

Abstract
Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) is identified by newborn screening (NBS). The natural history of MCADD includes metabolic decompensation with hypoglycemia, hyperammonemia, seizures, coma, and death. NBS enables expectant management thus severe symptoms are rare in managed patients. We report premature birth of an MCADD affected infant and resultant management challenges. Nutritional support advanced from parenteral nutrition at 24 hours to enteral feeds. A NBS sample was collected day 2, positive results for MCADD was reported day six, and diagnostic tests were performed day seven. Lab results confirmed MCADD; however, representation of pathologic analytes was so extreme that ingestion of medium chain triglycerides was suspected and subsequently confirmed. Diet was adjusted and reflected in moderation of pathologic analytes. This case emphasizes the need for prompt review NBS results in premature infants. Implementing informatic intervention within electronic medical records, when a disorder requiring special nutritional intervention is identified, will protect premature infants in this vulnerable setting. Standard of care management provided premature infants may be contraindicated in the context of a comorbid inborn error of metabolism.
Key words: MCAD deficiency, prematurity, nutritional support, decompensation, medium chain triglycerides
Introduction
Newborn screening (NBS) is among the most successful public health initiatives in modern medicine.1 NBS has identified MCAD deficiency as the most common inborn error of fatty acid oxidation, approaching the incidence of PKU in the United States and Western Europe.2 Prior to the availability of pre-symptomatic testing, MCAD deficiency was often diagnosed after presentation with devastating clinical outcomes. Indeed, ~30% of affected individuals historically were identified post mortem.3 With newborn screening, MCAD deficiency remains a serious but manageable disorder with an extraordinarily low death rate and normal neurodevelopmental outcomes. As NBS programs seek to assess the complete birth cohort, premature infants and infants with a plethora of comorbid medical issues (e.g. low birth weight) will be assessed and these conditions may influence screening tests.4-7 Further, intensive nutritional support, which may also impact results of screening tests, is an early and important intervention to assure optimum outcome for premature infants.8,9 Should a premature infant be affected with a disorder identified by newborn screening, the support provided to manage prematurity may be contraindicated in regard to an inborn error of metabolism. Management of premature infants with phenylketonuria, galactosemia, medium chain acyl-CoA dehydrogenase deficiency and other inborn errors of metabolism may require management strategies that deviate from standard protocols to avoid adverse outcomes.
Herein we report a case involving a screen positive, premature infant that was determined to be affected by MCAD deficiency. Appropriate means of nutritional support for the baby’s prematurity, prior to return of the NBS results, led to extraordinarily high intermediates of medium chain fat metabolism in blood and urine recognized by diagnostic testing, placing the patient at risk for metabolic decompensation. Removal of medium chain triglycerides, a nutrient common in enteral formula designed for premature infants, returned metabolites to a safe level.
Case Report
A G2 P1 woman gave birth at 33 weeks to a phenotypically normal male, approximately 25 hours after premature membrane rupture, following an otherwise uneventful pregnancy. An intramuscular injection of betamethasone was administered just prior to delivery to induce lung maturation. APGAR scores were 7 at one minute and 9 at five minutes. Birth weight was 2405 grams (70th centile), length was 50 centimeters (98th centile), and head circumference was 30 centimeters (27th centile). The newborn was noted to have tachypnea and moderate retractions with normal color and tone. He was admitted to Neonatal Intensive Care to monitor respiratory status and provide nasal CPAP and oxygen supplementation during the first three days of life after which he was weaned off the oxygen. An infectious disease workup was performed, doses of ampicillin and gentamicin were administered but ultimately cultures were negative. Basic chemistries, including liver and renal function, were unremarkable. Physical and neurological exams were normal and no dysmorphic features were noted. Feeding was deferred on day one of life until cardiorespiratory status stabilized. On day two of life parenteral nutrition was provided which transitioned to enteral feeds on day 3 of life with Similac Special Care Premature OptiGRO 24 calories/ounce and later breast milk supplemented with Similac Human Milk Fortifier. Feeds were advanced thereafter. A newborn screening sample, collected on day two of life, was reported as screen-positive for MCAD deficiency day 6 of life, and on day seven of life a genetics consult was performed which included collection of blood and urine samples for quantitative acylcarnitine profiling and qualitative urine organic acid screening respectively to be performed by the Biochemical Genetics Laboratory at Children’s Hospital of Pittsburgh of UPMC. The feeding regimen was not immediately changed.
