

Abstract
Ribosome assembly is an evolutionarily conserved and energy intensive process required for cellular growth, proliferation, and maintenance. In yeast, assembly of the small ribosomal subunit (SSU) requires approximately 75 assembly factors that act in coordination to form the SSU processome, a 6 MDa ribonucleoprotein complex. The SSU processome is required for processing, modifying, and folding the preribosomal RNA (rRNA) to prepare it for incorporation into the mature SSU. Although the protein composition of the SSU processome has been known for some time, the interaction network of the proteins required for its assembly has remained poorly defined. Here, we have used a semi-high-throughput yeast two-hybrid (Y2H) assay and coimmunoprecipitation validation method to produce a high-confidence interactome of SSU processome assembly factors (SPAFs), providing essential insight into SSU assembly and ribosome biogenesis. Further, we used glycerol density-gradient sedimentation to reveal the presence of protein subcomplexes that have not previously been observed. Our work not only provides essential insight into SSU assembly and ribosome biogenesis, but also serves as an important resource for future investigations into how defects in biogenesis and assembly cause congenital disorders of ribosomes known as ribosomopathies.
Keywords: small subunit (SSU) processome, interactome, protein–protein interaction (PPI), subcomplexes, glycerol-gradient sedimentation, ribosome biogenesis
INTRODUCTION
In the budding yeast Saccharomyces cerevisiae, ribosome assembly requires the coordination of many cellular resources. Ribosome biogenesis requires all three RNA polymerases (RNA polymerase I, RNA polymerase II, and RNA polymerase III), 76 small nucleolar RNAs (snoRNAs), 78 ribosomal proteins, and over 200 assembly factors that process, modify, cleave, and fold the preribosomal RNA (pre-rRNA) (Rodríguez-Galán et al. 2013; Woolford and Baserga 2013). Approximately 60% of cellular transcription and 90% of mRNA splicing in an actively growing budding yeast cell are devoted to ribosome biogenesis and assembly, resulting in over 2000 ribosomes being assembled per minute (Warner 1999). Additionally, >50% of RNA transcripts in a growing cell may be ribosomal RNA (rRNA, transcribed by RNA polymerase I), depending on the stage of growth and growth conditions (Hamperl et al. 2013). Indeed, a significant proportion of resources in an actively growing yeast cell are devoted to ribosome assembly.
Ribosome biogenesis begins in the nucleolus, a non-membrane bound nuclear compartment that forms around the tandem array of ribosomal DNA (rDNA) genes (Farley et al. 2015; McStay 2016; Mangan et al. 2017). In budding yeast, there are approximately 150–200 rDNA repeats located on chromosome XII (Woolford and Baserga 2013; Turowski and Tollervey 2015). Each repeat is comprised of the genes encoding the 18S, 5.8S, and 25S rRNAs separated by internally transcribed spacer (ITS) sequences and flanked by external transcribed spacer (ETS) sequences (Fig. 1). Transcribed as a polycistronic precursor by RNA polymerase I, the pre-rRNAs are cotranscriptionally processed by the small subunit (SSU) processome, a 6 MDa ribonucleoprotein complex that is required for ribosome assembly (Dragon et al. 2002; Grandi et al. 2002; Osheim et al. 2004; Chaker-Margot et al. 2015; Turowski and Tollervey 2015).
FIGURE 1.
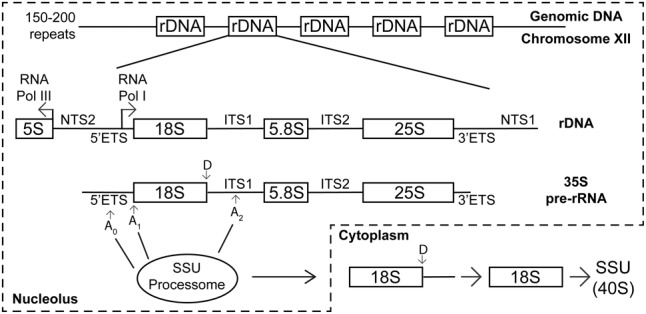
Yeast ribosome biogenesis is a complex nucleolar process. Yeast rDNA, arranged in 150–200 tandem repeats on Chromosome XII, is transcribed as a polycistronic precursor pre-rRNA containing the 18S, 5.8S, and 25S mature rRNAs by RNA polymerase I (RNA Pol I). The SSU processome is required for cleaving at A0 and A1 in the 5′ external transcribed spacer (ETS) sequence and A2 in the internal transcribed spacer 1 (ITS1) sequence, thereby separating SSU and LSU biogenesis. The nascent SSU is exported to the cytoplasm, where the final cleavage (at site D) occurs. At this point, the SSU is considered the mature 40S containing the 18S rRNA. The 5S rRNA is transcribed separately by RNA polymerase III (RNA Pol III).
The SSU processome is essential for pre-rRNA processing and mediates cleavages at sites A0, A1, and A2 on the pre-rRNA (Fig. 1; Phipps et al. 2011; Woolford and Baserga 2013; Fernández-Pevida et al. 2015; Turowski and Tollervey 2015). Specifically, the cleavage in ITS1 (A2; Fig. 1) separates SSU biogenesis from large ribosomal subunit (LSU) biogenesis, compartmentalizing what will become the mature 18S rRNA from what will later become the 25S and 5.8S rRNAs contained in the mature LSU. The 5S rRNA is transcribed separately by RNA polymerase III and is also incorporated into the LSU. After export to the cytoplasm, the mature LSU and SSU subunits come together to form the mature 80S ribosome, which translates mRNAs into proteins.
The SSU processome is comprised of approximately 75 assembly factors that are required to cleave, process, and modify the pre-rRNA (Table 1; Phipps et al. 2011; Rodríguez-Galán et al. 2013; Woolford and Baserga 2013). A subset of these SSU processome assembly factors (SPAFs) joins together to form five known protein subcomplexes: tUtp/UtpA, UtpB, UtpC, Mpp10, and the U3 snoRNP. Interestingly, fewer than half of the approximately 75 SPAFs have been assigned to subcomplexes, making the search for novel subcomplexes containing the remaining SPAFs particularly important to establish a better understanding of the assembly of both the SSU processome and the mature SSU (Phipps et al. 2011; Woolford and Baserga 2013).
TABLE 1.
