

Abstract
Objective:
To describe electroclinical features and outcome of 6 patients harboring KCNB1 mutations.
Methods:
Clinical, EEG, neuropsychological, and brain MRI data analysis. Targeted next-generation sequencing of a 95 epilepsy gene panel.
Results:
The mean age at seizure onset was 11 months. The mean follow-up of 11.3 years documented that 4 patients following an infantile phase of frequent seizures became seizure free; the mean age at seizure offset was 4.25 years. Epilepsy phenotypes comprised West syndrome in 2 patients, infantile-onset unspecified generalized epilepsy, myoclonic and photosensitive eyelid myoclonia epilepsy resembling Jeavons syndrome, Lennox-Gastaut syndrome, and focal epilepsy with prolonged occipital or clonic seizures in each and every one. Five patients had developmental delay prior to seizure onset evolving into severe intellectual disability with absent speech and autistic traits in one and stereotypic hand movements with impulse control disorder in another. The patient with Jeavons syndrome evolved into moderate intellectual disability. Mutations were de novo, 4 missense and 2 nonsense, 5 were novel, and 1 resulted from somatic mosaicism.
Conclusions:
KCNB1-related manifestations include a spectrum of infantile-onset generalized or focal seizures whose combination leads to early infantile epileptic encephalopathy including West, Lennox-Gastaut, and Jeavons syndromes. Long-term follow-up highlights that following a stormy phase, seizures subside or cease and treatment may be eased or withdrawn. Cognitive and motor functions are almost always delayed prior to seizure onset and evolve into severe, persistent impairment. Thus, KCNB1 mutations are associated with diffuse brain dysfunction combining seizures, motor, and cognitive impairment.
The KCNB1 gene encodes the pore-forming and voltage-sensing β subunit of the voltage-gated potassium (K+) channel subfamily 2 (Kv2.1) that is expressed throughout the brain1–3 and plays essential roles in regulating neuronal excitability, contributing to action potential repolarization4 and dynamic modulation of neuronal activity.5–7 De novo heterozygous missense (number = 13) and nonsense (number = 1) mutations of KCNB1 have been reported in 14 patients with neurodevelopmental disorders, including epilepsy of variable severity in 13 of them.8–15 Probands carrying KCNB1 mutations resulting in severe epilepsy phenotypes have been classified as epileptic encephalopathy–early infantile 26 (OMIM: EIEE26, *600397). Table 1 summarizes the clinical and genetic features of previously reported patients.
Table 1.
Clinical and genetic summary of previously published patients
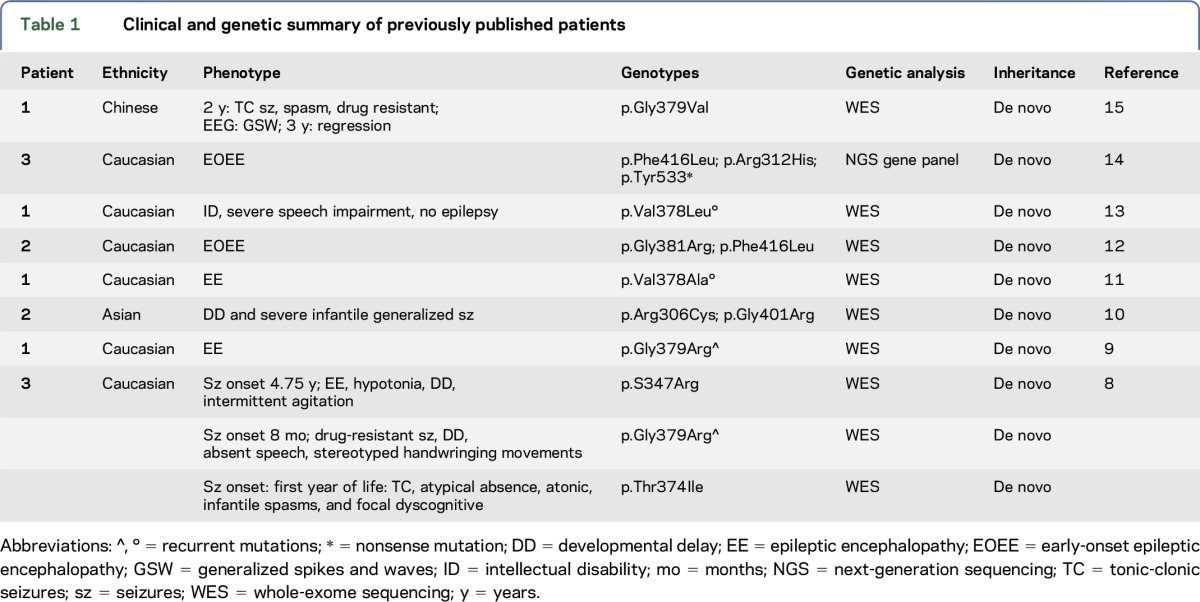
Here, we describe the electroclinical features and long-term follow-up of 6 novel patients carrying de novo missense and nonsense KCNB1 mutations and provide an overall view of the spectrum of epilepsy phenotypes associated with KCNB1 mutations, ranging from Lennox-Gastaut syndrome to remitting infantile spasms and to mild generalized epilepsy with eyelid myoclonia. Our small cohort of patients also suggests that the KCNB1 EIEE26 manifests infantile-onset seizures with a tendency to attenuate or resolve completely over time in some patients. Concomitant impairment of cognitive and motor functions, constant and often severe, persists over time and is at times combined with absent speech, autism spectrum disorder (ASD), and psychiatric problems. Mutations in this gene, which are not rare, confirm that genes coding for potassium channels are important genetic contributors to epilepsies and more broadly to neurodevelopmental disorders.
