

Abstract

Drug resistant tuberculosis (TB) infections are on the rise and antibiotics that inhibit Mycobacterium tuberculosis through a novel mechanism could be an important component of evolving TB therapy. Protein kinase A (PknA) and protein kinase B (PknB) are both essential serine-threonine kinases in M. tuberculosis. Given the extensive knowledge base in kinase inhibition, these enzymes present an interesting opportunity for antimycobacterial drug discovery. This study focused on targeting both PknA and PknB while improving the selectivity window over related mammalian kinases. Compounds achieved potent inhibition (Ki ≈ 5 nM) of both PknA and PknB. A binding pocket unique to mycobacterial kinases was identified. Substitutions that filled this pocket resulted in a 100-fold differential against a broad selection of mammalian kinases. Reducing lipophilicity improved antimycobacterial activity with the most potent compounds achieving minimum inhibitory concentrations ranging from 3 to 5 μM (1–2 μg/mL) against the H37Ra isolate of M. tuberculosis.
Keywords: Protein kinase A (PknA), protein kinase B (PknB), Mycobacterium tuberculosis, dual targeting, structure activity relationship (SAR)
Tuberculosis (TB) remains a major unmet medical need due to widespread drug resistance, HIV coinfection, and ineffective healthcare management with approximately two billion latent infections, ten million new cases, and two million deaths each year.1 Antibacterial compounds that act through a novel mechanism of inhibition may offer advantages to treat infections with drug-resistant strains. Of the 11 serine/threonine (Ser/Thr) protein kinases described in Mycobacterium tuberculosis (Mtb),2,3 protein kinase A (PknA) and protein kinase B (PknB) are especially attractive drug targets because they are essential for bacterial growth both in culture medium and in Mtb-infected host macrophages.4−6 The availability of protein crystal structures for PknB7−9 will enable the design of potent and selective compounds. PknA and PknB are crucial for cell wall formation, resuscitation from dormancy, and performing multiple regulatory functions in metabolism and adaptation to environmental stress.10−13 Antimycobacterial activity through PknB inhibition has been explored by others.14,15 However, only moderate antibacterial activity was achieved, and no data on PknA was reported. We expand on these results by targeting both PknA and PknB. Dual targeting has the potential to significantly lower the frequency at which resistance develops. However, broadening the inhibitor specificity may increase the risk of introducing adverse side effects through the inhibition of mammalian kinases. We report here the optimization of a lead series that achieved potent inhibition of both PknA and PknB with Ki values in the single digit nanomolar range. Structures of compound-enzyme cocrystals allowed the identification of a unique feature in the binding site of PknB. Compound substitutions that exploit this feature achieved a 100-fold selectivity window over a range of mammalian kinases. For many of the lead compounds, improved enzymatic potency translated to improved antimycobacterial activity. Several compounds achieved a minimum inhibitory concentration (MIC) around 3–5 μM (1–2 μg/mL).
The project was initiated by screening a collection of 1078 compounds representing kinase inhibitor chemical diversity against the PknA and PknB kinase domains. Compounds with Ki < 10 μM were modeled into the active site of PknB in complex with phosphomethylphosphonic acid adenylate ester (PDB ID: 1O6Y)9 and into a homology model of PknA to get an approximation of dual targeting. Hit compounds that were able to dock into both sites with similar poses and reasonable strain energies were prioritized for follow-up. Quinazoline 1 was selected as our starting point because it showed relatively strong PknB inhibition (Ki = 150 nM) and a good MIC against Mtb H37Ra (33 μM). Although 1 did not show inhibition of PknA, the sequence similarity between PknA and PknB (43% identity in the kinase domains; 56% identical in the ATP binding pocket) made it likely that we would be able to build in inhibition for both isoforms.
