

Abstract
G protein-coupled receptors (GPCRs) are increasingly recognized to operate from intracellular membranes as well as the plasma membrane. The β2-adrenergic GPCR can activate Gs-linkedcyclic AMP (cAMP) signaling from endosomes. We show here that the homologous human β1-adrenergic receptor initiates an internal Gs-cAMP signal from the Golgi apparatus. By developing a chemical method to acutely squelch G protein coupling at defined membrane locations, we demonstrate that Golgi activation contributes significantly to the overall cellular cAMP response. Golgi signalling utilizes a pre-existing receptor pool rather than receptors delivered from the cell surface, requiring separate access of extracellular ligands. Epinephrine, a hydrophilic endogenous ligand, accesses the Golgi-localized receptor pool by facilitated transport requiring the organic cation transporter 3 (OCT3) whereas drugs can access the Golgi pool by passive diffusion according to hydrophobicity. We demonstrate marked differences among both agonist and antagonist drugs in Golgi-localized receptor access, and show that β-blocker drugs presently used in the clinic differ markedly in ability to antagonize the Golgi signal. We propose ’location bias’ as a new principle for achieving functional selectivity of GPCR-directed drug action.
Introduction
GPCRs regulate diverse aspects of cellular and physiological responses and are considered therapeutic targets for intervening in numerous pathophysiological processes. A long-standing goal has been to understand how signalling specificity is achieved by a given GPCR. One way to achieve signalling specificity is through differential ligand affinity for different GPCRs, the long-recognized principle of receptor selectivity. Alternatively, ligands that bind the same GPCR can produce different effects by preferentially driving some downstream transduction pathways more strongly than others, the more recently recognized principle of functional selectivity. The present understanding of how functional selectivity occurs mechanistically is through differential stabilization of different GPCR conformational state(s) that shift receptor coupling from one G protein or arrestin mediator to another, the mechanism of agonist bias1,2.
A new cellular principle for generating functional selectivity has emerged from the discovery that various GPCRs initiate signalling from more than one subcellular location. For example, β2-adrenoceptors (β2ARs) stimulate a Gs-mediated cAMP response both from the plasma membrane and from endosomes after ligand-induced internalization3, with the endosome-initiated cAMP signal preferentially coupling to downstream transcriptional control4. A number of other GPCRs also signal after endocytosis, and there is now considerable evidence supporting the hypothesis that the location of signal initiation can affect both the specificity and timing of downstream events5-7. All presently known examples of such location bias are based on signalling after ligand-induced endocytosis of receptors from the plasma membrane. We wondered if this principle might extend further, beyond the realm of receptors that require physical transfer from the cell surface to endomembranes and, if so, whether this might reveal additional potential for generating functional selectivity in GPCR-directed drug action according to location.
Here we identify such an additional mechanism through study of the β1-adrenoceptor (β1AR), a close relative of the β2AR that, unlike the β2AR, internalizes poorly via clathrin-coated pits after ligand-induced activation 8. We initially assumed that the β1AR would provide a counterexample to β2AR, with β1AR initiating signalling only from the plasma membrane. We show here that β1ARs not only initiate signaling from the plasma membrane, but also from the Golgi apparatus. We directly demonstrate Golgi-localized β1AR and Gs activation using conformational biosensor technology and develop a novel strategy to acutely and specifically inactivate the Golgi signal, demonstrating that this internal membrane pool can contribute significantly to the overall cellular cAMP response elicited by β1AR agonists. A remarkable and distinguishing feature of Golgi-localized β1AR activation is that it is mediated by a pre-existing Golgi-localized receptor pool, further differentiating it from endosome signalling and requiring ligands to access separately. We describe two modes of alternate ligand access to the Golgi-localized membrane pool and find, through direct monitoring of β1AR activity, marked differences among drugs presently used in the clinic as β-blockers. Our findings reveal a discrete cellular basis for achieving functional selectivity in GPCR-directed drug action through location bias and differential ligand access.
Results
Golgi-localized β1AR activation by catecholamines
In adult cardiac myocytes endogenous β1AR but not β2AR has been shown to localize in a peri-nuclear distribution thought to represent nuclear membrane9. In these cells Golgi membranes are closely adjacent to the nuclear membrane, making them difficult to definitively resolve by light microscopy10,11, and a fraction of β1AR clearly localize to the Golgi in various cell types12. It is not known if the Golgi localized fraction of β1AR represents simply a reserve pool or if it also plays a role in catecholamine signalling. To address this question, we utilized a biosensor of activated β2AR derived from a conformation-specific single-domain camelid antibody (Nb80) 3,13,14. We reasoned that this nanobody, which selectively binds the agonist-occupied β2AR, might also recognize activated β1AR based on extensive sequence homology in the region recognized by Nb80 (Supplementary Results, Supplementary Fig 1a). This proved to be the case; in β1AR expressing cells maintained in the absence of agonist, Nb80 fused to GFP (Nb80–GFP) localized diffusely through out the cytoplasm (Fig. 1a, 0 min, top row). After application of the adrenergic endogenous ligand, epinephrine (10 μM) at 37°C, Nb80–GFP was rapidly recruited to the plasma membrane (Fig. 1a, 2.5 min, middle row). Nb80–GFP was recruited to internal membranes two to three minutes later. Co-localization analysis defined Golgi membranes as a major site of epinephrine-induced Nb80-GFP recruitment (Fig. 1a, 20min, bottom row, Pearson’s coefficient= 0.67, see also Supplementary Video 1), overlapping the pool of Golgi-localized β1AR (Fig 1c). These results suggest that the extracellular ligand promotes conformational activation of β1ARs not only the plasma membrane, but also in the Golgi apparatus.
Figure 1. Golgi localized β1AR achieve an activated conformation upon extracellular ligand application.
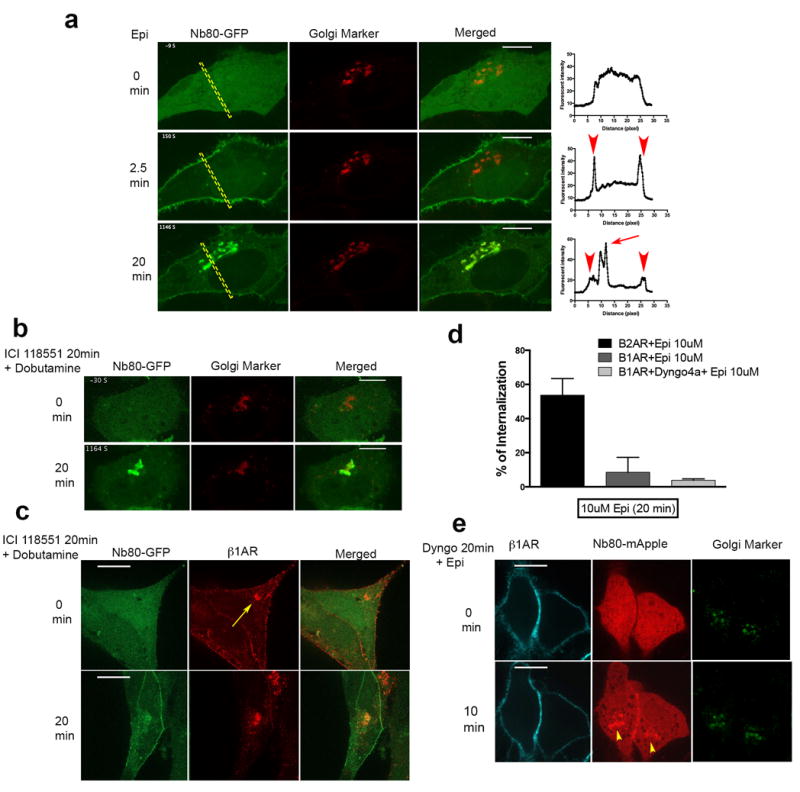
a, Representative β1AR expressing HeLa cell showing Nb80-GFP (green) and Golgi Marker (GalT-mRFP) (red) localization at the indicated time after 10 μM epinephrine addition (n = 18 cells, Pearson’s coefficient=0.67, 4 biological replicates, left); Representative individual Nb80–GFP line scans (arrowheads and arrow indicate plasma membrane and Golgi recruitment, respectively, right). b, Representative β1AR expressing HeLa cell showing Nb80–GFP (green) and Golgi Marker (GalT-mRFP) (red) localization at the indicated time after 10 μM dobutamine addition, cells were pre-treated with 10 μM ICI118551 (β2AR selective antagonist) 20 min before addition of dobutamine (n = 25 cells, Pearson’s coefficient=0.71, 7 biological replicates). c, Confocal images showing permeabilized cells labelled with conjugated Alexa-555 flag tagged-β1AR (red) and Nb80-GFP (green) localization at the indicated time after 10 μM dobutamine addition, cells were pre-treated with 10 μM ICI118551 (β2AR selective antagonist) 20 min before addition of dobutamine (n=19 cells, 2 biological replicates, arrow indicate Golgi-localized β1AR). d, Comparison of β2AR and β1AR internalization after 20 min 10 μM epinephrine treatment in HeLa cells by flow cytometry with and without Dyngo-4a treatment (bars represents mean ± s.e.m., 3 biological replicates). e, Representative surface-labelled β1AR expressing HeLa cell pre-treated with 30 μM Dyngo-4a (dynamin inhibitor) and labelled with conjugated Alexa-647 flag tagged-β1AR (cyan), Nb80-mApple (red) and Golgi Marker (GalT-GFP) (green) at the indicated time after 10 μM epinephrine addition (n = 10 cells, Pearson’s coefficient=0.5, 4 biological replicates, Yellow arrowheads indicates Golgi localization). Scale bars, 10 μm.