Newborn screening, biochemical genetic, and molecular genetic studies
Table 1 provides relevant acylcarnitine values including newborn screening and subsequent diagnostic assessments. Results of the initial diagnostic tests were returned the same day. The urine organic acid profile was significant for hexanoylglycine indicative of MCAD deficiency. Additionally, massive excretions of sebacic acid, suberic acid, adipic acid, octanoic acid, and 5- hydroxyhexanoic acid were observed. While excretion of dicarboxylic acids is anticipated in untreated MCAD deficiency, their representation was far in excess of that typically observed in a newly diagnosed patient. Low-level lactic aciduria was also noted. Quantitative assessment of acylglycines and dicarboxylic acids was not performed. Figure 1 displays the acylcarnitine profile from day 7 of life which is consistent with MCAD deficiency; however, the representation of pathological analytes (C8 25.42 μM; C10 1.65 μM, C10:1 0.87μM, and C6 4.22 μM) was far in excess of values typically observed. Combined observations from clinical biochemical genetic testing suggested the newborn was receiving medium chain triglycerides. Review of nutritional support to manage prematurity indicated medium chain triglycerides were major components of both the formula and the human breast milk fortifier, both of which are used on a large scale for early nutritional support in premature newborns. Medium chain triglycerides are contraindicated in a defect of medium chain fatty acid oxidation. Dietary management was subsequently modified to exclude medium chain triglycerides, facilitating a gradual moderation of medium chain metabolites in blood on days 11 and 14 of life (Table 1). The baby remained asymptomatic throughout the early NICU course and hypoglycemia was not noted during the course of enteral feeds given every three hours. ACADM gene assessment identified compound heterozygous mutations [c.985A>G (p.LYS304 GLU)]; c.347G>A (p.CYS116TYR) confirming the diagnosis.
Table 1.
Newborn screening and diagnostic acylcarnitine values.
| Days of life Indication | C8 | C10 | C10:1 | C6 | C8/C10 | C8/C10:1 | |
|---|---|---|---|---|---|---|---|
| 6# | NBS | 5.87 (nl <0.30 μM) | 0.39 (nl 0.40 μM) | ND | 0.97 (nl <0.20 μM) | 15.05+ | ND |
| 7 | Diagnostic | 25.42 (nl <0.44 μM) | 1.65 (nl <0.5 μM) | 0.87 (nl <0.45 μM) | 4.22 (nl <0.45 μM) | 15.41 (nl 0.26-1.32) | 29.31 (nl 0.1-1.51) |
| 11 | Diagnostic | 8.71 (nl <0.44 μM) | 0.57 (nl <0.5 μM) | 0.93 (nl <0.45 μM) | 1.58 (nl <0.45 μM) | 15.18 (nl 0.26-1.32) | 9.37 (nl 0.1-1.51) |
| 14 | Diagnostic | 4.57 (nl <0.44 μM) | 0.33 (nl <0.5 μM) | 0.78 (nl <0.45 μM) | 0.92 (nl <0.45 μM) | 13.84 (nl 0.26-1.32) | 5.85 (nl 0.1-1.51) |
#Sample collected day 2 of life. nl = normal range; ND = not determined; + no normal range for the NBS C8/C10 ratio reported.
Figure 1.
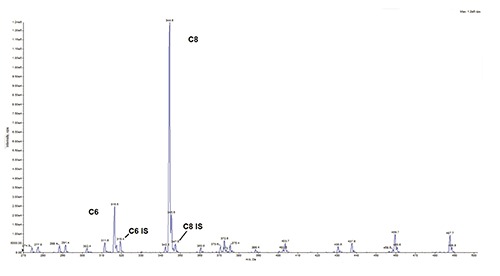
Acylcarnitine tandem mass spectrometry profile of MCAD-affected Patient at day 7 of life. Tandem mass spectrometry assessment of acylcarnitine esters. C8 = eight carbon acylcarnitine ester, C8 IS = stable isotope labeled eight carbon acylcarnitine internal standard, C6 = six carbon acylcarnitine ester, C6 IS = stable isotope labeled six carbon acylcarnitine internal standard.
Discussion
MCAD deficiency is essentially silent when patients are well; however, 20-25% of patients died during their first attack of coma, and as many as 40% of patients who survive episodes of coma suffer from chronic neurologic disabilities, ranging from mild developmental delay to seizures and cerebral palsy.10 The combination of a 33 week premature birth and MCAD deficiency led to a clash of circumstances that created the potential for an adverse outcome. Most enteral supplements tailored for premature infants are rich in medium chain triglycerides which promote growth and provide extra calories without stressing the underdeveloped gut; however, these are contraindicated in MCAD deficiency. The patient received a dietary regimen rich in medium chain triglycerides for five days (days 3-7 of life) including the day screen-positive results for MCAD deficiency were returned. The genetics consult on day 7 of life ordered diagnostic biochemical genetic assessments (acylcarnitine profile and urine organic acids) that were returned the same day, noting highly atypical representation of potentially toxic medium chain fat metabolites in blood and urine. A worse outcome may have been realized had follow-up of the screen positive result been delayed, with continued ingestion of medium chain triglycerides. In this setting, it is prudent to implement expectant dietary therapy even before diagnostic confirmation as the risk of moving even a premature infant with MCAD deficiency to standard formula containing long chain fats is negligible.
