

Abstract
Inhibition of hypoxia inducible factor prolyl hydroxylase (PHD) represents a promising strategy for the discovery of a next generation treatment for renal anemia. We identified several 5,6-fused ring systems as novel scaffolds of the PHD inhibitor on the basis of pharmacophore analysis. In particular, triazolopyridine derivatives showed potent PHD2 inhibitory activities. Examination of the predominance of the triazolopyridines in potency by electrostatic calculations suggested favorable π–π stacking interactions with Tyr310. Lead optimization to improve the efficacy of erythropoietin release in cells and in vivo by improving cell permeability led to the discovery of JTZ-951 (compound 14), with a 5-phenethyl substituent on the triazolopyridine group, which increased hemoglobin levels with daily oral dosing in rats. Compound 14 was rapidly absorbed after oral administration and disappeared shortly thereafter, which could be advantageous in terms of safety. Compound 14 was selected as a clinical candidate.
Keywords: JTZ-951, PHD inhibitors, prolyl hydroxylase inhibitors, hypoxia inducible factor, HIF, erythropoietin, EPO, anemia
Renal anemia occurs early in the development of chronic kidney disease (CKD), and the prevalence of anemia increases as the disease progresses. The primary cause is the depression of erythropoietin (EPO) production in the kidney associated with the kidney disorder.1 Current standard treatment for renal anemia is parenteral administration of erythropoiesis stimulating agents (ESAs) such as recombinant human erythropoietin (rhEPO), darbepoetin alfa, and epoetin beta pegol. Whereas ESAs have improved the quality of life in CKD patients, there still remain several problems to be improved, which include matters related to injection and adverse effects.2−4 Therefore, the development of small molecule alternatives to ESAs is highly anticipated.5,6
Hypoxia inducible factor (HIF), composed of an oxygen-regulated α subunit (HIFα) and a constitutively expressed β subunit, is a key transcription factor that plays a central role in response to hypoxia. HIF induces various genes related to erythropoiesis including EPO, angiogenesis, vascular tone, matrix metabolism, glucose metabolism, cell proliferation, and apoptosis.7 Levels of HIF are regulated metabolically via hydroxylation of HIFα at a proline residue by the prolyl hydroxylase (PHD) having three isoforms (PHD1–3).8 The PHD activity is dependent on intracellular oxygen concentrations, which means hydroxylation of HIFα by PHD is suppressed in the hypoxic state, resulting in stabilization of HIFα and adaptation to hypoxia.5−8 These mechanisms suggest that PHD inhibitors may increase internal EPO levels as a result of HIF stabilization and could serve as alternatives to ESAs. In fact, a PHD inhibitor FG-2216 was reported to increase the EPO levels in patients on hemodialysis.9 Currently, several compounds, including FG-4592 (roxadustat) and JTZ-951 (compound 14), are in clinical trials for renal anemia.6,10 We started our PHD inhibitor program from exploration of novel scaffolds and performed lead optimization, guided by the PHD2 enzyme assay and the EPO release assay from Hep3B cells. In this letter, we report our research efforts, which led to the discovery of compound 14.