To exclude a potential contribution of endogenous β2ARs we used the β1AR selective agonist, dobutamine, and pre-treated cells with a β2AR selective antagonist (ICI118551, 10μM). Robust Nb80-GFP recruitment to Golgi membranes was still observed, verifying that Golgi activation is mediated specifically by β1ARs (Fig. 1b, Pearson’s coefficient= 0.71, see also Supplementary Video 2).
The data so far suggest that Golgi localized β1AR achieve an activated conformation upon extracellular ligand application and shortly after receptor activation on the plasma membrane. We next investigated if activation of β1AR at the Golgi requires receptor activation on the plasma membrane and whether agonist is delivered to the internal membrane via receptor endocytosis. A number of agonists have been demonstrated to co-internalize with their cognate receptors into the same internal membrane compartment15-17. β1ARs, in contrast to β2ARs, display minimal internalization upon activation on the plasma membrane (Fig. 1d) 8,18 and this process has recently been proposed to require endophilin and dynamin but not clathrin19. To test whether activated β1AR on the Golgi is dependent on β1AR endocytosis from the plasma membrane, we used a dynamin inhibitor, Dyngo-4a, to acutely block all dynamin-dependent endocytosis mechanisms. Ligand-induced endocytosis of β1AR was undetectable in the presence of Dyngo-4a (Fig. 1d) but ligand-induced recruitment of Nb80-mApple to Golgi membranes was still observed (Fig. 1e, Pearson’s coefficient= 0.5). This suggests that β1AR activation at the Golgi is independent of its activation at the plasma membrane. Supporting this, we found that sotalol, amembrane-impermeant antagonist that blocks β1AR activation selectively at the plasma membrane, had no effect on receptor activation at the Golgi by the membrane-permeant agonist dobutamine (Supplementary Fig. 1b, Pearson’s coefficient=0.46). All together, these data suggest that β1AR resides on the Golgi at steady state and its activation by the extracellular ligand does not require receptor activation or internalization from the plasma membrane.
Golgi-localized β1ARs activate Gs
Because the Golgi-localized β1AR pool can clearly undergo conformational activation, we next asked if receptors can activate their cognate G protein, Gs, at the Golgi. Heterotrimeric G proteins and adenylyl cyclase can be observed at the Golgi as well as the plasma membrane, supporting the concept of Golgi-based G protein signalling20-24. More recently, it has been demonstrated that activation of KDEL receptor by cargo transport from the endoplasmic reticulum to the Golgi triggers a cascade that involves Gs and adenylyl cyclase activation at Golgi membranes25. We observed detectable localization of both over-expressed and endogenous Gs on the Golgi at steady state (Fig. 2a, Pearson’s coefficient=0.62 and 0.48 for overexpressed and endogenous Gs, respectively). To directly investigate β1AR-mediated Gs activation at the Golgi, we utilized Nb37-GFP, which specifically recognizes a transient intermediate in the process of Gs activation and thus serves as a local Gs activation biosensor3,26. Nb37–GFP localized in the cytoplasm of untreated cells and was rapidly recruited to the plasma membrane in response to agonist application and co-localized there with β1ARs (Fig. 2b, white inset shows higher magnification on the right). Nb37–GFP was then recruited to Golgi membranes, with a strong signal observed within 20 minutes after agonist application (Fig. 2b, yellow inset shows higher magnification on the right; Pearson’s coefficient= 0.33), see also Supplementary Video 3 and 4).Golgi-localized Gs activation was indeed mediated by β1ARs because it was elicited also by the β1AR-selective agonist dobutamine (Supplementary Fig. 2a, Pearson’s coefficient=0.6). All together, these data suggest that Gs associates with Golgi membranes at steady state and can be activated locally byβ1ARs in response to dobutamine.
Figure 2. Golgi-localized β1AR activates Gs-mediated cAMP response.
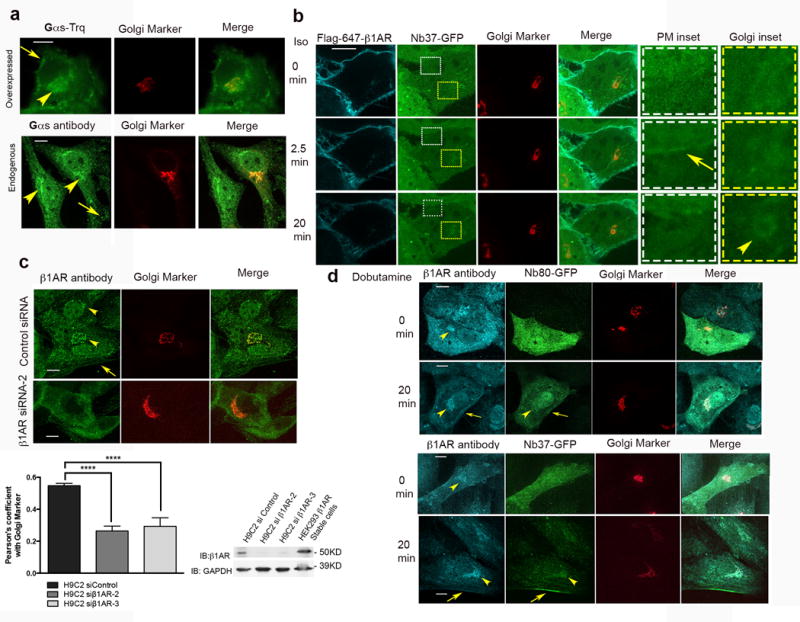
a, Confocal image of HeLa cell showing Gs-turquoise (top panel) or endogenous Gs (bottom panel) in green and Golgi marker (GalT-mRFP) in red (representative of n = 12 and 35 cells, Pearson’s coefficient=0.62 and 0.48 respectively, 2 biological replicates). Arrow and arrowheads indicate Gs plasma membrane and Golgi localization.b, Confocal images showings surface-labelled β1AR (cyan), Nb37–GFP (green) and GalT-mRFP (red) at indicated time points after agonist addition (representative of n = 12 cells, Pearson’s coefficient=0.33, 3 biological replicates). Arrow and arrowhead indicate Nb37-GFP recruitment to the PM and Golgi, respectively. Insets show higher magnification. c. Top, confocal images of H9C2 cells treated with control or β1AR siRNA and labelled with anti-β1AR (green) and GalT-mRFP (red) (representative of n = 44 and 46 cells respectively, Pearson’s coefficient=0.55 and0.26 from 5 and 3 biological replicates). Bottom-left, average Pearson’s coefficient of anti-β1AR antibody (green) and GalT-mRFP (red) in control or β1AR knockdown cells (bars represents mean ± s.e.m., 3 biological replicates; p<0.0001 by two-tailed unpaired t-test). Bottom-right, representative immunoblots from control and β1AR knockdown cells blotted for anti-β1AR antibody and GAPDH.Full images shown in Supplementary Figure 3a.Average knockdown was 82% of control (mean±SD, n=2). d. Confocal images of H9C2 cells expressing Nb80-GFP or Nb37-GFP before and after 10μM dobutamine treatment using anti-β1AR antibody (cyan) (representative of n = 24,16, 20 and 20 cells respectively, 3 biological replicates, arrows and arrowheads indicate plasma membrane and Golgi, respectively).Scale bar, 10 μm.
