

Abstract
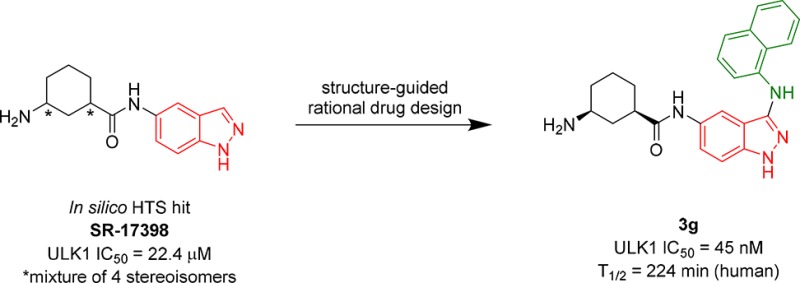
We present the outcome of an in silico high throughput screen (HTS) and optimization of a small molecule Unc-51-Like Kinase 1 (ULK1) inhibitor hit, SR-17398, with an indazole core. Docking studies guided design efforts that led to inhibitors with increased activity vs ULK1 (IC50 < 50 nM). The most advanced molecules in this inhibitor series (3a and 3g) hold promise for further development into selective ULK1 molecular probes to interrogate the biology of ULK1 and to assess whether selectively targeting autophagy is an effective anticancer strategy.
Keywords: In silico high-throughput screen, ULK1, inhibitor, indazole
Macroautophagy (hereafter autophagy) is an important cellular process that maintains energy homeostasis during periods of stress and starvation and plays a major role in controlling protein and organelle quantity and quality.1 Autophagy is an ancient, cannibalistic (literally “self-eating”) pathway, wherein cellular components including long-lived proteins, bulk cytoplasmic material, and aged or damaged organelles are encapsulated by a double membrane vesicle, coined the autophagosome.2 These vesicles then fuse with the lysosome, which degrades the delivered cargo to recoup building blocks and adenosine triphosphate (ATP) necessary for cell survival.3,4
Autophagy has been implicated in the pathology of various diseases such as neurodegeneration and cancer.5 The observed role of autophagy in cancer is complex and can be tumor-suppressive or tumor-promoting depending on context. Recent studies using genetically engineered mouse models have implicated autophagy in KRAS- and BRAF- driven cancers by demonstration of tumor suppression in response to inhibition of autophagy.6−8 Moreover, autophagy is protective for cancers experiencing a decrease in nutrient availability or damage caused by cancer therapeutics.9,10 Accordingly, blocking autophagy via small molecule inhibitors in autophagy-reliant cancers could increase the efficacy of current chemotherapeutics and may result in tumor suppression as a standalone chemotherapy.11,12
Unc-51-like kinase 1 (ULK1) is a 112 kDa ubiquitously expressed protein and is required for efficient stress-induced autophagy under most conditions.13 ULK1 is negatively regulated by mTOR under normal nutrient conditions and activated during periods of amino acid or glucose deprivation by AMPK through phosphorylation at multiple sites in the unstructured serine–proline-rich domain.14−16 ULK1 is also activated by the GSK3-TIP60 signaling pathway upon growth factor deprivation.17 Small molecule inhibition of ULK1 potentially provides an avenue for suppressing autophagy. Recently, X-ray crystal structures of ULK1 were elucidated by the Shokat group featuring ATP competitive inhibitors cocrystallized with the kinase; there are also reports of other early stage inhibitors in the literature.18−21
Physical HTS campaigns are useful for generating chemical starting points for drug discovery programs.22 Screening a large library of characterized ligands against a biochemical target provides insight into efficacious chemical scaffolds and structure–activity relationship (SAR) patterns. This approach has led to the generation of numerous therapeutic candidates following SAR optimization of screening hits.23 Experimental screens require expensive resources such as large chemical libraries, miniaturized assays, automated instruments, costly reagents, etc. By comparison, an in silico screen has far fewer requirements. The resources needed to carry out an in silico screen are minimal, including some that can be sourced freely. Suitable computational facilities are the largest physical asset needed. An additional benefit is that an in silico screen can be performed on any target as long as suitable crystal structures (or homology model) with three-dimensional coordinates of the protein target are available.24 We chose to perform an in silico HTS to identify ULK1 inhibitors due to the simplicity and cost-effective nature of this approach. There are several programs capable of executing in silico HTS campaigns available both free of charge and for purchase.25 Approaches utilizing in silico HTS campaigns have generated hits for numerous drug discovery projects.26
We employed Schrödinger’s Maestro software in our studies.27 Our protein target was a publicly available crystal structure of ULK1 with a bound ATP competitive inhibitor published by the Shokat group (PDB ID: 4WNP).19 The enzyme coordinates were obtained from the protein structure database (http://www.rcsb.org/pdb/). The protein was first prepared for docking studies via the Protein Prep application. Then, using the cocrystallized inhibitor as the center coordinate, we generated a grid with the Schrödinger Glide, Receptor Grid Generation task.27 The grid dimensions were 25 × 25 × 25 Å, encompassing the critical hinge-binding region residues Cys95, Tyr94, Glu93, and Met92. The grid also encompassed proximal solvent exposed and binding pocket areas, which could provide interactions with amino acids that are specific to ULK1.