List of SSU processome assembly factors (SPAFs)
SSU assembly proceeds in a stepwise, subcomplex-based manner. One of the first steps of ribosome biogenesis is mediated by tUtp/UtpA, which links pre-rRNA transcription with pre-rRNA processing and binds to the start of the 5′ ETS of the pre-rRNA (Fig. 1; Gallagher et al. 2004; Krogan et al. 2004; Pérez-Fernández et al. 2007; Poll et al. 2014; Sloan et al. 2014; Chaker-Margot et al. 2015; Zhang et al. 2016b). The U3 snoRNP also plays an important role in the earliest stages of formation of the SSU processome: The U3 snoRNA binds to complementary regions on the pre-rRNA and acts as a chaperone for pre-18S RNA folding and incorporation into the growing SSU processome (Beltrame and Tollervey 1992, 1995; Beltrame et al. 1994; Hughes 1996; Sharma and Tollervey 1999; Watkins et al. 2000; Dutca et al. 2011; Marmier-Gourrier et al. 2011; Phipps et al. 2011). Although the UtpB, UtpC, and Mpp10 subcomplexes have been less well characterized functionally, it has been proposed that they join the nascent SSU processome after the initial tUtp/UtpA and U3 snoRNP interactions occur with the nascent pre-rRNA (Pérez-Fernández et al. 2007, 2011; Chaker-Margot et al. 2015; Zhang et al. 2016b). Recent efforts have primarily focused on the structure of the SSU processome, providing important insights to the architecture of the complex, but none has systematically probed the protein–protein interaction (PPI) network of the SPAFs in S. cerevisiae (Poll et al. 2014; Hunziker et al. 2016; Kornprobst et al. 2016; Zhang et al. 2016a; Baßler et al. 2017; Chaker-Margot et al. 2017; Sun et al. 2017). While attempts to better understand the sequential involvement of the characterized subcomplexes have provided a basic schematic of assembly, the exact details of the SSU processome interaction network in S. cerevisiae have remained, to this point, undescribed.
In this study, we have defined the yeast SPAF interactome and have provided additional insights into SSU assembly. Using a semi-high-throughput yeast two-hybrid (Y2H) approach, we have identified 363 high-confidence interactions from a total of 5476 individual bait–prey pairs. We have established the robustness of our data set with 76% of tested interacting pairs validated by coimmunoprecipitation and with Markov clustering of the high-confidence interactome. The LSU processome biogenesis factor interactome was also recently mapped using a similar approach (McCann et al. 2015). Furthermore, we have identified the presence of previously unreported subcomplexes by glycerol-gradient sedimentation. Taken together, our results shed light on SSU assembly and reveal the complex network of interactions that occur among the SPAFs thus adding to our understanding of the vast coordination required for SSU biogenesis and providing a foundation for broadening our understanding of how defects in these factors may contribute to disease.
RESULTS
Generating an SSU processome interactome map by yeast two-hybrid (Y2H) analysis
To determine the SSU processome interactome map, we established the PPI network of 74 nucleolar proteins involved in SSU biogenesis (Lim et al. 2011) using a matrix-approach yeast two-hybrid analysis (Table 1). Each protein was individually queried against all 74 proteins. Ribosomal proteins were not included because of technical limitations. Of the selected proteins, 60 (81%) are essential in yeast and 69 (93%) are conserved to humans (Supplemental Table 1). When this work began, only 67 PPIs had been described between these 74 proteins, representing ∼18% of the total interactions we have identified in this study (Lim et al. 2011).
To establish the interactome map, we first generated plasmid libraries for the matrix-approach Y2H assays. Using Gateway cloning from either the MORF collection (Gelperin et al. 2005) or PCR amplification from yeast genomic DNA and cloning into a Gateway Entry vector (pDONR221), we cloned all 74 full-length ORFs into Y2H bait and prey vectors. Prior to screening, each bait was tested for autoactivation on SD-Trp-His media. Ltv1 autoactivated at concentrations of up to 200 mM 3-AT, and so was not included in our screen as a bait. Each bait vector was then screened against each of the 74 proteins in a directed Y2H. Previously, this matrix-approach Y2H assay was used to map the ribosomal large subunit (LSU) interactome (McCann et al. 2015).
Using Y2H assays, we were able to directly investigate pairwise PPIs between SPAFs. We assayed PPIs on media lacking amino acids (SD-Leu-Trp-His and SD-Leu-Trp-His-Ade) (Fig. 2A,B) as carried out previously (McCann et al. 2015). As a negative control, we assayed each bait vector against an empty prey vector. Growth was observed for 3 wk, and any growth greater than the growth of the negative control was scored as a positive PPI. The screen was carried out twice in the PJ69-4a/α strains (James et al. 1996) and once in the Y2H Gold/Y187 strains (Clontech) (Harper et al. 1993), for a total of three iterations.
FIGURE 2.
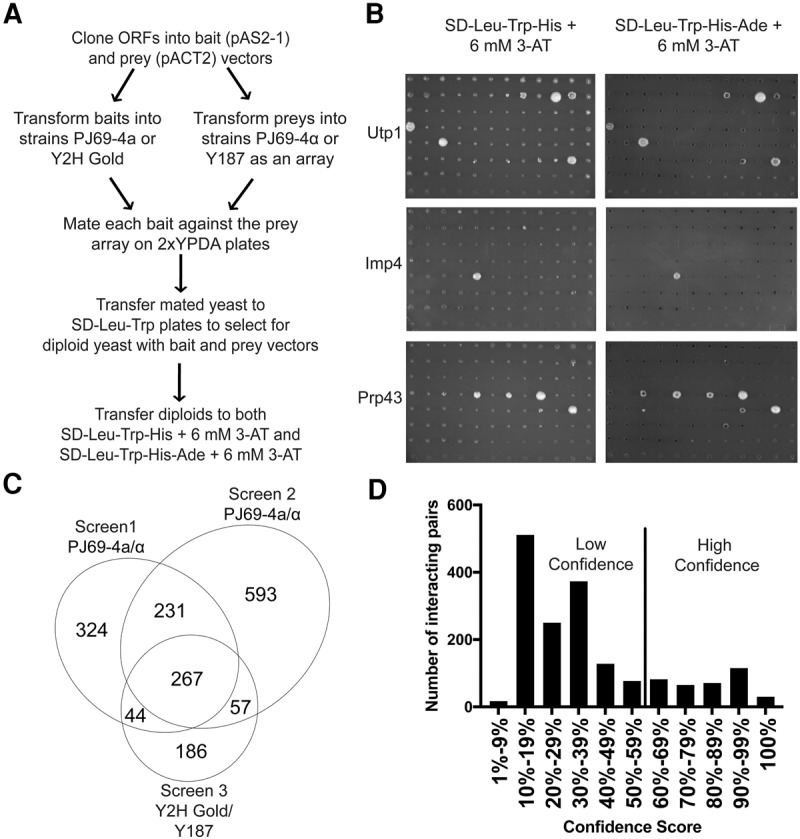
An array-based Y2H screen identifies novel interactions among SSU processome assembly factors (SPAFs). (A) Workflow of the array-based Y2H screen, as previously described in McCann et al. (2015). (B) Results from Y2H screens performed on SD-Leu-Trp-His + 6 mM 3-AT and SD-Leu-Trp-His-Ade + 6 mM 3-AT. Growth of a colony on either medium indicates a positive Y2H interaction. Negative controls were included on each plate in the top row, leftmost column. (C) A Venn diagram summarizing the interactions that were identified in each iteration of the screen. The screen was performed three times. Screens 1 and 2 were performed in PJ69-4a/α, and screen 3 was performed in Y2H Gold/ Y187 (Clontech) (Harper et al. 1993). The numbers on the Venn diagram indicate the number of PPIs that were identified in each screen and those that overlapped between screens. (D) Histogram of the confidence score distribution for all 1702 PPIs identified in this study. Interactions with a confidence score of ≥60% were identified as high-confidence PPIs and are listed in Supplemental Table 3. All interactions are listed in Supplemental Table 2. Confidence scores were graphed using GraphPad Prism 7.
