

Abstract
Structure-based drug design (SBDD) has become a powerful tool utilized by medicinal chemists to rationally guide the drug discovery process. Herein, we describe the use of SPROUT, a de novo-based program, to identify an indazole-based pharmacophore for the inhibition of fibroblast growth factor receptor (FGFR) kinases, which are validated targets for cancer therapy. Hit identification using SPROUT yielded 6-phenylindole as a small fragment predicted to bind to FGFR1. With the aid of docking models, several modifications to the indole were made to optimize the fragment to an indazole-containing pharmacophore, leading to a library of compounds containing 23 derivatives. Biological evaluation revealed that these indazole-containing fragments inhibited FGFR1–3 in the range of 0.8–90 μM with excellent ligand efficiencies of 0.30–0.48. Some compounds exhibited moderate selectivity toward individual FGFRs, indicating that further optimization using SBDD may lead to potent and selective inhibitors of the FGFR family.
Keywords: SBDD, de novo, FGFR, fragment, indazole
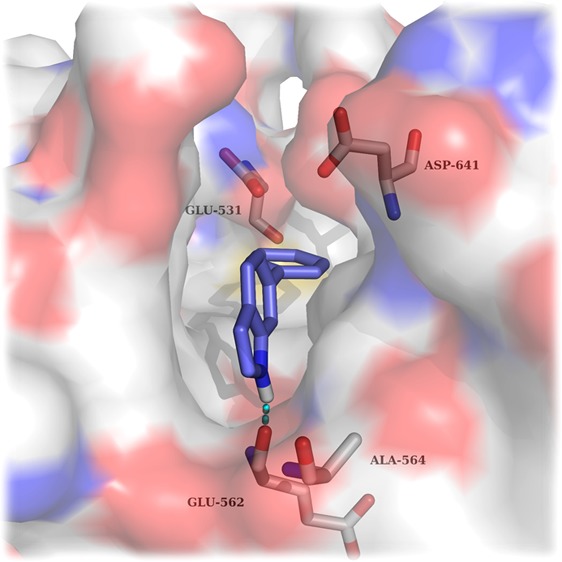
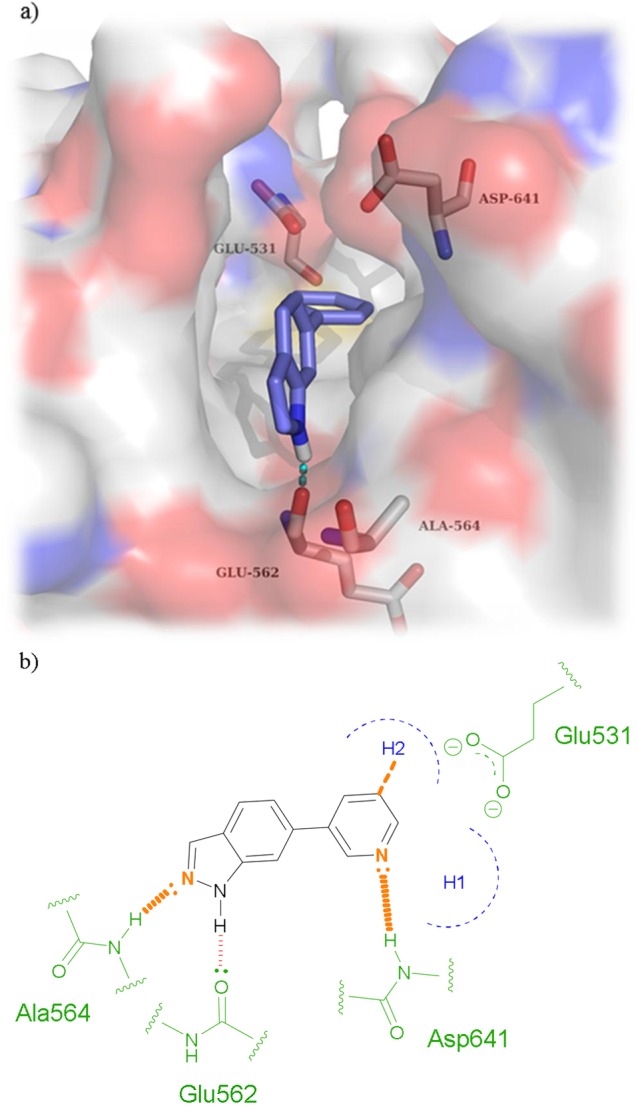
FGFRs are a subfamily of tyrosine kinases (TKs) that are involved in many cellular processes such as cell proliferation, cellular repair, and cell migration.1 Aberrant signaling within this class of kinase has many implications in cancer and particularly in bladder cancer.2 It has been shown that around 50% of upper-and lower-urinary tract tumors possess FGFR3 mutations.3 As well as mutations within FGFR3, overexpression in both FGFR3 and FGFR1 have been found in urothelial carcinomas at all grades and stages.4,5 Currently, several FGFR inhibitors are in clinical use or under clinical development with some acting as selective FGFR inhibitors while others are pan-kinase inhibitors (see Supplemental Section 1.0 (SI-1.0)). There are currently no examples of molecules that exhibit subtype selectivity for the FGFRs, and the role of subtype selectivity in the pharmacological profile of such anticancer agents has not been established. Development of a pan FGFR inhibitor may be more clinically beneficial than an inhibitor that only perturbs one FGFR subtype; however, unwanted inhibition of FGFR1 and FGFR3 leads to side effects such as hyperphosphatemia6 underlining the need for FGFR2 subtype selective inhibitors. CH5183284 is a potent and selective inhibitor of FGFR1–3 exhibiting IC50 values of 9.3, 7.6, and 22 nM,7 respectively, and this compound is currently under clinical investigation for the treatment of cancer patients that harbor FGFR genetic alterations. CH5183284 was discovered using the conventional approach of high throughput screening (HTS). As a prominent approach in lead discovery, HTS allows rapid screening of large compound libraries but is limited to the chemical diversity of that library. Typically, an HTS library may contain approximately one million compounds; a fraction of the total theoretical “drug-like” chemical space which is predicted to be between 1060–10100 compounds.8 SBDD is a useful addition tool for lead identification that can be used alone or in conjunction with HTS to initiate and facilitate a drug discovery program. The application of SBDD can take a number of different forms such as the use of virtual HTS, template matching, and de novo molecular design. A de novo approach can produce compounds “from scratch”. They are predicted to bind to a target and, in theory, access limitless untapped chemical space that may not be present in current compound libraries. De novo design was first used in the 1980s with numerous programs offering access to novel chemical entities from which a number of pharmacologically active molecules have resulted.9−11 One such program, SPROUT, was developed by Johnson et al. in the early 1990s and is designed for constrained structure generation.12 Structure generation can be simplified into two steps: the first part is formation of a molecular “skeleton” that must satisfy the steric constraints of a binding pocket; the second part is generation of a molecule(s) by atom-substitution to the skeleton.13 The constraints of the binding pocket are usually defined using the X-ray crystal data of a target. However, if X-ray crystal data is unavailable, it is possible to design novel inhibitors based purely on a pharmacophore hypothesis.13 Once the constraints have been defined, it is possible to choose interaction sites within the protein, e.g., amino acids within binding pockets for endogenous ligands. Complementary atoms or fragments from available libraries within the program are chosen to interact with these sites and are linked together using spacer fragments. The resulting solutions are clustered using a range of parameters including estimated binding affinity or molecular complexity.13 Further information on the different tools and modules within SPROUT that are used for structure generation can be found in the Supporting Information (SI-4.1). Here, we describe the use of SPROUT to generate an active indazole-based pharmacophore for the inhibition of FGFR kinases. The crystal structure of inhibitor CH5183284 cocrystallized within FGFR1 (PDB code: 3WJ6) was loaded into SPROUT (v6.4.10) and visualized using PyMol14 (Figure 1a). Compound CH5183284 occupies the adenosine triphosphate (ATP) binding site within FGFR1, and several interactions are observed. Two H-bonds form with