

Abstract
Elucidation of the folded structures that RNA forms in vivo is vital to understanding its functions. Chemical reagents that modify the Watson–Crick (WC) face of unprotected nucleobases are particularly useful in structure elucidation. Dimethyl sulfate penetrates cell membranes and informs on RNA base-pairing and secondary structure but only modifies the WC face of adenines and cytosines. We present glyoxal, methylglyoxal, and phenylglyoxal as potent in vivo reagents that target the WC face of guanines as well as cytosines and adenines. Tests on rice (Oryza sativa) 5.8S rRNA in vitro read out by reverse transcription and gel electrophoresis demonstrate specific modification of almost all guanines in a time- and pH-dependent manner. Subsequent in vivo tests on rice, a eukaryote, and Bacillus subtilis and Escherichia coli, Gram-positive and Gram-negative bacteria, respectively, showed that all three reagents enter living cells without prior membrane permeabilization or pH adjustment of the surrounding media and specifically modify solvent-exposed guanine, cytosine, and adenine residues.
Keywords: glyoxal, RNA structure, in vivo RNA probing
INTRODUCTION
Many essential biological processes require the presence of RNAs folded into complex structures. In some cases, such as translation (Noller et al. 1992; Yusupov et al. 2001), tRNA maturation by RNase P (Guerrier-Takada et al. 1983; Altman and Guerrier-Takada 1986; Evans et al. 2006), and self-splicing introns (Zaug and Cech 1986; Woodson 2005; Fedorova and Zingler 2007; Mitchell and Russell 2014), RNAs must attain a specific fold to perform functional steps. RNA structure also plays important roles in regulating gene expression, exemplified in bacteria by attenuation (Yanofsky 1981; Babitzke 1997; Naville and Gautheret 2010), ligand-binding riboswitches (Winkler et al. 2002; Peselis and Serganov 2014), and RNA thermometers that regulate translation (Altuvia et al. 1989; Kortmann et al. 2011; Barnwal et al. 2016). In eukaryotes, RNA structure allows miRNA maturation (Teixeira et al. 2004; West et al. 2004) and regulates mRNA turnover (Wan et al. 2012). These examples demonstrate the importance of studying RNA structure, particularly in vivo.
In vivo studies of RNA structure primarily use chemical reagents that permeate cells and covalently modify RNA nucleotides, with the RNA subsequently extracted and the locations of these modifications read out by reverse transcription, as is also typical with in vitro methods (Brunel and Romby 2000; Tijerina et al. 2007). SHAPE reagents have been used successfully to probe RNA structure in vivo (McGinnis et al. 2015; Smola et al. 2015; Lee et al. 2017). While modifying the ribose sugar may provide structural information (Wilkinson et al. 2005; Grohman et al. 2011; Smola et al. 2016), reagents that modify bases can directly inform on sequence-specific interactions such as base-pairing or protein interactions with bases. Among such reagents, dimethyl sulfate (DMS) has provided invaluable information. This reagent alkylates N1 of adenosines and N3 of cytidines (Peattie and Gilbert 1980; Schröder et al. 1998), and does not require a permeabilization agent to cross cell membranes (Ephrussi et al. 1985; Moazed et al. 1988; Zaug and Cech 1995; Méreau et al. 1997; Kwok et al. 2013; Ding et al. 2014; Talkish et al. 2014). Other base-modifying reagents such as kethoxal and 1-cyclohexyl-3-(2-morpholinoethyl)-carbodiimide methyl-p-toluenesulfonate (CMCT), while used extensively in vitro, are not amenable to in vivo work. They either require sonication or use of agents such as DMSO, high concentrations of CaCl2 and sodium borate to permeabilize cells—which may cause unintended cell damage—or require use in cell lysates that do not maintain biological context (Noller and Chaires 1972; Harris et al. 1995; Balzer and Wagner 1998; Antal et al. 2002; Incarnato et al. 2014). Therefore, currently only adenines and cytidines can be probed directly in vivo without needing to first damage the cell.
Here, we present evidence for glyoxal and glyoxal derivatives as chemical reagents that probe the Watson–Crick face of guanines, cytosines, and adenines in RNAs with high specificity, and within intact cells without the need for membrane permeabilization. Prior work has demonstrated specific glyoxal reactivity with free guanosine ribonucleotides and guanines within various RNAs in vitro, as well as weaker reaction with cytidines and adenosines (Staehelin 1959; Nakaya et al. 1968; Shapiro et al. 1969, 1970; Aubert et al. 1973; Carmichael and McMaster 1980; Grabowski et al. 1981). Similarly, methylglyoxal exhibits specific reactivity with poly(G) in vitro (Krymkiewicz 1973). Glyoxal and methylglyoxal were previously used to damage RNAs (Latham et al. 1990) rather than to probe them, and neither reagent had been previously identified as an in vivo RNA structure probe. Further, a third glyoxal derivative, phenylglyoxal, has not been evaluated regarding its propensity to modify RNA. All three of these glyoxals are readily available and inexpensive. We show that glyoxal, methylglyoxal, and phenylglyoxal can enter intact plant and Gram-positive and Gram-negative bacterial cells without previous disruption of either the cell wall or cell membrane and can covalently modify guanines at neutral pH, marking a novel use of these reagents as valuable in vivo RNA secondary structure probes.
RESULTS AND DISCUSSION
Prior to any in vivo examination of guanine modification by glyoxal and derivative reagents (hereafter termed glyoxylation), it is important to first assess their reactivity in an in vitro context. Here, we used primer extension and denaturing PAGE to analyze modification of rice (Oryza sativa) 5.8S rRNA in buffers ranging from pH 6 to 11 and at solution K+ and Mg2+ concentrations of 50 and 0.5 mM, respectively, mimicking typical cytoplasmic cation concentrations (Walker et al. 1996; Karley and White 2009; Maathuis 2009; Gout et al. 2014). Glyoxal, methylglyoxal, and phenylglyoxal each modified the majority of guanines at pH ≥8 within the examined region (G53 to G124), with high specificity for guanines (Fig. 1A–C; also see Supplemental Figs. S1–S6). Increasing reaction time or pH gave increased glyoxylation. No reliable data were obtained from G79, G101, and G120, as they abutted natural reverse transcriptase stops (observed in minus reagent control reactions), giving bands of near-equal intensity under all conditions and in sequencing lanes presumably due to reverse transcriptase dissociation at the preceding residue. At pH ≥8, N1 of guanine is more likely to be deprotonated, a prerequisite for the initial electrophilic attack of N1 on one of the aldehyde carbons of glyoxal and its derivatives (Fig. 2A; Nakaya et al. 1968). Glyoxal also reacted with three cytosines at pH 8 and 5 min reaction time (Fig. 1A), consistent with an early report that glyoxal reacts with cytosine and to a lesser extent adenine, in addition to guanine (Shapiro et al. 1970). Curiously, cytosine modifications disappeared upon increasing reaction time or pH, presumably due to hydroxide attack, offering a route to greater guanine specificity. Since adenine and cytosine each possesses an amidine moiety, N1 of adenine or N3 of cytosine (Fig. 2B), perhaps with elevated pKas or transiently made anionic by resonance, would allow glyoxal reactivity. The lack of an amidine moiety prevents glyoxal reactivity with uracil (Fig. 2C). Methylglyoxal and phenylglyoxal did not modify cytosines under any reaction condition (Fig. 1B,C), again offering greater specificity. A fourth reagent, dimethylglyoxal, was also examined but gave markedly reduced reactivity (Fig. 1D; also see Supplemental Figs. S7, S8).