Scheme 1 summarizes the diversification strategy used for quinazoline 1. Since 4-(aminopyrazolyl)-substituted quinazolines inhibit several mammalian host kinases as well as Mtb kinases by binding to the hinge region, we wanted to identify alternative hinge-binding elements in order to obtain selectivity. We synthesized an extensive set of amino heterocyclic replacements for the amino-pyrazole, including oxazole (2), triazole (3), thiadiazole (4), thiazole (5, 6), pyridine (7), and pyrimidine (8). Unfortunately, all these compounds had reduced PknB inhibition. Our hypothesis is that these compounds destabilized the preferred binding tautomer (in the case of the triazole), removed a key Ar–H interaction (in the case of the thiadiazole), or disturbed the overall geometry of interaction with the kinase hinge area (in the cases of pyridine and pyrimidine).
Scheme 1. Diversification Strategy and Initial Quinazoline SAR.

A small structure–activity relationship (SAR) study of the 6-position, replacing Cl with F (9), methyl (10), and methoxy (11), showed that it was possible to maintain PknB activity at this position with a variety of substituents, an opportunity that was later exploited for selectivity and cell-based activity (see below).
Next, an investigation of the quinazoline 2-position was conducted. One of our goals was to preserve the sp3 character of the azepane substituent, if possible, to retain more drug-like physicochemical properties.16 The neutral oxazepane analog 12 had an unchanged PknB Ki of 150 nM, but when a positive charge was introduced with the diazepane analog 13, we observed a six-fold loss of activity. Acylation of the amine to return it to the neutral state as either the amide or carbamate analog (14 and 15) recapitulated the original PknB potency of 150 and 230 nM, respectively. In accord with this developing SAR, the diazepanone analogs 16 and 17 also gave PknB Ki potencies of 190 and 310 nM, respectively. Contraction of the diazepane ring to the six-membered piperazine rings (18 and 19) gave weaker activity, but surprisingly so did the acylated version found in 20. We surmise that the much larger volume of the N-benzoyl group was responsible for its diminished activity.
To follow up on this result, a library of 48 amine compounds that were substituted at the quinazoline C2 position was generated. We selected a set of primary and secondary amines containing one or fewer aromatic rings, with the following constraints: MW < 200, PSA < 55 Å, and calculated logP < 2.5. Library members were found to have PknB Ki values ranging from >4 μM to <100 nM. The cyclobutylamino analog (21) was the most potent PknB inhibitor (Ki = 72 nM), while most of the other C2-amino analogs plateaued at approximately 100 nM PknB Ki without further improvement. These findings are summarized in Scheme 1.
In addition to our sp3-retaining C2-substitution strategy, we also explored aryl C2 substitutions. Starting from the baseline 2-phenyl analog (22: PknB Ki = 60 nM), we prepared approximately 140 other substituted 2-phenyl analogs. Substituents at the m- and p-position of the 2-phenyl ring offered excellent PknB potency, but showed no PknA activity. Table 1 shows SARs for a representative set of quinazoline 2-substituted phenyl compounds. The 4-cyanomethylphenyl analog 23 had a PknB Ki of 5 nM, while the 3-cyanomethylphenyl analog 24 had a PknB Ki of 38 nM. Additional polar functionalities such as 4-cyano (25), 4-CONH2 (26), and 4-hydroxymethyl (27) were examined, and additional 3-substituted analogs were made such as 3-acetamide 28 (Ki = 23 nM)), 3-CONH229 (Ki = 29 nM), and cyano 30 (Ki = 88 nM). Overall, the substituted phenyl analogs 24 to 30 maintained similar levels of PknB inhibitory activity compared to their parent phenyl analog 22. It was not until we prepared the 4-hydroxymethyl (27) and 3-acetamide (28) compounds that we began to observe inhibition of PknA (2.3 and 1.8 μM, respectively.) Further exploration of the H-bonding substituents at both 3- and 4-positions provided excellent inhibition of PknA and PknB with the primary sulfonamide in either position (compounds 31 and 32). The 4-SO2NH2 analog 31 reached Ki values of 24 and 22 nM for PknB and PknA, respectively. The 3-SO2NH2 analog 32 also demonstrated single-digit Ki values (6 and <8 nM) for PknB and PknA. The value of this substituent was further demonstrated when we changed the quinazoline 2-substituent from phenyl to a thiophene ring bearing a 5-sulfonamide group (33) and found even more potent dual PknB and PknA inhibition. These findings are summarized in Table 1.