Endogenous β1AR localizes at the Golgi and activates Gs
Endogenous β1AR has been shown to localize at the peri-nuclear regions in cardiac myocytes9. Additionally, previous studies using both biochemical fractionation and immunofluorescence microscopy, have provided evidence for Gs localization at Golgi membranes20,24. To ask if activation of endogenous β1AR and Gs can occur at the Golgi, we examined H9C2 cells, a rat myoblast cell line that expresses endogenous β1ARs 27. Using an antibody recognizing native β1AR, we detected localization both to the plasma membrane and to Golgi membranes in H9C2 cells (Fig. 2c top, Pearson’s coefficient=0.55). We verified specificity of endogenous receptor detection by knocking down β1AR using two different siRNAs (Fig 2c bottom, Pearson coefficient for β1AR si-2 and 3 = 0.26 and 0.29 respectively, full images of blots are shown in supplementary Fig 3a). Importantly, both Nb80-GFP and Nb37-GFP were recruited to Golgi upon addition of the β1AR-selective agonist dobutamine (Fig. 2d top and bottom panels, respectively; also see Supplementary Fig 2b), as verified quantitatively by Pearson’s correlation analysis of Nb80-GFP or Nb37-GFP localization relative to Golgi marker calculated before and after addition of dobutamine (Supplementary Fig 3 b and c). All together, these results indicate that endogenous β1ARs reside in the Golgi apparatus and can activate Gs from this location.
Golgi-localized β1ARs contribute tothe cAMP response
Nb80 can be used to block β2AR-mediated signalling when expressed in the cytoplasm at high levels, presumably through steric occlusion of the β2AR–Gs binding interface, although this requires Nb80 to be present at much higher concentration than when used as an activation biosensor28. Because Nb80 also recognizes activated β1AR, and β1AR engages Gs in a similar manner as β2AR, we reasoned that Nb80 might be useful for blocking β1AR–Gs coupling as well. We further reasoned that, because signal inhibition requires Nb80 to be present at high concentration, that it might be possible to use Nb80 as a location-specific quencher through enforced proximity mediated by rapamycin-induced heterodimerization of the FK506-binding protein (FKBP) with the FKBP-rapamycin binding domain of FRAP (FRB)29. We fused FRB to the N-terminus of Nb80 (FRB-Nb80) and FKBP to a plasma membrane localization sequence derived from Lyn30 (PM-FKBP) or to the Golgi resident protein GalT (Golgi-FKBP). We verified rapamycin-induced recruitment of FRB-Nb80 specifically to the plasma membrane in cells co-expressing PM-FKBP (Fig. 3a, top) and to Golgi membranes in cells co-expressing Golgi-FKBP (Fig. 3a, bottom). Recruiting FRB-Nb80 to the plasma membrane partially reduced the β1AR-Gs cellular cAMP response elicited by dobutamine (Fig. 3b, see also the time course in Supplementary Fig 4a). Chemical recruitment of FRB-Nb80 to Golgi membranes also produced a partial (and somewhat more pronounced) response inhibition (Fig. 3c, see also the time course in Supplementary Fig 4c). Verifying specificity, rapamycin produced no significant change in the cAMP response elicited by forskolin, a direct activator of adenylyl cyclase (Fig. 3d). Verifying that plasma membrane recruitment and Golgi recruitment of FRB-Nb80 suppress the cAMP response by inhibiting different pools of β1AR-Gs coupling, sotalol, a membrane-impermeant antagonist, produced an additive inhibitory effect in cells with FRB-Nb80 targeted to Golgi membranes but did not produce a significant additive effect in cells with FRB-Nb80 targeted to the plasma membrane (Fig. 3 b and c, see also Supplementary Fig 4a-c). All together, these data suggest that Golgi-localized β1ARs can contribute significantly to the overall cAMP response.
Figure 3. Rapamycin-inducible recruitment of Nb80 blocks β1AR-Gs coupling and inhibits Gs-mediated cAMP response at both the plasma membrane and the Golgi.
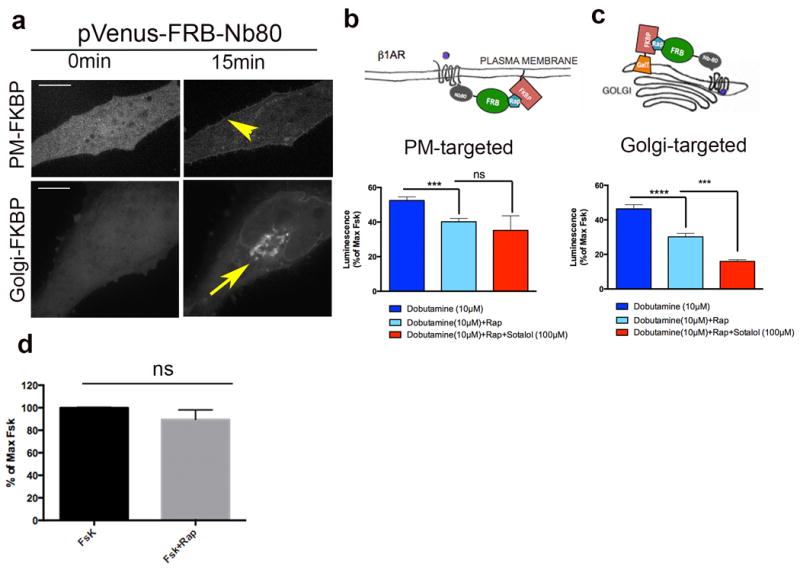
a, Representative images showing pVenus-FRB-Nb80 localization before and after rapamycin addition in HeLa cells expressing PM-FKPB (11-lyn-FKBP), top; or Golgi-FKBP (FKBP-GalT), bottom; (arrowhead and arrow shows plasma membrane and Golgi recruitment of pVenus-FRB-Nb80). b, Top, model for blocking β1AR-Gs coupling at the plasma membrane after recruitment of pVenus-FRB-Nb80. Bottom, forskolin-normalized β1AR-mediated cAMP response in the absence (dark blue bar) or presence (light blue bar) of 15 min pre-treatment of 1 μM rapamycin, rapamycin plus 100μM sotalol (red bar) (mean ± s.e.m., n = 4 biological replicates). Statistical significance was tested using two tailed unpaired t test (p =0.0008 and 0.3744 respectively). c, Top, model for blocking β1AR-Gs coupling at the Golgi membrane after recruitment of pVenus-FRB-Nb80. Bottom, forskolin-normalized β1AR-mediated cAMP response in the absence (dark blue bar) or presence (light blue bar) of 15 min pre-treatment of 1 μM rapamycin or rapamycin plus 100μM sotalol (red bar) (mean ± s.e.m., n = 7 biological replicates). Statistical significance was tested using two tailed unpaired t test (p <0.0001 and 0.0001, respectively). d. Effect of 1μM rapamycin on forskolin-mediated cAMP response (n=10 biological replicates). Scale bars, 10 μm.
Two modes of ligand access to the Golgi-localized β1AR pool
A key question raised by these observations is how ligands gain access to Golgi-localized β1ARs. Whereas dobutamine is a drug that is even more hydrophobic than alprenolol (logP 3.4 compared to 3.1; pubchem.ncbi.nlm.nih.gov), a membrane-permeant ligand31, epinephrine is a relatively hydrophilic ligand (log P -1.4) and is membrane-impermeant. Therefore, while it seems likely that dobutamine can access the Golgi-localized receptor pool by passive diffusion, this cannot explain the ability of epinephrine to activate the Golgi pool (Fig. 4a, table). A clue to resolving this conundrum came from the observation that epinephrine-induced activation of the Golgi-localized β1AR pool, assessed by Nb80-GFP recruitment, is blocked at low temperature (Supplementary Fig 5a-top row, Pearson’s coefficient=0.21) while dobutamine-induced recruitment still occurs (Supplementary Fig. 5a -bottom row, Pearson’s coefficient=0.51). Additionally, these ligands differ in the kinetics with which they activate the Golgi pool at 37˚C, with dobutamine driving Golgi-localized Nb80-GFP recruitment more rapidly (Supplementary Fig 5b). Together, these results support the hypothesis that epinephrine accesses the Golgi-localized β1AR pool in a different manner than dobutamine, requiring a time- and temperature-dependent process distinct from passive diffusion.
Figure 4. Epinephrine accesses the Golgi-localized β1AR pool via corticosterone-sensitive membrane transport.