FIGURE 1.

In vitro modification of rice 5.8S rRNA by glyoxal and derivatives analyzed by denaturing PAGE of cDNAs after reverse transcription. (A) Glyoxal reactions. Control reactions, reactions with 2.5 mM glyoxal, and dideoxy sequencing lanes are shown. Lanes 13, 22, and 26 were not loaded. Positions of all guanines within the examined range (G53 to G124) are marked on the sides of the gel with text, or between lanes 19 and 20 with asterisks, with red denoting glyoxalated guanines, violet denoting glyoxalated cytosines at pH 8 and 5 min time, and black denoting natural reverse transcriptase stops. (B) Methylglyoxal reactions. (C) Phenylglyoxal reactions. (D) Dimethylglyoxal reactions. In panels B–D, reactive guanines are shown in red text, unreactive guanines in black text, and natural reverse transcription stops in outlined text.
FIGURE 2.
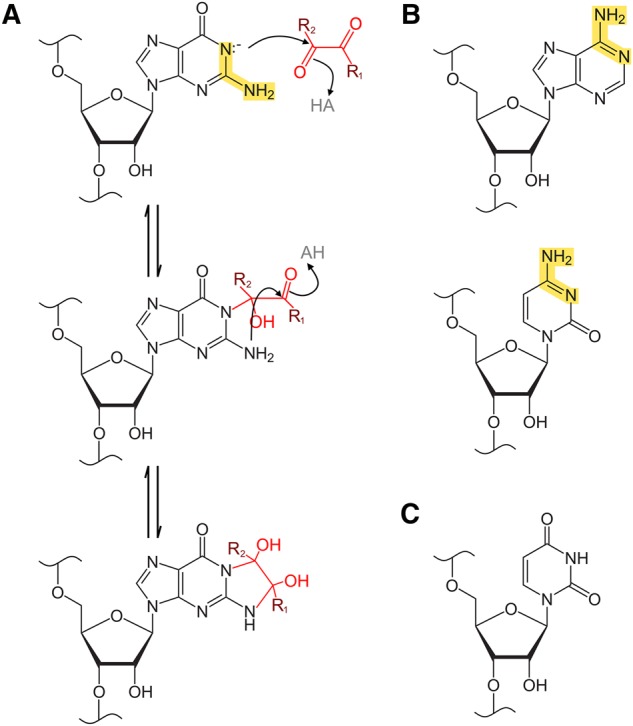
Reaction scheme for base glyoxalation by glyoxal and derivative reagents. (A) Reaction of a generalized glyoxal reagent with guanosine. R1 is H in glyoxal, CH3 in methylglyoxal, C6H5 in phenylglyoxal, and CH3 in dimethylglyoxal. R2 is H in glyoxal, methylglyoxal, and phenylglyoxal, and is CH3 in dimethylglyoxal. Highlighted in yellow is the amidine moiety of the guanine base, which is required for the glyoxalation reaction and is also present in adenine and cytosine bases but absent in uracil. In the first reaction step, deprotonated N1 attacks glyoxal, methylglyoxal, or phenylglyoxal at the more electronegative formyl carbon. In the following step, N2 then attacks at the keto carbon, leading to formation of a cyclic adduct between N1 and N2. In the case of dimethylglyoxal reactions, both carbons are keto carbons, but the generalized scheme is the same. (B) Adenine and cytosine also possess an amidine moiety, highlighted in yellow. (C) Uracil lacks an amidine moiety, which prevents its reactivity with glyoxal or similar reagents.
Having established specific modification of guanines by glyoxal, methylglyoxal, and phenylglyoxal in vitro, we then exposed rice tissue to glyoxal to examine its efficacy to probe RNA structure within living cells. We incubated excised shoots of 2-wk-old rice seedlings for 15 min in buffers containing 50 mM K+, 0.5 mM Mg2+, and 25 mM glyoxal. On the basis of our above in vitro studies, we reasoned that buffers of pH 8 and pH 9.2 would favor glyoxal reaction. Buffers of pH >9.2 were not tested due to excessive RNA degradation observed for 15 min reactions performed in vitro (Fig. 1A). In vivo, moderate glyoxalation was observed at G99 and weak glyoxalation was observed at G82 and G89 at both pHs (Fig. 3A). All three guanines are located in known loop regions of 5.8S rRNA (Fig. 3B; Cannone et al. 2002; Schmidt et al. 2016). While much of G99 is buried between ribosomal protein and 28S rRNA, its N1 and N2 atoms are solvent exposed and available to attack glyoxal (Fig. 4A). In both G82 and G89, N1 and N2 are partially buried by ribosomal protein (Fig. 4A) but may become transiently solvent accessible owing to breathing of the protein–RNA interface, resulting in weak glyoxalation. In that same vein, our data indicate that base-pairing or ribosomal proteins protect most guanines in 5.8S rRNA (e.g., G69, G72, G74, or G91) from glyoxal modification. Thus, glyoxal reveals both RNA base-pairing and protein footprinting in vivo.
FIGURE 3.

Glyoxal, methylglyoxal, and phenylglyoxal modification of rice 5.8S rRNA in vivo. (A) Denaturing PAGE analysis of cDNAs generated after reverse transcription. PAGE with glyoxal and methylglyoxal was run on a separate gel from phenylglyoxal and is indicated by gray brackets. Reaction conditions at buffer pH 8 or pH 9.2 for no reagent (NR), glyoxal (GX), methylglyoxal (MG), and phenylglyoxal (PG) are shown, along with dideoxy sequencing lanes. Red text indicates reactive guanines, and outlined text indicates natural reverse transcriptase stops. (B) Reactive guanines mapped as red discs onto the relevant portion of rice 5.8S rRNA comparative structure. (C) Fold change of methylglyoxal and phenylglyoxal reactivity at G82, G89, and G99 relative to glyoxal reactivity at these same residues.
FIGURE 4.