the benzimidazole moiety; one with the backbone nitrogen of Asp641 and the other with a side chain carboxy oxygen of Glu531 (Figure 1). Another H-bond forms between the pyrazole NH2 and the backbone carbonyl of Glu562. An H-bond can also be seen between the ketone moiety and the backbone NH of Ala564.7De novo design was carried out targeting the CH5183284 binding region within the crystal structure of FGFR1 using SPROUT. Three interaction sites (Asp641, Glu531, and Glu562) were chosen, and complementary fragments were selected (H-bond acceptors for Asp641 and H-bond donors for Glu531 and Glu562, respectively). Spacer templates consisting of aryl, alkyl, and amide groups were chosen to link these various moieties together consecutively. The resulting solutions were then considered visually in terms of perceived ease of synthesis of analogues, identifying 6-phenylindole as a particularly attractive fragment. As an independent check on the validity of 6-phenylindole as a starting point for further inhibitor design, 6-phenylindole was redocked into the FGFR1 crystal structure using Glide15 and the binding pose analyzed (Figure 2a). This fragment is predicted to bind in a similar way to that of CH5183284 with the indole NH forming an H-bond with the backbone carbonyl of Glu562. Further modeling indicated that several structural modifications could be made in order to increase the number of bonding interactions between the inhibitor and FGFR1. Specifically, substitution from an indole to an indazole would open up the opportunity for an H-bond to form between the indazole N-2 and the backbone NH of Ala564. Additionally, modification of the 6-phenyl ring to a 6-pyridyl derivative could also introduce an H-bond between the pyridine nitrogen and the backbone NH of Asp641. Furthermore, placement of a small hydrophobic group in the meta-position of the 6-phenyl ring was predicted to allow occupation of the subsite H2 pocket, hypothesized to further increase binding affinity (Figure 2). This led to us considering an initial fragment library of varying indole/indazole scaffolds, which appeared to offer modest binding potential in the enzyme and would also readily allow us to test the initial design hypotheses (Figure 3). Compounds 1–6 were readily prepared using classic Suzuki chemistry using available aryl bromides and boronic acids (SI-1.1).
Figure 1.
(a) Cocrystal structure of CH5183284 bound within FGFR1. H-bonding interactions are indicated using cyan dashes. (b) Schematic of binding pose of CH5183284 within FGFR1 showing intermolecular interactions. Amino acids, H-bonds, and hydrophobic pockets are shown in green, red, and blue, respectively.
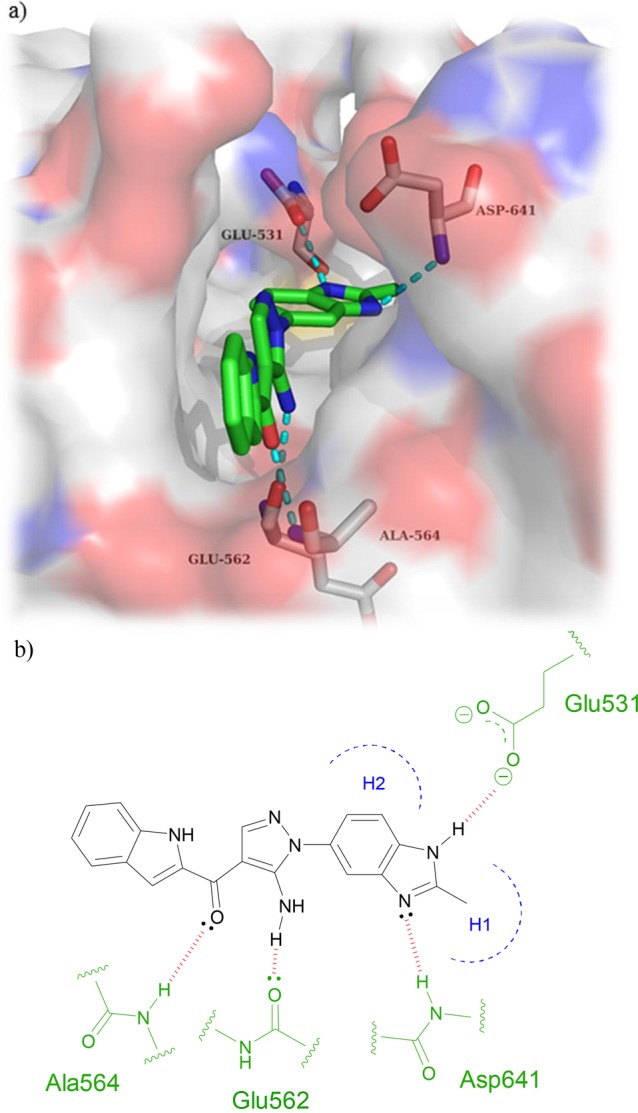
Figure 2.