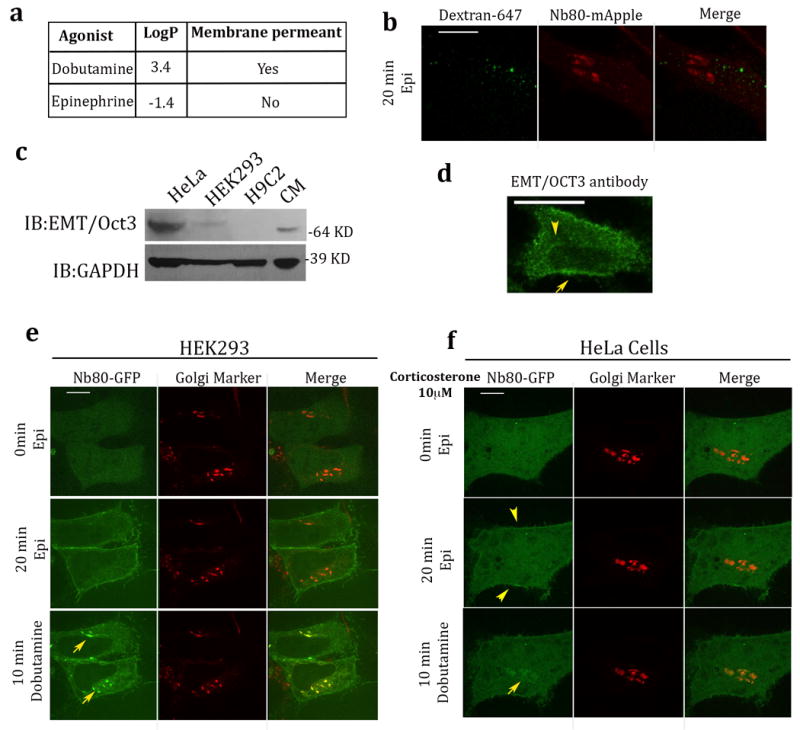
a, Examples of membrane permeant and membrane impermeant β adrenergic agonists. b, Confocal image of HeLa cell expressing β1AR and Nb80-mApple, showing distinct localization of Nb80-mApple (red) and Dextran-647 (green) after pulse-chased labelling with Dextran-647 for 20 min and 10μM epinephrine treatment at the same time (n = 25 cells, Pearson’s coefficient=0.18, 3 biological replicates).c. Lysates from indicated cells were probed with anti-SLC22A3 (anti-EMT/OCT3) and anti- GAPDH antibody to control for equal loading and expression. Full images shown in Supplementary Figure 6. d. Confocal image of HeLa cells stained with anti-SLC22A3 (anti-EMT/OCT3) showing plasma membrane (yellow arrow) and intracellular membrane (yellow arrowhead) localization (n = 26 cells, 3 biological replicates). e. Representative β1AR expressing HEK293 cell showing Nb80–GFP (green) and Golgi Marker (GalT-mRFP) (red) localization at the indicated time after 10 μM epinephrine (middle row, Pearson’s coefficient=0.26) and dobutamine (bottom row, Pearson’s coefficient=0.63) addition (n = 11 cells, 2 biological replicates). f. Representative β1AR expressing HeLa cell showing Nb80–GFP (green) and Golgi Marker (GalT-mRFP) (red) localization at the indicated time after 10 μM epinephrine (middle row, Pearson’s coefficient=0.15) and dobutamine (bottom row, Pearson’s coefficient=0.68) addition. Cells were pre-treated with 10μM corticoesterone for 15 min (n = 12 cells, 3 biological replicates). Scale bars, 10 μm.
We first considered the possibility that epinephrine accesses the Golgi β1AR pool by fluid phase uptake and vesicular transport. This appeared unlikely because fluorescent Dextran-647, a fluid phase uptake marker, was not detected in Golgi membranes and instead accumulated in distinct endosomal membranes (Fig. 4b, Pearson’s coefficient=0.18). We next considered the possibility that epinephrine accesses the Golgi pool through a membrane transporter. An extraneuronal monoamine transporter (EMT), also known as an organic cation transporter-3 (OCT3), has been shown to facilitate intracellular uptake of epinephrine and other catecholamines in cardiac cells32,33. EMT/OCT3 expression was clearly detected in HeLa cells, whereas it was undetectable in HEK293 cells when assessed at either the mRNA or protein level (Supplementary Fig 5c and Fig. 4c, full images of blots are shown in supplementary Fig 6). EMT/OCT3 is a bidirectional transporter protein that localizes to both the plasma membrane and intracellular membranes in cardiac cells 33, and we observed a similar localization in HeLa cells (Fig. 4d and Supplementary Fig. 5d). Interestingly, the strong Golgi recruitment signal elicited bye pinephrinein HeLa cells (Fig. 1a) was not observed in HEK293 cells (Fig. 4e, middle row, Pearson’s coefficient=0.26). Nevertheless, dobutamine remained able to stimulate Nb80-GFP recruitment to Golgi membranes in both cell types (Fig. 4e, bottom row, Pearson’s coefficient=0.63). A characteristic feature of EMT/OCT3 is that its transport activity is blocked by corticosterone 34 and, remarkably, pre-incubation of HeLa cells with corticosterone prevented epinephrine from driving visible Nb80-GFP recruitment to Golgi membranes while Nb80-GFP recruitment to the plasma membrane was unaffected (Fig. 4f, middle row, Pearson’s coefficient=0.15). By contrast, dobutamine-induced recruitment of Nb80-GFP to Golgi was not blocked in the same cells (Fig. 4f, bottom row, Pearson’s coefficient=0.68). All together, these data indicate that chemically distinct ligands can reach the Golgi membranes and activate Golgi-localized β1ARs using different mechanisms, diffusion in case of a hydrophobic agonist such as dobutamine and via extraneuronal monoamine transporters for the hydrophilic agonist epinephrine.
Epinephrine access to the Golgi-localized β1AR pool through a transporter-mediated process implies that this catecholamine is exposed to the cytoplasm for at least some time during transit. Cardiac cells express monoamine oxidase type A (MAO-A) on the mitochondrial outer membrane, and oxidative deamination catalysed by MAO-A is a major pathway of cytoplasmic epinephrine degradation35. HeLa cells are not known to express MAO-A, raising the question of whether the proposed route of epinephrine access to the Golgi-localized β1AR pool is plausible in cells that express this enzyme. To begin to investigate this, we overexpressed in HeLa cells a GFP-tagged MAO-A construct and then used Nb80-mApple as a biosensor to assess activation of the Golgi pool. Epinephrine was still able to produce clearly detectable activation of the Golgi-localized β1AR pool in cells that were verified (by GFP fluorescence) to express recombinant MAO-A (Supplementary Fig. 5e). This indicates that transporter-dependent activation of Golgi-localized β1ARs is plausible even in cells expressing MAO-A, but leaves unresolved if Golgi activation occurs in response to endogenous levels of catecholamine release.
Selective inhibition by β-blocker drugs
We next considered the possibility that antagonists also differ in access to the Golgi-localized β1AR pool. Differences between antagonists in hydrophobicity are known to affect drug access to the central nervous system according to drug permeability across the blood-brain endothelial barrier36,37. We hypothesized that a similar principle affects antagonist access to the Golgi-localized β1AR pool. If so, antagonist drugs could provide an independent means to investigate the contribution of Golgi-localized β1ARs to the overall cellular signalling response. To test this we compared the ability of metoprolol and sotalol, β1AR antagonists that differ ~50-fold in relative hydrophobicity and thus are anticipated to differ in membrane-permeance (Fig. 5a, table). We tested relative access to each receptor pool by assessing effects on dobutamine-stimulated recruitment of Nb80-GFP.Metoprolol fully reversed Nb80-GFP recruitment to both the plasma membrane and Golgi membranes (Fig. 5b top panel, also see Supplementary Video 5) whereas sotalol selectively blocked recruitment to the plasma membrane but not to Golgi membranes, even when applied at 10-fold higher concentration (Fig. 5b middle and bottom panel, also see Supplementary Video 6). We then compared the ability of these antagonists to block the overall dobutamine-stimulated cellular cAMP response. Preincubation of cells with either metoprolol or sotalol significantly inhibited the cellular response(Fig. 5c and d, first two bars), consistent with the ability of both drugs to block plasma membrane activation. However, sotalol produced less inhibition even when applied at very high concentration (Fig. 5c and d, light blue and grey bars 100μM and 5mM respectively). These findings further support the ability of Golgi-localized β1ARs to contribute to the overall cellular cAMP response and, as both metoprolol and sotalol are presently used as ‘β–blockers’, they suggest that differential access to the Golgi-localized pool may contribute to functional selectivity among clinically relevant antagonists.
