, Zhang Y, Zhang W, Reeves J, Wolfert MA, Boons GJ. Differential induction of innate immune responses by synthetic lipid a derivatives. J Biol Chem. 2010;285:29375–29386. doi: 10.1074/jbc.M110.115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zughaier SM, Tzeng YL, Zimmer SM, Datta A, Carlson RW, Stephens DS. Neisseria meningitidis lipooligosaccharide structure-dependent activation of the macrophage CD14/Toll-like receptor 4 pathway. Infect Immun. 2004;72:371–380. doi: 10.1128/IAI.72.1.371-380.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zahringer U, Lindner B, Seydel U, Rietschel ET, Naoki H, Unger FM, Imoto M, Kusumoto S, Shiba T. Structure of De-O-Acylated Lipopolysaccharide from the Escherichia-Coli Re Mutant Strain-F-515. Tetrahedron Lett. 1985;26:6321–6324. [Google Scholar]
- 111.Haunert F, Bolli MH, Hinzen B, Ley SV. Clean three-step synthesis of 4,5-dihydro-1H-pyrazoles starting from alcohols using polymer supported reagents. J Chem Soc, Perkin Trans 1. 1998:2235–2237. [Google Scholar]
- 112.Sakai Y, Oikawa M, Yoshizaki H, Ogawa T, Suda Y, Fukase K, Kusumoto S. Synthesis of Helicobacter pylori lipid A and its analogue using p-(trifluoromethyl)benzyl protecting group. Tetrahedron Lett. 2000;41:6843–6847. [Google Scholar]
- 113.Yu B, Tao HC. Glycosyl trifluoroacetimidates. Part 1: Preparation and application as new glycosyl donors. Tetrahedron Lett. 2001;42:2405–2407. [Google Scholar]
- 114.Shimoyama A, Saeki A, Tanimura N, Tsutsui H, Miyake K, Suda Y, Fujimoto Y, Fukase K. Chemical Synthesis of Helicobacter pylori Lipopolysaccharide Partial Structures and their Selective Proinflammatory Responses. Chem Eur J. 2011;17:14464–14474. doi: 10.1002/chem.201003581. [DOI] [PubMed] [Google Scholar]
- 115.Shimoyama A, Fujimoto Y, Fukase K. Stereoselective Glycosylation of 3-Deoxy-D-manno-2-octulosonic Acid with Batch and Microfluidic Methods. Synlett. 2011:2359–2362. [Google Scholar]
- 116.Fujimoto Y, Shimoyama A, Suda Y, Fukase K. Synthesis and immunomodulatory activities of Helicobacter pylori lipophilic terminus of lipopolysaccharide including lipid A. Carbohydr Res. 2012;356:37–43. doi: 10.1016/j.carres.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 117.Schmidt RR, Esswein A. Anomeric O-Alkylation .6. Simple Synthesis of Kdo Alpha-Glycosides by Anomerically Selective O-Alkylation. Angew Chem Int Ed. 1988;27:1178–1180. [Google Scholar]
- 118.Nielsen H, Birkholz S, Andersen LP, Moran AP. Neutrophil activation by Helicobacter pylori lipopolysaccharides. J Infect Dis. 1994;170:135–139. doi: 10.1093/infdis/170.1.135. [DOI] [PubMed] [Google Scholar]
- 119.Yokota S, Saito H, Kubota T, Yokosawa N, Amano K, Fujii N. Measles virus suppresses interferon-alpha signaling pathway: suppression of Jak1 phosphorylation and association of viral accessory proteins, C and V, with interferon-alpha receptor complex. Virology. 2003;306:135–146. doi: 10.1016/s0042-6822(02)00026-0. [DOI] [PubMed] [Google Scholar]
- 120.Suda Y, Ogawa T, Kashihara W, Oikawa M, Shimoyama T, Hayashi T, Tamura T, Kusumoto S. Chemical structure of lipid A from Helicobacter pylori strain 206–1 lipopolysaccharide. J Biochem. 1997;121:1129–1133. doi: 10.1093/oxfordjournals.jbchem.a021705. [DOI] [PubMed] [Google Scholar]