Table 1. Representative PknA and PknB Enzyme Inhibition (Ki): SAR of 2-Aryl-Substituted Quinazolines.

| compd | R1 | R2 | PknB Ki (nM) | PknA Ki (nM) |
|---|---|---|---|---|
| 23 | CH2CN | H | 5 | >4000 |
| 24 | H | CH2CN | 38 | ND |
| 25 | H | CN | 50 | >4000 |
| 26 | H | CONH2 | 15 | >4000 |
| 27 | H | CH2OH | 23 | 2300 |
| 28 | CH2CONH2 | H | 23 | 1800 |
| 29 | CONH2 | H | 29 | >4000 |
| 30 | CN | H | 88 | >4000 |
| 22 | H | H | 69 | >4000 |
| 31 | SO2NH2 | H | 24 | 22 |
| 32 | H | SO2NH2 | 6 | <8 |
| 33 | <1 | <8 |
We next investigated opportunities for achieving selectivity over mammalian host kinases. We chose GSK3β, CDK2, and SRC for selectivity assessment since each was known to be inhibited by compounds of this class. Overlay of the PknB hinge binding region with a selection of mammalian host kinases including GSK3β, CDK2, and SRC showed that the area immediately surrounding the 6-chloro substituent of 1 provided a distinct binding pocket that could be exploited for mycobacterial kinase selectivity. As shown in Figure 1, it can be seen that Gly-97 of PknB is “flipped” relative to the corresponding residues of the mammalian kinases, such that the backbone carbonyl of the adjacent residue, Asp-96, is pointed away from the inhibitor, creating additional space that accommodates the 6-Cl of 22.
Figure 1.
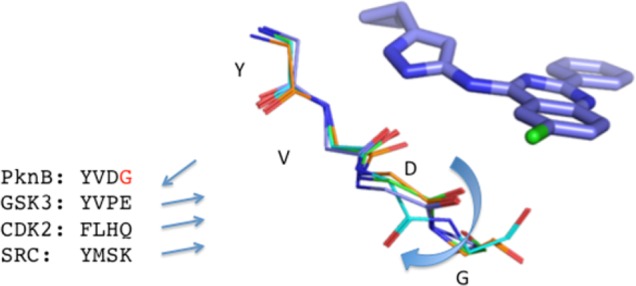
Compound 22 docked into the ATP binding site of PknB, and the mammalian kinase GSK3β, CDK2, and SRC. Labels identify PknB residue numbers corresponding to cyan structure. For clarity, amino acid side chains are not shown. Straight arrows indicate the direction of the C=O bond between the 3rd and 4th residues in the sequences shown, and the curved arrow indicates flipping of the peptide bond between D and G residues. Alignment was done using C-alpha atoms. Red G is the residue that allows PKnB to undergo conformational change.
To further substantiate the Mtb PknB selectivity of inhibitory compounds over mammalian kinases, 6-Cl quinazoline analogs were compared with 6-H compounds as summarized in Table 2. As predicted, we found similar PknB potency for both 6-Cl and 6-H quinazolines (34 vs 30 and 35 vs 25, respectively), while inhibition of the three mammalian kinases was reduced 5–50-fold with the 6-Cl compounds (30 and 25).
Table 2. 6-Substituted Quinazoline with Host Kinase Selectivity.

| compd |
||||
|---|---|---|---|---|
| kinase Ki (nM) | 34 | 30 | 35 | 25 |
| PknB | 40 | 88 | 46 | 50 |
| GSK3β | 9 | 190 | 70 | >4000 |
| CDK2 | 11 | 170 | 140 | 750 |
| SRC | 22 | 710 | 18 | 200 |
Thus far, we had been able to improve enzyme potency and selectivity for the quinazoline series. However, we were unable to improve antimycobacterial activity since, despite lowering PknB inhibition roughly 100-fold, the MIC against Mtb H37Ra remained at 33 μM or higher. It is possible that our inhibitors failed to penetrate the Mtb cell wall, or they were subject to rapid efflux.