METHODS
From a highly heterogenous cohort of 873 patients with pediatric epilepsies, either refractory or benign, with a supposed genetic etiology, recruited at the Meyer Children's Hospital or referred from other national or international epilepsy centers for genetic testing, we identified and studied 6 patients carrying de novo variants of the KCNB1 gene (GenBank Accession Number: NM_004975.2). Three patients were examined at the Neurology unit, and 3 were referred for genetic testing to our Neurogenetic Laboratory. Collection and analysis of retrospective clinical, EEG, neuropsychological, neuroimaging data were performed using a specific format filled by the treating specialist aiming to obtain accurate and homogenous information. We classified seizure types and epilepsy/syndromes according to the International League Against Epilepsy guidelines.16,17
Molecular genetic testing.
Genomic DNA was extracted from peripheral blood leukocytes using a QIAsymphony SP robot (Qiagen, Hilden, Germany) according to the manufacturer's protocol. High-quality DNA was quantified using a QuantiFluor Fluorometer (Promega, Madison, WI). Targeted resequencing of a panel including 95 genes associated with epilepsy was performed in all 6 patients. Target enrichment and library preparation were performed using a custom-designed Nextera Rapid Capture assay (Illumina, San Diego, CA), and sequencing was performed on an Illumina MiSeq (Illumina) with a 2 × 150-bp paired-end protocol as previously described.18 Variants were annotated and filtered using the ANNOVAR tool.19 Variants localized in intronic regions outside the 10-bp exon-flanking boundaries and in the 5′- and 3′-UTR regions were excluded. Variants reported in the Exome Aggregation Consortium (ExAC) database (exac.broadinstitute.org/) and/or in the 1000 Genomes Project (in the National Heart, Lung, and Blood Institute Exome Sequencing Project [ESP], ESP6500 database, evs.gs.washington.edu/EVS), in the Genome Aggregation Database (gnomAD) (gnomad.broadinstitute.org/), with a minor allele frequency >0.01 (1%) were dropped out. In silico prediction of mutations' pathogenicity was obtained using the dbNSFP database (v3.3), which provides functional prediction scores on more than 20 different algorithms (sites.google.com/site/jpopgen/dbNSFP). Putative causative variants were analyzed by Sanger sequencing to confirm the next-generation sequencing (NGS) results in probands and investigated in the parents to check the inheritance status. We classified variants according to the international guidelines of the American College of Medical Genetics and Genomics (ACMG) Laboratory Practice Committee Working Group.20
Standard protocol approvals, registrations, and patient consents.
Written informed consent to disclose clinical information was obtained from all parents/guardians of the participants. Written informed consent for genetic testing was obtained according to the Paediatric Ethic Committee of the Tuscany Region (approval no. 2014-0000559) in the context of the DESIRE project (grant agreement no. 602531).