Active site inhibitors of PHD are known to have three common pharmacophores, which consist of a bidentate coordination site to an iron atom, a carboxylic acid forming a salt bridge to the Arg383 side chain, and a hydrogen bond acceptor for the phenolic hydroxyl of Tyr303. Several compounds including FG-2216 have an additional interaction with Tyr310 by a π–π stacking (Figure 1a).11,12 On the basis of this pharmacophore model, many scaffold hopping efforts have been made and reviewed.12,13 We synthesized various scaffolds and tested them. Several compounds showed potent PHD inhibitory activities but failed to show adequate activity in the cell or in vivo. However, with sustained efforts of exploration, we finally identified 5,6-fused ring systems 1–5 as promising lead compounds (Table 1). The imidazopyridine derivative 1 and the pyrazolopyridine derivative 2 showed PHD2 inhibitory activity comparable to FG-2216, and moreover, the triazolopyridine derivatives 3–5 were more potent than 1 and 2. To discuss the differences in potency, we first analyzed the binding modes by modeling studies. Although there are two possible binding modes in which the 5,6-fused ring is flipped relative to the amide carbonyl (I and II in Figure 1b), the modeling studies suggested that only one binding mode I was accessible. Coordination of the iron ion is achieved through the azole nitrogen atom, which allows a π–π stacking between the fused pyridine rings and the phenol ring of Tyr310 (I in Figure 1b). However, the azole ring will make steric clashes with Tyr303 in another binding mode (II in Figure 1b). Then, we analyzed the electrostatic potential maps of the 5,6-fused rings in 1–5. Triazolopyridines had electron-deficient fused pyridine rings compared to imidazopyridine and pyrazolopyridine (Figure 1c, region within the green box). In general, π–π stacking is preferentially located between an electron-deficient ring and an electron-rich one.14 Therefore, triazolopyridines are considered more favorable as a stacking counterpart with the electron-rich phenol of tyrosine, assuming that the difference in the electronic property of the fused pyridines has an impact upon the PHD2 inhibitory activity.
Figure 1.
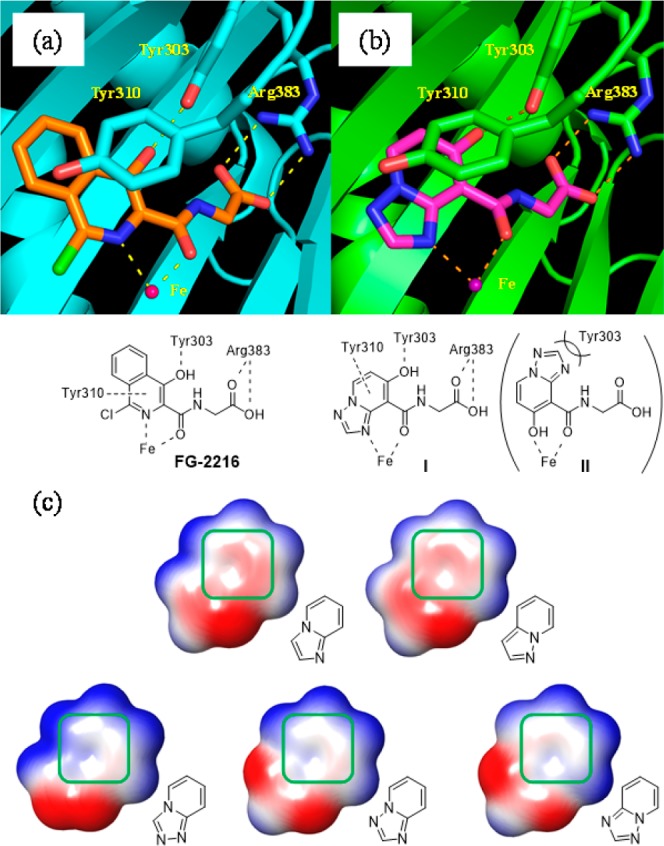
(a) Crystal structure of PHD2 in complex with FG-2216 and a representative pharmacophore common to typical PHD inhibitors (PDB code 2HBT). (b) Two binding models I and II of compound 4 bound to the active site of PHD2. (c) Electrostatic potential map of 5,6-fused ring systems. Positive, negative, and neutral charges are colored in blue, red, and white, respectively. The pyridine moiety is outlined by the green box.
Table 1. SAR of 5,6-Fused Ring Systems.