Some antibacterial compounds can cross the bacterial cell wall through channel-forming porins that enable the diffusion of small, hydrophilic solutes.17 Efflux pumps, while not well studied in Mtb, tend to be particularly active on lipophilic substrates. Hence, reducing lipophilicity can be a successful strategy both for improving permeability through the bacterial cell wall and for avoiding efflux. We set about reducing the lipophilicity of our most potent kinase inhibitors while also minimizing their size. Similar to the approach described by Chapman et al.,14 we truncated the quinazoline ring into a simple monocyclic pyrimidine ring. In addition, we incorporated the sulfonamide substituent to provide potent PknA inhibition. Toward this objective, we synthesized the pyrimidine analogs 36, 37, and 38 (Scheme 2). The preparation of 37 and 38 is described in Scheme 2. Treatment of 5-chloropyrimidine-2,4-diol (39) with POBr3 gave the 2,4-dibromo-5-chloro-pyrimidine (40), which reacted with 5-cyclopropyl-1H-pyrazol-3-amine to afford 41. Suzuki coupling of 41 with 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene-sulfonamide then produced target compound 37. For 38, the synthesis started with the reaction of 5-bromothiophene-2-sulfonyl chloride (42) with 2-methylpropan-2-amine to form 5-bromo-N-(tert-butyl)thiophene-2-sulfonamide (43), which then was converted to the pinacol-boron ester 44. Suzuki coupling, followed by removal of the t-butyl group with BCl3 provided 38.
Scheme 2. Compounds 36–38 and 46–49 and the Representative Syntheses of 37, 38, and 49.

N,N-Dimethylaniline (2 equiv), POBr3 (3 equiv), toluene, 90 °C, 2 h, 83%.
5-Cyclopropyl-1H-pyrazol-3-amine, EtOH, RT, 4 h, 78%.
3- or 4-(Pinacolatoboranyl)-benzenesulfonamide for 36 and 37, or 44 for 38 and 56, Pd(dppf)Cl2 (0.1 equiv), Na2CO3 (4 equiv), dioxane/H2O (4:1), 100 °C, 15 h.
t-Butylamine, dioxane, 20 °C, 4 h, 94%.
Bis-pinacolatodiboron (1.2 equiv), Pd(dppf)Cl2 (0.1 equiv), CH3COOK (4.0 equiv), dioxane, 100 °C, 3 h, 70%.
BCl3, DCM, 20 °C, 6 h.
NIS, AcOH, RT, 7 h, 95%.
TMS-acetylene (1.2 equiv), (Ph3P)2PdCl2 (0.25 equiv), CuI (0.25 equiv), EtOAc, RT, 4 h, 46%.
N,N-Dimethylaniline, POBr3, 100 °C, 50 min, toluene, 51%.
Each of the three sulfonamide analogs 36, 37, and 38 were potent PknB and PknA inhibitors, while 37 and 38 demonstrated single-digit micromolar MIC against Mtb H37Ra. Additional 5-substituted pyrimidine analogs 46, 47, and 48 were prepared and all showed excellent PknB and PknA potency that translated to improved Mtb H37Ra MIC values (Table 3).
Table 3. Enzyme Inhibition and MIC (μM) of Pyrimidines against Mycobacterium tuberculosis.
| compd | PknB Ki (nM) | PknA Ki (nM) | Mtb MIC (μM) |
|---|---|---|---|
| 36 | 13 | 2900 | >100 |
| 37 | 17 | <8 | 8.3 |
| 38 | <1.3 | 9 | 6.25 |
| 46 | 4 | 18 | 4.2 |
| 47 | <1 | 12 | 3.1 |
| 48 | 4 | 18 | 4.7 |
To confirm our proposed binding modes and to understand the SAR observed for PknA and PknB, we used X-ray crystallography to solve the structure of compound 38 complexed with PknA and also complexed with PknB. Figure 2 shows that the aminopyrazole provides the key hinge-binding interactions, with the cyclopropyl group interacting in the small hydrophobic pocket provided by Met95 and Val27 in PknA and by the analogous residues Met92 and Val25 in PknB. These structures also provide some clues to understanding why the SO2NH2 group provides enhanced inhibition of PknA. The −SO2NH2 group bound to PknA shows three relatively close polar contacts, to the side chains of Lys42, Thr158, and Asn146 (Figure 3).