RESULTS
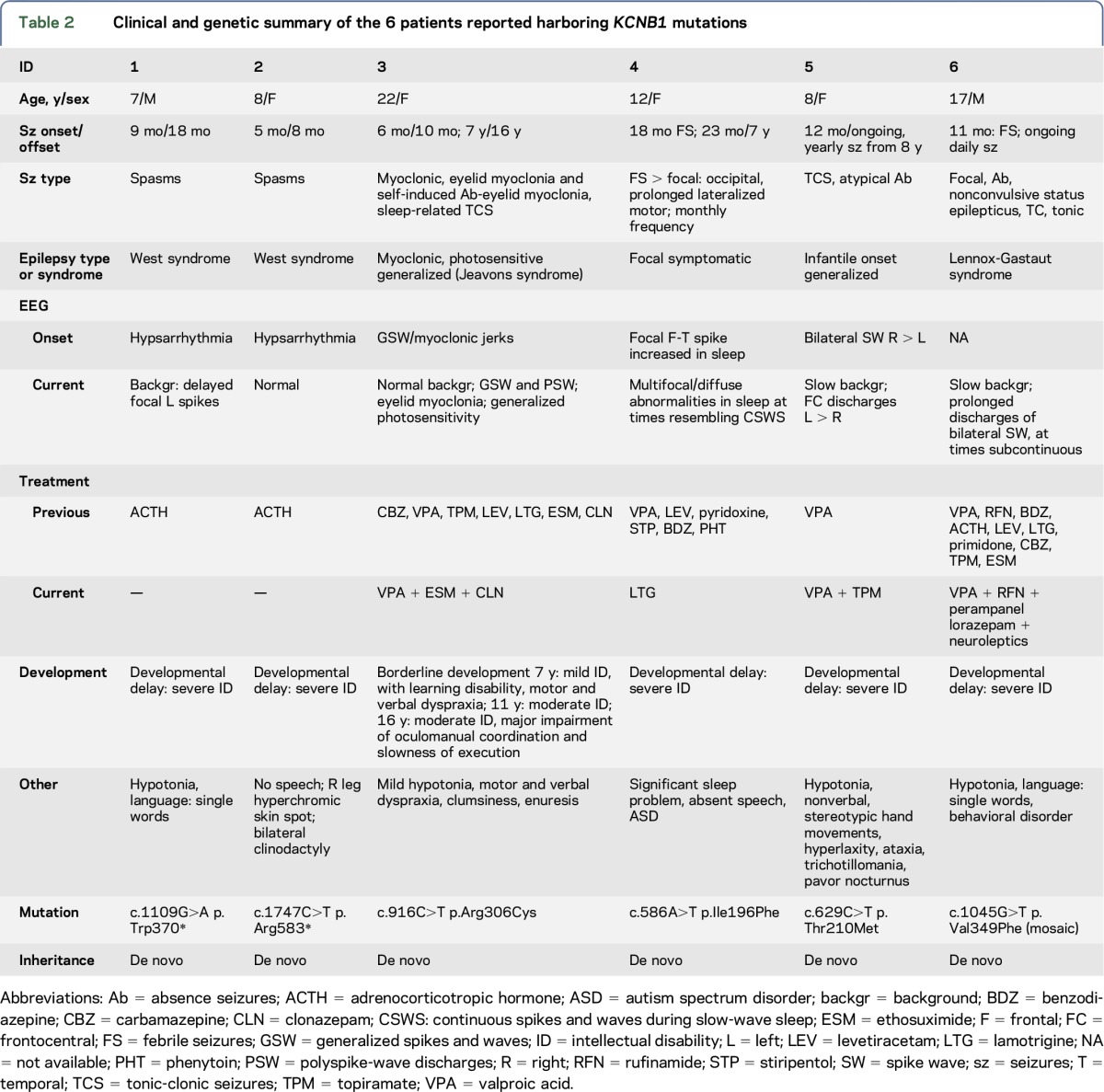
The 6 patients (4 females and 2 males) harboring KCNB1 mutations had a mean age at the time of the study of 12 years (range 7–22 years). They represented the 0.75% (6 of 873) of a highly heterogeneous cohort of patients with potentially genetic epilepsies, studied with our target resequencing gene panel.18 Table 2 summarizes the main clinical features of each KCNB1 mutation–positive patient.
Table 2.
Clinical and genetic summary of the 6 patients reported harboring KCNB1 mutations
Epilepsy.
The mean age at seizure onset was 11 months (median 9, range 5–23 months). Four of the 6 patients (66.6%) following an initial phase of active epilepsy with frequent seizures became seizure free at a median age of 4.25 years; the mean duration of seizure remission at the last follow-up was 6 years (range 8 months–16 years).
Seizure types (table 2) included infantile spasms (patients 1 and 2), myoclonic seizures with eyelid myoclonia (table 2, patient 3), febrile seizures (FS) followed by focal seizures (patients 4 and 5), absences and nonconvulsive status epilepticus (patient 6), and afebrile tonic-clonic seizures (TCS) followed by other generalized seizure types (patients 5 and 6). For each patient, we considered age of symptom appearance and age at the time of the study and calculated a mean follow-up of 11.3 years when last seen (median 8.5 years, range 6–21 years).
Two patients with infantile spasms (patients 1 and 2), classified as West syndrome due to their hypsarrhythmic EEG and co-occurring developmental delay, exhibited an identical phenotype and outcome. Both patients were immediately treated with adrenocorticotropic hormone (ACTH) after the onset of spasms, with a prompt remission of spasms and EEG improvement. After 6–7 years of follow-up, both patients with “remitting infantile spasms” remain seizure free and off antiepileptic drugs (AEDs). Yet, their development was delayed and currently sets on severe impairment of cognitive and motor functions including absent speech.