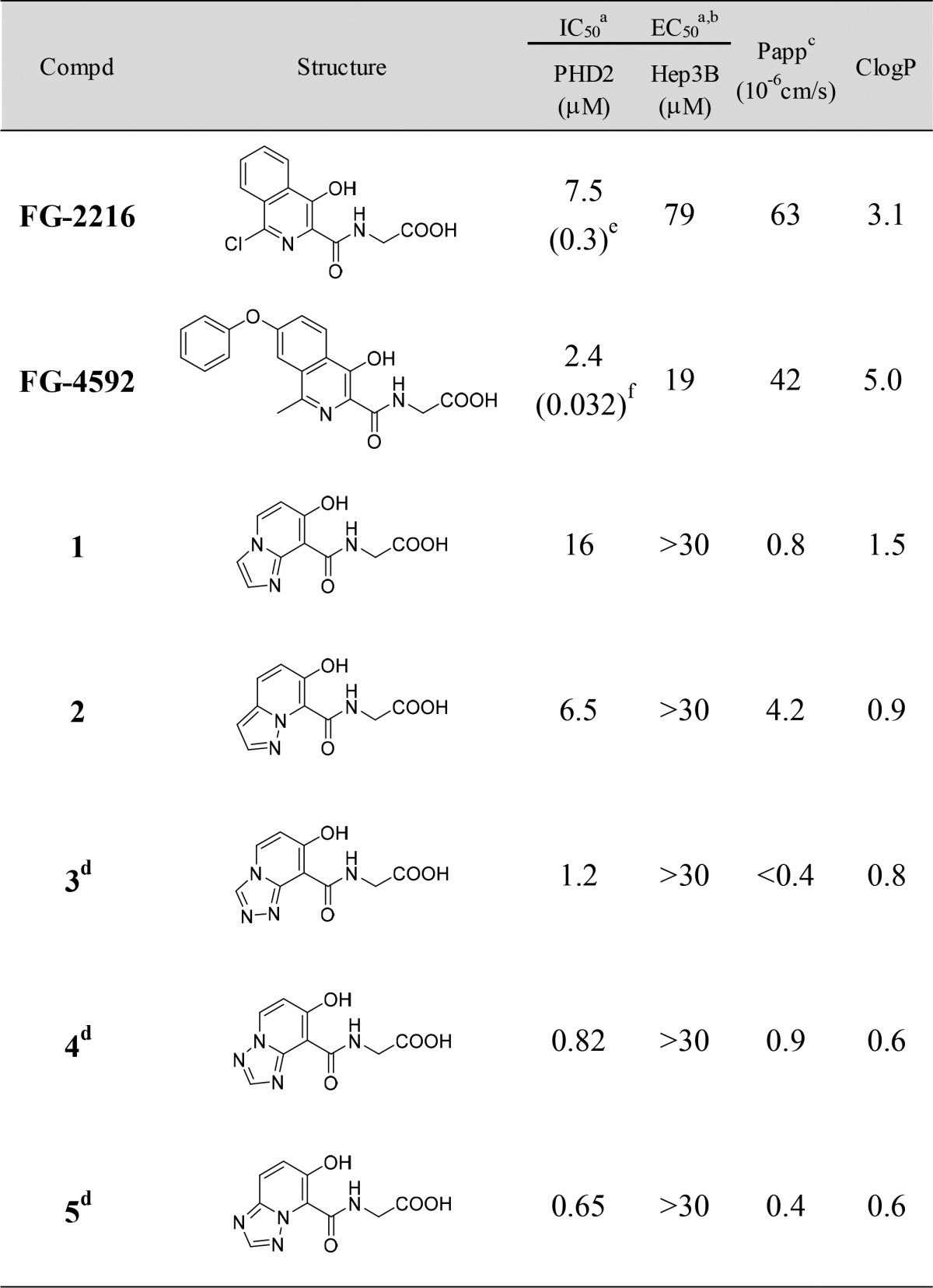
Mean value determined from four replicates. Standard deviations are reported in the Supporting Information.
The concentration showing 50% increase in EPO release from Hep3B cells relative to the positive control. See the Supporting Information for assay details.
Apparent permeability coefficient.
HCl salt.
Value reported in ref (15).
Value reported in ref (16).