Of the 5402 assayed PPIs, a total of 1702 interactions (32% of total possible interactions) were observed across the three screens (Fig. 2C,D; Supplemental Table 2). A total of 1103 interactions were observed in one screen, while 267 interactions were observed in all three. The remaining 332 interactions were observed in two screens. As has previously been reported (McCann et al. 2015), screens in the PJ69-4a/α strains revealed a greater number of interacting partners than in the Y2H Gold/Y187 strains (Screen 1 [PJ69-4a/α strain]: 866 interacting partners; Screen 2 [PJ69-4a/α strain]: 1148 interacting partners; Screen 3 [Y2H Gold/Y187 strain]: 554 interacting partners) (Fig. 2C).
Using a calculated weighted average confidence score, we sought to quantify each of the PPIs (McCann et al. 2015). Confidence scores for each of the 1702 PPIs were calculated on a scale of 0%–100% (Fig. 2D; Supplemental Tables 2, 3). Of the 1702 interactions, 363 had a high-confidence score (60% to 100%) (Supplemental Table 3). The remaining interactions (1339) fell between 0% and 59%, and were labeled low confidence. However, it is likely that there are many biologically relevant interactions in this group that were not reproducible across strains. Taken together, these findings represent an approximately five-and-a-half-fold increase from the 67 previously identified PPIs among approximately 70 SPAFs that had been reported at the start of this work.
Validation of the high-confidence SSU processome interactome
To validate the robustness of our data set, we analyzed a subset of the high-confidence interactions by coimmunoprecipitation. This method has been widely used to validate Y2H data sets (Suter et al. 2007; Wang et al. 2011; Hegele et al. 2012; McCann et al. 2015). ORFs were shuttled into either Gateway-converted p414GPD-3xFLAG or Gateway-converted p414GPD-3xHA (Mumberg et al. 1995; McCann et al. 2015). Sixty-seven coimmunoprecipitations were performed with anti-HA conjugated to Sepharose beads, and the copurifying proteins were visualized by western blotting with an anti-FLAG-HRP antibody (Fig. 3A,B). A smaller subset (21) was performed with an anti-FLAG resin, and the copurifying proteins were visualized by western blotting with an anti-HA-HRP antibody. In total, we analyzed 88 of the 363 high-confidence interactions (24%) by coimmunoprecipitation. Of these, 67 (76%) validated as positive interactions (Fig. 3C; Supplemental Table 4), a percentage similar to what has been reported in previous Y2H studies (Wang et al. 2011; McCann et al. 2015). As a negative control, we also tested an additional 15 interactions with confidence scores of 0. There was no detection of these interactions in any iteration of the screen, and these interactions had not been previously reported in the literature. Testing these 15 protein pairs for interactions served to identify the false positive rate of the Y2H assay. Of the 15 tested, 14 were, in fact, true negatives (Fig. 3C; Supplemental Table 5), indicating a 7% false-positive rate, which is even lower than what has been previously reported (McCann et al. 2015). Taken together, these results further establish the robustness of our data set for the yeast SPAF interactome.
FIGURE 3.
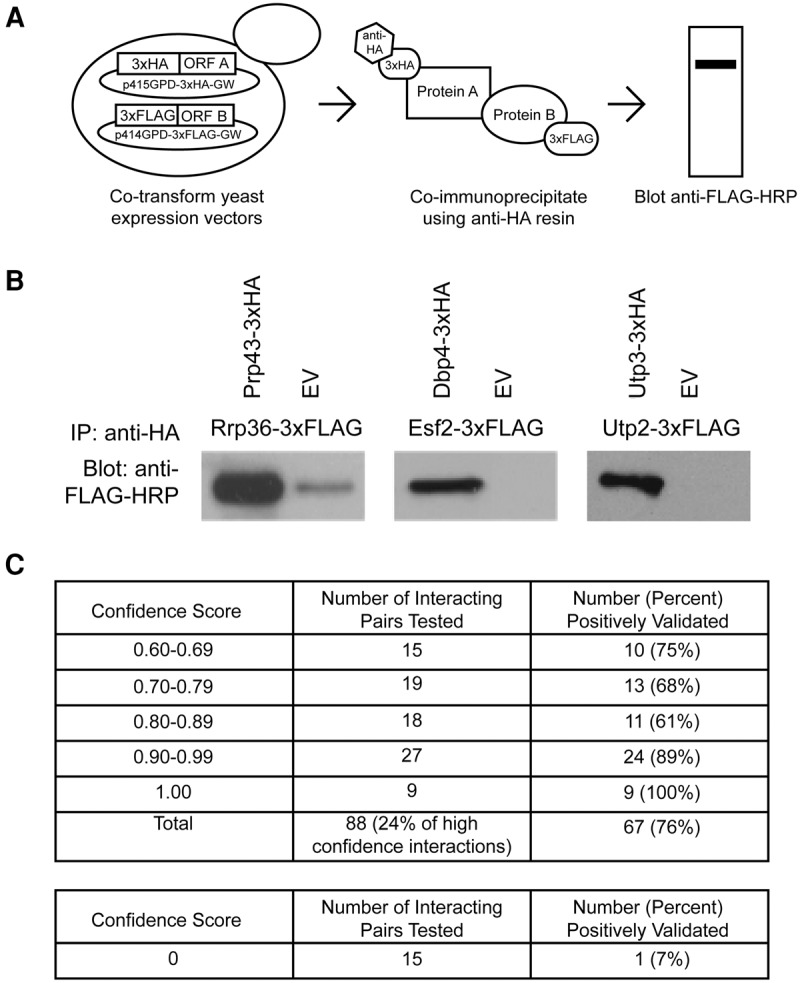
Validation of the high-confidence SSU processome interactome by coimmunoprecipitation. (A) Schematic depicting the coimmunoprecipitation approach used to validate novel PPIs. (B) Validation examples of high-confidence PPIs. (EV) Empty vector. (C) Tables summarizing the results of the high-confidence validation by coimmunoprecipitation. Approximately 25% of all high-confidence interactions were assayed by coimmunoprecipitation. Additionally, PPIs with confidence scores of 0 were included as negative controls.