Figure 5. Pharmacological manipulations differentially regulate β1AR compartmentalized signalling.
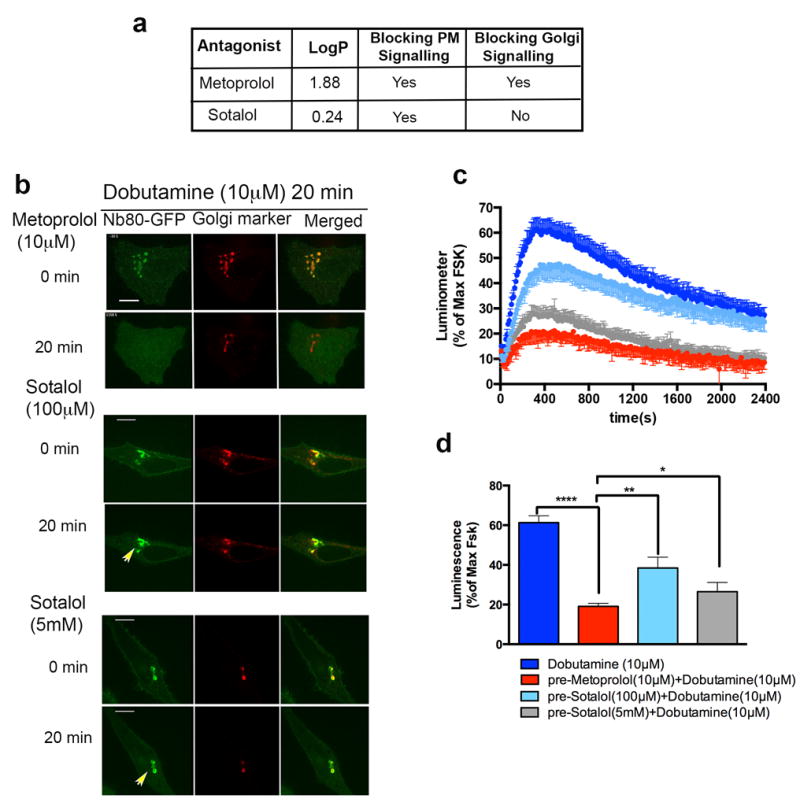
a, Examples of membrane permeant and membrane imperment β adrenergic antagonist and their effects on plasma membrane and Golgi signalling. b, Confocal image frames of activated β1AR expressing HeLa cells with 10 μM dobutamine showing Nb80-GFP (green) and Golgi marker colocalization followed by reversal with 10 μM Metoprolol (top) (n= 3 cells, 3 biological replicates) or 100μM Sotalol (middle) (n= 5 cells, 3 biological replicates) or 5mM Sotalol (bottom) addition at indicated time points (arrowheads indicate Golgi localization of Nb80-GFP after addition of 100μM and 5mM Sotalol for 20min). c, Time course and d. maximum forskolin-normalized β1-AR-mediated cAMP response by 10 μM dobutamine (dark blue), pre-treated with 10 μM Metoprolol (red), 100 μM sotalol (light blue), 5mM sotalol (grey) (mean ± s.e.m., n=3 biological replicates). Statistical significance was tested using two tailed unpaired t test (p <0.0001, p =0.0035 and p =0.034 respectively). Scale bars, 10 μm.
The Golgi β1AR pool can locally activate PKA
Protein Kinase A (PKA) holoenzymes are localized to distinct subcellular locations through their association to A-kinase anchoring proteins (AKAPs). Major PKA holoenzymes, type I and type II, are distinguished by the identity of their regulatory subunits (RI or RII isoforms)38, with type II holoenzymes known to localize to the Golgi apparatus39.PKA activation by cAMP causes the Cα catalytic subunit to dissociate into the cytoplasm while RII regulatory subunits remain Golgi-associated40-42. To test whether activation of Golgi-localized β1AR can cause PKA activation on the Golgi membrane, we imaged both subunits of a type II holoenzyme (Cα-YFP and RIIβ-mCherry) in living cells. Dobutamine induced the rapid translocation of the Cα subunit from Golgi membranes to the cytoplasm in β1AR-expressing HeLa cells, whereas the RII subunit remained Golgi-localized (Fig 6a and d, blue line; Supplementary Video 7). We then asked if this translocation can be elicited specifically by activation of the Golgi-localized β1AR pool. To test this we compared the effect of metoprolol and sotalol on dobutamine-induced PKA Cαtranslocation. The membrane-permeant antagonist metoprolol fully blocked PKA Cα Golgi translocation (Fig 6c and d, red line; supplementary Video 8) whereas sotalol did not, even when applied at very high concentration (5mM) (Fig 6b and d, grey line; supplementary Video 9). Further, dobutamine-induced dissociation of Cα from Golgi membranes was delayed in sotalol-treated cells, suggesting that plasma membrane and Golgi-localized β1AR pools promote activation of Golgi-localized PKA with distinct kinetics.
Figure 6. Pharmacological manipulations differentially regulate β1AR-mediated PKA activation on the Golgi.
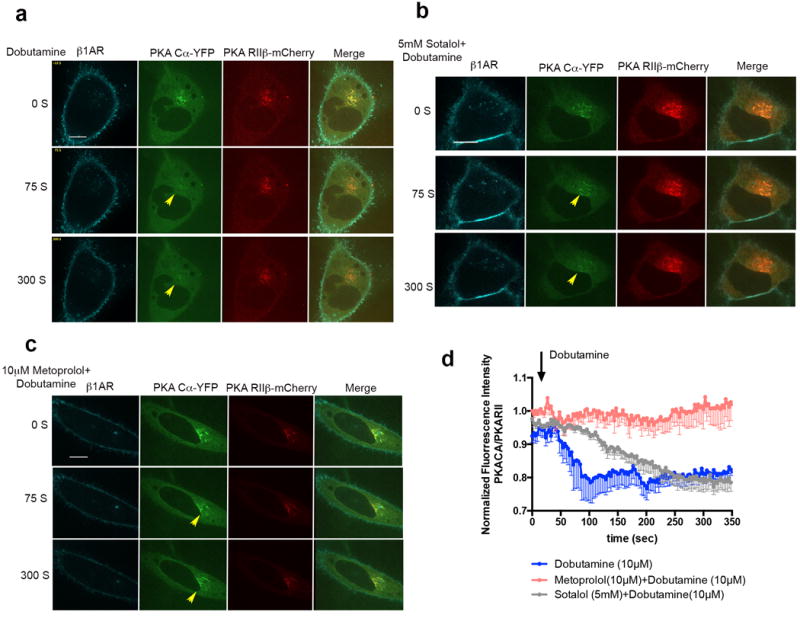
a. Confocal image frames of β1AR expressing HeLa cells treated with 10 μM dobutaine at indicated time points (n= 7 cells, 4 biological replicates). b. Confocal image frames of β1AR expressing HeLa cells pre-treated with 5mM sotalol and upon addition of 10μM dobutaine at indicated time points (n= 9 cells, 4 biological replicates). c. Confocal image frames of β1AR expressing HeLa cells pre-treated with 10μM metoprolol and upon addition of 10μM dobutamine at indicated time points (n= 5 cells, 4 biological replicates). d. Kinetics of PKA Cα-YFP translocation from the Golgi membrane in cells treated with 10μM dobutamine (blue), 10μM metoprolol+10μM dobutamine (red) or 5mM sotalol+ 10μM dobutamine; the fluorescent intensity of PKA Cα-YFP was normalized by PKA RIIβ-mCherry fluorescent (n= 7, 5 and 9, 4 biological replicates) (see methods).Scale bars, 10 μm.
Discussion
The present findings provide direct evidence for adrenergic signalling from the Golgi apparatus and demonstrate that catecholamines can reach the internal pool to initiate canonical β1AR-Gs signalling from this location. Thus Golgi-localized β1ARs do not simply represent intermediates for later delivery to the plasma membrane, but they also comprise a functional signalling pool at this location. Selective inhibition of β-AR-Gs coupling by chemical recruitment of nanobody independently verifies the signalling potential of the Golgi-localized β1AR pool and introduces a new approach for functionally interrogating compartmentalized GPCR signalling more generally.