Although triazolopyridine derivatives 3–5 exhibited potent PHD2 inhibitory activities, these compounds failed to stimulate EPO release from Hep3B cells at concentrations up to 30 μM. We assumed that this lack of effect was due to the poor cell permeability (Papp) of the compounds and that increasing the lipophilicity could stimulate EPO release. Since the triazolopyridine derivatives 3–5 had high LLEs (5.1–5.6), we thought there would be sufficient space to introduce appropriate lipophilic substituents.17 We selected compounds 4 and 5 as lead compounds, excluding 3, which had extremely low permeability, and conducted lead optimization (Table 2).
Table 2. SAR of Scaffold A and Scaffold B.


Mean value determined from four replicates. Standard deviations are reported in the Supporting Information.
The concentration showing 50% increase in EPO release from Hep3B cells relative to the positive control. See the Supporting Information for assay details.
Apparent permeability coefficient.
Mean value determined from three replicates. Standard deviations are reported in the Supporting Information.
Free form.
Initially, various alkyl chains were introduced to compound 4 (scaffold A) at the R1 position to verify the expected improvement on the cell permeability and the activity in cells. The introduction of n-propyl (6), n-butyl (7), and n-pentyl groups (8) had no influence on PHD2 inhibitory activity. Cell permeability (Papp) was improved in these compounds along with ClogP as expected. Encouragingly, compounds 6, 7, and 8 stimulated EPO release from Hep3B cells with EC50 values equivalent to or more potent than that of FG-4592. Moreover, compounds 7 and 8 elicited significant increases of EPO levels in mice (plasma EPO > 2000 pg/mL at 8 h after oral administration at a dose of 10 mg/kg). We next synthesized compounds 9 and 10 with substituents at the R2 position. These compounds showed weak PHD2 inhibitory activity and were not effective in mice at 10 mg/kg. These results indicated that the spatial tolerance around the R2 position is narrow. Therefore, we focused the introduction of lipophilic substituents on the R1 position. We investigated several different types of lipophilic substitution at the R1 position (11–15). The substituents having a phenyl ring showed higher PHD2 inhibitory activities (compounds 12–15), while all the compounds exhibited similar potency in cells as obtained with 7 and 8. In particular, compounds 13 and 14 showed significant EPO production activities in mice. Introduction of a methyl group at the R3 position (compound 16) resulted in a large decrease in PHD2 inhibitory activity. Having these results, another lead compound 5 (scaffold B) was examined. Introduction of the same substituents as compounds 11–14 gave similar results both in the enzyme assay and the cell assay (compounds 17–20). However, they were not active in mice at 10 mg/kg, suggesting that scaffold A was superior to scaffold B in terms of EPO production in mice.
The compounds 7, 8, 13, and 14, which significantly increased plasma EPO levels in mice, were then tested in rats. They significantly stimulated EPO production in rats as well. Unfortunately, a slight anorectic effect was observed in rats orally administered 100 mg/kg of compounds 8 and 13. However, compounds 7 and 14 did not show anorectic effects. These results encouraged us to advance compounds 7 and 14 for further investigation.
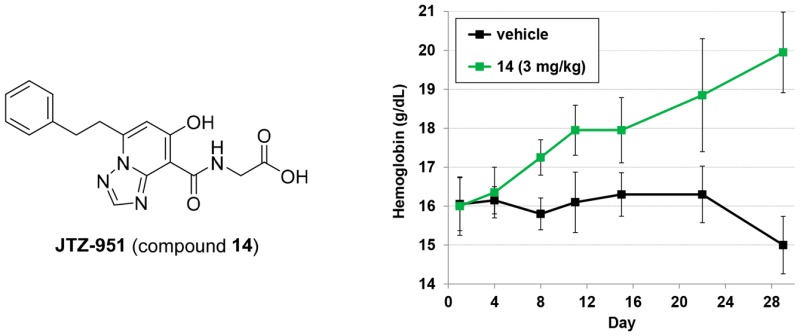
The effect of compounds 7 and 14 on hemoglobin levels in rats was examined. Oral dosing of compound 14 at 1 and 3 mg/kg daily resulted in dose-dependent increases of hemoglobin levels (Figure 2). No increase in hemoglobin levels was observed at 3 mg/kg dose of compound 7, but administration at 10 mg/kg increased these levels.