(a) De novo designed fragment 6-phenylindole (1) docked within the active site of FGFR1 using Glide. An H-bond is predicted to form between the indole NH and the backbone carbonyl of Glu562. (b) Schematic of binding pose of 6-phenylindole core within FGFR1 with modifications outlined in orange.
Figure 3.

Initial fragment compound library.
Biological Evaluation
Compounds 1–6 were screened against FGFR1 at an initial concentration of 500 μM using a fluorescence resonance energy transfer (FRET)-based Z’-lyte assay16 (SI-3.1). IC50 measurements for compounds 1–6 were determined using a 10-point titration experiment with three-fold serial dilutions starting from a concentration of 500 μM. The results are outlined (Table 1).
Table 1. Biological Results for Compounds 1–6 When Screened against FGFR1.
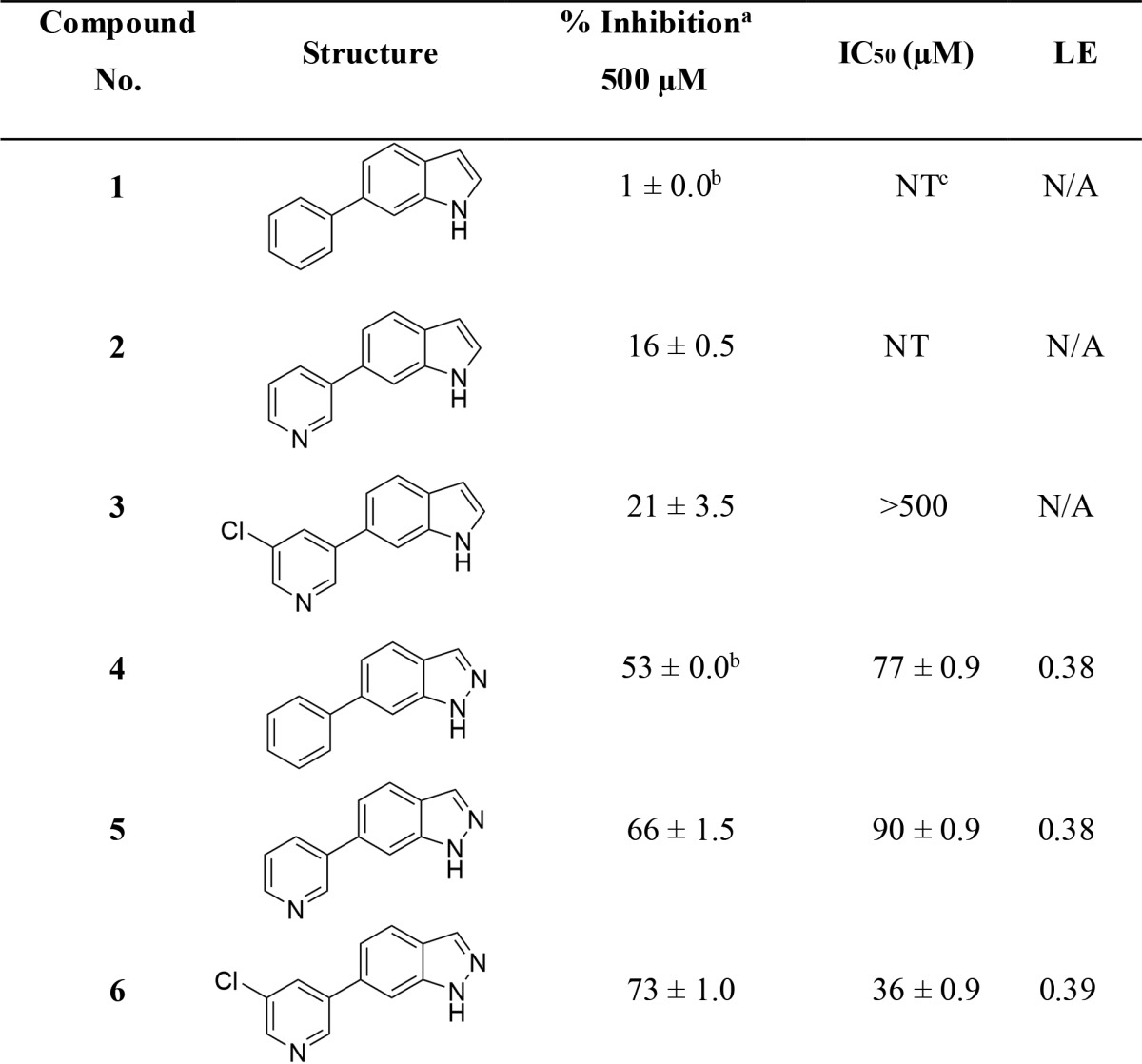
Percent inhibition and IC50 values are given as the mean ± SD of all data points.