Using this structure, we performed an in silico HTS campaign employing the molecular structures contained in the ∼650,000 Scripps HTS library (Figure 1a). The Scripps Molecular Library Screening Center hosts this library and is maintained by the Lead Identification Department at Scripps Florida. This library comprises primarily commercially available compounds but also includes small molecules developed in-house. We prepared the digital screening ligand library using the LigPrep workflow incorporating the following parameters: OPLS2005 force field, pH 7, and generation of tautomers. This process produced a digital file containing ∼1.2 million tautomers of the original structures. The prepared compounds were subsequently docked in a standard precision (SP) protocol, as this method has been observed to proceed with a balance of speed and accuracy.28 The output of this docking campaign was analyzed by inspection of the top 500 hits selected according to the Glide docking score.
Figure 1.

(a) Workflow for in silico HTS and structure of SR-17398 selected from analysis of the best candidates. (b,c) Docking images of SR-17398 and optimized scaffold 3g in the ULK1 crystal structure.
Screening hits were initially prioritized by their H-bonding interactions with the hinge-binding region of the ULK1 ATP pocket. Next, the top hits were grouped into common cores based on repeat scaffolds observed in the top tier.
While performing this evaluation, hits containing promiscuous binding groups or PAINS were eliminated.29 In this way, we identified a variety of cores and purchased a small set of representative compounds from ChemNavigator. The purchased compounds were selected by using substitution patterns and functional groups observed in screening hits with the highest Glide scores. The molecules purchased were >90% similar, if not identical, to the molecules identified in the docking studies. Structures of a subset of the hits obtained from this screen are provided in Figure S1.
The purchased hits were evaluated in a biochemical assay for ULK1.30 This assay used full-length ULK1 and full-length human Atg13 tagged with Flag. DMSO solutions (10 mM) of assayed compounds were prepared for IC50 determination. Of these compounds, only one (SR-17398) displayed ULK1 inhibitory activity <30 μM (the IC50 for SR-17398 was 22.4 μM). IC50 values are reported with a standard deviation <±5 nM based on repeat experiments carried out with control compounds. We used this compound as a starting point for subsequent structure based design and SAR studies.
We critically evaluated the docking pose of SR-17398 in the ULK1 crystal structure. The two nitrogen atoms contained in the indazole ring system make two H-bonding interactions with the amide backbone of the hinge region, specifically with Glu93 and Cys95 (Figure 1b). The amide carbonyl at the 5-position of the indazole participates in a H-bonding interaction with Lys46 mediated by a water molecule in the active site. Lastly, the primary amine attached to the cyclohexyl ring makes a H-bonding interaction with Asn143. This residue is located in an oxygen-rich portion of the active site made up of an amino acid backbone orienting amide carbonyls toward the binding pocket.