Figure 2.
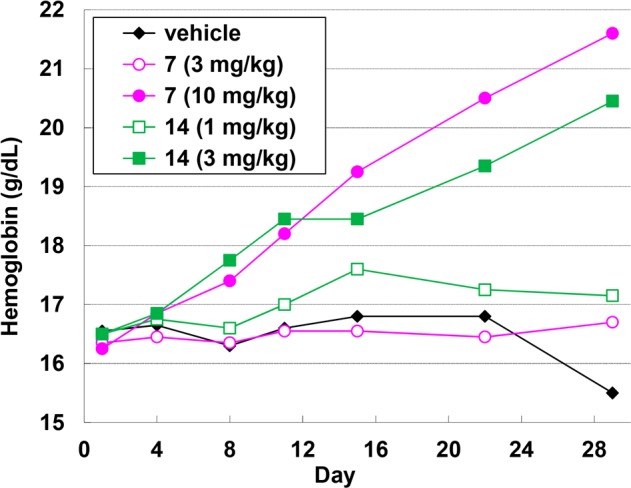
Changes in hemoglobin levels following 29-day once-daily dosing of 7 (magenta) and 14 (green) in rats. Standard deviations are reported in the Supporting Information.
In terms of pharmacokinetic (PK) profiles, compound 14 was rapidly absorbed after oral administration in rats and disappeared shortly thereafter (Figure 3a,b). As Vachal and others have mentioned, the short-acting characteristics could be beneficial in reducing unpredictable adverse effects in light of the HIF-PHD mechanism.18 Compound 14 also had excellent solubility and metabolic stability (Figure 3c). In addition, it showed neither CYP (IC50 > 100 μM; CYP3A4/5, CYP2C9, CYP2D6, CYP1A2, CYP2A6, CYP2C19, CYP2C8, CYP2B6) nor hERG (IC50 > 100 μM) inhibition. Having these results, compound 14 was selected for a clinical candidate.
Figure 3.
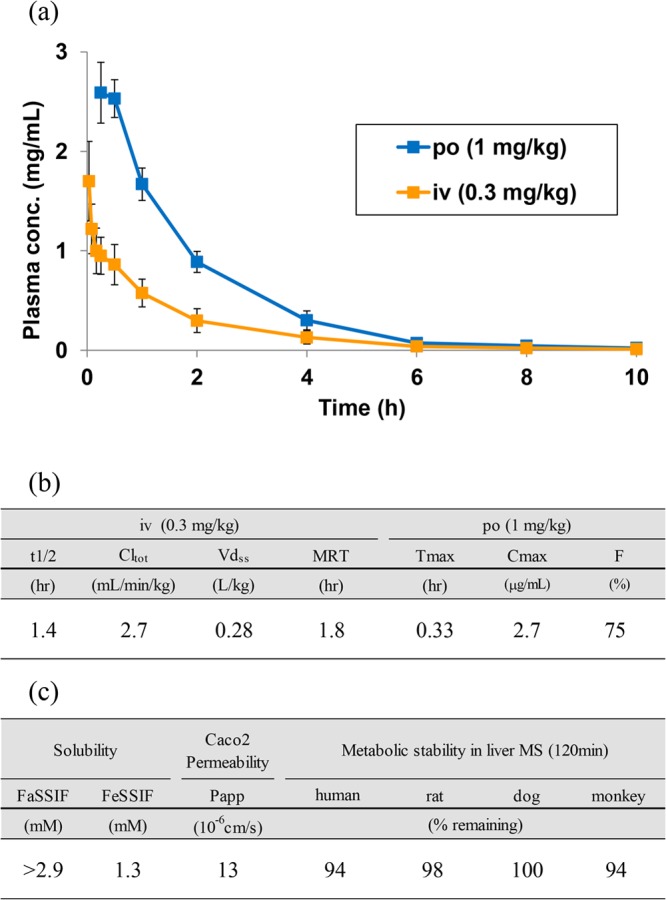
(a) Pharmacokinetic curves of 14 in rats. Each data point represents the mean ± SD (n = 3). (b) PK parameters of 14 in rats. (c) PK related parameters of 14.