The remaining 4 patients manifested different and heterogeneous phenotypes. Patient 3 (table 2) showed an interesting and peculiar phenotype consisting of myoclonic seizures and absences with eyelid myoclonia with persistent generalized photosensitivity. Her epilepsy started at 6 months, with bilateral myoclonic jerks, and was very active for 3 months when treatment with valproic acid (VPA) prompted a prolonged, yet temporary, resolution of symptoms. Indeed, at age 7 years, during primary school age, myoclonic seizures and absences with eyelid myoclonia with photosensitivity were observed (figure 1A). Episodes of absences with eyelid myoclonia occurred frequently on eye closure or were autoinduced like seen in Jeavons syndrome.21 Two generalized TCS during sleep were also reported around age 15 years. The patient was trialed with several AEDs, but only a combination of VPA, ethosuximide, and clonazepam was effective at age 16 years. At the time of the study, this patient was 22 years old and is still seizure free on AEDs. Patients 4 and 5 (table 2) exhibited infantile-onset multifocal and generalized seizures preceded by global developmental delay. Then, patient 4 manifested monthly seizures from age 2 to 5 years when seizures ceased, she is currently 12, still on AEDs, and seizure free. Despite 5 years of seizure freedom, her cognitive and motor functions are yet severely impaired with absent speech and autistic traits. Patient 5 has ongoing yearly seizures and severe cognitive impairment. The last patient (patient 6) exhibited the most refractory epilepsy with infantile-onset FS followed by focal seizures, frequent absences, nonconvulsive status, tonic, and TCS configuring a phenotype resembling Lennox-Gastaut syndrome. He is currently 17 and has daily atypical absences and sleep-related TCS. Concomitant with such refractory epilepsy, he manifested early developmental delay evolving into moderate to severe intellectual disability (ID) with a behavioral disorder requiring antipsychotic drugs.
Figure 1. Representative EEG findings in 3 patients.

(A) Ictal videopolygraphic EEG recording of patient 3 (table 2), showing discharges of diffuse polyspike waves concomitant with myoclonic jerks evident on the leads recording from both deltoid muscles. (B) Ictal videopolygraphic EEG recording of patient 3 (table 2), showing generalized paroxysmal activity evoked by eye closure with concomitant eyelid myoclonia (video available as supplementary material). (C) Interictal EEG recording of patient 5, showing bifrontal single complexes of spikes and waves with left predominance. (D) Interictal sleep EEG recording of patient 4, showing very frequent paroxysmal activity over the fronto-centro-temporal regions (continuous spikes and waves during the slow-wave sleep pattern).
EEG recordings.
All patients underwent repeated EEG recordings from seizure onset to current age. According to their epilepsy phenotype, at onset, 2 patients presented a severely abnormal EEG with hypsarrhythmia that overtime evolved into a normal EEG in 1 (patient 2) and slow background activity with rare left temporal spikes in the second patient (patient 1). Video-EEG polygraphic recordings of the patient with Jeavons syndrome (patient 3) showed, at onset and during follow-up, generalized spike- and polyspike-wave discharges with a prominent generalized photoparoxysmal response (figure 1B). Several episodes of myoclonia and absences with eyelid myoclonia were recorded (figure 1, A and B). Patient 4 (table 2), with infantile-onset multifocal epilepsy, consistently showed slow background with multifocal or diffuse paroxysmal activity in sleep and semiperiodic posterior fast activity runs on awakening. Some EEG recordings in this patient showed, in early childhood, a relevant increase in paroxysmal activity during sleep, resembling the continuous spike and wave during the slow-wave sleep (CSWS) EEG pattern (figure 1C). The EEG recordings in patient 5 (table 2) exhibited slow background activity and intermittent bilateral frontocentral spike and wave discharges with left predominance (figure 1B). EEG recordings of patient 6 showed slow background and frequent, at times almost continuous multifocal and diffuse spike and wave discharges.
Cognition and behavior.
Five of the 6 patients (table 2, patients 1, 2, and 4–6) had clear-cut developmental delay prior to seizure onset, involving both motor and cognitive functions. Their cognitive outcome evolved into severe impairment with absent speech and autistic traits in 1 (table 2, patient 4). In addition, one of them (table 2, patient 5) manifested stereotypic hand movements and impulse control disorder consisting of trichotillomania, and patient 6 (table 2) developed a psychiatric disorder requiring treatment with neuroleptics.
The girl with Jeavons syndrome instead had a delayed early development (sitting: 8 months, ambulation: 15 months; and language: 2 years) evolving, in childhood, into mild cognitive impairment with motor and verbal dyspraxia and poor coordination. Despite such a cognitive profile, she attended primary school unassisted. While during secondary school, she required a special needs teacher, and further neuropsychological testing documented moderate ID (Wechsler Intelligence Scale for Children—Revised: full-scale IQ = 45). Yet, she was able to reach secondary school graduation.
Five of the 6 patients acquired independent walking, with a variable degree of hypotonia, hyperlaxity, and mild ataxia in 3. One patient was using a wheelchair (patient 1).
MRI.
All patients underwent normal brain MRI.
Genetic.