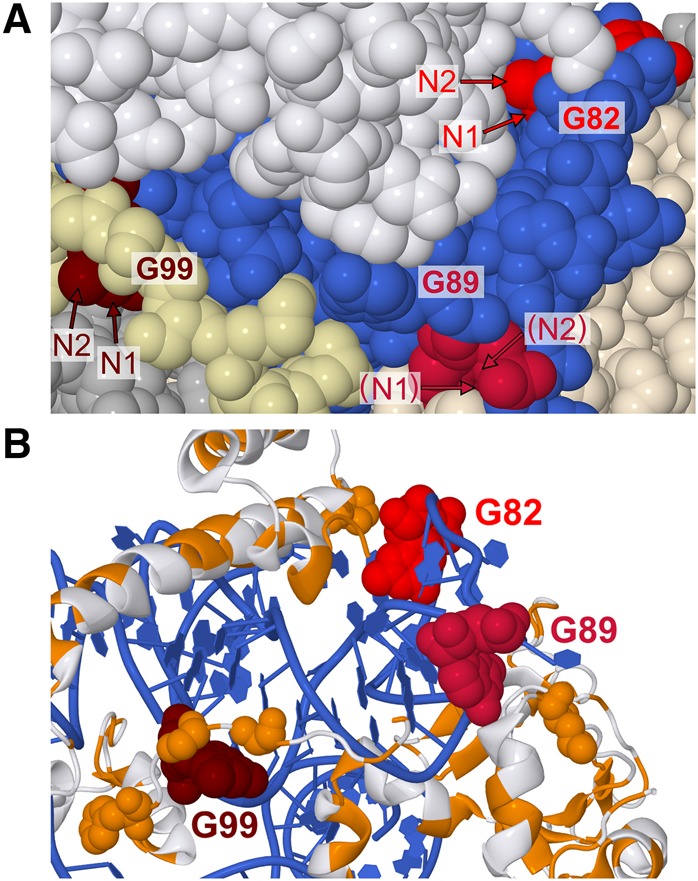
Cryo-EM structure of Saccharomyces cerevisiae 60S subunit (PDB: 5GAK), a homolog of rice 60S subunit used here as no rice ribosome structure currently exists. (A) Relative orientations of N1 and N2, the atoms responsible for glyoxal reactivity. 5.8S rRNA is blue, G82 is scarlet, G89 is crimson, and G99 is maroon. 28S rRNA and other RNAs are silver. Ribosomal proteins are white, except for two ribosomal proteins in the immediate vicinity of G89 and G99, which are tan (L26-A) and pale yellow (L37-A). Arrows indicate N1 and N2 atoms, with parentheses indicating N1 or N2 obscured by other atoms. (B) Proximity of G82, G89, and G99 to hydrophobic protein residues. Only 5.8S rRNA and ribosomal proteins L26-A, L35-A, L37-A, are shown. Hydrophobic residues are orange and nonhydrophobic residues are white while G82, G89, and G99 are colored as in panels A and B. Hydrophobic residues within 10 Å of N1 or N2 atoms in reactive guanines are shown as spheres.
The above in vivo experiments were performed with glyoxal. We then performed the same in vivo structure-probing assay with 25 mM methylglyoxal and 25 mM phenylglyoxal with the idea that they would react with guanines but might exhibit improved cell penetration owing to their greater hydrophobicity. Similar to glyoxal, both methylglyoxal and phenylglyoxal modified G82, G89, and G99 (Fig. 3A), but strikingly, the extent of modification at all three guanines increased with methylglyoxal and even more so with phenylglyoxal (Fig. 3C). Glyoxalation of G99 increased 3.8-fold with methylglyoxal and 3.3-fold with phenylglyoxal relative to modification by glyoxal. Furthermore, G82 and G89 glyoxalation increased with methylglyoxal by 1.5-fold for G82 and fourfold for G89, and with phenylglyoxal by twofold for G82 and 5.7-fold for G89. As anticipated, increased hydrophobicity conferred by the methyl and phenyl groups may enhance the ability of methylglyoxal and phenylglyoxal to traverse the cell membrane. Another putative explanation for greater reactivity of methylglyoxal and phenylglyoxal is interaction between the hydrophobic moieties of these reagents and hydrophobic residues of nearby ribosomal proteins, providing a platform for holding in place the electrophilic glyoxal carbon. The N1 atoms of every reactive guanine are within 10 Å of a hydrophobic amino acid (Fig. 4B). Further, the larger bulk of phenylglyoxal may enable it to better position a carbonyl near N1 of guanine relative to methylglyoxal.
Excessively alkaline intracellular conditions can denature RNA structure by deprotonating atoms involved in hydrogen bonding, preventing accurate probing of native in vivo RNA structures. As such, we tested whether glyoxal and its derivatives can glyoxalate guanines within cells at neutral external buffer pH by performing the in vivo structure-probing assay in pH 7 buffer. Remarkably, all three glyoxal reagents glyoxalated G82, G89, and G99 in the pH 7 buffer, with each reagent giving almost identical modification as in pH 9.2 external buffer (Fig. 5). As the aforementioned tests indicated that pH ≥8 is required for reactivity by glyoxal and derivatives in vitro, this result implies that N1 of guanine is at least partially deprotonated in vivo by means of a shifted pKa, possibly via electrostatic interactions with nearby protein residues or cations.
FIGURE 5.
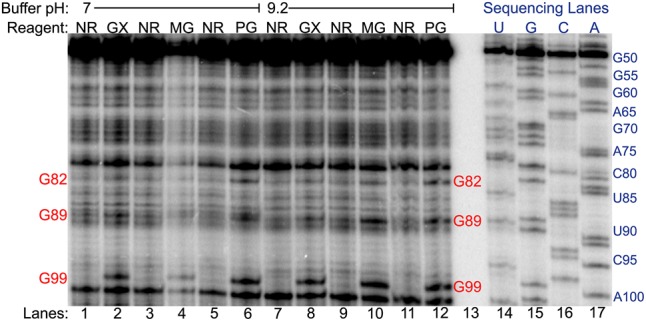
Glyoxal, methylglyoxal, and phenylglyoxal modification of rice 5.8S rRNA in vivo analyzed by denaturing PAGE of cDNAs generated by reverse transcription. Reaction conditions at buffer pH 7 or pH 9.2 for no reagent (NR), glyoxal (GX), methylglyoxal (MG), and phenylglyoxal (PG) are shown, along with dideoxy sequencing lanes. Red text indicates reactive guanines.
To determine the universal applicability of glyoxal and derivative reagents as structural probes, we extended our in vivo structure-probing assays to another domain of life. We used two different bacterial species, the Gram-positive Bacillus subtilis strain PLBS338 (Yakhnin et al. 2004) and Gram-negative Escherichia coli strain MG1655, and probed 16S and 23S rRNAs in B. subtilis and 5S rRNA in both species. Glyoxal modified no nucleotides within the examined range of B. subtilis 5S rRNA (Supplemental Fig. S9). However, glyoxal modified three guanines (G74, G85, and G91) and one adenine (A75) in B. subtilis 16S rRNA (Fig. 6A). In an effort to understand the reactivity of these residues, we examined the cryo-EM structure of 16S rRNA from B. subtilis. All glyoxalated nucleotides in 16S rRNA are located in helix 6, in a stretch of three consecutive non-Watson–Crick base pairs (Fig. 6B), which is expected to be unstable and thus increase the probability of solvent exposure and subsequent glyoxal reactivity. Examination of the B. subtilis 70S ribosome cryo-EM structure (Sohmen et al. 2015) shows that the sugar edge of G91 potentially forms a sheared base pair with the Hoogsteen face of A75, an interaction that would increase the pKa of A75 enough for its N1 to react with glyoxal (Fig. 7A). Glyoxal also modified three guanines (G63, G92, and G142) and one adenine (A91) in B. subtilis 23S rRNA, though we only examined here the portion of 23S rRNA similar in structure to eukaryotic 5.8S rRNA (Fig. 6C). All four modified nucleotides are located in loops (Fig. 6D), and the cryo-EM structure shows they are situated on the exterior of the ribosome (Fig. 7B). Interestingly, A91 and G92 are relatively more solvent exposed than G63, yet both residues give fourfold lower reactivity compared to G63. On the other end of the examined range of nucleotides, G142 is located within 6 Å of residues in the ribosomal protein L23, which may account for its reduced reactivity when compared to G63.