- 121.Moran AP, Lindner B, Walsh EJ. Structural characterization of the lipid A component of Helicobacter pylori rough- and smooth-form lipopolysaccharides. J Bacteriol. 1997;179:6453–6463. doi: 10.1128/jb.179.20.6453-6463.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kobayashi S, Yoneda A, Fukuhara T, Hara S. Deoxyfluorination of alcohols using N,N-diethyl-alpha,alpha-difluoro-(m-methylbenzyl)amine. Tetrahedron. 2004;60:6923–6930. [Google Scholar]
- 123.Suda Y, Tochio H, Kawano K, Takada H, Yoshida T, Kotani S, Kusumoto S. Cytokine-inducing glycolipids in the lipoteichoic acid fraction from Enterococcus hirae ATCC 9790. FEMS Immunol Med Microbiol. 1995;12:97–112. doi: 10.1111/j.1574-695X.1995.tb00181.x. [DOI] [PubMed] [Google Scholar]
- 124.Ogawa T, Asai Y, Sakai Y, Oikawa M, Fukase K, Suda Y, Kusumoto S, Tamura T. Endotoxic and immunobiological activities of a chemically synthesized lipid A of Helicobacter pylori strain 206–1. FEMS Immunol Med Microbiol. 2003;36:1–7. doi: 10.1016/S0928-8244(03)00093-2. [DOI] [PubMed] [Google Scholar]
- 125.Tanaka SI, Goi T, Tanaka K, Fukase K. Highly efficient alpha-sialylation by virtue of fixed dipole effects of N-phthalyl group: Application to continuous flow synthesis of alpha(2–3)- and alpha(2–6)-Neu5Ac-Gal motifs by microreactor. J Carbohydr Chem. 2007;26:369–394. [Google Scholar]
- 126.Whitfield C. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu Rev Biochem. 2006;75:39–68. doi: 10.1146/annurev.biochem.75.103004.142545. [DOI] [PubMed] [Google Scholar]
- 127.Han W, Wu B, Li L, Zhao G, Woodward R, Pettit N, Cai L, Thon V, Wang PG. Defining function of lipopolysaccharide O-antigen ligase WaaL using chemoenzymatically synthesized substrates. J Biol Chem. 2012;287:5357–5365. doi: 10.1074/jbc.M111.308486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Woodward R, Yi W, Li L, Zhao G, Eguchi H, Sridhar PR, Guo H, Song JK, Motari E, Cai L, Kelleher P, Liu X, Han W, Zhang W, Ding Y, Li M, Wang PG. In vitro bacterial polysaccharide biosynthesis: defining the functions of Wzy and Wzz. Nat Chem Biol. 2010;6:418–423. doi: 10.1038/nchembio.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhao G, Wu B, Li L, Wang PG. O-Antigen polymerase adopts a distributive mechanism for lipopolysaccharide biosynthesis. Appl Microbiol Biotechnol. 2014;98:4075–4081. doi: 10.1007/s00253-014-5552-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Danishefsky SJ, Allen JR. From the laboratory to the clinic: A retrospective on fully synthetic carbohydrate-based anticancer vaccines. Angew Chem Int Ed. 2000;39:836–863. doi: 10.1002/(sici)1521-3773(20000303)39:5<836::aid-anie836>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 131.Guo ZW, Wang QL. Recent development in carbohydrate-based cancer vaccines. Curr Opin Chem Biol. 2009;13:608–617. doi: 10.1016/j.cbpa.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen YA, Star A, Vidal S. Sweet carbon nanostructures: carbohydrate conjugates with carbon nanotubes and graphene and their applications. Chem Soc Rev. 2013;42:4532–4542. doi: 10.1039/c2cs35396b. [DOI] [PubMed] [Google Scholar]