Heterozygous KCNB1 mutations occurred de novo (table 2, figure 2) and were classified as pathogenic according to the ACMG guidelines. Both patients 1 and 2 carried a novel de novo nonsense mutation, including the c.1109G>A [p.(Trp370*)] variant that is located in the pore domain of the protein and the c.1747C>T [p.(Arg583*)] variant, which falls in the C-terminal domain. Patient 3 harbored the c.916C>T [p.(Arg306Cys)] missense variant, which is located in the S4 segment of the voltage-sensor domain of the protein. This variant had previously been reported in a patient with developmental delay and infantile-onset seizures.10 Patient 4 carried the novel c.586A>T [p.(Ile196Phe)] missense variant, located in the S1 segment of the protein, and patient 5 carried the novel c.629C>T [p.(Thr210Met)] missense variant, located in the first extracellular domain, between S1 and S2. Patient 6 carried the novel c.1045G>T [p.(Val349Phe)] missense variant, located in the S5 segment of the pore; both NGS and Sanger sequencing data showed evidence of somatic mosaicism (figure e-1, http://links.lww.com/NXG/A1). None of the KCNB1 variants were reported in the public available allele frequency databases (table e-1, http://links.lww.com/NXG/A0), and the tools included in the dbNSFP, comprising the MetaSVM and MetaLR ensemble scores, predicted the 4 missense variants to be damaging (table e-1, http://links.lww.com/NXG/A0).
Figure 2. Schematic representation of the Kv2.1 protein with mutation distribution.

Structure of the human Kv2.1 channel including previously published mutations and those reported here (specified under each mutation). The protein topology was performed using the Protter online tool (29) with the Uniprot accession Q14721 (KCNB1_HUMAN).
DISCUSSION
We describe the electroclinical features and outcome of 6 patients harboring de novo heterozygous KCNB1 mutations, confirming that mutations in this gene are not rare. Indeed, in our highly heterogenous cohort of pediatric epilepsies with a supposed genetic etiology, nearly 1% of patients carried mutations in this gene.
Genetic studies, from the “old” linkage analyses studies to the modern era of whole-exome sequencing/whole-genome sequencing techniques, have demonstrated that heterozygous mutations in several genes coding for different K+ channels, including KNCQ2, KCNQ3, KCNA2, KCNA1, KCNC1, KCNH1, KCNMA1, HCN2, and KCNT1, are involved in epileptic disorders whose severity varies from benign forms to early infantile epileptic encephalopathy (EIEE).22–32 The list of K+ channel genes implicated in epilepsy includes KCNB1 that was, in 2014, linked to the EIEE26.8 At present, there are 14 patients on record carrying mutations in this gene, and 13 of them exhibit epilepsy.8–15
All patients, including the 6 reported here, present with early motor and cognitive delay and, thenceforth, with different types of infantile-onset seizures. Review of previously published patients shows that both focal and generalized seizures including tonic-clonic, tonic, atonic, absences, myoclonic seizures, and infantile spasms can be equally observed (figure 3A). Epilepsy, often classified as early-onset epileptic encephalopathy, has been reported as severe and refractory to AEDs in most patients. In our small series, 2 of the 6 patients were diagnosed as West syndrome (figure 3B). Of the remaining 4, 2 had generalized epilepsy with myoclonic seizures and eyelid myoclonia with photosensitivity, resembling Jeavons syndrome in 1 (figure 3B). Focal epilepsy with prolonged occipital seizures or focal clonic, possibly frontal, seizures was documented only in 1, and the last, sixth patient's, had a phenotype consistent with Lennox-Gastaut syndrome (figure 3B).
Figure 3. Graphic representation of seizure type distribution in patients with KCNB1 mutations.

(A) Previously published patients (data from 10 of the 13 patients of previously published KCNB1 mutations and epilepsy) and (B) in our series of 6 patients. AB = absences; CSWS = continuous spikes and waves during slow-wave sleep; FS = febrile seizures; MY = myoclonia; NCSE = nonconvulsive status epilepticus; TCS = tonic-clonic seizures.
Combining the analysis of the seizure types of all known patients harboring KCNB1 mutations (including our 6 probands and the 14 previously published, for a total of 20, of whom 19 with seizures), we can observe that (1) TCS are the commonest seizure type observed in 57.8% (11/19) of patients; (2) focal seizures are the second most common type, reported in 47% (9/19); (3) infantile spasms are also a relatively common manifestation occurring in 31.5% (6/19); (4) approximately 15% (3–4/19) of patients have either absences, atonic, or tonic seizures; (5) single patients might have peculiar and unusual phenotypes consisting of CSWS, nonconvulsive status epilepticus (NCSE) or myoclonic seizures or eyelid myoclonia, and photosensitive seizures. Of course, most patients manifest more than 1 seizure type that combined the result in unspecified EE or more rarely to known epilepsy syndromes including West, Lennox-Gastaut, and Jeavons syndromes.
Likewise, previously published patients, in our small series, seizures started in infancy, around 1 year of age, and were also drug resistant at onset. Yet, over time in 4 of our 6 patients (66.6%), seizure frequency attenuated and disappeared, still being under remission after a seizure-free period of 6 years. Two patients are also at present off AEDs. The reason why there is such a wide spectrum of epilepsy severity in this and in other EIEE remains a true mystery at present. Thus, further speculative discussion is better postponed until we improve our understanding about the underlying pathophysiologic mechanism and how individual genetic backgrounds influence outcomes.