FIGURE 6.

In vivo glyoxal modification of B. subtilis and E. coli rRNAs. (A) Denaturing PAGE analysis of cDNAs generated after reverse transcription (RT) for B. subtilis 16S rRNA. Shown are reactions in glyoxal from 0 to 100 mM. Reactive nucleotides are shown in red text. (B) Reactive nucleotides mapped as red discs onto the relevant portion of B. subtilis 16S rRNA comparative secondary structure. Portions of the structure not relevant to this study were removed and replaced with dotted lines. (C) Denaturing PAGE after RT for B. subtilis 23S rRNA. Shown are reactions in glyoxal from 0 mM to 100 mM and dideoxy sequencing lanes. Reactive nucleotides are shown in red text. (D) Reactive nucleotides mapped as red discs onto the relevant portion of B. subtilis 23S rRNA comparative secondary structure. (E) Denaturing PAGE after RT for E. coli 5S rRNA. Shown are reactions in glyoxal from 0 to 120 mM. Reactive nucleotides are shown in red text. (F) Reactive nucleotides mapped as orange and red discs onto the E. coli 5S rRNA comparative secondary structure. Orange discs indicate moderate glyoxalation of a given nucleotide while red discs indicate strong glyoxalation at that nucleotide.
FIGURE 7.

Cryo-EM structures of B. subtilis strain 168 70S ribosome (PDB: 3J9W) and E. coli 70S ribosome (PDB: 5UYL) to show glyoxal-reactive nucleotides. (A) B. subtilis 16S rRNA is shown as blue sticks, with glyoxalated nucleotides colored red. Proteins are in cartoon form and are colored yellow. (B) B. subtilis 23S rRNA is shown in cartoon form and is colored pale blue, except for nucleotides within the examined range, which are shown as sticks and are colored darker blue. Reactive nucleotides are colored red. Proteins are shown in cartoon form and are colored yellow. (C) E. coli 5S rRNA. All RNAs are shown as space-filling spheres, with 5S rRNA in blue and 23S rRNA in gray. Proteins are in cartoon form and are colored yellow. G13 and G41 are red, G81 is magenta, all reactive Cs (C19, C35–C38, and C47) are orange, and A52 is olive.
Glyoxal modifications within E. coli 5S rRNA revealed reactivity at guanine, cytosine, and adenine (Fig. 6E). It modified a stretch of four consecutive cytosines (C35–C38), as well as three guanines (G13, G41, and G81), two other cytosines (C19 and C47), and one adenine (A52). The two most intensely glyoxalated guanines, G13 and G41, are located in Loop A and in Loop C, respectively (Fig. 6F). Residue G81, while modified to a lesser extent relative to the other two guanines, forms a G•U wobble pair with U95 and neighbors another G•U wobble in U80•G96. Five of the six modified cytosines, C35–C38 and C47, are located in the base of Loop C and give 10- to 20-fold lower modification relative to G41. The E. coli 70S cryo-EM structure (Loveland et al. 2017) shows that these cytosines are all unpaired but form stacking interactions with nearby nucleotides, with C35–C36 and C37–C38 each forming a two-residue stack on the exterior of the ribosome surface (Fig. 7C). The weakly reactive A52 is located within a 2-nt bulge of Helix III and stacks atop G51, and transient disruption of this stacking interaction may allow reactivity. The decreased reactivity of cytosine and adenine residues may be due to preferential glyoxal modification of guanines relative to adenines and cytosines (Shapiro et al. 1970; Krymkiewicz 1973). Overall, these results demonstrate the extensive applicability of glyoxal as an RNA structural probe in vivo.
In summary, we present the novel use of glyoxal, methylglyoxal, and phenylglyoxal to provide highly specific, molecular-level probing of the Watson–Crick face of unpaired guanines, and to a lesser extent cytosines and adenines, within structured RNAs in intact cells at neutral pH. These glyoxal reagents are sensitive to base-pairing and protein binding. Specificity for guanine, if desired, can be enhanced by working with methylglyoxal or phenylglyoxal, or extending time. As current in vivo methods to probe the Watson–Crick face of RNA bases rely on DMS, which targets only adenines and cytosines, the availability of glyoxal and its derivatives as guanine-targeting reagents allows for more accurate and extensive elucidation of RNA structures. These base-reactive reagents, together with sugar-reactive SHAPE reagents, will be useful for examining RNA structures in a chemically comprehensive fashion. Glyoxals in particular should facilitate in vivo genome-wide, high-throughput methods to examine RNA structures, including G-quadruplexes (Kwok and Balasubramanian 2015; Guo and Bartel 2016), and their dynamic modulation by changing cellular conditions.
MATERIALS AND METHODS
Plant materials and growth conditions
Standard 100 mm × 15 mm or 150 mm × 15 mm petri dishes were inverted and the lids (now on the bottom) were lined with filter paper (VWR) prior to the addition of ∼30–40 Oryza sativa seeds per 100 mm dish or ∼50–60 seeds per 150 mm dish. Approximately 100 mL water was added and the seeds were covered with the bottom of the dish. The seeds were incubated in a 30°C–37°C greenhouse under light of intensity ∼500 µmol photons m−2 s−1 supplied by natural daylight supplemented with 1000 W metal halide lamps (Philips Lighting Co.) for 7–8 d. Seedlings then were transferred to premoistened Sunshine LC1 RSi potting soil (SunGro Horticulture) in 15-cm-tall pots so that the seeds were ∼1 cm below the soil surface and the radicle or roots were completely buried within the soil. Water was added to an underlying plastic tray to ∼6 cm depth and the level allowed to drop during the course of the growth incubation. Note that excessive watering of the seedlings can inhibit growth. A spoonful (∼0.5–1 g) of Sprint 330 powdered iron chelate (BASF) was added to the water to prevent iron deficiency within the seedlings. The seedlings were illuminated with ∼500 µmol photons m−2 s−1 light intensity as above for another 7–8 d until attaining a height of ∼8–12 cm.
B. subtilis growth conditions
A single colony from B. subtilis strain PLBS338 (Yakhnin et al. 2004) plated on LB agar was inoculated in liquid tryptophan-supplemented ACH growth media [0.5% glucose, 0.2% acid casein hydroxylate, 0.01% L-tryptophan, 17 mM K2HPO4, 8 mM KH2PO4, 3 mM (NH4)2SO4, 1.2 mM Na3C6H5O7·2H2O, 0.16 mM MgSO4] and incubated in a standing culture overnight at 37°C. The overnight culture was diluted 1:50 into 45 mL of fresh tryptophan-supplemented ACH media and incubated in a shaking water bath at 37°C until attaining a Klett value of 75 (mid-exponential growth phase).