- 133.Kaiser A, Gaidzik N, Becker T, Menge C, Groh K, Cai H, Li YM, Gerlitzki B, Schmitt E, Kunz H. Fully Synthetic Vaccines Consisting of Tumor-Associated MUC1 Glycopeptides and a Lipopeptide Ligand of the Toll-like Receptor 2. Angew Chem Int Ed. 2010;49:3688–3692. doi: 10.1002/anie.201000462. [DOI] [PubMed] [Google Scholar]
- 134.Buskas T, Ingale S, Boons GJ. Towards a fully synthetic carbohydrate-based anticancer vaccine: Synthesis and immunological evaluation of a lipidated glycopeptide containing the tumor-associated Tn antigen. Angew Chem Int Ed. 2005;44:5985–5988. doi: 10.1002/anie.200501818. [DOI] [PubMed] [Google Scholar]
- 135.Wang Q, Zhou Z, Tang S, Guo Z. Carbohydrate-monophosphoryl lipid a conjugates are fully synthetic self-adjuvanting cancer vaccines eliciting robust immune responses in the mouse. ACS Chem Biol. 2012;7:235–240. doi: 10.1021/cb200358r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pan YB, Chefalo P, Nagy N, Harding C, Guo ZW. Synthesis and immunological properties of N-modified GM3 antigens as therapeutic cancer vaccines. J Med Chem. 2005;48:875–883. doi: 10.1021/jm0494422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Markham RB, Pier GB, Schreiber JR. The Role of Cytophilic Igg3 Antibody in T-Cell-Mediated Resistance to Infection with the Extracellular Bacterium, Pseudomonas-Aeruginosa. J Immunol. 1991;146:316–320. [PubMed] [Google Scholar]
- 138.Gavin AL, Barnes N, Dijstelbloem HM, Hogarth PM. Cutting edge: Identification of the mouse IgG3 receptor: Implications for antibody effector function at the interface between innate and adaptive immunity. J Immunol. 1998;160:20–23. [PubMed] [Google Scholar]
- 139.Kulshin VA, Zahringer U, Lindner B, Frasch CE, Tsai CM, Dmitriev BA, Rietschel ET. Structural characterization of the lipid A component of pathogenic Neisseria meningitidis. J Bacteriol. 1992;174:1793–1800. doi: 10.1128/jb.174.6.1793-1800.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhou Z, Liao G, Mandal SS, Suryawanshi S, Guo Z. A Fully Synthetic Self-Adjuvanting Globo H-Based Vaccine Elicited Strong T Cell-Mediated Antitumor Immunity. Chem Sci. 2015;6:7112–7121. doi: 10.1039/c5sc01402f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mandal SS, Liao G, Guo Z. Chemical Synthesis of the Tumor-Associated Globo H Antigen. RSC Adv. 2015;5:23311–23319. doi: 10.1039/C5RA00759C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liao G, Zhou Z, Suryawanshi S, Mondal MA, Guo Z. Fully synthetic self-adjuvanting α-2,9-oligosialic acid-based conjugate vaccines against group C meningitis. ACS Cent Sci. 2016;2:210–218. doi: 10.1021/acscentsci.5b00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Liao G, Zhou Z, Guo Z. Synthesis and immunological study of α-2,9-oligosialic acid conjugates as anti-group C meningitis vaccines. Chem Commun. 2015;51:9647–9650. doi: 10.1039/c5cc01794g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kaeothip S, Paranjape G, Terrill SE, Bongat AFG, Udan ML, Kamkhachorn T, Johnson HL, Nichols MR, Demchenko AV. Development of LPS antagonistic therapeutics: Synthesis and evaluation of glucopyranoside-spacer-amino acid motifs. RSC Adv. 2011;1:83–92. [Google Scholar]