The first objective was to increase the activity of SR-17398 by adding functional groups capable of engaging in additional binding interactions with ULK1. The first modification was the addition of an amino group at the 3-position of the indazole core, as in structure 1a. Our docking models predicted this amine would make a new H-bonding interaction with the amide carbonyl of Cys95 in the ATP binding domain. A second aim was to modify the 3-aminocyclohexane unit by eliminating stereocenters. We also considered using other amines capable of maintaining the interactions that we identified as important for binding in the oxygen-rich portion of the active site. To achieve this goal, we examined the docking pose of various structures capable of maintaining two H-bonding interactions, one with Lys46 and the second with Asn134 or a proximal residue. The best candidates based in these criteria were selected for synthesis. Lastly, we designed compounds predicted to interact with Tyr94. The aryl ring system of this residue overhangs the active site. We envisioned projecting an aryl unit from the 3-amino position of the indazole that could engage in a π-stacking or edge-to-face π-interaction with Tyr 94. Aryl substituents capable of H-bonding interaction with Tyr94 were also considered. We used a combination of biochemical data along with docking poses to assist in validating hypotheses about structural changes capable of improving the ULK1 inhibitory activity in this compound series.
The screening hit SR-17398 was synthesized via a coupling reaction of 5-aminoindazole and 1-N-Boc-3-carboxyl-cyclohexane followed by deprotection of the Boc group (Scheme 1a). This allowed us to verify its structure and activity. Multiple unsubstituted 3-aminoindazole derivatives (Table 1) were synthesized following the sequence in Scheme 1a, featuring 1a as a representative example. This compound was synthesized by first Boc protection of 1 followed by reduction of the 5-nitro substituent to yield 2. The same amide coupling and deprotection sequence described above was used to complete the synthesis of 1a from the 5-aminoindazole intermediate (2), as well as for all other compounds in Table 1.
Scheme 1. Synthesis of Representative ULK1 Inhibitors.

Reagents and conditions: (a) Boc2O, Et3N, DMAP, THF, 0 °C; (b) Pd/C, H2, MeOH, 22 °C; (c) 3-(Boc-amino)cyclohexanecarboxylic acid, HATU, Et3N, CH2Cl2, 22 °C; (d) 10% TFA in CH2Cl2, 22 °C; (e) 1-naphthylamine, DIPEA, CH2Cl2, 0 to 22 °C; (f) Lawesson’s reagent, toluene, 110 °C; (g) hydrazine, EtOH, 78 °C; (h) Br2, AcOH, 80 °C; (i) 3,4-dihydro-2H-pyran, p-TsOH, EtOAc, 77 °C; (j) 5-aminoisoquinoline, Pd2(dba)3, [(t-Bu)3PH]BF4, NaOtBu, 1,4-dioxane; (k) 4 M HCl in dioxane, 22 °C.
Table 1. Structure–Activity Relationship for ULK1 Inhibitors with Various Amide Units.
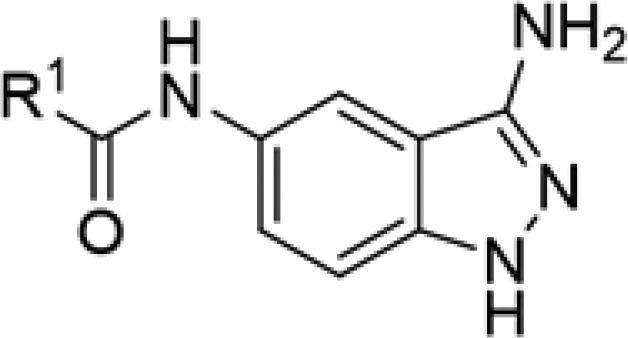
| compd | SR # | R1 | IC50 ULK1 (μM) |
|---|---|---|---|
| la | SR-18938 | 3-aminocyclohexanea | 0.368 |
| 1b | SR-18939 | 3-aminophenyl | 18.10 |
| 1c | SR-19557 | trans-4-aminocyclohexane | 13.96 |
| 1d | SR-19558 | cis-4-aminocyclohexane | 1.49 |
| 1e | SR-19559 | propylamine | >33 |
| 1f | SR-19778 | 4-piperidine | 0.560 |
| 1g | SR-19779 | 3-piperidinea | 0.670 |
| 1h | SR-19780 | 2-piperidinea | 0.840 |
| 1i | SR-19874 | (1S,3R)-3-aminocyclohexane | 0.330 |
Compounds tested as a mixture of stereoisomers.