E. coli growth conditions
A single colony from E. coli (strain MG1655) plated on LB agar was inoculated in liquid LB media and incubated overnight at 37°C. The overnight culture was diluted 1:50 into 6 mL of fresh LB media and incubated at 37°C until attaining an OD600 of 1 (mid-exponential growth phase).
Total RNA extraction from rice
Rice seedlings grown for 14–16 d total were cut ∼5–10 mm from the soil line, quickly frozen in liquid nitrogen, and ground to fine powder using a mortar and pestle precleaned with RNase Zap (Ambion). In an Eppendorf tube, 80–100 mg of powder was added to 350 mL of lysis buffer (Macherey-Nagel) and 35 mL of 500 mM dithiothreitol (DTT), then centrifuged for 1 min at >11,000 rpm. This lysis buffer is at pH 6.5, which helps stabilize the glyoxal adduct (Carmichael and McMaster 1980). The supernatant then was subjected to total RNA extraction following the protocol described in the NucleoSpin RNA Plant kit (Macherey-Nagel).
In vitro glyoxal probing of rice RNA
All reactions involving glyoxal were performed in a chemical fume hood. Reaction buffer was added to 1 µg total extracted rice seedling RNA to give a final total volume of 9 µL containing 50 mM pH buffer (MES for pH 6, HEPES for pH 7–8, CHES for pH 9.2–10, or CAPS for pH 11), 50 mM KCl, and 0.5 mM MgCl2. The reaction was mixed thoroughly and incubated at room temperature for 15 min to allow equilibration. Glyoxal (40% v/v in water, giving 8.8 M) stock solution (Sigma-Aldrich; 50649-100ML) was diluted to 25 mM in deionized water and added to the reaction mixture to a final concentration of 2.5 mM in a final reaction volume of 10 µL. In the control (–glyoxal) treatment, an equivalent volume of deionized water was added to the reaction mixture in place of glyoxal. Glyoxal and control reactions proceeded for 5 min or 15 min at room temperature (∼22°C) before being quenched by the addition of 3 µL of 1 M sodium acetate (pH 6), 0.5 µL glycogen, and 25 µL 95% ethanol, followed immediately by freezing on dry ice for 1 h and subsequent ethanol precipitation of the RNA.
In vitro methylglyoxal probing of rice RNA
All reactions involving methylglyoxal were performed in a chemical fume hood. Reaction buffer was added to 1 µg total extracted rice seedling RNA to give a final total volume of 9 µL containing 50 mM pH buffer (HEPES for pH 7–8 or CHES for pH 9.2–10), 50 mM KCl, and 0.5 mM MgCl2. The reaction was mixed thoroughly and incubated at room temperature for 15 min to allow equilibration. Methylglyoxal (40% v/v in water, giving 8.8 M) stock solution (Sigma-Aldrich; M0252-25ML) was diluted to 25 mM in deionized water and added to the reaction mixture to a final concentration of 2.5 mM in a final reaction volume of 10 µL. In a control (–methylglyoxal) treatment, an equivalent volume of deionized water was added to the reaction mixture in place of methylglyoxal. Methylglyoxal and control reactions proceeded for 15 min at room temperature before being quenched by the addition of 3 µL of 1 M sodium acetate (pH 6), 0.5 µL glycogen, and 25 µL 95% ethanol, followed immediately by freezing on dry ice for 1 h and subsequent ethanol precipitation of the RNA.
In vitro phenylglyoxal probing of rice RNA
All reactions involving phenylglyoxal were performed in a chemical fume hood. Reaction buffer was added to 1 µg total extracted rice seedling RNA to give a final total volume of 9 µL containing 50 mM pH buffer (HEPES for pH 7–8 or CHES for pH 9.2–10), 50 mM KCl, and 0.5 mM MgCl2. The reaction was mixed thoroughly and incubated at room temperature for 15 min to allow equilibration. Phenylglyoxal stock (Alfa Aesar; A10677-06) was dissolved in dimethyl sulfoxide (DMSO) to 1 M concentration, further serial diluted in DMSO to a concentration of 25 mM, and added to the reaction mixture to a final concentration of 2.5 mM in a final reaction volume of 10 µL. In a control (–phenylglyoxal) treatment, an equivalent volume of DMSO was added to the reaction mixture in place of phenylglyoxal. Phenylglyoxal and control reactions proceeded for 15 min at room temperature before being quenched by the addition of 3 µL of 1 M sodium acetate (pH 5.9), 0.5 µL glycogen, and 25 µL 95% ethanol, followed immediately by freezing on dry ice for 1 h and subsequent ethanol precipitation of the RNA.
In vitro dimethylglyoxal probing of rice RNA
All reactions involving dimethylglyoxal were performed in a chemical fume hood. Reaction buffer was added to 1 µg total extracted rice seedling RNA to give a final total volume of 9 µL containing 50 mM pH buffer (HEPES for pH 7–8 or CHES for pH 9.2–10), 50 mM KCl, and 0.5 mM MgCl2. The reaction was mixed thoroughly and incubated at room temperature for 15 min to allow equilibration. Dimethylglyoxal (2,3-butanedione; 40% v/v in water, giving 8.8 M) stock solution (Sigma-Aldrich; B85307-5ML) was diluted to 25 mM in deionized water and added to the reaction mixture to a final concentration of 2.5 mM in a final reaction volume of 10 µL. In a control (–dimethylglyoxal) treatment, an equivalent volume of deionized water was added to the reaction mixture in place of dimethylglyoxal. Dimethylglyoxal and control reactions proceeded for 15 min at room temperature before being quenched by the addition of 3 µL of 1 M sodium acetate (pH 6), 0.5 µL glycogen, and 25 µL 95% ethanol, followed immediately by freezing on dry ice for 1 h and subsequent ethanol precipitation of the RNA.
In vivo glyoxal probing of rice
All reactions involving glyoxal were performed in a chemical fume hood. Rice seedlings grown for 14–16 d as described above were cut 5–10 mm above the soil line. For reactions in 25 mM glyoxal, 4–6 cut seedlings were placed in a 50 mL Falcon tube that contained 51.3 mM buffer (HEPES [pH 7] HEPES [pH 8] or CHES [pH 9.2]), 51.3 mM KCl, and 0.513 mM MgCl2 in a total volume of 9.75 mL. Next, 0.25 mL of glyoxal diluted to 1 M in deionized water was added to the reaction mixture to a final total volume of 10 mL containing 50 mM pH buffer, 50 mM KCl, 0.5 mM MgCl2, and 25 mM glyoxal. In control (–glyoxal) reactions, equivalent volumes of deionized water were added in place of glyoxal. For all experimental and control conditions, the reactions occurred for 15 min at room temperature with periodic shaking and swirling. The reaction buffer was decanted and the seedlings were washed six times with ∼20 mL deionized water before immediate drying and freezing in liquid N2. Frozen seedlings then were subjected to total RNA extraction as described above, with separate mortars and pestles used for each treatment.
In vivo methylglyoxal probing of rice
All procedures for in vivo methylglyoxal probing were identical to those for in vivo glyoxal probing, except that only 25 mM methylglyoxal was used.