Analysis of the high-confidence SSU processome interactome map
The SSU processome interactome is also highly supported by PPIs that have been previously reported. Using the Saccharomyces Genome Database (http://www.yeastgenome.org), Uniprot (uniprot.org), IntAct (www.ebi.ac.uk/intact/) and BioGRID (thebiogrid.org), we collected published Y2H interactions and compared them to our high-confidence interactome data set. We found 95 PPIs that have been reported in the literature, which represents an increase from the 67 that were previously reported in Lim et al. (2011). Of the 95, 59 were recapitulated as part of our high-confidence PPI interactome data set (Fig. 4A; Supplemental Table 6). Within this set of recapitulated interactions, we observed previously characterized subcomplexes of the SSU processome (Fig. 4A), including tUtp/UtpA (blue), UtpB (gray), UtpC (yellow), the Mpp10 subcomplex (green), and the Box C/D snoRNP (purple). The presence of these previously published PPIs in our data set validates the ability of our screen to identify novel SSU subcomplexes.
FIGURE 4.

Computational analysis of the high-confidence SSU processome interactome. (A) High-confidence PPIs previously reported by Y2H and protein complementation (PCA) assays. Characterized subcomplexes implicated in SSU assembly are labeled as follows: tUtp/UtpA (blue); UtpB (gray); UtpC (yellow); Mpp10 (green); Box C/D snoRNP (purple). Previously published interactions are listed in Supplemental Table 6. (B) Markov clustering (MCL) of the high-confidence SSU processome interactome reveals a large tightly interconnected network and also reveals interactions found in previously characterized subcomplexes: tUtp/UtpA (Utp4–Utp5–Utp8–Utp9–Utp10–Utp15–Utp17); UtpB (Utp12–Utp13); UtpC (Cka2–Ckb1); Nop1–Nop56–Snu13 (Box C/D snoRNP). Two lines between proteins indicate a reciprocal interaction. The interaction networks were created using Cytoscape, and MCL clustering was performed using the clusterMaker app in Cytoscape. (Hexagon) GTPase; (diamond) helicase; (octagon) kinase; (square) methyltransferase; (parallelogram) nuclease; (circle) no known enzymatic function.
To better characterize the SPAF interactome, we constructed a map of the high-confidence interactions in Cytoscape (Supplemental Fig. 1). Of the 73 bait proteins we tested in our screens, 57 interacted with a prey with high confidence. Each of the baits interacted with an average of 6.4 prey partners, with the total number of interacting partners ranging from 1 to 22. It has been estimated that the average protein has approximately 3.5 interacting partners (Blow 2009; Lim et al. 2011). The higher than expected mean of interacting partners per bait in our data set suggests a high level of interconnectivity among proteins, and the presence of hub proteins, which interact with far more than the average number of interacting partners and often play an important structural role in complex formation. Additionally, 26 of the baits in the interactome network self-interacted.
We also used Markov clustering (MCL option in the clusterMaker app in Cytoscape) to predict additional subcomplexes that make up the building blocks of the SSU processome (Shannon et al. 2003; Morris et al. 2011). Markov clustering is a computational method that has been shown to be particularly effective in predicting subcomplex formation from larger complex interaction networks (Pereira-Leal et al. 2004; Krogan et al. 2006; Hart et al. 2007; Pu et al. 2007; Nastou et al. 2014). Although Markov clustering did not reveal novel subcomplexes from our interaction network, it did reveal elements of known subcomplexes. Sixty-one of the 63 baits that interacted with preys with high confidence were clustered into six modules (Fig. 4B). The largest of these modules, which contains 50 SPAFs, includes many of the proteins that have the highest number of high confidence interacting partners. Interestingly, the components of the tUtp/UtpA subcomplex can be detected in the larger network (Fig. 4B). Additionally, the Utp12–Utp13 reciprocal pairing represents an isolated interaction from the UtpB subcomplex that has been previously identified as a dimer module of UtpB (Poll et al. 2014; Hunziker et al. 2016; Zhang et al. 2016a). Markov clustering also identified Nop1–Nop56–Snu13 (Box C/D snoRNP) interactions within an isolated set that also includes Nsr1. The emergence of these known sets of interactions that play important roles in building SSU subcomplexes further emphasizes the robustness of the SSU processome interactome map, but suggests that Y2H, while a valuable tool for identifying PPI networks, may not be as valuable in identifying previously undiscovered protein complexes when coupled with MCL analysis.
Biochemical analysis of the SSU processome subcomplexes
Although several SSU processome subcomplexes have previously been described (Wehner et al. 2002; Gallagher et al. 2004; Krogan et al. 2004; Pérez-Fernández et al. 2011; Phipps et al. 2011) and our Y2H results support their presence, half of the proteins in the SSU processome have not yet been assigned to subcomplexes (Woolford and Baserga 2013). Protein subcomplexes are the building blocks of SSU assembly; the discovery of the presence of novel protein subcomplexes will help to better illustrate the modular architecture of the SSU processome. To ascertain the existence of novel subcomplexes, we used glycerol density-gradient sedimentation, which separates protein complexes in a native state by density (mass and volume). We determined the approximate mass in kilodaltons (kDa) of these particles using protein standards (Supplemental Fig. 2).
Glycerol-gradient analysis of 42 of the 74 (57%) SPAFs revealed three main classes of sedimentation profiles visualized by western blotting of the tagged proteins. The first class of proteins sedimented in discrete peaks smaller than the SSU processome (examples and full list in Fig. 5A; additional examples in Supplemental Fig. 3; Supplemental Table 7). These proteins likely form building blocks of the SSU processome. The second class of proteins sedimented in only the fastest sedimenting fractions, which are the fractions in which the mature SSU processome sediments (examples in Fig. 5B; additional examples in Supplemental Fig. 3; Supplemental Table 7). The third class of proteins sedimented in all fractions of the glycerol gradient (examples in Supplemental Fig. 3; Supplemental Table 7). These proteins did not form discrete subcomplexes, and were excluded from further analysis (Supplemental Table 7).
FIGURE 5.