No difference in measured data points (N = 2).
NT = not tested.
Interestingly, indole-based fragments 1–3 were found to be essentially inactive with >50% less inhibition against FGFR1 than their corresponding indazole counterparts 4–6. This was confirmed following IC50 measurements with the indazole derivatives having a potency in the range of 36–90 μM against FGFR1. This is consistent with the design hypothesis, which requires the 2-position nitrogen present in the indazole compounds to be involved in H-bonding with the backbone NH of Ala564 and would appear to be crucial for inhibition of FGFR1. These results are also consistent with those demonstrated by Liu et al., who have recently reported inhibitors of FGFR1 containing indazole cores.17 The indazole ligands 4–6 possess ligand efficiencies (LE) of >0.35, which is an excellent start for fragment optimization.18 Compound 6 is the most active with an IC50 value of 36 μM. Compound 4 is more active than compound 5 suggesting that the pyridine nitrogen has a detrimental effect upon the binding affinity of these fragments to FGFR1. Rationalization of these observed activities was investigated using docking models (SI-4.2).
Library Expansion
In order to expand the structure–activity relationships (SARs) for the active indazole pharmacophore 7, a larger focus library of target compounds was developed. Compounds were readily synthesized using the conditions outlined in SI-1.1 and subjected to biological evaluation (Table 2).
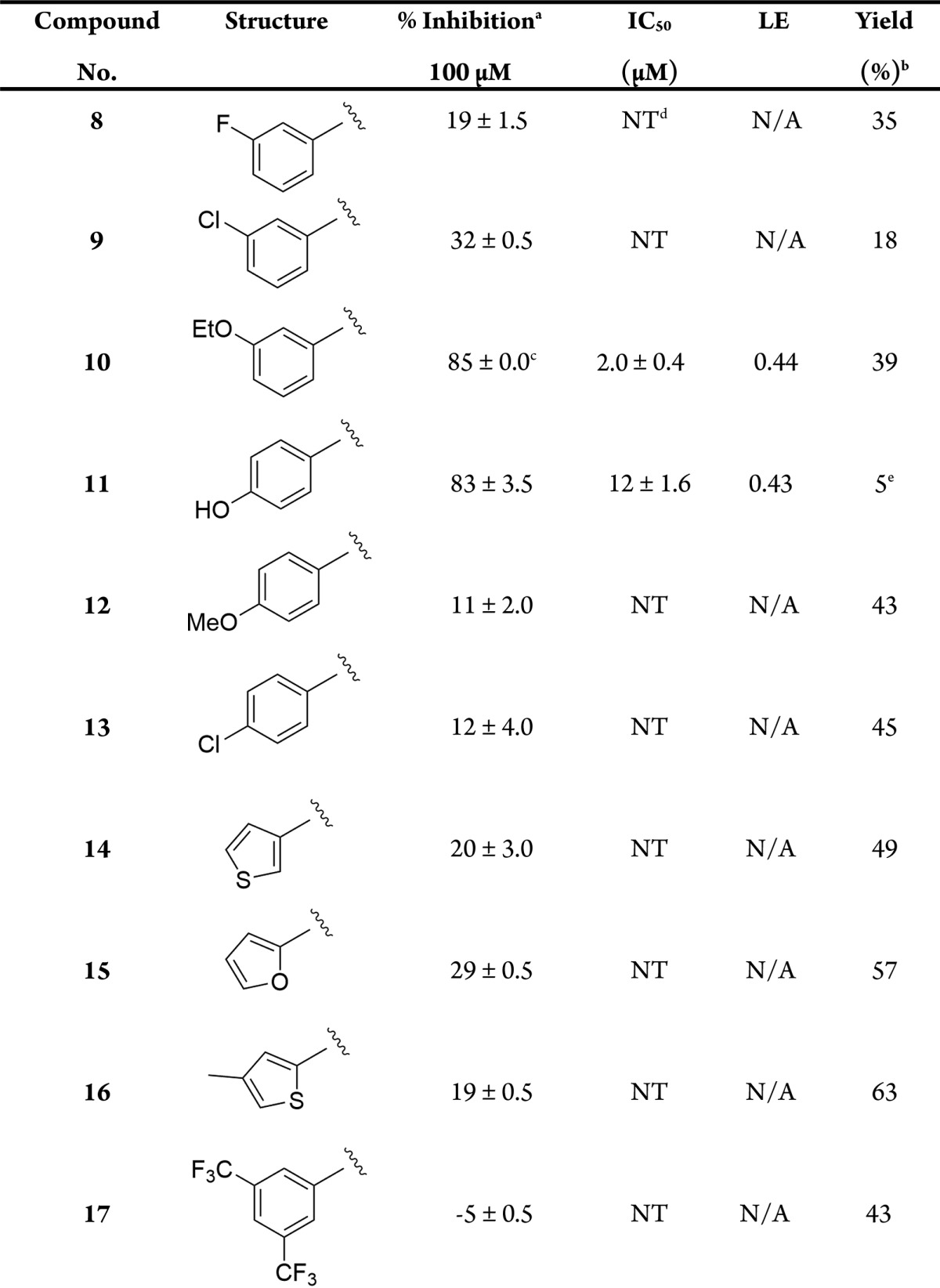
Table 2. Reaction Yields and Biological Results for Compounds 8–17 when Screened against FGFR1.