Figure 2.
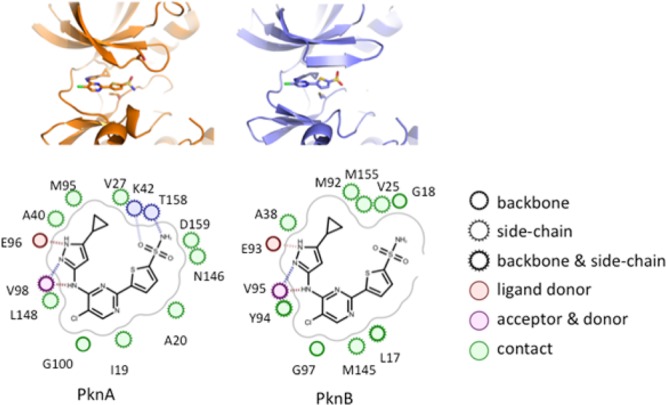
Compound 38 bound to PknA (left) and PknB (right), and with key amino acid residues that interact with 38.
Figure 3.
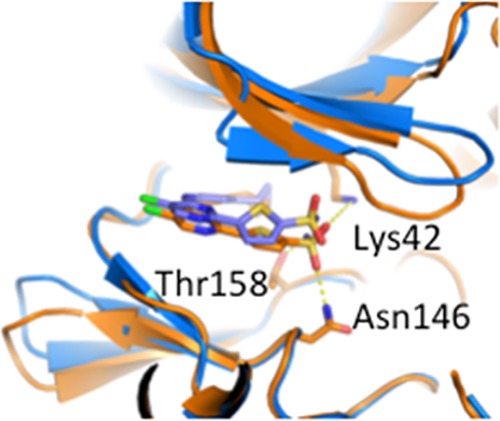
X-ray crystal structures for 38 complexed to PknA (orange) and PknB (blue.) Structures were overlaid using PyMol for alignment. Dotted lines represent close polar contacts discussed in the text.
We then investigated the C5-position on the pyrimidine ring to determine if we could again exploit the “flipped glycine” pocket for selectivity over mammalian kinases. Toward this effort, the 5-acetylenyl compound 49 was made as shown in Scheme 2. The 5-H analog 46 only showed two-fold selectivity versus CDK2, while the 5-Cl compound 38 gave at least a 15-fold improvement in Mtb PknB selectivity vs CDK2. The sterically more demanding 5-acetylenyl compound 49 demonstrated greater than 100-fold selectivity vs CDK2, with increased PknB potency (Table 4) while maintaining Mtb H37Ra MIC at 4.7 μM.
Table 4. Selectivity of PknB Inhibitors with Reduced Inhibition of the Mammalian Kinase CDK2.
| compd | PknB Ki (nM) | PknA Ki (nM) | CDK2 Ki (nM) | selectivity |
|---|---|---|---|---|
| 46 | 4 | 18 | 7 | 2 |
| 38 | <1.3 | 9 | 17 | >13 |
| 49 | <2 | 11 | 147 | >74 |
Finally, we wanted to demonstrate a clear link between kinase inhibition and the observed antimycobacterial activity. For this purpose, we designed the N-methyl version of compound 48, where the kinase hinge-binding nitrogen is blocked. Compound 57 was synthesized as shown in Scheme 3. The starting material 5-cyclopropyl-1-methyl-1H-pyrazol-3-amine 58 was prepared from simple methylation of 3-cyclopropyl-1H-pyrazol-5-amine. Reaction with 2,4-dibromo-5-methylpyrimidine followed by Suzuki coupling with intermediate 44 and N-t-butyl removal gave compound 57. Consistent with our expectations, compound 57 lost both PknA and PknB enzyme activities (Ki both >4 μM) as well as antimycobacterial activity (Mtb H37Ra MIC > 100 μM).