The present study provides evidence for two routes of ligand access to the Golgi localized β1AR pool. Drugs such as dobutamine access the Golgi pool by passive diffusion, determined largely by degree of lipophilicity43. The catecholamine agonist epinephrine is relatively hydrophilic and membrane-impermeantlig and whose access to the Golgi-localized β1AR pool requires OCT3, a member of the extraneuronal monoamine transporter (EMT) / organic cation transporter (OCT) family that is already known to function in cellular catecholamine uptake32. OCT3 exists both in the plasma membrane and internal membranes and can operate in a bidirectional manner43. Whether OCT3 is fully responsible for epinephrine uptake to the Golgi-localized β1AR pool, or if additional transporter(s) are also required or contribute, is an interesting question for future study. Another interesting question that remains to be answered is whether the transporter-dependent ligand access route is relevant to native β1AR signalling elicited by endogenously produced catecholamines or represents a drug-specific target.
The effect of different antagonists on the Golgi-localized signal has interesting implications for pharmacology.We speculate that differential drug access to the Golgi pool contributes to diversity in the clinical effects of existing β blocker drugs. Evidence suggests that metoprolol and propranolol, both hydrophobic antagonists, cause reduction of chronotropic (heart rate) and inotropic (force of contraction) responses, whereas sotalol can only cause a reduction in the heart rate44,45. The present study has focused on cAMP signalling but we note that heterotrimeric G proteins are thought to also regulate membrane trafficking processes, both at the Golgi and in endosomes11,25,46,47. Thus it is possible that differential drug effects on Golgi-localized β1AR activation may also have trafficking consequences.
The β2AR, a closely homologous adrenergic receptor, can mediate Gs signalling from endosomes as well as the plasma membrane 3, with the endosome-initiated signal preferentially promoting downstream cAMP-dependent transcription4. It remains unknown if Golgi-localized β1ARs also have distinct functions compared to plasma membrane localized β1ARs. Although both β1ARs and β2ARs couple to Gs and activate adenylyl cyclase, they have different effects on physiological responses such as regulating heart rate (chronotropic), contractility (inotropic) and the rate of relaxation (lusitropic) in cardiac myocytes8. β1AR plays a major role in regulating, inotropic and lusitropic effects, whereas β2AR mainly regulates chronotropic effects and have significantly smaller inotropic effect and no lusitropic effect48,49. The underlying reasons for these distinctions remain unclear but there is already evidence that differences in downstream consequences of β1AR versus β2AR activation can result from differences in receptor localization between lateral microdomains of the plasma membrane50. The present results expand this concept by revealing that β1AR and β2AR initiate signalling from different internal membrane compartments and these are differentially accessible by ligands.
ONLINE METHODS
Cell Culture, cDNA constructs and transfection
HeLa, HEK293 and H9C2 cells (purchased from ATCC as authenticated lines CCL-2, CRL-1573 and CRL 1446 respectively) were grown in Dulbecco’s minimal essential medium supplemented with 10% Fetal Bovine Serum (FBS)(UCSF Cell Culture Facility) without antibiotics. Cell cultures were free of mycoplasma contamination. pVenus-FRB-Nb80 was created by amplifying Nb80 and FRB from Nb80-GFP3, and pC4-RHE plasmid (ARIAD Pharmaceuticals), using 5’-CGAATCTCAAAGGATAGTGCTGGTAGTGCTGGTGGACAGGTGCA -3’; 5’- GGATCCTCATGAGGAGACGGTGACCTGGGT -3’ and 5’- GAATTCAATCCTCTGGCATGAGATGTGGCATGAAGGCCTGGAAGA -3’; 5’- CGAATCTCAAAGGATAGTGCTGGTAGTGCTGGTGGACAGGTGCA -3 primers respectively, such that it contained the linker sequence GATAGTGCTGGTAGTGCTGGTGGAC, and inserted into the pVenus-C1 vector using EcoRI and BamH1. FKBP-GalT-mApple was created by amplifying FKBP and GalT from KDELr-FKBP47 and GalT-mCherry plasmids (a generous gift from Dr.Farese lab), using 5’- CATGCTAGCGCCGCCACCATGGGAGTGCAGGTGGAAACCAT-3’, 5’-GAGCTCGAGACCAGCACTACCAGCACTATCCTCCAGCTTCAGCAGCTCCACG3’ and 5’- GCTCAAAGCTTGCCGCCACCGGAAGGCTTCGGGAGCCG-3’, 5’- ACCGGATCCTTAGGCCCCTCCGGTCCGGAGCTCCCCG-3’ primers, respectively and inserted into the pmApple-N1 vector using NheI, XhoI for FKBP and HindIII and BamHI for GalT. Mouse PKA Cα-YFP and PKA RIIβ-mCherry constructs were generous gits from Dr. Xiang lab. Transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. ADRB1-2 (5’-CGUCCGUCGUCUCCUUCUATT-3’) and 3 (5’GGGUGUUCCGCGAGGCCCATT-3’) siRNAs (Qiagen) were used to knockdown β ARs. siRNA transfections were performed using RNAiMax (Invitrogen) according to the manufacturer’s instructions. Flag tagged human βAR constructs were labelled with Alexa-647 or 555-conjugated M1 anti-FLAG monoclonal antibody (Sigma) as described previously 51. Anti-SLC22A3 antibody (abcam-ab124826) and anti-beta 1 adrenergic receptor antibody (abcam-ab3442) were used to detect endogenous levels of EMT/OCT3 and β1AR, respectively. Anti Gαs/olf antibody (SC-383) was used to detect endogenous Gαs in HeLa cells.
Live-cell confocal imaging
Live cell imaging was carried out using Yokagawa CSU22 spinning disk confocal microscope with a 100 ×, 1.4 numerical aperture, oil objective and a CO2 and 37 °C temperature-controlled incubator. A 488, 568 nm and 640 Coherent OBIS laser were used as light sources for imaging GFP and Flag signals, respectively. Cells expressing both Flag-tagged receptor (2μg) and the indicated nanobody–GFP (100ng) were plated onto glass coverslips. Receptors were surface labelled by addition of M1 anti-Flag antibody (1:1000, Sigma) conjugated to Alexa 555 (A10470, Invitrogen) to the media for 10 min, as described previously. Indicated agonist (isoprenaline-Sigma, epinephrine-Sigma, dobutamine-Sigma), or antagonist (ICI 118551- TOCRIS, metoprolol-Sigma, sotalol-Sigma) were added and cells were imaged every 3 s for 20 min in DMEM without phenol red supplemented with 30 mM HEPES, pH 7.4 (UCSF cell culture facility). Time-lapse images were acquired with a Cascade II EM-charge-coupled device (CCD) camera (Photometrics) driven by Micro-Manager 1.4 (http://www.micro-manager.org).
Fixed-cell confocal imaging
Cells were permeabilized with saponin to reduce the cytoplasmic background, as described previously52. Briefly, transfected cells with indicated constructs or non-transfected H9C2 cells were permeabilized with 0.05% saponin (Sigma) in PEM buffer (80mM K-Pipes, pH 6.8, 5 mM EGTA, 1mM MgCl2) for 5 min on ice. Cells were then fixed with 3% paraformaldehyde in PBS for 10 min and then quenched with 50mM Nh4Cl in PBS for 15 min. Primary antibodies (β1AR antibody (abcam-ab3442) 1:100, M1 anti-Flag mouse antibody (1:1000, Sigma) conjugated to Alexa 647, anti GFP mouse antibody (1:500, Sigma) and anti GFP Rabbit antibody (1:500, Life Technologies) were diluted in PBS supplemented with 0.05% saponin. Confocal images were taken using Yokagawa CSU22 spinning disk confocal microscope with a 100x or 60x 1.4 numerical aperture, oil objective.
Image analysis and statistical analysis
Images were saved as 16 bit TIFF files. Quantitative image analysis was carried out on unprocessed images using ImageJ software (http://rsb.info.nih.gov/ij). Analysis of Nb80–GFP intensity profile along the segmented was carried out using the ImageJ plot profile function. For estimating kinetics of Nb80–GFP recruitment at the Golgi membrane over time in confocal images, individual cells were selected manually and average fluorescence intensity was measured at the Golgi area in each frame and normalized to the fluorescence intensity of the Golgi marker. A blank area of the image lacking cells was used to estimate background fluorescence. For estimating kinetics of PKACα YFP translocation from the Golgi membrane over time in confocal images, individual cells were selected manually and average fluorescence intensity was measured at the Golgi area in each frame and normalized to the fluorescence intensity of the PKA RIIβ-mCherry. A blank area of the image lacking cells was used to estimate background fluorescence. Co-localization analysis at the Golgi was estimated by calculating the Pearson’s coefficient between the indicated image channels with the Golgi marker channel, using the co-localization plug-in for ImageJ (Coloc2).For visual presentation (but not quantitative analysis) image series were processed using Kalman stack filter in ImageJ.P values are from two-tailed unpaired Student’s t-tests calculated using Prism 6.0 software (GraphPad Software).