Long-term follow-up of our 6 patients showed that following delay of milestone acquisition and despite seizure freedom, they evolved into moderate to severe impairment of cognitive functions including psychiatric symptoms and autistic traits. Overall, these findings indicate that Kv2.1 impaired function causes diffuse brain dysfunction expressing, in humans, as epilepsy and developmental or behavioral disorders. Thus, it might be more appropriate to define patients with KCNB1 mutations as having developmental encephalopathy with epilepsy. An intermingled pathophysiologic relationship between the triad of seizures/ASD/ID and potassium channel genes is further supported by the identification of mutations in KCNJ10, a gene coding for the inwardly rectifying potassium channel Kir4.1, in several probands exhibiting seizures, ASD, and ID.33
All mutations reported here were de novo and included 4 missense and 2 nonsense variants. None of the mutations appeared in allele frequency databases (table e-1, http://links.lww.com/NXG/A0), and all were predicted to be damaging by several prediction tools and ensemble scores (table e-1, http://links.lww.com/NXG/A0) and novel, with the exception of the p.Arg306Cys falling in the S4 of the voltage-sensor domain and demonstrated to be damaging by Saitsu et al.10 Patients carrying the p.Arg306Cys mutation appear to have different phenotypes. The patient we are reporting in this study has generalized epilepsy with myoclonic seizures and eyelid myoclonia, while the patient described by Saitsu et al.10 had a more severe phenotype with earlier seizure onset and heterogeneous, drug-resistant, seizures including infantile spasms, myoclonic, tonic-clonic, and focal seizures. The latter patient manifested also severe ID and macrocephaly unlike the one we are reporting, who had a normal head size and moderate ID.
While assessing genotype-phenotype correlations, we noted that both p.Trp370* and p.Arg583* nonsense mutations occurred in the 2 patients with remitting infantile-onset spasms and severe cognitive impairment. At present, there is a single previously reported nonsense mutation14 in a patient with EE, yet detailed clinical features are not available for further correlations.
The remaining 2 mutations, p.Ile196Phe and the p.Thr210Met, both predicted to be damaging, are missense variants (table 1) located in the S1 and in the S1–S2 extracellular loop. The S1–S4 segments serve as the voltage-sensing module, and the voltage sensitivity of the channel is conferred by a series of highly conserved basic side chains in the S4 segment. These side chains are stabilized in a transmembrane configuration by the formation of fully solvated salt bridges with a set of highly conserved acidic side chains in S1–S3.34 The p.Ile196Phe and p.Thr210Met mutations are located in the S1 conserved acidic side chains and are therefore likely to impair the structural signature of voltage-sensing motion between S1–S3 and S4 segments.35
NGS and Sanger sequencing showed that the p.Val349Phe variant was present as mosaicism although associated with the most refractory epilepsy type in our cohort. This might correlate with the location of this mutation in the pore-forming region of the channel, thus supposedly causing a severe alteration of its function. A missense mutation located very closely, at position 347, causing severe impairment of the channel function and associated with refractory epilepsy strikingly resembling our patient's phenotype, had previously been reported.8
Patch-clamp studies have demonstrated that some KCNB1 variants cause significant alterations of Kv2.1 channel function and increased excitability of cortical neurons. Functional studies suggested different mechanisms including abolished voltage-activated gating, loss of ion selectivity,11 gain of depolarizing inward cation,8 defects in expression and subcellular localization,8 disruption of sensitivity and cooperativity of the sensor,10 and abolished endogenous Kv2.1 currents.10 The role of K+ channels in the neurobiology of both epilepsy and cognitive impairment has been investigated also using animal models. Mice lacking Kv2.1 are strikingly hyperactive, defective in spatial learning, hypersensitive to convulsants, and exhibit accelerated seizure progression.36 Moreover, the Kcnb1−/− mice model showed neuronal hyperexcitability and confirmed the critical role of Kv2.1 in hippocampal neuronal network homeostatic regulation.36
Thus, KCNB1-related manifestations comprise a wide spectrum of infantile onset generalized or focal seizures whose combination leads to EIEE, including West, Lennox-Gastaut, and Jeavons syndromes. Long-term follow-up highlights that following a stormy phase, seizures cease and AEDs might be withdrawn at least in some patients. Instead, cognitive and motor functions are almost always delayed prior to seizure onset and evolve into severe, persistent impairment. Altogether, these findings suggest that KCNB1 mutations are associated with diffuse brain dysfunction and that the KCNB1-related clinical picture might also be defined as a developmental “encephalopathy” with seizures. Further understanding of the underlying pathophysiology by experimental studies including animal models is required to improve treatment and prognosis of these disorders.
