

Abstract
A series of novel selenides bearing benzenesulfonamide moieties was synthesized and investigated for the inhibition of five human (h) isoforms of zinc enzyme carbonic anhydrase (CA, EC 4.2.1.1), hCA I, II, IV, VII, and IX. These enzymes are involved in a variety of diseases, including glaucoma, retinitis pigmentosa, epilepsy, arthritis, and tumors. The investigated compounds showed potent inhibitory action against hCA II, VII, and IX, in the low nanomolar range, thus making them of interest for the development of isoform-selective inhibitors and as candidates for biomedical applications.
Keywords: Carbonic anhydrase, inhibitor, metalloenzymes, selenium, selenides, organoselenium compounds
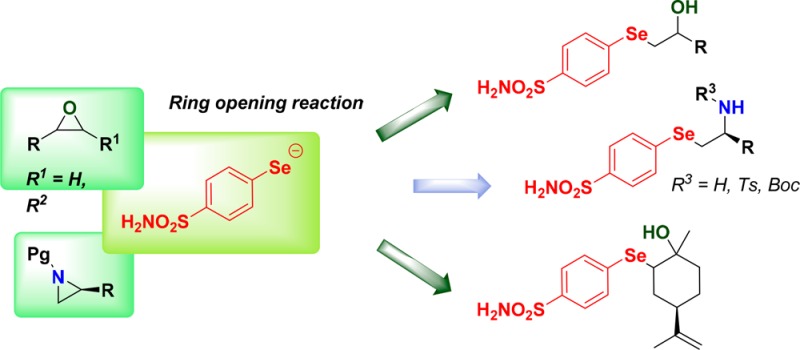
Selenium has a long history of association with human health and disease.1,2 Interest in the potential biological, pharmacological, and therapeutic exploitation of synthetic organoselenium compounds started several decades ago. Organochalcogen derivatives played a crucial role in identifying free radical scavengers or antioxidants that can inhibit or retard oxidative damage.3,4 Oxidative stress, induced by the generation of reactive oxygen species (ROS), is considered a major causative factor of many serious conditions, including diabetes, cardiovascular diseases, cancer, and several neurodegenerative diseases.5,6 Furthermore, organoselenium derivatives showed inhibitory effects on a variety of enzymes such as nitric oxide synthase (NOS),7−10 lipoxygenases (LOX),11 and carbonic anhydrases12−14 (CAs, EC 4.2.1.1). CAs are metalloenzymes that catalyze a very simple reaction: the hydration of carbon dioxide to bicarbonate and protons.15 This reaction plays an important role in many physiological and pathological processes associated with pH control, ion transport, fluid secretion, biosynthetic reactions, etc.16,17 For this reason, we continued to investigate a new type of organoselenium derivatives as human (h) CA inhibitors (CAIs). Our long-standing interest in the reactivity of strained heterocycles with chalcogen-containing nucleophiles led us to disclose novel procedures for the synthesis of a wide variety of functionalized selenium- and tellurium-containing organic small molecules.18−21 Some of these structures exhibited interesting catalytic antioxidant activity.22−24 With the aim of synthesizing a new series of hydroxy- and amino-functionalized selenium containing CAI, we sought to exploit the reactivity of the three-membered ring, such as epoxides and aziridines, with a suitable selenolate, bearing the benzenesulfonamide moiety (as CA inhibiting chemotype),25 generated from the corresponding diselenide 3. In the present study, we investigated different selenides incorporating a benzenesulfonamide moiety as CAI. We began our investigation with the synthesis of diselenide 3, as shown in Scheme 1. The diazonium salt of sulfanilamide was prepared by reaction of 1 with sodium nitrite in the presence of acid (Sandmeyer reaction) and used as the key intermediate for the synthesis of compound 2. Successively, the selenocyanate derivative 2 was converted easily into the diselenide 3 by reaction with NaBH4 in ethanol, as outlined in Scheme 1.