Luminescence-based cAMP assay
HEK 293 cells stably expressing β1AR were transfected with a plasmid encoding a cyclic-permuted luciferase reporter construct, (pGloSensor-20F, Promega) and luminescence values were measured, as described previously3. Briefly, cells were plated in 24-well dishes (~200,000 cells per well) in 500 μl DMEM without phenol red/no serum and equilibrated to 37 °C in a light-proof cabinet and images were collected every 10 s using a CCD camera (Hamamatsu C9100-13) and Micro-Manager (http://www.micro-manager.org).MATLAB (MathWorks)scripts were used to calculate integrated luminescence intensity and background subtraction.In rapamycin heterodimerization experiments, cells were pre-incubated with 1μM rapamycin (Sigma) for 15 min. 5μM forskolin was used as a reference value in each multi-well plate and for each experimental condition. The average luminescence value (measured across duplicate wells) was normalized to the maximum luminescence value measured in the presence of 5μM forskolin. For rapamycin treated cells, the average luminescence value was normalized to the maximum luminescence value measured in the presence of 5μM forskolin and 1μM Rapamycin.
Quantitative real-time PCR
Total RNA was extracted from samples with RNeasy Mini Kit (Qiagen). Reverse transcription was carried out using SuperScript III RT (Invitrogen), mix of oligo (dT). The resulting cDNA was used for quantitative PCR with StepOnePlus (ABI). SYBER Select Mastermix (Invitrogen) was used for EMT/OCT3 expression measurements using 5’CCTTGTCTGTGTCAATGCGTGG3’ and 5’CCAACACCAAGGCAGGATAGCA3’ primers. All levels were normalized to the levels of a housekeeping gene, GAPDH.
Western blotting
Cells from HEK293, HeLa, H9C2 and neonatal cardiac myocytes were lysed in extraction buffer (0.2% Triton X-100, 50mM NaCl, 5mM EDTA, 50mM Tris at pH 7.4 and complete EDTA-free protease inhibitor cocktail (Roche). Extracts were mixed with SDS sample buffer for degradation. The proteins were resolved by SDS-PAGE and transferred to nitrocellulose membrane and blotted for anti-β1AR (ab-3442- 1:1000), anti-SLC22A3 (ab124826-1:1000) or GAPDH (1:10,000) antibodies to detect β1AR, EMT/OCT3 and GAPDH expression by horseradish-peroxidase-conjugated rabbit IgG, sheep anti-mouse and rabbit IgG (1:10,000 Amersham Biosciences) and SuperSignal extended duration detection reagent (Pierce).
Internalization Assay by Flow Cytometry
Internalization assay was carried out on HeLa cells transiently expressing Flag-tagged β1AR or β2AR. Cells were treated either with 10μM isoproterenol alone for 20min to promote endocytosis or pre-treated with 30μM Dyngo-4a for 15min to block endocytosis and then 10μM isoproterenol for 20 min at 37°C. Surface receptors were labelled by M1 anti-Flag antibody conjugated to Alexa 647 (Sigma, 1:1000) for 45min at 4°C. Cells were then collected in cold PBS and the fluorescence intensity profiles of cell populations (10,000 cells per sample) were measured using a FACS-Calibur instrument (BD Biosciences). In each experiment, triplicate samples were analysed for each condition. Experiments were carried out three times on three separate days.
Data availability
Our research resources, including methods, cells and protocols are available upon request. All reagents developed, such as FRB and FKBP constructs, as well as detailed methods will be available upon request. The corresponding author adheres to the NIH Grants Policy and Sharing of Unique Research Resources.
Supplementary Material
Acknowledgments
We thank B. Kobilka, J. Steyaert, H. Bourne,N.G. Tsvetanova, G. Peng, B. Lobingier, D. Larsen and K. Thorn for assistance, advice and valuable discussion. These studies were supported by the National Institute on Drug Abuse (DA012864 and DA010711 to M.v.Z.), the National Heart, Lung and Blood Institute (HL122508 to R.I.), the National Institute of Biomedical Imaging and Bioengineering (EB022798 to B.H.), the National Institute of General Medicine (GM056444 to P.B.W) and the National Heart, Lung and Blood Institute (HL0927088 to M.C.) of the U.S. National Institutes of Health and the American Heart Association (15PRE21770003 to V.P.).
Footnotes
Author Contributions
R.I. designed experimental strategy, carried out all of the experiments and analysis, and took a lead role in writing the manuscript. V.P contributed to the PKA experiments and analysis. D.M. contributed to the cAMP experimental design and analysis. B.H. provided essential reagents. P.B.W. contributed to the beta-blockers experimental design and data interpretation. M.C. contributed to overall experimental strategy and interpretation, and provided essential reagents. M.v.Z. together with R.I. designed the experimental strategy, contributed to interpreting the results and writing the paper.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Galandrin S, Oligny-Longpre G, Bouvier M. The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol Sci. 2007;28:423–430. doi: 10.1016/j.tips.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Maudsley S, Martin B, Luttrell LM. The origins of diversity and specificity in g protein-coupled receptor signaling. J Pharmacol Exp Ther. 2005;314:485–494. doi: 10.1124/jpet.105.083121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irannejad R, et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature. 2013;495:534–538. doi: 10.1038/nature12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsvetanova NG, von Zastrow M. Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol. 2014;10:1061–1065. doi: 10.1038/nchembio.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mullershausen F, et al. Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat Chem Biol. 2009;5:428–434. doi: 10.1038/nchembio.173. [DOI] [PubMed] [Google Scholar]
- 6.Calebiro D, et al. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 2009;7:e1000172. doi: 10.1371/journal.pbio.1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrandon S, et al. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol. 2009;5:734–742. doi: 10.1038/nchembio.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki T, et al. Distinct regulation of beta 1- and beta 2-adrenergic receptors in Chinese hamster fibroblasts. Mol Pharmacol. 1992;41:542–548. [PubMed] [Google Scholar]
- 9.Boivin B, et al. Functional beta-adrenergic receptor signalling on nuclear membranes in adult rat and mouse ventricular cardiomyocytes. Cardiovasc Res. 2006;71:69–78. doi: 10.1016/j.cardiores.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 10.Zhang L, et al. Phospholipase Cepsilon hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell. 2013;153:216–227. doi: 10.1016/j.cell.2013.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malik S, et al. G protein betagamma subunits regulate cardiomyocyte hypertrophy through a perinuclear Golgi phosphatidylinositol 4-phosphate hydrolysis pathway. Mol Biol Cell. 2015;26:1188–1198. doi: 10.1091/mbc.E14-10-1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu WZ, et al. Heterodimerization of beta1- and beta2-adrenergic receptor subtypes optimizes beta-adrenergic modulation of cardiac contractility. Circ Res. 2005;97:244–251. doi: 10.1161/01.RES.0000176764.38934.86. [DOI] [PubMed] [Google Scholar]
- 13.Rasmussen SG, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steyaert J, Kobilka BK. Nanobody stabilization of G protein-coupled receptor conformational states. Curr Opin Struct Biol. 2011;21:567–572. doi: 10.1016/j.sbi.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ziemek R, et al. Fluorescence- and luminescence-based methods for the determination of affinity and activity of neuropeptide Y2 receptor ligands. Eur J Pharmacol. 2006;551:10–18. doi: 10.1016/j.ejphar.2006.08.075. [DOI] [PubMed] [Google Scholar]
- 16.Brand F, Klutz AM, Jacobson KA, Fredholm BB, Schulte G. Adenosine A(2A) receptor dynamics studied with the novel fluorescent agonist Alexa488-APEC. Eur J Pharmacol. 2008;590:36–42. doi: 10.1016/j.ejphar.2008.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stoddart LA, Kilpatrick LE, Briddon SJ, Hill SJ. Probing the pharmacology of G protein-coupled receptors with fluorescent ligands. Neuropharmacology. 2015;98:48–57. doi: 10.1016/j.neuropharm.2015.04.033. [DOI] [PubMed] [Google Scholar]