MCAD deficiency is one of many screened disorders that require specialized dietary management that also includes phenylketonuria, galactosemia, organic acidemias, disorders of long chain fatty acid oxidation, and urea cycle disorders. The reported newborn received appropriate management regarding prematurity as neither clinical nor other findings could have suggested MCAD deficiency until NBS results were returned. Individually, screened disorders are rare but the combined total of 2-3/1000 live births is significant thus a busy tertiary care referral NICU is likely to encounter cases such as that described. As screened disorders are typically asymptomatic in the first few days of life, newborn screen results should be reviewed prior to prescribing a nutritional regimen for premature infants. In this regard, it is reasonable to build alerts into the electronic medical record to cross reference newborn screen results to nursery feeding orders to identify and alert clinicians to potentially contraindicated nutrients or supplements. Similar precautions are being suggested regarding pharmacogenetics issues primarily in adult patients. Modification of the premature infant’s diet should immediately follow a positive NBS, in spite of the possibility of a false positive result. Briefly placing a healthy infant on medical diet leads to little or no risk to the baby and can be relieved as appropriate when definitive testing has returned.
Conclusions
Survival of premature infants often requires intensive management. The premature birth of an infant affected with a rare inborn error of metabolism may lead to unanticipated life threatening complications brought on by standard management of prematurity. To avoid adverse outcomes during the interim between screen positive results and diagnostic evaluation, we suggest building tools into electronic medical records to alert neonatal intensive care units of a positive NBS result and cross check nutritional orders to determine if modification is necessary. Definitive diagnostic studies will ultimately determine if a NBS-positive infant is truly affected, but early modification of the nutritional regimen will serve to avoid the possibility of metabolic complications in the newborn.
References
- 1.Wilcken B, Wiley V. Newborn screening. Pathology 2008;40:104-15. [DOI] [PubMed] [Google Scholar]
- 2.Andresen BS, Dobrowolski SF, O’Reilly L, et al. Medium-chain acyl- CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am J Hum Genet 2001;68:1408-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iafolla AK, Thompson RJ Jr, Roe CR. Medium-chain acylcoenzyme A dehydrogenase deficiency: clinical course in 120 affected children. J Pediatr 1994;124:409-15. [DOI] [PubMed] [Google Scholar]
- 4.Oladipo OO, Weindel AL, Saunders AN, Dietzen DJ. Impact of premature birth and critical illness on neonatal range of plasma amino acid concentrations determined by LC-MS/MS. Mol Genet Metab 2011;104:476-9. [DOI] [PubMed] [Google Scholar]
- 5.Clark RH, Kelleher AS, Chace DH, Spitzer AR. Gestational age and age at sampling influence metabolic profiles in premature infants. Pediatrics 2014;134:e37-46. [DOI] [PubMed] [Google Scholar]
- 6.Ryckman KK, Berberich SL, Shchelochkov OA, et al. Clinical and environmental influences on metabolic biomarkers collected for newborn screening. Clin Biochem 2013;46:133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimozawa K, Matsumoto M, Okada K, et al. Analysis of blood spot 17 alpha-hydroxyprogesterone concentration in neonates. Horm Res 1988;30:246-51. [DOI] [PubMed] [Google Scholar]
- 8.Ditzenberger G. Nutritional support for premature infants in the neonatal intensive care unit. Crit Care Nurs Clin North Am 2014;26:181-98. [DOI] [PubMed] [Google Scholar]
- 9.Chen P, Zhang Y, Li LY. Current perspectives for management of acute respiratory insufficiency in premature infants with acute respiratory syndrome. Cell Biochem Biophys 2014;70:73-6. [DOI] [PubMed] [Google Scholar]
- 10.Bentler K, Zhai S, Elsbecker SA, et al. 221 newborn-screened neonates with medium-chain acyl-coenzyme A dehydrogenase deficiency: Findings from the Inborn Errors of Metabolism Collaborative. Mol Genet Metab 2016;119:75-82. [DOI] [PMC free article] [PubMed] [Google Scholar]