Scheme 1. Synthesis of Selenocyanate and Diselenide Bearing Benzenesulfonamide Moiety.

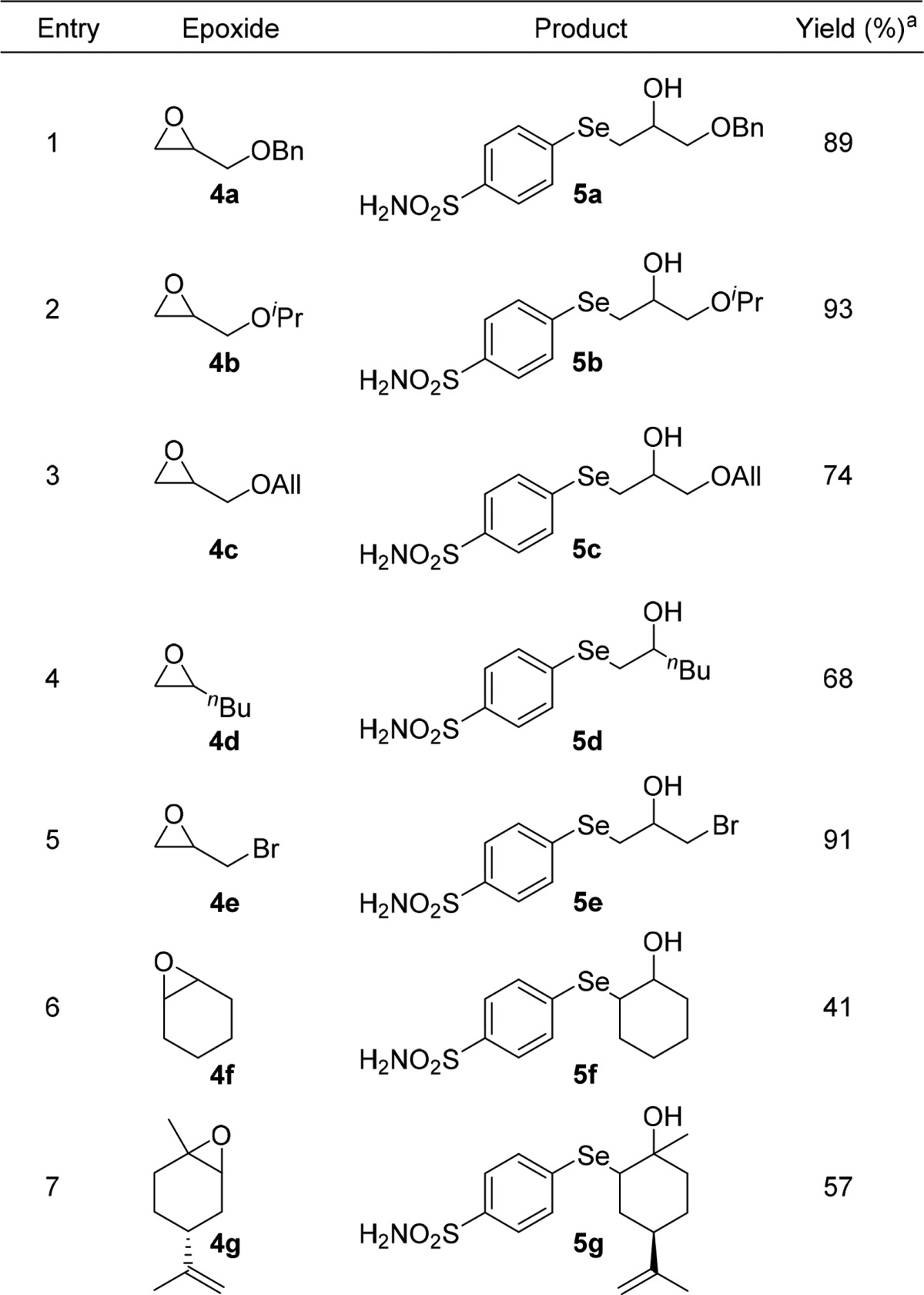
Having obtained diselenide 3, we evaluated the possibility to access β-hydroxy selenides by using the ring opening reaction of this compound with epoxides.26−28 Thus, 3 was reduced with NaBH4 to the corresponding selenolate, which was treated in situ with benzyl glycidyl ether 4a, affording the β-hydroxyselenide 5a in good yield (Table 1, entry 1). The process proved to be highly regioselective, as only the isomer arising from the nucleophilic attack at the less hindered carbon of the oxirane was observed. On the basis of these results, and in order to study the generality of such a procedure, a series of epoxides was reacted with 3 under the same conditions, as reported in Table 1. Thus, differently substituted hydroxyl selenides 5b–g were obtained from the corresponding epoxides 4b–g through a regioselective ring opening route (Table 1, entries 2–4). Interestingly, epibromohydrin 4e was smoothly converted into 5e in excellent yields, the nucleophilic attack occurred exclusively on the epoxide, and the halide was preserved on the side chain (Table 1, entry 5). Disubstituted hydroxy selenides 5f,g were obtained by reacting 3 with cyclohexene oxide 4f and limonene oxide 4g (Table 1, entries 6,7).
Table 1. Synthesis of β-Hydroxy Selenides Bearing Benzenesulfonamide Moiety.
Yields refer to isolated products.