In vivo phenylglyoxal probing of rice
All procedures for in vivo phenylglyoxal probing were identical to those for in vivo glyoxal probing, except that only 25 mM phenylglyoxal was used and anhydrous DMSO (Sigma-Aldrich) was used for the control in place of deionized water.
In vivo glyoxal probing of B. subtilis
All reactions involving glyoxal were performed in a chemical fume hood. B. subtilis grown to 75 Klett in tryptophan-supplemented ACH growth media as described above was dispensed into smaller flasks each containing 6 mL of culture. Glyoxal (40% v/v in water, giving 8.8 M) and DMSO were mixed together to give final concentrations of glyoxal ranging from 5 to 100 mM in 10% DMSO. The reactions occurred for 5 min at 37°C with continuous shaking. The reaction was stopped and cell growth was arrested by quenching in 6 mL of frozen ice crash buffer (10 mM Tris–HCl [pH 7.2], 5 mM MgCl2, 25 mM NaN3, 1.5 mM chloramphenicol, and 12.5% ethanol). After spinning down at 10,000 rpm for 10 min to pellet the cells, the liquid was decanted and the cells were washed two additional times with ice crash buffer (spinning down each time) before cooling on ice. Glyoxal-reacted B. subtilis cells then were subjected to total RNA extraction following the protocol described in the RNeasy Mini kit (QIAGEN).
In vivo glyoxal probing of E. coli
All reactions involving glyoxal were performed in a chemical fume hood. Glyoxal diluted to 1 M in deionized water was added to the LB media containing E. coli grown as described above to a final volume of 6 mL containing 10 mM to 120 mM glyoxal. The reactions occurred for 5 min at 37°C with continuous shaking, at which time the cells were pelleted by centrifugation. The LB media was decanted and the cells were washed three times with buffer containing 10 mM Tris–HCl (pH 7.2), 5 mM MgCl2, 25 mM NaN3, 1.5 mM chloramphenicol, and 12.5% ethanol before cooling on ice. Glyoxal-reacted E. coli cells then were subjected to total RNA extraction following the protocol described in the RNeasy Mini kit (QIAGEN).
Gene-specific reverse transcription for rice
In vitro or in vivo rice total RNA was extracted as described above and combined with RNase-free water to give 0.5–1 µg RNA in 5.5 µL total volume. Then, 1 µL of ∼500,000 cpm/µL 32P-radiolabeled primer targeting 5.8S rRNA (5′-GCGTGACGCCCAGGCA-3′) was added to the mixture. The solution was incubated at 75°C for 3 min, then cooled to 35°C whereupon 3 µL of reverse transcription reaction buffer was added to a final concentration of 20 mM Tris–HCl (pH 7.5), 1 mM DTT, 100 mM KCl, 8 mM MgCl2, and 1 mM dNTPs. Annealing was allowed to proceed at 35°C for 5 min, after which the solution was heated to 55°C for 1 min, 0.5 µL of Superscript III reverse transcriptase (Invitrogen; 100 U total) was added to the reaction, and the reverse transcription reaction was allowed to proceed at 55°C for 15 min. Next, 1 µL of 5 M NaOH was added to the solution, which was heated to 95°C for 7 min and then cooled to 4°C for ≥3 min to hydrolyze all RNAs and denature reverse transcriptase. Finally, 1 µL of 5 M HCl was added to neutralize the solution, and an equal volume of 2× stop solution was added which contained 100% deionized formamide, 20 mM Tris–HCl (pH 7.5), 40 mM EDTA, 0.1% xylene cyanol and 0.025% bromophenol blue. The mixture was then loaded onto a 10% denaturing polyacrylamide gel (8.3 M urea) and run at 80 W for ∼2 h. The resulting data then were analyzed using semi-automated footprinting analysis software (SAFA) (Das et al. 2005).
Gene-specific reverse transcription for B. subtilis
In vivo B. subtilis total RNA extracted as described above was combined with RNase-free water to give 0.5–1 µg RNA in 5.5 µL total volume. Then, 1 µL of ∼250,000 cpm/µL 32P-radiolabeled primer targeting 5S rRNA (5′-GCTTGGCGGCGTCCTACTCTC-3′), 16S rRNA (5′-CCCGGAGTTATCCCAGTCTTACAGGCAGG-3′), or 23S rRNA (5′-CTGCCTTCTCATATCCTATGAATTCAGATATGG-3′) was added to the mixture in addition to 1 µL of 10× reverse transcription reaction buffer containing 200 mM Tris–HCl (pH 7.5) and 500 mM KCl. The solution was incubated at 95°C for 1 min, then cooled to 35°C whereupon 1 µL of 80 mM MgCl2, 1 µL of 100 mM DTT, and 1 µL of 10 mM dNTPs were added to the solution to a final concentration of 20 mM Tris–HCl (pH 7.5), 1 mM DTT, 50 mM KCl, 8 mM MgCl2, and 1 mM dNTPs. Annealing was allowed to proceed at 35°C for 5 min, after which the solution was heated to 55°C for 1 min, 0.5 µL of Superscript III reverse transcriptase (Invitrogen; 100 U total) was added to the reaction, and the reverse transcription reaction was allowed to proceed at 55°C for 15 min. Next, 0.5 µL of 2 M NaOH was added to the solution, which was heated to 95°C for 5 min and then cooled to 4°C for ≥3 min to hydrolyze all RNAs and denature reverse transcriptase. Finally, an equal volume of 2× stop solution was added, which contained 100% deionized formamide, 20 mM Tris–HCl, 40 mM EDTA, 0.1% xylene cyanol and 0.025% bromophenol blue. The mixture was then loaded onto a 10% denaturing polyacrylamide gel (8.3 M urea) and run at 80 W for ∼2 h. The resulting data then were analyzed using semi-automated footprinting analysis software (SAFA) (Das et al. 2005).
Gene-specific reverse transcription for E. coli
In vivo E. coli total RNA extracted as described above was combined with 5× First Strand buffer (Invitrogen) and RNase-free water to give 0.5–1 µg RNA in 4.5 µL total volume. Then, 1 µL of ∼500,000 cpm/µL 32P-radiolabeled primer targeting 5S rRNA (5′-ATGCCTGGCAGTTCCCTACTCTC-3′) was added to the mixture. The solution was incubated at 80°C for 1 min, then quick-cooled on ice whereupon 2 µL of reverse transcription reaction buffer was added to the solution to a final concentration of 20 mM Tris–HCl (pH 7.5), 15 mM DTT, 5 mM MgCl2, and 2 mM dNTPs. The solution was heated to 50°C for 1 min, 0.5 µL of Superscript III reverse transcriptase (Invitrogen; 100 U total) was added to the reaction, and the reverse transcription reaction was allowed to proceed at 50°C for 30 min. Next, 1 µL of 1 M NaOH was added to the solution, which was heated to 95°C for 10 min and then cooled to 4°C for ≥3 min to hydrolyze all RNAs and denature reverse transcriptase. Finally, an equal volume of 2× stop solution was added, which contained 100% deionized formamide, 20 mM Tris–HCl, 40 mM EDTA, 0.1% xylene cyanol and 0.025% bromophenol blue. The mixture was then loaded onto a 10% denaturing polyacrylamide gel (8.3 M urea) and run at 80 W for ∼2 h. The resulting data then were analyzed using semi-automated footprinting analysis software (SAFA) (Das et al. 2005).