The synthetic route of compound 14 is shown in Scheme 1. tert-Butyl esterification of 2,4-dichloronicotinic acid 21 and subsequent regioselective nucleophilic aromatic substitution with benzyl alcohol gave 22. After displacement of chlorine with hydrazine, sequential cyclization with ortho-ester afforded the [1,2,4]triazolo[4,3-a]pyridine derivative 23. Dimroth rearrangement proceeded smoothly in the presence of morpholine in refluxing AcOEt to give the [1,2,4]triazolo[1,5-a]pyridine derivative 24, which was converted by regioselective iodination into the coupling precursor 25. Sonogashira coupling with phenylacetylene and removal of the tert-butyl group by treating with TFA provided the carboxylic acid 26. Amide formation with glycine ethyl ester followed by hydrogenation and hydrolysis of the ester afforded compound 14.
Scheme 1. Synthesis of 14.

Reagents and conditions: (a) tert-butyl 2,2,2-trichloroacetimidate, BF3·Et2O, THF; (b) benzyl alcohol, NaH, DMF, 0 °C; (c) hydrazine hydrate, 1,4-dioxane, 100 °C; (d) p-TsOH·H2O, trimethyl orthoformate, 60 °C; (e) morpholine, AcOEt, reflux; (f) I2, LHMDS, THF, −60 °C; (g) phenylacetylene, PdCl2(PPh3)2, CuI, TEA, toluene; (h) MsOH, toluene–AcOEt; (i) glycine ethyl ester hydrochloride, EDC, HOBt, TEA, DMF; (j) H2, Pd–C, THF–MeOH; (k) 4 N NaOH aq., EtOH, reflux.
In conclusion, following the identification of novel triazolopyridine derivatives as potent scaffolds of PHD2 inhibitor, our lead optimization to improve cell permeability led to the discovery of compound 14 (JTZ-951), a potent and orally active PHD inhibitor. Compound 14 at 1 and 3 mg/kg dose-dependently increased hemoglobin levels with daily oral dosing in rats. The short-acting PK profile of compound 14 is expected to be advantageous in terms of safety. Compound 14 has been advanced to phase II clinical trials for renal anemia.
Acknowledgments
We thank Dr. Hisashi Shinkai and Dr. Susumu Kato for helpful discussions. We are also grateful to Dr. Hisashi Kawasaki and Dr. Hiromasa Hashimoto for suggestions on writing this letter.
Glossary
ABBREVIATIONS
- SD
standard deviation
- BF3·Et2O
boron trifluoride diethyl ether complex
- p-TsOH·H2O
p-toluenesulfonic acid monohydrate
- LHMDS
lithium hexamethyldisilazan
- MsOH
methanesulfonic acid
- EDC
1-(3-(dimethylamino)propyl)-3-ethylcarbodiimide hydrochloride
- HOBt
1-hydroxybenzotriazole monohydrate
- TEA
triethylamine
- CYP
cytochrome P450
- hERG
human ether-a-go-go-related gene
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00404.
Synthetic schemes, procedures, experimental data of all compounds, assay procedures, and supporting tables with standard deviation (PDF)
Author Present Address
# Toxicology Research Laboratories Central Pharmaceutical, Central Pharmaceutical Research Institute, Japan Tobacco Inc., 23 Naganuki, Hadano, Kanagawa 257-0024, Japan.
Author Contributions
Y.O., T.M., I.M., M.Y., M.T., D.M., K.U., T.H., T.I., and H.A. contributed to design and synthesis of JTZ-951. Y.H. contributed to the modeling studies. K.F., K.D., and H.Y. contributed to the biological assay of all compounds in vitro and in vivo. S.I. contributed to the PK studies.