In order to access benzenesulfonamide-substituted selenides bearing the amino group, the procedure was extended to differently N-protected aziridines 6,29,30 synthesized from natural amino acids. As reported in t he Scheme 2, enantioenriched N-tosyl and N-Boc selenides 7a–c were obtained in good yields from 6a–c through a regio- and stereoselective reaction.
Scheme 2. Synthesis of N-Protected β-Amino Selenides Bearing Benzenesulfonamide Moiety.

Finally, the free selenoamine 8 was obtained from the N-Boc derivative 7c by the acetyl chloride promoted cleavage of the protecting group (Scheme 3).31
Scheme 3. Synthesis of β-Amino Selenide Bearing Benzenesulfonamide Moiety.

As a further investigation, in order to propose an alternative way to access the target compounds, we sought to achieve β-hydroxy- and β-amino-selenides from the selenocyanate 2, thus avoiding the synthesis of the diselenide 3. After having optimized the reaction conditions, we were pleased to observe that selenides 5a,b and 7a were obtained by ring opening of epoxides 4a,b and aziridine 6a with the selenolate, in situ generated by reducing 2, as reported in Scheme 4.
Scheme 4. Synthesis of Selenides Bearing Benzenesulfonamide Moiety.

We investigated the CA inhibitory proprieties of compounds 2, 3, 5a–g, 7a–c, and 8 against the physiologically relevant hCA isoforms I, II, IV, VII, and IX by means of the stopped-flow carbon dioxide hydration assay32 after a period of 15 min of incubation of the enzyme and inhibitor solutions.33−38 Their activities were compared to the standard CAI acetazolamide (AAZ) (Table 2).
Table 2. Inhibition Data of Human CA Isoforms I, II, IV, VII, and IX with Compounds 2, 3, 5a–g, 7a–c and 8 and AAZ by a Stopped-Flow CO2 Hydrase Assay32.