Scheme 3. Synthesis of 57.

t-BuOK, 18-crown-6, CH3I, Et2O, 0 °C, 3 h, 43%.
2,4-Dibromo-5-methylpyrimidine, EtOH, RT, 8 h, 70%.
44, Pd(dppf)Cl2 (0.1 equiv), Na2CO3 (4 equiv), dioxane/H2O (4:1), 100 °C, 15 h, 28%.
BCl3, DCM, RT, 30 min, 71%.
In conclusion, starting from a 1078-compound screen of a focused collection of diverse kinase-inhibitory compounds, we identified a weak and nonselective quinazoline-based inhibitor of PknB with a quinazoline core that lacked activity against PknA and showed weak antimycobacterial activity. We successfully optimized this series to achieve potent, dual-targeting inhibitors with activity against both PknB and PknA. We identified a structural pocket unique to the mycobacterial kinases that we exploited for two related but distinct series. Compound modifications that filled this pocket gave a significant selectivity window over mammalian kinases. Reducing compound lipophilicity (for example, quinazoline 33 has a calculated logP of 4.1, while for the analogous pyrimidine 38, 2.7) greatly improved the antibacterial activity, with the best compounds in the series active against Mtb H37Ra with MIC in the range of 3–5 μM (1–2 μg/mL) and potency commensurate with clinically used antibiotics. Thus, pending further lead optimization efforts, kinase inhibition could be a valuable approach to the discovery of much needed new TB therapeutics.
Acknowledgments
We would like to express our gratitude to ChemPartner’s scientists Jianguo Chen, Xiaoming Li, Zhongtai Piao, and Jingwen Zang for their synthetic (J.C., X.L., Z.P.) and biological (J.Z.) assistance. We thank Barry Davis and Brian Ledford, both of Vertex, for HRMS and NOE studies.
Glossary
ABBREVIATIONS
- Mtb
Mycobacterium tuberculosis
- PknA
Protein kinase A
- PknB
Protein kinase B
- SAR
structure–activity relationship
- GSK 3-beta
glycogen synthase kinase 3 beta
- CDK2
cyclin-dependent kinase 2
- MIC
minimum inhibitory concentration
- PSA
polar surface area
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00239.
Accession Codes
PDB codes for the X-ray crystal structures described in this study have been deposited in the PDB under the accession codes 6B2P (PknA) and 6B2Q (PknB).
Author Present Address
§ Hennessy Research Associates, 12700 Johnson Drive, Shawnee, Kansas 66216, United States. E-mail: bhanzelka@hennessyresearch.com.
Author Present Address
∥ Versatope Therapeutics, MassChallenge Design and Innovation Center, 21 Drydock Lane, Boston, Massachusetts 02210, United States. E-mail: Christopher.locher@versatope.com.
Author Present Address
⊥ GlaxoSmithKline R&D China, No. 3 Building, 898 Halei Road, Shanghai 201203, China. E-mail: shaohui.wang@gsk.com.
Author Present Address
# P.O. Box 2241, Acton, Massachusetts 01720, United States. E-mail: jtexvertex@yahoo.vom.
Author Present Address
∇ Department of Microbiology, University of Iowa, 51 Newton Road, Iowa City, Iowa 52242, United States. E-mail: ute-muh@uniowa.edu.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO. Global tuberculosis report, 2013.