The analogs presented in Tables 2 and 3 were synthesized using one of two processes. First, compounds 2c–h and all the compounds in Table 3 were synthesized by building an indazole core from a substituted benzene derivative. This process is illustrated in Scheme 1b using the intermediates for the synthesis of 3a as an example. The first reaction involves amide formation between 1-naphthylamine and 5-fluoro-3-nitrobenzoyl chloride (5). The amide was converted into thioamide 6 using Lawesson’s reagent. Subsequent treatment of 6 with hydrazine forms the 5-nitroindazole ring system 7. Compounds 2j and 2k were synthesized from 7 by using a Buchwald–Hartwig coupling reaction.31 The synthesis of 2k is found in Scheme 1 for the cis representative. The sequence begins with bromination of 5-nitroindazole (6) followed by THP protection to generate 3-bromoindazole 7. This intermediate was then coupled with 5-aminoisoquinoline using Buchwald–Hartwig coupling conditions and subsequently THP deprotected to yield indazole derivative 8. The 5-nitroindazole intermediates of type 5 and 8 were then subjected to acylation reactions as described in Scheme 1. The synthesis of 2a, 2b, and 2i utilized modified procedures that are described in the Supporting Information.
Table 2. Structure–Activity Relationship for ULK1 Inhibitors with Various Aryl Groups at the 3-Amino Position.
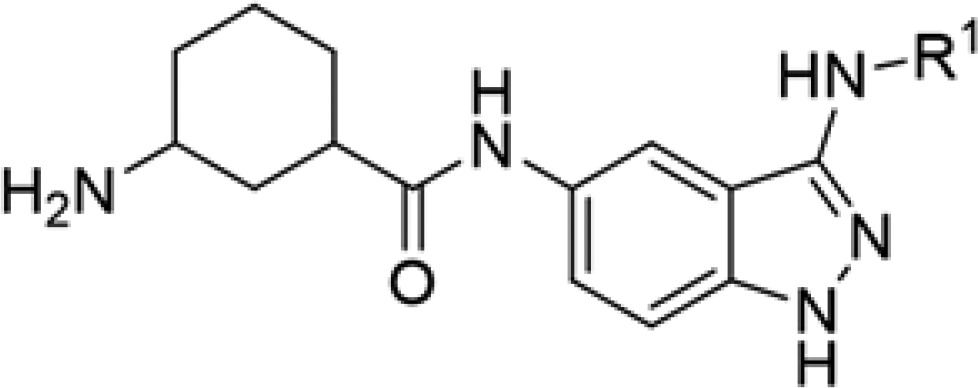
| compd | SR # | R1 | IC50 ULK1 (μM) |
|---|---|---|---|
| 2a | SR-18937 | benzamide | 0.242 |
| 2b | SR-19398 | 4-methoxybenzyl | 0.477 |
| 2c | SR-19399 | phenyl | 2.73 |
| 2d | SR-19401 | 4-bromophenyl | 0.416 |
| 2e | SR-19403 | 2-methoxyphenyl | 0.110 |
| 2f | SR-19873 | 4-trifluoromethylphenyl | 0.840 |
| 2g | SR-20077 | 3-trifluoromethylphenyl | 0.288 |
| 2h | SR-20078 | methyl-3-benzoate | 0.183 |
| 2i | SR-20080 | 4-quinoline | 0.239 |
| 2j | SR-20291 | 8-quinoline | 0.589 |
| 2k | SR-20296 | 5-isoquinoline | 0.040 |
Table 3. Structure–Activity Relationship for ULK1 Inhibitors with a 3-Aminonaphthyl Group and Various Amides.