|
Kia (nM) |
|||||
|---|---|---|---|---|---|
| Compound | hCA I | hCA II | hCA IV | hCA VII | hCA IX |
| 2 | 95.6 | 53.1 | 30.6 | 7.1 | 9.3 |
| 3 | 1522.7 | 7.9 | 298.4 | 40.5 | 2.7 |
| 5a | 193.8 | 1.4 | 377.7 | 1.9 | 10.1 |
| 5b | 73.2 | 4.4 | 403.1 | 0.71 | 15.9 |
| 5c | 8084.3 | 920.8 | 8133.0 | 74.2 | 11.9 |
| 5d | 228.8 | 8.8 | 429.2 | 0.85 | 5.6 |
| 5e | 127.2 | 4.9 | 319.3 | 7.4 | 6.5 |
| 5f | 148.6 | 7.4 | 458.2 | 0.77 | 8.3 |
| 5g | 8.4 | 0.18 | 34.8 | 0.68 | 2.4 |
| 7a | 881.1 | 14.0 | 435.2 | 0.35 | 10.1 |
| 7b | 4365.5 | 90.2 | 5601.0 | 3.4 | 2.4 |
| 7c | 1471.2 | 15.9 | 2825.0 | 3.5 | 2.3 |
| 8 | 93.0 | 0.51 | 2321.0 | 36.2 | 2.4 |
| AAZ | 250 | 12.1 | 74 | 6 | 25.8 |
Mean from three different assays, by a stopped-flow technique (errors were in the range of ±5–10% of the reported values).
The following structure–activity relationships (SARs) may be noted regarding the inhibition data of Table 2:
-
(i)
The ubiquitous cytosolic hCA I was inhibited by all compounds with Ki spanning between low nanomolar (Ki 8.4 nM) to the high micromolar range (Ki 8084.3 nM). Selenocyanate derivative 2 inhibited hCA I in the medium nanomolar range (Ki 95 nM), but the diselenide 3 showed a decreased potency of inhibition by almost 15-fold. The β-hydroxy selenide 5g showed the best inhibition potency, with a Ki of 8.4 nM. Moreover, a less bulky tail moiety such as in the cyclohexane derivative (5f) decreased the activity 16-fold. Compound 8 inhibited this isoform in medium nanomolar range with a Ki of 93 nM. Compounds with different N-protecting groups, such as 7a and 7c, led to a decrease of the inhibitory activity of nearly nine times (with the tosyl group in 7a) and 18 times (for the Boc derivative 7c) compared to 8.
-
(ii)
The dominant cytosolic human isoform hCA II was inhibited in the low-medium nanomolar range by all compounds investigated here, except for derivative 5c, which acted in the high nanomolar range (Ki 920.8 nM). Selenocyanate derivate 2 showed a six-fold loss of activity compared to the diselenide 3. β-Hydroxy selenides 5a–g proved to be potent inhibitors of this isoform, with Kis ranging between 0.18 and 8.8 nM, except for 5c mentioned earlier. In addition, the β-amino selenide 8 showed a very potent inhibition profile of hCA II (Ki of 0.51 nM). The introduction of N-protecting groups as in 7a and 7c led to a decrease of the inhibition potency of nearly 29 times compared to 8.
-
(iii)
The last cytosolic human isoform studied, hCA VII, was inhibited by all compounds in the subnanomolar–nanomolar range (Kis of 0.35–74.2 nM, Table 2). Many of the new selenium-containing derivatives, such as 5b,d,f,g and 7a were subnanomolar hCA VII inhibitors, making them of great interest for further studies, considering that this isoform was shown to be involved in oxidative stress.39,40 The presence of N-protecting groups for compounds 7a and 7c increased the efficacy 10 times for the Boc moiety and 100 times for tosyl moiety, with respect to the compound without such moieties (8).
-
(iv)
Almost all compounds investigated here possessed low inhibitory activity for the membrane-bound hCA IV with Kis spanning between the high nanomolar range to the micromolar range. Compound 2 showed the best activity against this isoform with a Ki of 30.6 nM, but the efficacy decreased for the diselenide derivative 4 (Ki 298.4 nM). Different substituents on the β-hydroxy selenides 5a–g did not influence significantly the inhibition activity, except for 5c, which had a decrease of efficacy (Ki 8133 nM). Compound 7a, with a tosyl moiety as a protecting group, proved to have a better inhibition profile compared to the other β-amino selenides investigated here.