The authors declare no competing financial interest.
Supplementary Material
References
- Babitt J. L.; Lin H. Y. Mechanisms of Anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634. 10.1681/ASN.2011111078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall I. C.; Casadevall N.; Locatelli F.; Combe C.; London G. M.; Paolo S. D.; Kribben A.; Fliser D.; Messner H.; McNeil J.; Stevens P.; Santoro A.; Francisco A. L. M.; Percheson P.; Potamianou A.; Foucher A.; Fife D.; Mérit V.; Vercammen E. Incidence of erythropoietin antibody-mediated pure red cell aplasia: the Prospective Immunogenicity Surveillance Registry (PRIMS). Nephrol., Dial., Transplant. 2015, 30, 451–460. 10.1093/ndt/gfu297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer M. A.; Burdmann E. A.; Chen C.-Y.; Cooper M. E.; de Zeeuw D.; Eckardt K.-U.; Feyzi J. M.; Ivanovich P.; Kewalramani R.; Levey A. S.; Lewis E. F.; McGill J. B.; McMurray J. J. V.; Parfrey P.; Parving H.-H.; Remuzzi G.; Singh A. K.; Solomon S. D.; Toto R. A Trial of Darbepoetin Alfa in Type 2 Diabetes and Chronic Kidney Disease. N. Engl. J. Med. 2009, 361, 2019–2032. 10.1056/NEJMoa0907845. [DOI] [PubMed] [Google Scholar]
- Singh A. K.; Szczech L.; Tang K. L.; Barnhart H.; Sapp S.; Wolfson M.; Reddan D. Correction of Anemia with Epoetin Alfa in Chronic Kidney Disease. N. Engl. J. Med. 2006, 355, 2085–2098. 10.1056/NEJMoa065485. [DOI] [PubMed] [Google Scholar]
- Higashijima Y.; Tanaka T.; Nangaku M. Structure-based drug design for hypoxia-inducible factor prolyl-hydroxylase inhibitors and its therapeutic potential for the treatment of erythropoiesis-stimulating agent-resistant anemia: raising expectations for exploratory clinical trials. Expert Opin. Drug Discovery 2013, 8, 965–976. 10.1517/17460441.2013.796358. [DOI] [PubMed] [Google Scholar]
- Maxwell P. H.; Eckardt K.-U. HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nat. Rev. Nephrol. 2016, 12, 157–168. 10.1038/nrneph.2015.193. [DOI] [PubMed] [Google Scholar]
- Ke Q.; Costa M. Hypoxia-Inducible Factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- Epstein A. C. R.; Gleadle J. M.; McNeill L. A.; Hewitson K. S.; O’Rourke J.; Mole D. R.; Mukherji M.; Metzen E.; Wilson M. I.; Dhanda A.; Tian Y.-M.; Masson N.; Hamilton D. L.; Jaakkola P.; Barstead R.; Hodgkin J.; Maxwell P. H.; Pugh C. W.; Schofield C. J.; Ratcliffe P. J. C. elegans EGL-9 and Mammalian Homologs Define a Family of Dioxygenases that Regulate HIF by Prolyl Hydroxylation. Cell 2001, 107, 43–54. 10.1016/S0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Bernhardt W. M.; Wiesener M. S.; Scigalla P.; Chou J.; Schmieder R. E.; Günzler V.; Eckardt K.-U. Inhibition of Prolyl Hydroxylases Increases Erythropoietin Production in ESRD. J. Am. Soc. Nephrol. 2010, 21, 2151–2156. 10.1681/ASN.2010010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besarab A.; Provenzano R.; Hertel J.; Zabaneh R.; Klaus S. J.; Lee T.; Leong R.; Hemmerich S.; Yu K.-H. P.; Neff T. B. Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrol., Dial., Transplant. 