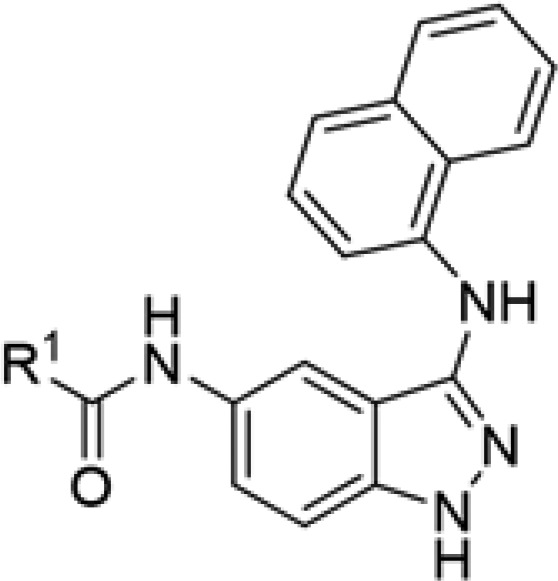
| compd | SR # | R1 | IC50 ULK1 (μM) |
|---|---|---|---|
| 3a | SR-19871 | 3-aminocyclohexanea | 0.011 |
| 3b | SR-20079 | 3-dimethylaminocyclohexane | 4.23 |
| 3c | SR-20290 | 4-piperidine | 0.315 |
| 3d | SR-20292 | (1S,3R)-3-aminocyclohexane | 3.05 |
| 3e | SR-20293 | 3-hydroxycyclohexane | 2.22 |
| 3f | SR-20294 | 4-tetrahydropyran | 1.83 |
| 3g | SR-20295 | (1R,3S)-3-aminocyclohexane | 0.045 |
| 3h | SR-20297 | trans-3-aminocyclohexanea | 0.024 |
| 3i | SR-20298 | 2-azetidine | 4.94 |
Compounds tested as a mixture of stereoisomers.
Analogs of screening hit SR-17398 were evaluated for ULK1 inhibitory activity using the previously described biochemical assay.30 As predicted based on the modeling efforts, addition of an amino group at the 3-position of the indazole ring system led to a significant increase in ULK1 inhibition potency (368 nM, 1a, Table 1). Next, we attempted to alter the cyclohexane ring of 1a. Our goal was to eliminate the stereocenters while also retaining the H-bonding interactions believed to be important for activity. Analog 1b with a 3-aminophenyl group in place of the 3-aminocyclohexane had markedly reduced inhibitory activity (18.1 μM). 3-Aminopropyl functionalization also resulted in a loss of activity (1e, >33 μM). 4-Aminocyclohexane derivatives (cis and trans, 1c and 1d) proved to be much less active than the 3-amino variant.
However, 2-, 3-, and 4-piperidine derivatives retained moderate activity against ULK1 (1f, 1g, 1h). The 4-pipderidine analog 1f was the most potent in this series (560 nM) apart from 1a. As noted in Table 1, many of these derivatives were synthesized as a mixture of stereoisomers, but in the case of 1a, we purposefully examined the effect of stereochemistry on ULK1 activity. Inhibitor 1i has (1R,3S) configuration in the cyclohexane ring and proved to have about equivalent potency when compared to the mixture of isomers 1a (330 nM).
Derivatives functionalized with various aryl units at the 3-amino position of the indazole ring were examined next (Table 2). Appending a benzamide to this position led to retention of activity (2a, 242 nM), while using reductive amination to incorporate a 4-methoxybenzyl unit retained comparable activity to 1a (2b, 477 nM). Appending an aromatic six-membered ring directly to this position with varying methoxy or trifluoromethyl substituent groups resulted in a retention of submicromolar inhibition (2e, 2f, 2g); the ortho-methoxy derivative displayed the most potent activity (2f, 110 nM). 4-Bromo and 3-methoxycarbonyl substitution did not provide additional activity (2d, 2h). Introduction of 1-naphthyl unit as in 3a (Table 3) resulted in a significant increase in activity, generating an inhibitor with 11 nM ULK1 inhibition potency. When using a quinoline derivative with the nitrogen at the 4 or 8 position (analogs 2i, 2j), a reduction in activity occurred compared to 3a. However, a 5-isoquinoline derivative yielded comparable activity to the 1-naphthyl functionalized system (2k, 40 nM). We hypothesize that potent activity is retained in 2k (compared to that in 2i, 2j) because the quinolone nitrogen of 2k is not conjugated to the 3-amino group in the indazole core.