-
(v)
The transmembrane, tumor-associated hCA IX was effectively inhibited by all compounds investigated here, in the low nanomolar range (Kis of 2.3–154.9 nM), all of them being more effective inhibitors compared to the clinically used standard acetazolamide (AAZ), Table 1. As for the other membrane isoform, hCA IV, the substituents on the β-hydroxy selenides 5a–g did not influence significantly the inhibitory efficacy in this small series of derivatives. N-Protection for compounds 7a and 7c did not change significantly the inhibition profile compared to the β-amino selenide 8, a special mention regarding the important difference of inhibitory activity of 5b and 5c against all isoforms except CA IX. In fact, the two compounds only differ by the presence of an allyl instead of an iso-propyl moiety. Although these structural differences are minor, in many similar cases when the X-ray structures were reported in complex with various CA isoforms,41,42 important differences in the orientation within the active site were observed, which may explain the difference of inhibitory power of these quite similar derivatives.
In conclusion, we have developed methods for the synthesis of a novel series of selenoethers as inhibitors on five α-carbonic anhydrases (CAs, EC 4.2.1.1) of pharmacologic relevance, i.e., hCA I, II, IV, VII, and IX. These isoforms are drug targets for antiglaucoma (hCA I, II and IV), antiepileptic (hCA VII), or antitumor (hCA IX) agents. β-Hydroxy 5a–g and N-protected β-amino selenides prove to be potent inhibitors for hCA VII. Indeed, β-amino selenide 8 showed a potent inhibition against hCA II. In this contest, the investigated selenoether compounds showed potent inhibitory action, thus making them interesting leads for the development of more potent and more isoform-selective inhibitors.
Glossary
ABBREVIATIONS
- CAI(s)
carbonic anhydrase inhibitor(s)
- AAZ
acetazolamide
- (h)CA
(human) carbonic anhydrase
- KI
inhibition constant
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00387.
Synthetic procedures, characterization of compounds, and in vitro kinetic procedure (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Olcott H. S.; Brown W. D.; Van Derveen J. Selenomethionine as an Antioxidant. Nature 1961, 191, 1201. 10.1038/1911201a0. [DOI] [PubMed] [Google Scholar]
- Walter R.; Schwartz I. L.; Roy J. Can selenoamino acids act as reversible biological antioxidants?. Ann. N. Y. Acad. Sci. 1972, 192, 175–180. 10.1111/j.1749-6632.1972.tb52588.x. [DOI] [PubMed] [Google Scholar]
- Halliwel B.; Gutteridge J. M. C.. Free Radicals in Biology and Medicine, 4th ed.; Oxford University Press: Oxford, 2007. [Google Scholar]
- Halliwell B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol. 2006, 141, 312. 10.1104/pp.106.077073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J. K. Oxidative stress in neurodegeneration: cause or consequence?. Nat. Rev. Neurosci. 2004, 5, S18–25. 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- Seet R. C. S.; Lee C.-Y. J.; Lim E. C. H.; Tan J. J. H.; Quek A. M. L.; Chong W.-L.; Looi W.-F.; Huang S.-H.; Wang H.; Chan Y.-H.; Halliwell B. Oxidative damage in Parkinson disease: Measurement using accurate biomarkers. Free Radical Biol. Med. 2010, 48, 560. 10.1016/j.freeradbiomed.2009.11.026. [DOI] [PubMed] [Google Scholar]