2015, 30, 1665–1673. 10.1093/ndt/gfv302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshakoon N. C.; Wu S.; Boyer A.; Kawamoto R.; Sheville J.; Bhatt R. T.; Renock S.; Xu K.; Pokross M.; Zhou S.; Walter R.; Mekel M.; Evdokimov A. G.; East S. Design and synthesis of substituted pyridine derivatives as HIF-1α prolyl hydroxylase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 5616–5620. 10.1016/j.bmcl.2006.08.026. [DOI] [PubMed] [Google Scholar]
- Yan L.; Colandrea V. J.; Hale J. J. Prolyl hydroxylase domain-containing protein inhibitors as stabilizers of hypoxia-inducible factor: small molecule-based therapeutics for anemia. Expert Opin. Ther. Pat. 2010, 20, 1219–1245. 10.1517/13543776.2010.510836. [DOI] [PubMed] [Google Scholar]
- Rabinowitz M. H. Inhibition of Hypoxia-Inducible Factor Prolyl Hydroxylase Domain Oxygen Sensors: Tricking the Body into Mounting Orchestrated Survival and Repair Responses. J. Med. Chem. 2013, 56, 9369–9402. 10.1021/jm400386j. [DOI] [PubMed] [Google Scholar]
- Bissantz C.; Kuhn B.; Stahl M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–5084. 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R.; Candela-Lena J. I.; Chan M. C.; Greenald D. J.; Yeoh K. K.; Tian Y.-M.; McDonough M. A.; Tumber A.; Rose N. R.; Conejo-Garcia A.; Demetriades M.; Mathavan S.; Kawamura A.; Lee M. K.; van Eeden F.; Pugh C. W.; Ratcliffe P. J.; Schofield C. J. Selective Small Molecule Probes for the Hypoxia Inducible Factor (HIF) Prolyl Hydroxylases. ACS Chem. Biol. 2013, 8, 1488–1496. 10.1021/cb400088q. [DOI] [PubMed] [Google Scholar]
- Debenham J. S.; Madsen-Duggan C.; Clements M. J.; Walsh T. F.; Kuethe J. T.; Reibarkh M.; Salowe S. P.; Sonatore L. M.; Hajdu R.; Milligan J. A.; Visco D. M.; Zhou D.; Lingham R. B.; Stickens D.; DeMartino J. A.; Tong X.; Wolff M.; Pang J.; Miller R. R.; Sherer E. C.; Hale J. J. Discovery of N-[Bis(4-methoxyphenyl)methyl]-4-hydroxy-2-(pyridazin-3-yl)pyrimidine-5-carboxamide (MK-8617), an Orally Active Pan-Inhibitor of Hypoxia-Inducible Factor Prolyl Hydroxylase 1–3 (HIF PHD1–3) for the Treatment of Anemia. J. Med. Chem. 2016, 59, 11039–11049. 10.1021/acs.jmedchem.6b01242. [DOI] [PubMed] [Google Scholar]
- LE and LLE were calculated by the following calculation formulas: LE = −1.37 log IC50/number of heavy atoms. LLE = pIC50 – ClogP.
- Vachal P.; Miao S.; Pierce J. M.; Guiadeen D.; Colandrea V. J.; Wyvratt M. J.; Salowe S. P.; Sonatore L. M.; Milligan J. A.; Hajdu R.; Gollapudi A.; Keohane C. A.; Lingham R. B.; Mandala S. M.; DeMartino J. A.; Tong X.; Wolff M.; Steinhuebel D.; Kieczykowski G. R.; Fleitz F. J.; Chapman K.; Athanasopoulos J.; Adam G.; Akyuz C. D.; Jena D. K.; Lusen J. W.; Meng J.; Stein B. D.; Xia L.; Sherer E. C.; Hale J. J. 1,3,8-Triazaspiro[4.5]decane-2,4-diones as Efficacious Pan-Inhibitors of Hypoxia-Inducible Factor Prolyl Hydroxylase 1–3 (HIF PHD1–3) for the Treatment of Anemia. J. Med. Chem. 2012, 55, 2945–2959. 10.1021/jm201542d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.