Percent inhibition and IC50 values are given as the mean ± SD of all data points.
Reaction yields for Suzuki chemistry (SI-1.1).
No difference in measured data points (N = 2).
NT = not tested.
Low yield explained by propensity of 2/4-hydroxyphenylboronic acids undergoing rapid protodeboronation.19
The results obtained for single-point analyses were carried out at a concentration of 100 μM. The observed activities for compounds 8–10 indicate that substituting larger groups in the 3-position of the phenyl ring increases potency, with compound 10 having an IC50 value of 2.0 μM and an excellent LE of 0.44. The increase in potency could be due to the ethoxy group occupying the H1 pocket as predicted using modeling (SI-4.2). The phenol moiety present in compound 11 has the potential to be an H-bond donor to residue Glu531 that lies deep within the ATP binding pocket, whereas compounds 12–13 lack this ability. Interestingly, compound 11 has an IC50 of 12 μM, whereas compounds 12–13 are inactive. This suggests that the H-bond donor potential of the phenol is crucial for inhibition of FGFR1, and modeling reinforces this hypothesis (SI-4.2). Substitution at the 6-position substituent on the indazole core from a six-membered ring to a five-membered ring results in a loss of activity, shown by the results obtained for compounds 15–16. Compound 17 is completely inactive; revealing that a 3,5-substitution pattern around the 6-position phenyl ring is detrimental to activity suggesting there may be a steric limit to what can be tolerated around this ring.