- Wang J.-F.; Komarov P.; Sies H.; de Groot H. Inhibition of superoxide and nitric oxide release and protection from reoxygenation injury. Hepatology 1992, 15, 1112. 10.1002/hep.1840150623. [DOI] [PubMed] [Google Scholar]
- Wang J.-F.; Komarov P.; Sies H.; de Groot H. Contribution of nitric oxide synthase to luminol-dependent chemiluminescence generated by phorbol-ester-activated Kupffer cells. Biochem. J. 1991, 279, 311. 10.1042/bj2790311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zembowicz A.; Hatchett R. J.; Radziszewski W.; Gryglewski R. J. Inhibition of endothelial nitric oxide synthase by ebselen. Prevention by thiols suggests the inactivation by ebselen of a critical thiol essential for the catalytic activity of nitric oxide synthase. J. Pharmacol. Exp. Therap. 1993, 267, 1112. [PubMed] [Google Scholar]
- Southan G. J.; Salzman A. L.; Szabó C. Potent inhibition of the inducible isoform of nitric oxide synthase by aminoethylisoselenourea and related compounds. Life Sci. 1996, 58, 1139. 10.1016/0024-3205(96)00072-0. [DOI] [PubMed] [Google Scholar]
- Schewe C.; Schewe T.; Wendel A. Strong inhibition of mammalian lipoxygenases by the antiinflammatory seleno-organic compound ebselen in the absence of glutathione. Biochem. Pharmacol. 1994, 48, 65. 10.1016/0006-2952(94)90224-0. [DOI] [PubMed] [Google Scholar]
- Angeli A.; Carta F.; Bartolucci G.; Supuran C. T. Synthesis of novel acyl selenoureido benzensulfonamides as carbonic anhydrase I, II, VII and IX inhibitors. Bioorg. Med. Chem. 2017, 25, 3567. 10.1016/j.bmc.2017.05.014. [DOI] [PubMed] [Google Scholar]
- Angeli A.; Tanini D.; Viglianisi C.; Panzella L.; Capperucci A.; Menichetti S.; Supuran C. T. Evaluation of selenide, diselenide and selenoheterocycle derivatives as carbonic anhydrase I, II, IV, VII and IX inhibitors. Bioorg. Med. Chem. 2017, 25, 2518. 10.1016/j.bmc.2017.03.013. [DOI] [PubMed] [Google Scholar]
- Angeli A.; Peat T. S.; Bartolucci G.; Nocentini A.; Supuran C. T.; Carta F. Intramolecular oxidative deselenization of acylselenoureas: a facile synthesis of benzoxazole amides and carbonic anhydrase inhibitors. Org. Biomol. Chem. 2016, 14, 11353–11356. 10.1039/C6OB02299E. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discovery 2008, 2, 168–181. 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. How many carbonic anhydrase inhibition mechanisms exist?. J. Enzyme Inhib. Med. Chem. 2016, 31, 345–360. 10.3109/14756366.2015.1122001. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. 10.1042/BCJ20160115. [DOI] [PubMed] [Google Scholar]
- Tanini D.; Grechi A.; Dei S.; Teodori E.; Capperucci A. An easy one-step procedure for the synthesis of novel β-functionalised tellurides. Tetrahedron 2017, 73, 5646–5653. 10.1016/j.tet.2017.07.061. [DOI] [Google Scholar]
- Tanini D.; Capperucci A.; Degl’Innocenti A. Bis(trimethylsilyl)selenide in the Selective Synthesis of β-Hydroxy, β-Mercapto, and β-Amino Diorganyl Diselenides and Selenides Through Ring Opening of Strained Heterocycles. Eur. J. Org. Chem. 2015, 2015, 357–369. 10.1002/ejoc.201403015. [DOI] [Google Scholar]
- Capperucci A.; Tanini D.; Borgogni C.; Degl’Innocenti A. Thiosilane- and Organoselenosilane-Mediated Novel Access to 3,7-Disubstituted-1,2,5- trithiepanes and −1,2,5-dithiaselenepanes. Heteroat. Chem. 2014, 25, 678–683. 10.1002/hc.21157. [DOI] [Google Scholar]