GLOSSARY
- ACMG
American College of Medical Genetics and Genomics
- AED
antiepileptic drug
- ASD
autism spectrum disorder
- CSWS
continuous spikes and waves during slow-wave sleep
- EIEE
early infantile epileptic encephalopathy
- ESP
Exome Sequencing Project
- ExAC
Exome Aggregation Consortium
- FS
febrile seizures
- ID
intellectual disability
- NGS
next-generation sequencing
- TCS
tonic-clonic seizures
- VPA
valproic acid
AUTHOR CONTRIBUTIONS
Carla Marini, Michele Romoli, Elena Parrini, and Paolo Prontera: study design, data collection and analysis, and manuscript preparation. Francesco Mari, Cinzia Costa, Lucio Parmeggiani, Tiziana Metitieri, and Mattia Gentile: data collection and analysis and manuscript revision. Davide Mei, Elena Cellini, Simona Virdò, and Dalila De Vita: genetic study, data collection and analysis, and manuscript revision. Paolo Calabresi and Renzo Guerrini: study design, data analysis, and manuscript revision.
STUDY FUNDING
This work was supported by a grant from the EU Seventh Framework Programme FP7 under the project DESIRE (grant agreement no. 602531) (to R.G.) and by the Italian Ministry of Health (grant agreement no. RF-2013-02355240) (to R.G.).
DISCLOSURE
Carla Marini has received travel funding and/or speaker honoraria at VII CONGRESSO NAZIONALE SIMMESN–Firenze, December 16–18, 2015, from SOBI, Swedish Orphan Biovitrum; has served on the editorial boards of Epilepsia and Epileptic Disorders; and has received research support from the Italian Ministry of Health and Cariplo Foundation. Michele Romoli, Elena Parrini, and Cinzia Costa report no disclosures. Davide Mei has received funding for travel or speaker honoraria from BioMarin Pharmaceutical and serves on the editorial board of Epilepsia. Francesco Mari, Lucio Parmeggiani, Elena Procopio, Tiziana Metitieri, Elena Cellini, Simona Virdò, Dalila De Vita, Mattia Gentile, Paolo Prontera, and Paolo Calabresi report no disclosures. Renzo Guerrini has served on scientific advisory boards of Eisai Inc., Novartis, Zogenix, and BioMarin; has received funding for travel and/or speaker honoraria from EISAI, Novartis, UCB, and BioMarin; has served on the editorial boards of Epilepsia, Progress in Epileptic Disorders, Neuropediatrics, Journal of Child Neurology, Seizure, BMC Medical Genetics, Topics in Epilepsy, Journal of Pediatric Epilepsy, Epileptic Disorders, European Neurological Journal, Neurology, and Journal of Embryology & Developmental Biology; receives/has received royalties from the publication of Epilepsy and Movements disorders (Cambridge University Press, 2002), Aicardi's Epilepsy in Children (Lippincott Williams & Wilkins, 2004), Progress in Epileptic Spasms and West Syndrome (John Libbey Eurotext, 2007), Epilepsy and Migraine (John Libbey Eurotext, 2009), The Causes of Epilepsy (Cambridge University Press, 2011), Dravet syndrome (John Libbey Eurotext, 2011), and Epilepsy and epileptic seizures (Oxford University Press, 2012); and has received research support from Tuscany Region Research Department, EU and EC Programs, the Italian Ministry of Health, and the PISA Foundation. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Hwang PM, Fotuhi M, Bredt DS, Cunningham AM, Snyder SH. Contrasting immunohistochemical localizations in rat brain of two novel K+ channels of the Shab subfamily. J Neurosci 1993;13:1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maletic-Savatic M, Lenn NJ, Trimmer JS. Differential spatiotemporal expression of K+ channel polypeptides in rat hippocampal neurons developing in situ and in vitro. J Neurosci 1995;15:3840–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Du J, Tao-Cheng JH, Zerfas P, et al. . The K+ channel, Kv2.1, is apposed to astrocytic processes and is associated with inhibitory postsynaptic membranes in hippocampal and cortical principal neurons and inhibitory interneurons. Neuroscience 1998;84:37–48. [DOI] [PubMed] [Google Scholar]
- 4.Liu PW, Bean BP. Kv2 channel regulation of action potential repolarization and firing patterns in superior cervical ganglion neurons and hippocampal CA1 pyramidal neurons. J Neurosci 2014;34:4991–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Misonou H, Mohapatra DP, Park EW, et al. . Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci 2004;7:711–718. [DOI] [PubMed] [Google Scholar]
- 6.Misonou H, Thompson SM, Cai X. Dynamic regulation of the Kv2.1 voltage-gated potassium channel during brain ischemia through neuroglial interaction. J Neurosci 2008;28:8529–8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fox PD, Loftus RJ, Tamkun MM. Regulation of Kv2.1 K(+) conductance by cell surface channel density. J Neurosci 2013;3:1259–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Torkamani A, Bersell K, Jorge BS, et al. . De novo KCNB1 mutations in epileptic encephalopathy. Ann Neurol 2014;76:529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srivastava S, Cohen JS, Vernon H, et al. . Clinical whole exome sequencing in child neurology practice. Ann Neurol 2014;76:473–483. [DOI] [PubMed] [Google Scholar]