SAR Exploration of Compounds 10 and 11
Compounds 10 and 11 were taken forward to further optimize focusing on substitution around the phenyl ring. Based on modeling (SI-4.2), it became apparent that further substitution of small hydrophobic groups in addition to the existing substituent on the 6-position phenyl ring could result in tighter binding to FGFR1. A further library was therefore developed, synthesized using the conditions outlined in SI-1.1, and biologically evaluated (Table 3). Compounds 21–24 were synthesized using an alternative route (SI-1.1.1). In addition to FGFR1, the activity of these compounds against FGFR2/3 was also evaluated. Inspection of the data in Table 3 reveals some interesting points. In addition to FGFR1, compound 10 was also found to inhibit FGFR2/3 with IC50 values of 0.8 and 4.5 μM, respectively. In general, addition of small hydrophobic substituents to the phenyl ring of compound 10 results in a decrease in potency against FGFR1–3. Compound 18 exhibits a complete loss of activity for FGFR3, a 42-fold decrease in activity for FGFR1, and a 15-fold decrease for FGFR2 when compared to compound 10. A similar trend can be seen for compounds 19 and 20. This suggests that the requirements to inhibit FGFR3 are more stringent than FGFR1/2 with FGFR2 being the most tolerant to further substitution around the 6-position phenyl ring. Compound 11 was found to exhibit IC50 measurements of 3.0 and 51 μM against FGFR2/3, respectively. Addition of small hydrophobic groups onto the 6-position phenyl ring in compound 11 generally results in an increase in potency with a preference for substitution in the 2-position. Compounds 21 and 23 show an increase in size of the 2-position substituent from fluorine to methyl, respectively, which results in an increase in potency against FGFR1; this trend is also seen for compounds 22 and 24. Substitution of small hydrophobic groups in the 3-position of the 6-position phenyl ring is less well tolerated when compared to 2-position substituted rings. These fragments show small selectivity differences for individual subtype FGFRs. Compound 10 exhibits a 2.5-fold selectivity preference for FGFR2 over FGFR1, and compound 18 shows that, although the potency has dropped, the difference in selectivity has increased to ∼7-fold by addition of a fluorine atom in the 5-position of the 6-position phenyl ring. As these compounds are fragments (MW ≈ 250), we hypothesize that structural expansion of these molecules will result in an increase in the small selectivity difference between FGFR1/2 that compounds 10 and 18 currently exhibit.
Table 3. Biological Results for Compounds 10–24 When Screened against FGFR1-3.

Conclusion
We have identified an indazole-based pharmacophore that shows encouraging levels of inhibition against FGFR kinases using a de novo-based design approach to identify the initial hit. Optimization of this hit led to a library of fragments whereby SARs were established and specific structural aspects of the molecules upon which subtype selectivity appear to depend have been identified. Current efforts are focused on designing larger compounds in order to increase potency and, more importantly, selectivity for the individual FGFR subtypes.
Acknowledgments
The authors thank Dr. Martin McPhillie, Medical Research Council and Life Technologies Ltd.
Glossary
ABBREVIATIONS
- ATP
adenosine triphosphate
- FGFR
fibroblast growth factor receptor
- FRET
fluorescence resonance energy transfer
- HTS
high-throughput screening
- LE
ligand efficiency
- SAR
structure–activity relationship
- SBDD
structure-based drug design
- TK
tyrosine kinase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00349.
Current FGFR inhibitors. Synthetic routes. All experimental details: general procedures and instrumentation, general methods, compound characterization, 1H NMR and 13C NMR spectra for all compounds. FRET-based Z′ lyte assay conditions and IC50 curves. Computational details: SPROUT and docking models (PDF)
Author Contributions
‡ These authors contributed equally through experimental lab work. The manuscript was written through contributions of L.D.T., M.A.K., and C.W.G.F. M.A.K. and C.W.G.F. provided supervisory support throughout this work. Project development and experimental work were carried out principally by L.D.T. All authors have given approval to the final version of the manuscript.
Funded by a Medical Research Council-doctoral training grant.
The authors declare no competing financial interest.