- Capperucci A.; Tiberi C.; Pollicino S.; Degl’Innocenti A. Fluoride Ion Induced Thiophilic Reactivity of Organosilanes with Sulfines: Regiospecific Access to Allyl and Benzyl Sulfoxides. Tetrahedron Lett. 2009, 50, 2808–2810. 10.1016/j.tetlet.2009.03.167. [DOI] [PubMed] [Google Scholar]
- Tanini D.; D’Esopo V.; Chen D.; Barchielli G.; Capperucci A. Novel sulfur and selenium-containing antioxidants: Synthesis and evaluation of their GPx-like activity. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 166–168. 10.1080/10426507.2016.1252365. [DOI] [Google Scholar]
- Menichetti S.; Capperucci A.; Tanini D.; Braga A. L.; Botteselle G. V.; Viglianisi C. A One-Pot Access to Benzo[b][1,4]selenazines from 2-Aminoaryl Diselenides. Eur. J. Org. Chem. 2016, 2016, 3097–3102. 10.1002/ejoc.201600351. [DOI] [Google Scholar]
- Tanini D.; Panzella L.; Amorati R.; Capperucci A.; Pizzo E.; Napolitano A.; Menichetti S.; D’Ischia M. Resveratrol-based benzoselenophenes with an enhanced antioxidant and chain breaking capacity. Org. Biomol. Chem. 2015, 13, 5757–5764. 10.1039/C5OB00193E. [DOI] [PubMed] [Google Scholar]
- Capperucci A.; Tanini D. Silicon-assisted synthesis and functionalization of sulfurated and selenated compounds. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 1320–1338. 10.1080/10426507.2015.1024790. [DOI] [Google Scholar]
- Silva P. C.; Borges E. L.; Lima D. B.; Jacob R. G.; Lenardão E. J.; Perin G.; Silva M. S. A simple and non-conventional method for the synthesis of selected β-arylalkylchalcogeno substituted alcohols, amines and carboxylic acids. Arkivoc 2016, 5, 376–389. [Google Scholar]
- Ganesh V.; Chandrasekaran S. One-Pot Synthesis of β-Amino/β-Hydroxy Selenides and Sulfides from Aziridines and Epoxides. Synthesis 2009, 19, 3267–3278. 10.1055/s-0029-1216960. [DOI] [Google Scholar]
- Tiecco M.; Testaferri L.; Marini F.; Sternativo S.; Santi C.; Bagnoli L.; Temperini A. intramolecular addition of carbon radicals to aldehydes: synthesis of enantiopure tetrahydrofuran-3-ols. Tetrahedron 2007, 63, 5482–5489. 10.1016/j.tet.2007.04.047. [DOI] [Google Scholar]
- Tanini D.; Barchielli G.; Benelli F.; Degl’Innocenti A.; Capperucci A. Aziridines Ring Opening by Silyl Chalcogenides: a Stereoselective Access to Polyfunctionalized Molecules as Precursor of Sulfurated and Selenated Heterocycles. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 1265–1270. 10.1080/10426507.2014.1002615. [DOI] [Google Scholar]
- Braga A. L.; Schneider P. H.; Paixao M. W.; Deobald A. M.; Peppe C.; Bottega D. P. Chiral Seleno-Amines from Indium Selenolates. A Straightforward Synthesis of Selenocysteine Derivatives. J. Org. Chem. 2006, 71, 4305–4307. 10.1021/jo060286b. [DOI] [PubMed] [Google Scholar]
- Charrat C.; Biscotti A.; Godeau G.; Greiner J.; Vierling P.; Guigonis J. M.; Di Giorgio C. Formulation of highly functionalizable DNA nanoparticles based on 1,2-dithiolane derivatives. ChemBioChem 2015, 16, 792–804. 10.1002/cbic.201402657. [DOI] [PubMed] [Google Scholar]
- Khalifah R. G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561. [PubMed] [Google Scholar]