- 10.Saitsu H, Akita T, Tohyama J, et al. . De novo KCNB1 mutations in infantile epilepsy inhibit repetitive neuronal firing. Sci Rep 2015;5:15199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiffault I, Speca DJ, Austin DC, et al. . A novel epileptic encephalopathy mutation in KCNB1 disrupts Kv2.1 ion selectivity, expression, and localization. J Gen Physiol 2015;146:399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen NM, Conroy J, Shahwan A, et al. . Unexplained early onset epileptic encephalopathy: exome screening and phenotype expansion. Epilepsia 2016;57:e12–e17. [DOI] [PubMed] [Google Scholar]
- 13.Latypova X, Matsumoto N, Vinceslas-Muller C, et al. . Novel KCNB1 mutation associated with non-syndromic intellectual disability. J Hum Genet 2017;62:569–573. [DOI] [PubMed] [Google Scholar]
- 14.de Kovel CGF, Brilstra EH, van Kempen MJA, et al. . Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients. Mol Genet Genomic Med 2016;4:568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miao P, Peng J, Chen C, et al. . A novel mutation in KCNB1 gene in a child with neuropsychiatric comorbidities with both intellectual disability and epilepsy and review of literature. Zhonghua Er Ke Za Zhi 2017;55:115–119. [DOI] [PubMed] [Google Scholar]
- 16.Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389–399. [DOI] [PubMed] [Google Scholar]
- 17.Berg AT, Berkovic SF, Brodie MJ, et al. . Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010;51:676–685. [DOI] [PubMed] [Google Scholar]
- 18.Parrini E, Marini C, Mei D, et al. . Diagnostic targeted resequencing in 349 patients with drug-resistant pediatric epilepsies identifies causative mutations in 30 different genes. Hum Mutat 2017;38:216–225. [DOI] [PubMed] [Google Scholar]
- 19.Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc 2015;10:1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards S, Aziz N, Bale S, et al. . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caraballo RH, Fontana E, Darra F, et al. . A study of 63 cases with eyelid myoclonia with or without absences: type of seizure or an epileptic syndrome? Seizure 2009;18:440–445. [DOI] [PubMed] [Google Scholar]
- 22.Biervert C, Schroeder BC, Kubisch C, et al. . A potassium channel mutation in neonatal human epilepsy. Science 1998;279:403–406. [DOI] [PubMed] [Google Scholar]
- 23.Charlier C, Singh NA, Ryan SG, et al. . A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet 1998;18:53–55. [DOI] [PubMed] [Google Scholar]
- 24.Singh NA, Charlier C, Stauffer D, et al. . A novel potassium channel gene KCNQ2 is mutated in an inherited epilepsy of newborns. Nat Genet 1998;18:25–29. [DOI] [PubMed] [Google Scholar]
- 25.Benarroch EE. Potassium channels: brief overview and implications in epilepsy. Neurology 2009;72:664–669. [DOI] [PubMed] [Google Scholar]
- 26.Muona M, Berkovic SF, Dibbens LM, et al. . A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet 2015;47:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simons C, Rash LD, Crawford J, et al. . Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat Genet 2015;47:73–77. [DOI] [PubMed] [Google Scholar]
- 28.Du W, Bautista JF, Yang H, et al. . Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet 2005;37:733–778. [DOI] [PubMed] [Google Scholar]
- 29.Nava C, Dalle C, Rastetter A, et al. . De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat Genet 2014;46:640–645. [DOI] [PubMed] [Google Scholar]
- 30.Barcia G, Fleming MR, Deligniere A, et al. . De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 2012;44:1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Syrbe S, Hedrich UB, Riesch E, et al. . De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat Genet 2015;47:393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eunson LH, Rea R, Zuberi SM, et al. . Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol 2000;48:647–656. [PubMed] [Google Scholar]
- 33.Sicca F, Imbrici P, D'Adamo MC, et al. . Autism with seizures and intellectual disability: possible causative role of gain-of-function of the inwardly-rectifying K+ channel Kir4.1. Neurobiol Dis 2011;43:239–247. [DOI] [PubMed] [Google Scholar]
- 34.Wood ML, Freites JA, Tombola F, Tobias DJ. Atomistic modeling of ion conduction through the voltage-sensing domain of the Shaker K+ ion channel. J Phys Chem B 2017;121:3804–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freites JA, Tobias DJ. Voltage sensing in membranes: from macroscopic currents to molecular motions. J Membr Biol 2015;248:419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Speca DJ, Ogata G, Mandikian D, et al. . Deletion of the Kv2.1 delayed rectifier potassium channel leads to neuronal and behavioral hyperexcitability. Genes Brain Behav 2014;13:394–408. [DOI] [PMC free article] [PubMed] [Google Scholar]