Glycerol-gradient sedimentation analysis of SPAFs. (A) Novel subcomplexes revealed by glycerol-gradient sedimentation. Western blot of tagged SPAFs from glycerol gradient (10%−30%) fractions (1–19). (T) 1% total extract. Estimated masses of subcomplexes were determined using protein standards (Supplemental Figure 2). (B) Glycerol-gradient sedimentation reveals “late joiner” SPAFs. Western blot analysis of tagged SPAFs from glycerol-gradient (10%–30%) fractions (1–19). (T) 1% total extract.
The tagged proteins that formed discrete peaks sedimented in particles between 140 and 470 kDa in mass, larger than would be predicted from the individual tagged protein's predicted molecular mass, indicating association with other proteins and likely membership in a protein complex rather than a monomeric protein (Fig. 5A; Supplemental Fig. 3; Supplemental Table 7). These proteins are also often found in the fastest sedimenting fractions, indicating their association with the mature SSU processome. Thus, the SSU processome is likely composed of these additional previously undescribed subcomplexes (Fig. 5A; Supplemental Fig. 3; Supplemental Table 7). Indeed, we were able to detect the presence of proteins that have already been assigned to subcomplexes on glycerol gradients, and they sedimented in discrete peaks as well (Utp5, Utp6, Utp15; Fig. 5A; Supplemental Fig. 3).
The sedimentation profile for SSU processome-only associated proteins is distinct from proteins that are members of subcomplexes. These proteins do not form discrete peaks, but instead are found only in the fastest sedimenting fractions, and so likely join the nascent SSU processome in later stages of maturation as individual proteins (Fig. 5B; Supplemental Fig. 3). Interestingly, this implies that the SSU processome is not entirely comprised of subcomplexes, but that some proteins do join on an individual basis to perform their function as needed.
We asked whether the new protein complexes observed in Figure 5A formed independently of nucleic acids. Previously, it has been shown that some subcomplexes required for SSU biogenesis exist in the absence of RNA polymerase I transcription of rDNA and in the absence of SSU processome formation (Gallagher et al. 2004). To answer this question, whole cell yeast extract was treated with Benzonase, a nonspecific nuclease, positing that subcomplexes not dependent on the presence of nucleic acids would remain intact. To confirm nucleic acid degradation in Benzonase-treated extracts, whole cell yeast extract was treated with Benzonase, and the RNA was analyzed on a 1% TAE gel, using degradation of the mature 25S and 18S rRNAs as a readout for degradation of cellular nucleic acids. After treatment with Benzonase, there were no mature 25S or 18S rRNAs present, as expected (Fig. 6A).
FIGURE 6.

Some novel SPAF subcomplexes persist in the absence of RNA. (A) Benzonase treatment of yeast extract confirms degradation of RNA. Total RNA was isolated from YPH499, treated with or without Benzonase, and run on a 1% (w/v) TAE gel. (−B) No Benzonase; (+B) Benzonase treated. The mature rRNAs, 18S and 25S, are indicated in the untreated lane. (B) Some SSU processome subcomplexes do not change sedimentation profile after treatment with Benzonase. Glycerol-gradient analysis of the tagged proteins as in Figure 5. (−) No Benzonase; (+) Benzonase treated. (C) A Nop9-containing subcomplex is released from the SSU processome after treatment with Benzonase. Glycerol-gradient analysis of tagged Nop9 as in Figure 5. (−) No Benzonase; (+) Benzonase treated.
We tested five complexes that sedimented in discrete peaks from Figure 5A to determine if their formation is dependent on the presence of nucleic acids. The five tagged proteins that were tested showed no change in their sedimentation profiles with and without Benzonase (Fig. 6B; Supplemental Fig. 4). We did, however, observe a decrease in the signal of the tagged protein in the fastest sedimenting fractions in the Benzonase-treated samples and a corresponding increase in the signals in the discrete peaks (Fig. 6B; Supplemental Fig. 4). Importantly, this suggests not only that the discrete peaks are not mediated by the presence of nucleic acids, but also that they can be released from the mature SSU processome as that complex is degraded in the presence of Benzonase.
We then tested Nop9, a SPAF that sedimented in only the fastest sedimenting fractions (Fig. 5B; Supplemental Fig. 3) to determine whether its association with the SSU processome is dependent on the presence of nucleic acids. After treating Nop9-tagged yeast extract with Benzonase, we observed the appearance of a discrete peak of ∼270 kDa by western blot (Fig. 6C). This result suggests that Nop9 exists in a small complex that is maintained as part of the mature SSU processome, likely mediated by RNA. In addition to the discrete peak in fraction 6, there is a noticeable reduction in tagged Nop9 in the fastest sedimenting fractions relative to the control extract that was not treated with Benzonase (Fig. 6C, Fraction 19). Moreover, a slower migrating band appears in the lightest fractions (Fig. 6C, arrowhead; fractions 1–3) that may represent a modified form of Nop9 that is excluded from the observed subcomplex and the mature SSU processome (fastest sedimenting fractions).
Taken together, these results support the existence of an additional set of protein subcomplexes comprised of SSU biogenesis factors that have been previously undescribed and remain uncharacterized. These include Dbp8, Dhr2, Has1, Prp43, Emg1, Enp1, Esf1, Esf2, Nsr1, Rrp5, Utp1, Utp5, Utp6, Utp8, Utp15, Utp16, Utp17, Utp19, and Utp25 (Fig. 5A; Supplemental Fig. 3; Supplemental Table 7) Importantly, these results suggest the existence of novel building blocks that may be implicated in the stepwise assembly of the SSU processome.
DISCUSSION
We have characterized the S. cerevisiae SSU processome interactome using a matrix-based Y2H assay. When this study began, fewer than 70 PPIs had been described among approximately 70 SPAFs (Lim et al. 2011). Our recently updated database mining revealed 95 S. cerevisiae PPIs that have been reported in the literature among the 74 proteins we included in our study (Supplemental Table 6). Here, we have described 363 high-confidence interactions among 74 SPAFs, representing an approximately five-and-a-half-fold increase in the number of known interactions. Using Y2H and an orthogonal coimmunoprecipitation validation method, we have provided insight into the organization of the SSU processome. Using glycerol-gradient sedimentation, we have also performed the first systematic biochemical analysis of subcomplex formation among these SPAFs. These results further our understanding of the building blocks of SSU processome assembly.
Although it is possible that some of the observed binary PPIs are mediated by a third “bridging” protein, it remains unlikely. The 363 high-confidence interactions we observed represent fewer than 7% of the total number of interactions assayed. If cellular SSU processome proteins were bridging these binary PPIs, we would have recovered a higher number of high-confidence PPIs. Additionally, the plasmids used in the Y2H system express genes at very high levels; a cellular protein would have to be expressed at comparably high levels, and in the correct cellular compartment, to adequately bridge the interaction. Although the existence of bridging proteins remains a distinct possibility, the yield from the repeated Y2H assays and the subsequent validation step suggests that we are observing few, if any, bridging proteins.