Calculation of significant glyoxalation
In all plots constructed from SAFA results, significant glyoxalation was calculated in the following manner. The background-corrected band intensity for all residues within the examined nucleotide range, except for guanosine residues and the largest and smallest values for each reaction condition, were averaged and their standard deviation was calculated. For in vitro glyoxal reactions at buffer pH 8 and pH 9.2, C95, C103, and C112 (see Supplemental Fig. S1) were also excluded from this total. Next, the value for significant glyoxalation (S) for a number of reaction conditions n was calculated as the grand average of the averages (Ai) plus three times the standard deviation for each reaction condition (σi), as shown below:
Significant glyoxalation was defined as bands with intensity greater than S. Here, as most reaction conditions give bands of light intensity even in the absence of modification by a reagent, three standard deviations from the mean ensures sufficient separation between such background bands and true bands caused by modified nucleotides.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation Plant Genome Research Program under NSF-IOS-1612170 and NSF-IOS-1339282, and by the Charles E. Kaufman Foundation of the Pittsburgh Foundation under KA2016-85222.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.064014.117.
REFERENCES
- Altman S, Guerrier-Takada C. 1986. M1 RNA, the RNA subunit of Escherichia coli ribonuclease P, can undergo a pH-sensitive conformational change. Biochemistry 25: 1205–1208. [DOI] [PubMed] [Google Scholar]
- Altuvia S, Kornitzer D, Teff D, Oppenheim AB. 1989. Alternative mRNA structures of the cIII gene of bacteriophage λ determine the rate of its translation initiation. J Mol Biol 210: 265–280. [DOI] [PubMed] [Google Scholar]
- Antal M, Boros E, Solymosy F, Kiss T. 2002. Analysis of the structure of human telomerase RNA in vivo. Nucleic Acids Res 30: 912–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert M, Bellemare G, Monier R. 1973. Selective reaction of glyoxal with guanine residues in native and denatured Escherichia coli 5S RNA. Biochimie 55: 135–142. [DOI] [PubMed] [Google Scholar]
- Babitzke P. 1997. Regulation of tryptophan biosynthesis: Trp-ing the TRAP or how Bacillus subtilis reinvented the wheel. Mol Microbiol 26: 1–9. [DOI] [PubMed] [Google Scholar]
- Balzer M, Wagner R. 1998. A chemical modification method for the structural analysis of RNA and RNA-protein complexes within living cells. Anal Biochem 256: 240–242. [DOI] [PubMed] [Google Scholar]
- Barnwal RP, Loh E, Godin KS, Yip J, Lavender H, Tang CM, Varani G. 2016. Structure and mechanism of a molecular rheostat, an RNA thermometer that modulates immune evasion by Neisseria meningitidis. Nucleic Acids Res 44: 9426–9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunel C, Romby P. 2000. Probing RNA structure and RNA-ligand complexes with chemical probes. Methods Enzymol 318: 3–21. [DOI] [PubMed] [Google Scholar]
- Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Müller KM, et al. 2002. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 3: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael GG, McMaster GK. 1980. The analysis of nucleic acids in gels using glyoxal and acridine orange. Methods Enzymol 65: 380–391. [DOI] [PubMed] [Google Scholar]
- Das R, Laederach A, Pearlman SM, Herschlag D, Altman RB. 2005. SAFA: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA 11: 344–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, Assmann SM. 2014. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505: 696–700. [DOI] [PubMed] [Google Scholar]
- Ephrussi A, Church GM, Tonegawa S, Gilbert W. 1985. B lineage–specific interactions of an immunoglobulin enhancer with cellular factors in vivo. Science 227: 134–140. [DOI] [PubMed] [Google Scholar]
- Evans D, Marquez SM, Pace NR. 2006. RNase P: interface of the RNA and protein worlds. Trends Biochem Sci 31: 333–341. [DOI] [PubMed] [Google Scholar]
- Fedorova O, Zingler N. 2007. Group II introns: structure, folding and splicing mechanism. Biol Chem 388: 665–678. [DOI] [PubMed] [Google Scholar]
- Gout E, Rébeillé F, Douce R, Bligny R. 2014. Interplay of Mg2+, ADP, and ATP in the cytosol and mitochondria: unravelling the role of Mg2+ in cell respiration. Proc Natl Acad Sci 111: E4560–E4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski PJ, Zaug AJ, Cech TR. 1981. The intervening sequence of the ribosomal RNA precursor is converted to a circular RNA in isolated nuclei of tetrahymena. Cell 23: 467–476. [DOI] [PubMed] [Google Scholar]
- Grohman JK, Kottegoda S, Gorelick RJ, Allbritton NL, Weeks KM. 2011. Femtomole SHAPE reveals regulatory structures in the authentic XMRV RNA genome. J Am Chem Soc 133: 20326–20334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. 1983. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35: 849–857. [DOI] [PubMed] [Google Scholar]
- Guo JU, Bartel DP. 2016. RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science 353: aaf5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KA Jr, Crothers DM, Ullu E. 1995. In vivo structural analysis of spliced leader RNAs in Trypanosoma brucei and Leptomonas collosoma: a flexible structure that is independent of cap4 methylations. RNA 1: 351–362. [PMC free article] [PubMed] [Google Scholar]
- Incarnato D, Neri F, Anselmi F, Oliviero S. 2014. Genome-wide profiling of mouse RNA secondary structures reveals key features of the mammalian transcriptome. Genome Biol 15: 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karley AJ, White PJ. 2009. Moving cationic minerals to edible tissues: potassium, magnesium, calcium. Curr Opin Plant Biol 12: 291–298. [DOI] [PubMed] [Google Scholar]
- Kortmann J, Sczodrok S, Rinnenthal J, Schwalbe H, Narberhaus F. 2011. Translation on demand by a simple RNA-based thermosensor. Nucleic Acids Res 39: 2855–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krymkiewicz N. 1973. Reactions of methylglyoxal with nucleic acids. FEBS Lett 29: 51–54. [DOI] [PubMed] [Google Scholar]
- Kwok CK, Balasubramanian S. 2015. Targeted detection of G-quadruplexes in cellular RNAs. Angew Chem Int Ed Engl 54: 6751–6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok CK, Ding Y, Tang Y, Assmann SM, Bevilacqua PC. 2013. Determination of in vivo RNA structure in low-abundance transcripts. Nat Commun 4: 2971. [DOI] [PubMed] [Google Scholar]
- Latham JA, Zaug AJ, Cech TR. 1990. Self-splicing and enzymatic cleavage of RNA by a group I intervening sequence. Methods Enzymol 181: 558–569. [DOI] [PubMed] [Google Scholar]