- Av-Gay Y.; Jamil S.; Drews S. J. Expression and characterization of the Mycobacterium tuberculosis serine/threonine protein kinase PknB. Infect. Immun. 1999, 67, 5676–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Av-Gay Y.; Everett M. The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol. 2000, 8, 238–244. 10.1016/S0966-842X(00)01734-0. [DOI] [PubMed] [Google Scholar]
- Sassetti C. M.; Boyd D. H.; Rubin E. J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Sassetti C. M.; Rubin E. J. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 12989–12994. 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez P.; Saint-Joanis B.; Barilone N.; Jackson M.; Gicquel B.; Cole S. T.; Alzari P. M. The Ser/Thr protein kinase PknB is essential for sustaining mycobacterial growth. J. Bacteriol. 2006, 188, 7778–7784. 10.1128/JB.00963-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekely R.; Waczek F.; Szabadkai I.; Nemeth G.; Hegymegi-Barakonyi B.; Eros D.; Szokol B.; Pato J.; Hafenbradl D.; Satchell J.; Saint-Joanis B.; Cole S. T.; Orfi L.; Klebl B. M.; Keri G. A novel drug discovery concept for tuberculosis: inhibition of bacterial and host cell signalling. Immunol. Lett. 2008, 116, 225–231. 10.1016/j.imlet.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Young T. A.; Delagoutte B.; Endrizzi J. A.; Falick A. M.; Alber T. Structure of Mycobacterium tuberculosis PknB supports a universal activation mechanism for Ser/Thr protein kinases. Nat. Struct. Biol. 2003, 10, 168–174. 10.1038/nsb897. [DOI] [PubMed] [Google Scholar]
- Ortiz-Lombardia M.; Pompeo F.; Boitel B.; Alzari P. M. Crystal structure of the catalytic domain of the PknB serine/threonine kinase from Mycobacterium tuberculosis. J. Biol. Chem. 2003, 278, 13094–13100. 10.1074/jbc.M300660200. [DOI] [PubMed] [Google Scholar]
- Prisic S.; Dankwa S.; Schwartz D.; Chou M. F.; Locasale J. W.; Kang C. M.; Bemis G.; Church G. M.; Steen H.; Husson R. N. Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 7521–7526. 10.1073/pnas.0913482107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou M. F.; Prisic S.; Lubner J. M.; Church G. M.; Husson R. N.; Schwartz D. Using bacteria to determine protein kinase specificity and predict target substrates. PLoS One 2012, 7, e52747. 10.1371/journal.pone.0052747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C. M.; Abbott D. W.; Park S. T.; Dascher C. C.; Cantley L. C.; Husson R. N. The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: substrate identification and regulation of cell shape. Genes Dev. 2005, 19, 1692–1704. 10.1101/gad.1311105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega C.; Liao R.; Anderson L. N.; Rustad T.; Ollodart A. R.; Wright A. T.; Sherman D. R.; Grundner C. Mycobacterium tuberculosis Ser/Thr protein kinase B mediates an oxygen-dependent replication switch. PLoS Biol. 2014, 12, e1001746. 10.1371/journal.pbio.1001746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman T. M.; Bouloc N.; Buxton R. S.; Chugh J.; Lougheed K. E.; Osborne S. A.; Saxty B.; Smerdon S. J.; Taylor D. L.; Whalley D. Substituted aminopyrimidine protein kinase B (PknB) inhibitors show activity against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2012, 22, 3349–3353. 10.1016/j.bmcl.2012.02.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lougheed K. E.; Osborne S. A.; Saxty B.; Whalley D.; Chapman T.; Bouloc N.; Chugh J.; Nott T. J.; Patel D.; Spivey V. L.; Kettleborough C. A.; Bryans J. S.; Taylor D. L.; Smerdon D. J.; Buxton R. S. Tuberculosis (Oxford, U. K.) 2011, 91, 277–286. 10.1016/j.tube.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from flatland: increasing saturation as an approach to improvimng clinical success. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- Vargas J. R.; Stanzl E. G.; Teng N. N. H.; Wender P. A. Cell-penetrating, guanidinium-rich molecular transporters for overcoming efflux-mediated multidrug resistance. Mol. Pharmaceutics 2014, 11, 2553–2565. 10.1021/mp500161z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.