Recently, there have been efforts to characterize the temporal involvement of SPAFs in SSU biogenesis (Chaker-Margot et al. 2015; Zhang et al. 2016b). Using pre-rRNA transcripts of various lengths to identify proteins associated with each transcript in vivo, both Chaker-Margot and coworkers and Zhang and coworkers produced a relative temporal map of the involvement of SPAFs in SSU assembly. Interestingly, proteins that we observed to sediment the fastest are likely the “late joiners” that both Chaker-Margot and colleagues and Zhang and colleagues have observed. Seven out of 11 tested proteins with this sedimentation profile (Krr1, Nop9, Rrp7, Rok1, Utp2, Utp20, and Utp30; Fig. 5B; Supplemental Fig. 3; Supplemental Table 7) were also found to be associated with the nascent pre-rRNA transcript at later stages of transcription and processing (Chaker-Margot et al. 2015; Zhang et al. 2016b). Additionally, Utp20 and Utp30 were also found to be late joining factors in a cryo-electron microscopy structure of the SSU processome. (Sun et al. 2017).
Baßler et al. (2017) reported the results of an extensive Y2H screen that included approximately 180 ribosome AFs from the thermophilic fungus Chaetomium thermophilum. Only 57 of the 74 proteins in this study were included in the screen of Baßler and colleagues (Supplemental Table 8). Among these 57 proteins, Baßler and coworkers reported 133 total interactions and 45 robust interactions (the equivalence of high-confidence interactions in this study). Comparatively, we observed 1241 total interactions and 268 high-confidence interactions in this same set of 57 proteins. There are, however, 25 robust PPIs that Baßler and colleagues identified that we also identified as high-confidence interactions (for full list, see Supplemental Table 8). Importantly, these interactions were not included in our previously published interactions (Supplemental Table 6), as the PPIs included in our curated list are solely from S. cerevisiae.
There are important methodological differences between this study and that of Baßler and colleagues that may explain the differences in the results of our similar screens. The Baßler et al. (2017) screen used proteins from C. thermophilum, which only has protein homologs for 73% of the proteins that are found in S. cerevisiae, which we used in this study (Amlacher et al. 2011). Additionally, while Baßler and coworkers’ screen scored growth at 4 and 7 d, we scored growth at 7, 14, and 21 d, which permits the detection of interacting pairs that do not grow within the first few days (McCann et al. 2015). Although one possibility is that permitting the yeast to grow for longer periods of time produced a higher degree of artifact PPIs, our results show that this is not the case: The 76% validation rate of the Y2H PPIs emphasizes the robustness of the data set. At the same time, we maintained a low rate of false positives. The current study has, therefore, provided increased coverage of the SPAF interactome, recovering more high-confidence interactions across a larger subset of proteins.
The Baßler et al. (2017) study and other recent studies have also focused on the structure of the SSU processome and its known subcomplexes (Poll et al. 2014; Zhang et al. 2014, 2016a; Hunziker et al. 2016; Kornprobst et al. 2016; Baßler et al. 2017; Chaker-Margot et al. 2017; Sun et al. 2017). While these studies have provided important structural insights to our understanding of SSU processome architecture, they lack specific details of the intricate interconnectedness of all proteins involved in the process: Even the most recent structures are incomplete and lack many SPAFs (Chaker-Margot et al. 2017; Sun et al. 2017). Moreover, it is not possible to identify binary PPIs from these structures, as it is possible to do using a matrix-based Y2H assay; that is, proximity in a structure does not always equal direct interaction.
This study, however, does provide evidence for novel subcomplexes containing several SPAFs, including Emg1, Utp25, Enp1, Nsr1, Dhr2, Utp16, Dbp8, Esf2, Prp43, Has1, Rrp5, and Esf1 (Fig. 5A; Supplemental Fig. 3; Supplemental Table 7). Additionally, we hypothesize that we have identified previously unreported modules of novel subcomplexes. As supporting evidence for this hypothesis, we have identified modules of known protein subcomplexes. Previous efforts to structurally characterize tUtp/UtpA and UtpB identified the presence of stable modules that interact to form the full subcomplex (Poll et al. 2014; Hunziker et al. 2016). For example, UtpB has been described as being composed of a core complex of Utp1, Utp21, Utp12, and Utp13, and formed of three heterodimers: Utp1–Utp21, Utp12–Utp13, and Utp6–Utp18 (Chaker-Margot et al. 2015; Hunziker et al. 2016). Other studies have described the core complex as being comprised of Utp1, Utp6, Utp18, and Utp21, with the Utp12–Utp13 dimer associating separately (Poll et al. 2014). In this study, Utp1 sediments at ∼165 kDa (Fig. 5A; Supplemental Fig. 3; Supplemental Table 7), which likely represents the Utp1–Utp21 dimer (expected mass of ∼210 kDa). Additionally, tUtp/UtpA has been described as being composed of a core complex of Utp4, Utp5, Utp8, Utp9, and Utp15 (the t-Utp pentamer) (Poll et al. 2014), with a Utp10–Utp17 dimer associating separately (Poll et al. 2014; Hunziker et al. 2016). Our Y2H results reveal that Utp10 and Utp17 interact with high confidence (confidence scores = 1 in both directions; Supplemental Tables 2, 3). Our hypothesis that the Utp17 peak (270 kDa) represents the Utp10–Utp17 dimer (expected mass ∼300 kDa) is, therefore, strongly supported by the literature (Poll et al. 2014; Hunziker et al. 2016) and by our Y2H results, as is our hypothesis that the Utp15 peak we have observed at 320 kDa is the t-Utp pentamer core complex (expected mass ∼360 kDa) (Poll et al. 2014). We further hypothesize that the Nop9 peak we observe after treatment with Benzonase (Fig. 6C) is one of these functional modules that has effectively “popped off” the mature SSU processome. Therefore, we have not only provided evidence for novel subcomplexes, but also for novel protein modules that act as building blocks of larger subcomplexes.
Although this study has provided important findings for SSU assembly, questions remain. Future studies will be required to identify the components of the protein subcomplexes that have been observed in our glycerol-gradient sedimentation experiments (Fig. 5; Supplemental Fig. 3). In particular, the protein complexes will need to be purified and analyzed using mass spectrometry to identify complex members. As the novel subcomplexes are defined, it will be possible to study their involvement in and impact on SSU biogenesis. It will also be important to discern at what stages the various subcomplexes act on SSU assembly. Through an understanding of the organization of the SSU processome and the function of its building blocks, a fully functional understanding of SSU biogenesis will emerge.