- 18.Shiina T, Kawasaki A, Nagao T, Kurose H. Interaction with beta-arrestin determines the difference in internalization behavor between beta1- and beta2-adrenergic receptors. J Biol Chem. 2000;275:29082–29090. doi: 10.1074/jbc.M909757199. [DOI] [PubMed] [Google Scholar]
- 19.Boucrot E, et al. Endophilin marks and controls a clathrin-independent endocytic pathway. Nature. 2015;517:460–465. doi: 10.1038/nature14067. [DOI] [PubMed] [Google Scholar]
- 20.Denker SP, McCaffery JM, Palade GE, Insel PA, Farquhar MG. Differential distribution of alpha subunits and beta gamma subunits of heterotrimeric G proteins on Golgi membranes of the exocrine pancreas. J Cell Biol. 1996;133:1027–1040. doi: 10.1083/jcb.133.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng H, Farquhar MG. Presence of adenylate cyclase activity in Golgi and other fractions from rat liver. II. Cytochemical localization within Golgi and ER membranes. J Cell Biol. 1976;70:671–684. doi: 10.1083/jcb.70.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng H, Farquhar MG. Presence of adenylate cyclase activity in Golgi and other fractions from rat liver. I. Biochemical determination. J Cell Biol. 1976;70:660–670. doi: 10.1083/jcb.70.3.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michaelson D, Ahearn I, Bergo M, Young S, Philips M. Membrane trafficking of heterotrimeric G proteins via the endoplasmic reticulum and Golgi. Mol Biol Cell. 2002;13:3294–3302. doi: 10.1091/mbc.E02-02-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maier O, Ehmsen E, Westermann P. Trimeric G protein alpha subunits of the Gs and Gi families localized at the Golgi membrane. Biochem Biophys Res Commun. 1995;208:135–143. doi: 10.1006/bbrc.1995.1315. [DOI] [PubMed] [Google Scholar]
- 25.Cancino J, et al. Control systems of membrane transport at the interface between the endoplasmic reticulum and the Golgi. Dev Cell. 2014;30:280–294. doi: 10.1016/j.devcel.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 26.Westfield GH, et al. Structural flexibility of the G alpha s alpha-helical domain in the beta2-adrenoceptor Gs complex. Proc Natl Acad Sci U S A. 2011;108:16086–16091. doi: 10.1073/pnas.1113645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Branco AF, et al. Isoproterenol cytotoxicity is dependent on the differentiation state of the cardiomyoblast H9c2 cell line. Cardiovasc Toxicol. 2011;11:191–203. doi: 10.1007/s12012-011-9111-5. [DOI] [PubMed] [Google Scholar]
- 28.Staus DP, et al. Regulation of beta2-adrenergic receptor function by conformationally selective single-domain intrabodies. Mol Pharmacol. 2014;85:472–481. doi: 10.1124/mol.113.089516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crabtree GR, Schreiber SL. Three-part inventions: intracellular signaling and induced proximity. Trends Biochem Sci. 1996;21:418–422. doi: 10.1016/s0968-0004(96)20027-1. [DOI] [PubMed] [Google Scholar]
- 30.Inoue T, Heo WD, Grimley JS, Wandless TJ, Meyer T. An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat Methods. 2005;2:415–418. doi: 10.1038/nmeth763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Staehelin M, Simons P, Jaeggi K, Wigger N. CGP-12177. A hydrophilic beta-adrenergic receptor radioligand reveals high affinity binding of agonists to intact cells. J Biol Chem. 1983;258:3496–3502. [PubMed] [Google Scholar]
- 32.Wright CD, et al. Nuclear alpha1-adrenergic receptors signal activated ERK localization to caveolae in adult cardiac myocytes. Circ Res. 2008;103:992–1000. doi: 10.1161/CIRCRESAHA.108.176024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Connell TD, Jensen BC, Baker AJ, Simpson PC. Cardiac alpha1-adrenergic receptors: novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacol Rev. 2014;66:308–333. doi: 10.1124/pr.112.007203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grundemann D, et al. Activation of the extraneuronal monoamine transporter (EMT) from rat expressed in 293 cells. Br J Pharmacol. 2002;137:910–918. doi: 10.1038/sj.bjp.0704926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arai R, et al. Differential subcellular location of mitochondria in rat serotonergic neurons depends on the presence and the absence of monoamine oxidase type B. Neuroscience. 2002;114:825–835. doi: 10.1016/s0306-4522(02)00351-2. [DOI] [PubMed] [Google Scholar]
- 36.Yang Y, et al. Biopharmaceutics classification of selected beta-blockers: solubility and permeability class membership. Mol Pharm. 2007;4:608–614. doi: 10.1021/mp070028i. [DOI] [PubMed] [Google Scholar]
- 37.Neil-Dwyer G, Bartlett J, McAinsh J, Cruickshank JM. Beta-adrenoceptor blockers and the blood-brian barrier. Br J Clin Pharmacol. 1981;11:549–553. doi: 10.1111/j.1365-2125.1981.tb01169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 39.Nigg EA, Schafer G, Hilz H, Eppenberger HM. Cyclic-AMP-dependent protein kinase type II is associated with the Golgi complex and with centrosomes. Cell. 1985;41:1039–1051. doi: 10.1016/s0092-8674(85)80084-2. [DOI] [PubMed] [Google Scholar]
- 40.Nigg EA, Hilz H, Eppenberger HM, Dutly F. Rapid and reversible translocation of the catalytic subunit of cAMP-dependent protein kinase type II from the Golgi complex to the nucleus. EMBO J. 1985;4:2801–2806. doi: 10.1002/j.1460-2075.1985.tb04006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin BR, Deerinck TJ, Ellisman MH, Taylor SS, Tsien RY. Isoform-specific PKA dynamics revealed by dye-triggered aggregation and DAKAP1alpha-mediated localization in living cells. Chem Biol. 2007;14:1031–1042. doi: 10.1016/j.chembiol.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 42.Mavillard F, Hidalgo J, Megias D, Levitsky KL, Velasco A. PKA-mediated Golgi remodeling during cAMP signal transmission. Traffic. 2010;11:90–109. doi: 10.1111/j.1600-0854.2009.01007.x. [DOI] [PubMed] [Google Scholar]
- 43.Roth M, Obaidat A, Hagenbuch B. OATPs, OATs and OCTs: the organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br J Pharmacol. 2012;165:1260–1287. doi: 10.1111/j.1476-5381.2011.01724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holubarsch C, et al. Positive and negative inotropic effects of DL-sotalol and D-sotalol in failing and nonfailing human myocardium under physiological experimental conditions. Circulation. 1995;92:2904–2910. doi: 10.1161/01.cir.92.10.2904. [DOI] [PubMed] [Google Scholar]
- 45.Seipel L, Hoffmeister HM. Inotropic and haemodynamic effects of d- and d,l-sotalol: comparison with other antiarrhythmics. Eur Heart J. 1993;14(Suppl H):36–40. doi: 10.1093/eurheartj/14.suppl_h.36. [DOI] [PubMed] [Google Scholar]
- 46.Rosciglione S, Theriault C, Boily MO, Paquette M, Lavoie C. Galphas regulates the post-endocytic sorting of G protein-coupled receptors. Nat Commun. 2014;5:4556. doi: 10.1038/ncomms5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Irannejad R, Wedegaertner PB. Regulation of constitutive cargo transport from the trans-Golgi network to plasma membrane by Golgi-localized G protein betagamma subunits. J Biol Chem. 2010;285:32393–32404. doi: 10.1074/jbc.M110.154963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Devic E, Xiang Y, Gould D, Kobilka B. Beta-adrenergic receptor subtype-specific signaling in cardiac myocytes from beta(1) and beta(2) adrenoceptor knockout mice. Mol Pharmacol. 2001;60:577–583. [PubMed] [Google Scholar]
- 49.Brodde OE. Beta 1- and beta 2-adrenoceptors in the human heart: properties, function, and alterations in chronic heart failure. Pharmacol Rev. 1991;43:203–242. [PubMed] [Google Scholar]
- 50.Nikolaev VO, et al. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010;327:1653–1657. doi: 10.1126/science.1185988. [DOI] [PubMed] [Google Scholar]
- 51.Nikolaev VO, et al. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010;327:1653–1657. doi: 10.1126/science.1185988. [DOI] [PubMed] [Google Scholar]
- 52.Lobert VH, Stenmark H. The ESCRT machinery mediates polarization of fibroblasts through regulation of myosin light chain. J Cell Sci. 2012;125:29–36. doi: 10.1242/jcs.088310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Our research resources, including methods, cells and protocols are available upon request. All reagents developed, such as FRB and FKBP constructs, as well as detailed methods will be available upon request. The corresponding author adheres to the NIH Grants Policy and Sharing of Unique Research Resources.