Supplementary Material
References
- Eswarakumar V. P.; Lax I.; Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16 (2), 139–149. 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- di Martino E.; Tomlinson D. C.; Knowles M. A. A Decade of FGF Receptor Research in Bladder Cancer: Past, Present, and Future Challenges. Adv. Urol 2012, 2012, 429213. 10.1155/2012/429213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rhijn B. W. G.; Montironi R.; Zwarthoff E. C.; Jobsis A. C.; van der Kwast T. H. Frequent FGFR3 mutations in urothelial papilloma. J. Pathol 2002, 198 (2), 245–251. 10.1002/path.1202. [DOI] [PubMed] [Google Scholar]
- Tomlinson D. C.; Baldo O.; Harnden P.; Knowles M. A. FGFR3 protein expression and its relationship to mutation status and prognostic variables in bladder cancer. J. Pathol. 2007, 213 (1), 91–98. 10.1002/path.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson D. C.; Lamont F. R.; Shnyder S. D.; Knowles M. A. Fibroblast Growth Factor Receptor 1 Promotes Proliferation and Survival via Activation of the Mitogen-Activated Protein Kinase Pathway in Bladder Cancer. Cancer Res. 2009, 69 (11), 4613–4620. 10.1158/0008-5472.CAN-08-2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz D. M.; Itoh N. The Fibroblast Growth Factor signaling pathway. Wires Dev Biol. 2015, 4 (3), 215–266. 10.1002/wdev.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi Y.; Akiyama N.; Tsukaguchi T.; Fujii T.; Sakata K.; Sase H.; Isobe T.; Morikami K.; Shindoh H.; Mio T.; Ebiike H.; Taka N.; Aoki Y.; Ishii N. The Fibroblast Growth Factor Receptor Genetic Status as a Potential Predictor of the Sensitivity to CH5183284/Debio 1347, a Novel Selective FGFR Inhibitor. Mol. Cancer Ther. 2014, 13 (11), 2547–2558. 10.1158/1535-7163.MCT-14-0248. [DOI] [PubMed] [Google Scholar]
- Simmons K. J.; Chopra I.; Fishwick C. W. G. Structure-based discovery of antibacterial drugs. Nat. Rev. Microbiol. 2010, 8 (7), 501–510. 10.1038/nrmicro2349. [DOI] [PubMed] [Google Scholar]
- Honma T. Recent advances in De novo design strategy for practical lead identification. Med. Res. Rev. 2003, 23 (5), 606–632. 10.1002/med.10046. [DOI] [PubMed] [Google Scholar]
- Babine R. E.; Bleckman T. M.; Kissinger C. R.; Showalter R.; Pelletier L. A.; Lewis C.; Tucker K.; Moomaw E.; Parge H. E.; Villafranca J. E. Design, Synthesis and X-Ray Crystallographic Studies of Novel FKBP-12 Ligands. Bioorg. Med. Chem. Lett. 1995, 5 (15), 1719–1724. 10.1016/0960-894X(95)00290-A. [DOI] [Google Scholar]
- Iwata Y.; Naito S.; Itai A.; Miyamoto S. Protein structure-based de novo design and synthesis of aldose reductase inhibitors. Drug Des Discovery 2001, 17 (4), 349–59. [PubMed] [Google Scholar]
- Gillet V.; Johnson A. P.; Mata P.; Sike S.; Williams P. SPROUT - A Program for Structure Generation. J. Comput.-Aided Mol. Des. 1993, 7 (2), 127–153. 10.1007/BF00126441. [DOI] [PubMed] [Google Scholar]
- Gillet V. J.; Newell W.; Mata P.; Myatt G.; Sike S.; Zsoldos Z.; Johnson A. P. SPROUT - Recent Developments in the De-Novo Design of Molecules. J. Chem. Inf. Model. 1994, 34 (1), 207–217. 10.1021/ci00017a027. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.The PyMOL molecular graphics system; Schrödinger, LLC: New York, 2002. [Google Scholar]
- Small-Molecule Drug Discovery Suite 2015–4: Glide, version 6.9; Schrödinger, LLC: New York, 2015. [Google Scholar]
- Z′-Lyte Screening Protocol and Assay Conditions; Life Technologies: Paisley, Scotland, 2015. [Google Scholar]
- Liu J.; Peng X.; Dai Y.; Zhang W.; Ren S. M.; Ai J.; Geng M. Y.; Li Y. X. Design, synthesis and biological evaluation of novel FGFR inhibitors bearing an indazole scaffold. Org. Biomol. Chem. 2015, 13 (28), 7643–7654. 10.1039/C5OB00778J. [DOI] [PubMed] [Google Scholar]
- Schultes S.; de Graaf C.; Haaksma E. E. J.; de Esch I. J. P.; Leurs R.; Krämer O. Ligand efficiency as a guide in fragment hit selection and optimization. Drug Discovery Today: Technol. 2010, 7 (3), e157–e162. 10.1016/j.ddtec.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Lee C. Y.; Ahn S. J.; Cheon C. H. Protodeboronation of ortho- and para-Phenol Boronic Acids and Application to ortho and meta Functionalization of Phenols Using Boronic Acids as Blocking and Directing Groups. J. Org. Chem. 2013, 78 (23), 12154–12160. 10.1021/jo402174v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.