Our results provide an important and unique advancement in our understanding of the assembly of the intricate network of SPAFs required for SSU assembly. We have described, in binary detail, the interactions of these proteins that, in turn, will help inform future studies on the assembly and function of the yeast SSU processome. These results will also provide a more comprehensive framework for studies seeking to understand ribosomopathies, a family of diseases affecting ribosome assembly and function (McCann and Baserga 2013; Nakhoul et al. 2014; Sondalle and Baserga 2014; Danilova and Gazda 2015; Yelick and Trainor 2015). Several human diseases result from mutations in factors required for assembling the SSU (Sondalle and Baserga 2014). Understanding the role of these mutations in the formation of the subcomplexes of the SSU processome will be essential in further characterizing the etiologies of these diseases. A clear illustration of the interaction network in yeast will broaden our understanding of these human diseases and provide valuable insight into the way they are either managed or treated.
MATERIALS AND METHODS
Yeast two-hybrid (Y2H) assay
Open reading frames (ORFs) encoding small subunit (SSU) biogenesis factor proteins were either obtained in a Gateway vector (pBG1805 or pDONR221) from the moveable ORF (MORF) library (Gelperin et al. 2005) or PCR-amplified from yeast genomic DNA and cloned into pDONR221, a Gateway Entry vector. ORFs were shuttled into both bait (pAS2-1) and prey (pACT2) destination vectors for Y2H that had been adapted for Gateway cloning (Life Technologies) as previously described (Nakayama et al. 2002; Charette and Baserga 2010; McCann et al. 2015). All clones were fully sequenced by Genewiz, Inc. and those that were constructed for this study and not included in the MORF collection were deposited in AddGene.
Each bait vector was transformed into PJ69-4α (MATα trp1-901 leu2-3 112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ) (James et al. 1996) or Y2H Gold (MATa trp1-901 leu2-3 112 ura3-52 his3- 200 gal4Δ gal80Δ LYS2::GAL1UAS-Gal1TATA-His3 GAL2UASGal2TATA-Ade2 URA3::MEL1UAS-Mel1TATA AUR1-C MEL1) (Clontech). Each prey vector was transformed into PJ69-4a (MATa trp1-901 leu2-3 112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ) (James et al. 1996) or Y187 (MATα trp1-901 leu2-3 112 ura3-52 his3-200 gal4Δ gal80Δ met-, URA3::GAL1-LacZ, MEL1) (Harper et al. 1993) in a 96-well array format, and each bait was tested for autoactivation, as previously described (McCann et al. 2015). As a negative control, yeast transformed with an empty prey vector (pACT2) were included. Interactions that were previously identified by Y2H served as positive controls (Fig. 4A).
Each bait was mated against each prey in a semi-high-throughput Y2H matrix screen, as described in McCann et al. (2015) and de Folter and Immink (2011). In brief, the mated yeast were transferred to SD-Leu-Trp plates to select for diploid yeast containing both the bait and prey vectors. Diploids were then transferred to the selective media SD-Leu-Trp-His + 6 mM 3-AT and SD-Leu-Trp-His-Ade + 6 mM 3-AT. Growth on selective medium greater than that of the negative control after 3 wk was considered an interacting bait-prey pair. Each of the PPIs was assayed three times (twice in PJ69-4a/α and once in Y2H Gold/ Y187), and all observed interactions were assigned a confidence score.
A weighted average confidence score based on the following equation,
and as described in McCann et al. (2015) was calculated for each PPI, with the following changes: x2 is growth in screen 2 (PJ69-4a/α), x4 is growth on SD-Leu-Trp-His + 6 mM 3-AT in screen 1, x5 is growth on SD-Leu-Trp-His-Ade + 6 mM 3-AT in screen 1, x6 is growth on SD-Leu-Trp-His + 6 mM 3-AT in screen 2, x7 is growth on SD-Leu-Trp-His-Ade + 6 mM 3-AT in screen 2, x8 is growth on SD-Leu-Trp-His + 6 mM 3-AT in screen 3, and x9 is growth on SD-Leu-Trp-His-Ade + 6 mM 3-AT in screen 3. All observed interactions and the corresponding confidence scores are listed in Supplemental Table 2 and all observed high-confidence interactions and corresponding confidence scores are listed in Supplemental Table 3.
The high-confidence interactions from this study have been submitted to the International Molecular Exchange (IMEx) Consortium (http://www.imexconsortium.org) through IntAct (Orchard et al. 2014) and assigned the identifier IM-25795.
Y2H result validation
A subset of SSU biogenesis factor ORFs was shuttled from the Gateway Entry vector (pDONR221) into p414GPD-3xFLAG-GW (TRP1 marker) or p415GPD-3xHA-GW (LEU2 marker), both Gateway-modified yeast expression vectors (Mumberg et al. 1995; McCann et al. 2015). p414GPD-3xFLAG-ORF-containing vectors were individually transformed into the parental strain YPH499 (MATa ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1) (Sikorski and Hieter 1989). After appropriate growth and selection, p415GPD-3xHA-ORF-containing vectors were then sequentially transformed into yeast containing the appropriate p414GPD-3xFLAG-ORF vector, resulting in yeast strains containing individual plasmid pairs to be tested by coimmunoprecipitation. Yeast containing plasmid pairs were grown on SD-Leu-Trp (2% dextrose, lacking leucine and tryptophan) at 30°C, as previously described (McCann et al. 2015). For negative controls for HA pulldowns, strains were transformed with the p414GPD-3xFLAG-ORF-containing vector and p414GPD-3xHA-empty vector. For negative controls for FLAG pulldowns, strains were transformed with the p414GPD-3xFLAG-empty vector and p415GPD-3xHA-ORF. For coimmunoprecipitations, 50 mL of cells at an OD600 of 0.35–0.55 were harvested, washed with sterile water, and resuspended in NET2 (20 mM Tris–HCL pH 7.5, 150 mM NaCl, 0.01% Nonidet P-40) with protease inhibitors (Roche cOmplete Protease Inhibitor Cocktail). Cells were lysed using a glass bead extraction method, and the lysate was cleared, both according to McCann et al. (2015). Aliquots of 500 µL of lysate were then incubated with 12CA5 (α-HA) that had been conjugated to Protein A Sepharose CL-4B beads (GE Healthcare) by nutating at room temperature for 1 h or Anti-FLAG M2 Affinity Gel (Sigma-Aldrich).