- Mollica A.; Locatelli M.; Macedonio G.; Carradori S.; Sobolev A. P.; De Salvador R. F.; Monti S. M.; Buonanno M.; Zengin G.; Angeli A.; Supuran C.T. Microwave-assisted extraction, HPLC analysis, and inhibitory effects on carbonic anhydrase I, II, VA, and VII isoforms of 14 blueberry Italian cultivars. J. Enzyme Inhib. Med. Chem. 2016, 31, 1–6. 10.1080/14756366.2016.1214951. [DOI] [PubMed] [Google Scholar]
- De Vita D.; Angeli A.; Pandolfi F.; Bortolami M.; Costi R.; Di Santo R.; Suffredini E.; Ceruso M.; Del Prete S.; Capasso C.; Scipione L.; Supuran C. T. Inhibition of the α-carbonic anhydrase from Vibrio cholerae with amides and sulfonamide incorporating imidazole moieties. J. Enzyme Inhib. Med. Chem. 2017, 32, 798. 10.1080/14756366.2017.1327522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno E.; Buemi M. R.; Di Fiore A.; De Luca L.; Ferro S.; Angeli A.; Cirilli R.; Sadutto D.; Alterio V.; Monti S. M.; Supuran C. T.; De Simone G.; Gitto R. Probing Molecular Interactions between Human Carbonic Anhydrases (hCAs) and a Novel Class of Benzenesulfonamides. J. Med. Chem. 2017, 60, 4316. 10.1021/acs.jmedchem.7b00264. [DOI] [PubMed] [Google Scholar]
- Abdoli M.; Angeli A.; Bozdag M.; Carta F.; Kakanejadifard A.; Saeidian H.; Supuran C. T. Synthesis and carbonic anhydrase I, II, VII, and IX inhibition studies with a series of benzo[d]thiazole-5- and 6-sulfonamides. J. Enzyme Inhib. Med. Chem. 2017, 32, 1071–1078. 10.1080/14756366.2017.1356295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra C. B.; Kumari S.; Angeli A.; Monti S. M.; Buonanno M.; Tiwari M.; Supuran C. T. Discovery of Benzenesulfonamides with Potent Human Carbonic Anhydrase Inhibitory and Effective Anticonvulsant Action: Design, Synthesis, and Pharmacological Assessment. J. Med. Chem. 2017, 60, 2456–2469. 10.1021/acs.jmedchem.6b01804. [DOI] [PubMed] [Google Scholar]
- Abdel-Aziz A. A.; Angeli A.; El-Azab A. S.; Abu El-Enin M. A.; Supuran C. T. Synthesis and biological evaluation of cyclic imides incorporating benzenesulfonamide moieties as carbonic anhydrase I, II, IV and IX inhibitors. Bioorg. Med. Chem. 2017, 25, 1666–1671. 10.1016/j.bmc.2017.01.032. [DOI] [PubMed] [Google Scholar]
- Monti D. M.; De Simone G.; Langella E.; Supuran C. T.; Di Fiore A.; Monti S. M. Insights into the role of reactive sulfhydryl groups of Carbonic Anhydrase III and VII during oxidative damage. J. Enzyme Inhib. Med. Chem. 2017, 32, 5–12. 10.1080/14756366.2016.1225046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Giudice R.; Monti D. M.; Truppo E.; Arciello A.; Supuran C. T.; De Simone G.; Monti S. M. Human carbonic anhydrase VII protects cells from oxidative damage. Biol. Chem. 2013, 394, 1343–8. 10.1515/hsz-2013-0204. [DOI] [PubMed] [Google Scholar]
- De Simone G.; Langella E.; Esposito D.; Supuran C. T.; Monti S. M.; Winum J. Y.; Alterio V. Insights into the binding mode of sulphamates and sulphamides to hCA II: crystallographic studies and binding free energy calculations. J. Enzyme Inhib. Med. Chem. 2017, 32, 1002–1011. 10.1080/14756366.2017.1349764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinin S.; Kopylov S.; Tuccinardi T.; Sapegin A.; Dar’in D.; Angeli A.; Supuran C. T.; Krasavin M. Lucky Switcheroo: Dramatic Potency and Selectivity Improvement of Imidazoline Inhibitors of Human Carbonic Anhydrase VII. ACS Med. Chem. Lett. 2017, 8, 1105–1109. 10.1021/acsmedchemlett.7b00300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.