SAR data for analogs deriving from potent inhibitor 3a are presented in Table 3. The cis-(1S,3R) configuration of the cyclohexyl amine led to a significant reduction in activity (3d, 3 μM), while the cis-(1R,3S) enantiomer (3g, 45 nM) was about four-fold less active compared to the mixture of stereoisomers (3a). We evaluated a racemic mixture of the two trans isomers (3h; 24 nM), which proved to be only two-fold less active than the stereoisomeric mixture 3a. These data suggest that the most potent isomer is in the trans configuration. Replacement of the primary amine with a hydroxyl group or a dimethylamine resulted in a significant reduction in activity (3b, 3d). Tetrahydropyran or azetidine substituents led to a significant loss of activity (3f, 3i). Lastly, use of a 4-piperdine in place of the cyclohexane unit (3c) resulted in a modest drop in activity to 315 nM.
Assessment of in vitro drug metabolism was obtained on some of our most potent analogs (2e, 3a, 3c, and 3g). These compounds proved to have excellent stability in human, rat, and mouse microsomes, and they also exhibited negligible CYP inhibition (Supplementary Figure 2). These results are encouraging for our goal of using these (or further optimized) compounds in animal models of cancer.
In summary, using an in silico HTS campaign utilizing a published X-ray structure of ULK1 and the electronic coordinates of an in-house chemical library, we identified SR-17398 as a moderately active ULK1 inhibitor. Further optimization of SR-17398 using structure-guided rational drug design then led to the generation of significantly more potent ULK1 inhibitors. Utilizing two specific modifications: (1) addition of an amino group at the 3-position of the indazole and (2) substitution of the 3-amino unit with an aromatic 10-membered ring system [either naphthyl (3a) or 5-isoquinolyl (2k)]. Docking models suggest how these substituents potentially interact with active site residues (Figure 1c). The 3-amino group provides a third H-bonding interaction with the hinge region, while the 10-membered aromatic system can engage in π-interactions with Tyr94. SAR efforts for the 3-aminocyclohexane substituent have confirmed that it is essential for ULK1 inhibition.
Further studies on the development of this series of indazole-derived ULK1 inhibitors, for use in in vivo studies vs ULK1-dependent cancers, will be reported in due course.
Acknowledgments
This project was supported by grants from the National Institutes of Health.
Glossary
ABBREVIATIONS
- HTS
high throughput screening
- ULK1
Unc-51-Like Kinase 1
- ATP
adenosine triphosphate
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00344.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This research was supported by NIH grant R01 GM113972
The authors declare no competing financial interest.
Supplementary Material
References
- Klionsky D. J.; Emr S. D. Autophagy as a Regulated Pathway of Cellular Degradation. Science 2000, 290 (5497), 1717–1721. 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze S. A.; Yoshimori T. The Origin of the Autophagosomal Membrane. Nat. Cell Biol. 2010, 12 (9), 831–835. 10.1038/ncb0910-831. [DOI] [PubMed] [Google Scholar]