- Lee B, Flynn RA, Kadina A, Guo JK, Kool ET, Chang HY. 2017. Comparison of SHAPE reagents for mapping RNA structures inside living cells. RNA 23: 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveland AB, Demo G, Grigorieff N, Korostelev AA. 2017. Ensemble cryo-EM elucidates the mechanism of translation fidelity. Nature 546: 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maathuis FJ. 2009. Physiological functions of mineral macronutrients. Curr Opin Plant Biol 12: 250–258. [DOI] [PubMed] [Google Scholar]
- McGinnis JL, Liu Q, Lavender CA, Devaraj A, McClory SP, Fredrick K, Weeks KM. 2015. In-cell SHAPE reveals that free 30S ribosome subunits are in the inactive state. Proc Natl Acad Sci 112: 2425–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méreau A, Fournier R, Grégoire A, Mougin A, Fabrizio P, Luhrmann R, Branlant C. 1997. An in vivo and in vitro structure-function analysis of the Saccharomyces cerevisiae U3A snoRNP: protein-RNA contacts and base-pair interaction with the pre-ribosomal RNA. J Mol Biol 273: 552–571. [DOI] [PubMed] [Google Scholar]
- Mitchell D III, Russell R. 2014. Folding pathways of the Tetrahymena ribozyme. J Mol Biol 426: 2300–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed D, Robertson JM, Noller HF. 1988. Interaction of elongation factors EF-G and EF-Tu with a conserved loop in 23S RNA. Nature 334: 362–364. [DOI] [PubMed] [Google Scholar]
- Nakaya K, Takenaka O, Horinishi H, Shibata K. 1968. Reactions of glyoxal with nucleic acids. Nucleotides and their component bases. Biochim Biophys Acta 161: 23–31. [DOI] [PubMed] [Google Scholar]
- Naville M, Gautheret D. 2010. Transcription attenuation in bacteria: theme and variations. Brief Funct Genomic Proteomic 9: 178–189. [DOI] [PubMed] [Google Scholar]
- Noller HF, Chaires JB. 1972. Functional modification of 16S ribosomal RNA by kethoxal. Proc Natl Acad Sci 69: 3115–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noller HF, Hoffarth V, Zimniak L. 1992. Unusual resistance of peptidyl transferase to protein extraction procedures. Science 256: 1416–1419. [DOI] [PubMed] [Google Scholar]
- Peattie DA, Gilbert W. 1980. Chemical probes for higher-order structure in RNA. Proc Natl Acad Sci 77: 4679–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peselis A, Serganov A. 2014. Themes and variations in riboswitch structure and function. Biochim Biophys Acta 1839: 908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Becker T, Heuer A, Braunger K, Shanmuganathan V, Pech M, Berninghausen O, Wilson DN, Beckmann R. 2016. Structure of the hypusinylated eukaryotic translation factor eIF-5A bound to the ribosome. Nucleic Acids Res 44: 1944–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder AR, Baumstark T, Riesner D. 1998. Chemical mapping of co-existing RNA structures. Nucleic Acids Res 26: 3449–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro R, Cohen BI, Shiuey SJ, Maurer H. 1969. On the reaction of guanine with glyoxal, pyruvaldehyde, and kethoxal, and the structure of the acylguanines. A new synthesis of N2-alkylguanines. Biochemistry 8: 238–245. [DOI] [PubMed] [Google Scholar]
- Shapiro R, Cohen BI, Clagett DC. 1970. Specific acylation of the guanine residues of ribonucleic acid. J Biol Chem 245: 2633–2639. [PubMed] [Google Scholar]
- Smola MJ, Calabrese JM, Weeks KM. 2015. Detection of RNA-protein interactions in living cells with SHAPE. Biochemistry 54: 6867–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola MJ, Christy TW, Inoue K, Nicholson CO, Friedersdorf M, Keene JD, Lee DM, Calabrese JM, Weeks KM. 2016. SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across the Xist lncRNA in living cells. Proc Natl Acad Sci 113: 10322–10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohmen D, Chiba S, Shimokawa-Chiba N, Innis CA, Berninghausen O, Beckmann R, Ito K, Wilson DN. 2015. Structure of the Bacillus subtilis 70S ribosome reveals the basis for species-specific stalling. Nat Commun 6: 6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staehelin M. 1959. Inactivation of virus nucleic acid with glyoxal derivatives. Biochim Biophys Acta 31: 448–454. [DOI] [PubMed] [Google Scholar]
- Talkish J, May G, Lin Y, Woolford JL Jr, McManus CJ. 2014. Mod-seq: high-throughput sequencing for chemical probing of RNA structure. RNA 20: 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira A, Tahiri-Alaoui A, West S, Thomas B, Ramadass A, Martianov I, Dye M, James W, Proudfoot NJ, Akoulitchev A. 2004. Autocatalytic RNA cleavage in the human β-globin pre-mRNA promotes transcription termination. Nature 432: 526–530. [DOI] [PubMed] [Google Scholar]
- Tijerina P, Mohr S, Russell R. 2007. DMS footprinting of structured RNAs and RNA-protein complexes. Nat Protoc 2: 2608–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DJ, Leigh RA, Miller AJ. 1996. Potassium homeostasis in vacuolate plant cells. Proc Natl Acad Sci 93: 10510–10514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Qu K, Ouyang Z, Kertesz M, Li J, Tibshirani R, Makino DL, Nutter RC, Segal E, Chang HY. 2012. Genome-wide measurement of RNA folding energies. Mol Cell 48: 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West S, Gromak N, Proudfoot NJ. 2004. Human 5′−> 3′ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature 432: 522–525. [DOI] [PubMed] [Google Scholar]
- Wilkinson KA, Merino EJ, Weeks KM. 2005. RNA SHAPE chemistry reveals nonhierarchical interactions dominate equilibrium structural transitions in tRNAAsp transcripts. J Am Chem Soc 127: 4659–4667. [DOI] [PubMed] [Google Scholar]
- Winkler W, Nahvi A, Breaker RR. 2002. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature 419: 952–956. [DOI] [PubMed] [Google Scholar]
- Woodson SA. 2005. Structure and assembly of group I introns. Curr Opin Struct Biol 15: 324–330. [DOI] [PubMed] [Google Scholar]
- Yakhnin H, Zhang H, Yakhnin AV, Babitzke P. 2004. The trp RNA-binding attenuation protein of Bacillus subtilis regulates translation of the tryptophan transport gene trpP (yhaG) by blocking ribosome binding. J Bacteriol 186: 278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanofsky C. 1981. Attenuation in the control of expression of bacterial operons. Nature 289: 751–758. [DOI] [PubMed] [Google Scholar]
- Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. 2001. Crystal structure of the ribosome at 5.5 Å resolution. Science 292: 883–896. [DOI] [PubMed] [Google Scholar]
- Zaug AJ, Cech TR. 1986. The intervening sequence RNA of Tetrahymena is an enzyme. Science 231: 470–475. [DOI] [PubMed] [Google Scholar]
- Zaug AJ, Cech TR. 1995. Analysis of the structure of Tetrahymena nuclear RNAs in vivo: telomerase RNA, the self-splicing rRNA intron, and U2 snRNA. RNA 1: 363–374. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.