- Mizushima N.; Levine B.; Cuervo A. M.; Klionsky D. J. Autophagy Fights Disease through Cellular Self-Digestion. Nature 2008, 451 (7182), 1069–1075. 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P.; Reggiori F.; Codogno P. Emerging Regulation and Functions of Autophagy. Nat. Cell Biol. 2013, 15 (7), 713–720. 10.1038/ncb2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein D. C.; Codogno P.; Levine B. Autophagy Modulation as a Potential Therapeutic Target for Diverse Diseases. Nat. Rev. Drug Discovery 2012, 11 (9), 709–730. 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. Y.; Karsli-Uzunbas G.; Mathew R.; Aisner S. C.; Kamphorst J. J.; Strohecker A. M.; Chen G.; Price S.; Lu W.; Teng X.; Snyder E.; Santanam U.; DiPaola R. S.; Jacks T.; Rabinowitz J. D.; White E. Autophagy Suppresses Progression of K-Ras-Induced Lung Tumors to Oncocytomas and Maintains Lipid Homeostasis. Genes Dev. 2013, 27 (13), 1447–1461. 10.1101/gad.219642.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A.; Rajeshkumar N. V.; Wang X.; Yabuuchi S.; Alexander B. M.; Chu G. C.; Von Hoff D. D.; Maitra A.; Kimmelman A. C. Autophagy Is Critical for Pancreatic Tumor Growth and Progression in Tumors with p53 Alterations. Cancer Discovery 2014, 4 (8), 905–913. 10.1158/2159-8290.CD-14-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohecker A. M.; Guo J. Y.; Karsli-Uzunbas G.; Price S. M.; Chen G. J.; Mathew R.; McMahon M.; White E. Autophagy Sustains Mitochondrial Glutamine Metabolism and Growth of BrafV600E–Driven Lung Tumors. Cancer Discovery 2013, 3 (11), 1272–1285. 10.1158/2159-8290.CD-13-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K. S. Autophagy and Cancer. Exp. Mol. Med. 2012, 44 (2), 109–120. 10.3858/emm.2012.44.2.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. The Role for Autophagy in Cancer. J. Clin. Invest. 2015, 125 (1), 42–46. 10.1172/JCI73941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman A. C. The Dynamic Nature of Autophagy in Cancer. Genes Dev. 2011, 25 (19), 1999–2010. 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn A.; Thamm D. H.; Gustafson D. L. Autophagy and Cancer Therapy. Mol. Pharmacol. 2014, 85 (6), 830–838. 10.1124/mol.114.091850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E. Y. W.; Kir S.; Tooze S. A. siRNA Screening of the Kinome Identifies ULK1 as a Multidomain Modulator of Autophagy. J. Biol. Chem. 2007, 282 (35), 25464–25474. 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- Hosokawa N.; Hara T.; Kaizuka T.; Kishi C.; Takamura A.; Miura Y.; Iemura S.; Natsume T.; Takehana K.; Yamada N.; Guan J.-L.; Oshiro N.; Mizushima N. Nutrient-Dependent mTORC1 Association with the ULK1–Atg13–FIP200 Complex Required for Autophagy. Mol. Biol. Cell 2009, 20 (7), 1981–1991. 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrpour M.; Esclatine A.; Beau I.; Codogno P. Overview of Macroautophagy Regulation in Mammalian Cells. Cell Res. 2010, 20 (7), 748–762. 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- Egan D. F.; Shackelford D. B.; Mihaylova M. M.; Gelino S.; Kohnz R. A.; Mair W.; Vasquez D. S.; Joshi A.; Gwinn D. M.; Taylor R.; Asara J. M.; Fitzpatrick J.; Dillin A.; Viollet B.; Kundu M.; Hansen M.; Shaw R. J. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2011, 331 (6016), 456–461. 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.-Y.; Li T. Y.; Liu Q.; Zhang C.; Li X.; Chen Y.; Zhang S.-M.; Lian G.; Liu Q.; Ruan K.; Wang Z.; Zhang C.-S.; Chien K.-Y.; Wu J.; Li Q.; Han J.; Lin S.-C. GSK3-TIP60-ULK1 Signaling Pathway Links Growth Factor Deprivation to Autophagy. Science 2012, 336 (6080), 477–481. 10.1126/science.1217032. [DOI] [PubMed] [Google Scholar]
- Lazarus M. B.; Shokat K. M. Discovery and Structure of a New Inhibitor Scaffold of the Autophagy Initiating Kinase ULK1. Bioorg. Med. Chem. 2015, 23 (17), 5483–5488. 10.1016/j.bmc.2015.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus M. B.; Novotny C. J.; Shokat K. M. Structure of the Human Autophagy Initiating Kinase ULK1 in Complex with Potent Inhibitors. ACS Chem. Biol. 2015, 10 (1), 257–261. 10.1021/cb500835z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petherick K. J.; Conway O. J. L.; Mpamhanga C.; Osborne S. A.; Kamal A.; Saxty B.; Ganley I. G. Pharmacological Inhibition of ULK1 Kinase Blocks Mammalian Target of Rapamycin (mTOR)-Dependent Autophagy. J. Biol. Chem. 2015, 290 (18), 11376–11383. 10.1074/jbc.C114.627778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan D. F.; Chun M. G. H.; Vamos M.; Zou H.; Rong J.; Miller C. J.; Lou H. J.; Raveendra-Panickar D.; Yang C.-C.; Sheffler D. J.; Teriete P.; Asara J. M.; Turk B. E.; Cosford N. D. P.; Shaw R. J. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59 (2), 285–297. 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber S. L.; Kotz J. D.; Li M.; Aubé J.; Austin C. P.; Reed J. C.; Rosen H.; White E. L.; Sklar L. A.; Lindsley C. W.; Alexander B. R.; Bittker J. A.; Clemons P. A.; de Souza A.; Foley M. A.; Palmer M.; Shamji A. F.; Wawer M. J.; McManus O.; Wu M.; Zou B.; Yu H.; Golden J. E.; Schoenen F. J.; Simeonov A.; Jadhav A.; Jackson M. R.; Pinkerton A. B.; Chung T. D. Y.; Griffin P. R.; Cravatt B. F.; Hodder P. S.; Roush W. R.; Roberts E.; Chung D.-H.; Jonsson C. B.; Noah J. W.; Severson W. E.; Ananthan S.; Edwards B.; Oprea T. I.; Conn P. J.; Hopkins C. R.; Wood M. R.; Stauffer S. R.; Emmitte K. A.; Brady L. S.; Driscoll J.; Li I. Y.; Loomis C. R.; Margolis R. N.; Michelotti E.; Perry M. E.; Pillai A.; Yao Y. Advancing Biological Understanding and Therapeutics Discovery with Small-Molecule Probes. Cell 2015, 161 (6), 1252–1265. 10.1016/j.cell.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macarron R.; Banks M. N.; Bojanic D.; Burns D. J.; Cirovic D. A.; Garyantes T.; Green D. V. S.; Hertzberg R. P.; Janzen W. P.; Paslay J. W.; Schopfer U.; Sittampalam G. S. Impact of High-Throughput Screening in Biomedical Research. Nat. Rev. Drug Discovery 2011, 10 (3), 188–195. 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- Lavecchia A.; Di Giovanni C. Virtual Screening Strategies in Drug Discovery: A Critical Review. Curr. Med. Chem. 2013, 20 (23), 2839–2860. 10.2174/09298673113209990001. [DOI] [PubMed] [Google Scholar]
- Pagadala N. S.; Syed K.; Tuszynski J. Software for Molecular Docking: A Review. Biophys. Rev. 2017, 9, 91–102. 10.1007/s12551-016-0247-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villoutreix B. O.; Eudes R.; Miteva M. A. Structure-Based Virtual Ligand Screening: Recent Success Stories. Comb. Chem. High Throughput Screening 2009, 12 (10), 1000–1016. 10.2174/138620709789824682. [DOI] [PubMed] [Google Scholar]
- Schrödinger Release 2016–3: Glide; Schrödinger, LLC: New York, 2016. [Google Scholar]
- Zhou Z.; Felts A. K.; Friesner R. A.; Levy R. M. Comparative Performance of Several Flexible Docking Programs and Scoring Functions: Enrichment Studies for a Diverse Set of Pharmaceutically Relevant Targets. J. Chem. Inf. Model. 2007, 47 (4), 1599–1608. 10.1021/ci7000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53 (7), 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Rosenberg L. H.; Lafitte M.; Grant W.; Chen W.; Cleveland J. L.; Duckett D. R. Development of an HTS-Compatible Assay for the Discovery of Ulk1 Inhibitors. J. Biomol. Screening 2015, 20 (7), 913–920. 10.1177/1087057115579391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surry D. S.; Buchwald S. L. Dialkylbiaryl Phosphines in Pd-Catalyzed Amination: A User’s Guide. Chem. Sci. 2011, 2 (1), 27–50. 10.1039/C0SC00331J. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.