Coimmunoprecipitations were incubated for 1 h at 4°C. Beads were washed five times in NET2 and resuspended in 20 µL Laemmeli sample buffer. Eluates were run on 10% SDS-PAGE gels and transferred to PVDF membranes (Bio-Rad). Western blot analysis was carried out with a dilution of 1:10,000 α-3xFLAG-HRP (Sigma-Aldrich) or 1:10,000 α-3xHA-HRP (Roche).
Markov clustering analysis
The high-confidence SSU processome interactome map was imported into Cytoscape (Shannon et al. 2003), and the network was clustered using the MCL option in clusterMaker (Cytoscape app plug-in) (Morris et al. 2011) as previously described (McCann et al. 2015).
Yeast strains
Yeast strains containing a C-terminally tagged endogenous gene of interest were constructed in YPH499 (MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3-Δ200 leu2-Δ1) or NOY504 (MATα rpa12::LEU2 leu2-3, 112 ura3-1 trp-1 his3-11 CAN1-100) (Nogi et al. 1993) using primers complementary to the locus of the gene of interest (Supplemental Table 9). For TAP-tagged strains, the cassette from pBS1479 (Puig et al. 2001) was amplified by PCR. For 3xFLAG tagged strains, the cassette from p3X-FLAG-KanMX (Gelbart et al. 2001) was amplified by PCR. For V5 tagged strains, the cassette from pFA6a-6xGLY-V5-hphMX4 (Funakoshi and Hochstrasser 2009) was amplified by PCR. All C-terminally tagged strains were validated by NaOH extraction of yeast and western blot. All yeast strains used and constructed in this study are included in Supplemental Table 10.
Glycerol-gradient sedimentation analysis
To perform glycerol-gradient sedimentation analysis of YPH499 tagged strains, 50 mL of cells were grown to an OD600 = 0.35–0.55 at 30°C and harvested, washed with water, and resuspended in 750 µL 10% glycerol RNBP (20 mM HEPES at pH 7.5, 110 mM KOAc, 0.5% Triton, 0.1% Tween, 4 µg/mL pepstatin A, 180 µg/mL PMSF, 1:5000 anti-foam B [Sigma], 1:5000 protector RNase inhibitor [Roche]) (Oeffinger et al. 2007). Yeast cells were lysed using 0.55-mm glass beads, and 600 µL of cleared whole cell lysate was run on 10%−30% glycerol gradients (RNPB). The gradients were centrifuged on an SW-41 rotor at 35,000 RPM for 18 h at 4°C. Gradients were harvested from the top in 600 µL fractions. Fractions were then analyzed by western blot analysis using anti-FLAG-HRP (1:10,000; Sigma), anti-HA-HRP (1:10,000; Roche), PAP (1:6,000; Sigma), or V5-HRP (1:5,000; Invitrogen). For each gradient analysis, 3% of each fraction and 1% of total extract were loaded on SDS-PAGE gels for western blot analysis.
Glycerol-gradient sedimentation analysis was also performed for each of three standard size proteins: catalase from bovine liver (Sigma), apoferritin from equine spleen (Sigma), and thyroglobulin from bovine thyroid (Sigma). For catalase, 20 µL of 10 mg/mL catalase was added to 500 µL 10% glycerol RNPB and loaded on a 10%−30% glycerol gradient. Gradients were spun as described above, and fractions were analyzed by silver stain (Blum et al. 1987). For apoferritin, 10 µL was added to 500 µL 10% glycerol RNPB and loaded on a 10%−30% glycerol gradient. Gradients were spun as described above, and fractions were analyzed by silver stain (Blum et al. 1987). For thyroglobulin, 300 µL (in 5% glycerol) was brought to a final concentration of 10% glycerol, and 500 µL thyroglobulin with 10% glycerol was loaded on a 10%−30% glycerol gradient. Gradients were spun as described above, and fractions were analyzed by Coomassie staining.
RNA extraction and TAE gel
RNA was extracted from whole cell lysate yeast extracts using phenol chloroform extraction, and 5 µg of RNA were run on a 1% (w/v) Tris base (Sigma), acetic acid (JT Baker), and EDTA (americanBIO) (TAE) gel for analysis. Ethidium bromide staining was used to visualize the RNA.
Benzonase assay
Tagged yeast strains were grown to OD600 0.5–0.6, and glass bead whole cell lysates were extracted into 1200 µL 10% glycerol RNPB (lacking RNase inhibitor) as described above. Lysates were divided into two aliquots. One aliquot was treated with 2 µL Benzonase (Sigma), and both aliquots were nutated for 1 h at 4°C. Aliquots were then loaded onto 10%–30% glycerol gradients and spun and analyzed as described above.
For the RNA degradation analysis, YPH499 was grown to OD600 ∼ 0.4 and harvested into 800 µL 10% glycerol RNPB (lacking RNase inhibitor) as described above. Lysates were divided and treated also as described above. RNA was extracted from each aliquot and run on a 1% (w/v) TAE gel.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Baserga laboratory for their close readings of this manuscript and for their helpful discussions during its development. N.G.V. was supported by the Predoctoral Program in Cellular and Molecular Biology (T32 GM 007223). J.M.C. was supported by a postdoctoral National Institutes of Health–Ruth L. Kirchstein National Research Service Award and an Institutional Research Training Grant (Radiation Therapy, Biology, Physics T32 CA 009259). This work was supported by the National Institutes of Health (NIH) (R01 GM115710 to S.J.B.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.062927.117.
REFERENCES
- Amlacher S, Sarges P, Flemming D, van Noort V, Kunze R, Devos DP, Arumugam M, Bork P, Hurt E. 2011. Insight into structure and assembly of the nuclear pore complex by utilizing the genome of a eukaryotic thermophile. Cell 146: 277–289. [DOI] [PubMed] [Google Scholar]
- Baßler J, Ahmed YL, Kallas M, Kornprobst M, Calviño FR, Gnädig M, Thoms M, Stier G, Ismail S, Kharde S, et al. 2017. Interaction network of the ribosome assembly machinery from a eukaryotic thermophile. Protein Sci 26: 327–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrame M, Henry Y, Tollervey D. 1994. Mutational analysis of an essential binding site for the U3 snoRNA in the 5′ external transcribed spacer of yeast pre-rRNA. Nucleic Acids Res 22: 5139–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrame M, Tollervey D. 1992. Identification and functional analysis of two U3 binding sites on yeast pre-ribosomal RNA. EMBO J 11: 1531–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrame M, Tollervey D. 1995. Base pairing between U3 and the pre-ribosomal RNA is required for 18S rRNA synthesis. EMBO J 14: 4350–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow N. 2009. Systems biology: untangling the protein web. Nature 460: 415–418. [DOI] [PubMed] [Google Scholar]
- Blum H, Beier H, Gross HJ. 1987. Improved silver staining of plant-proteins, RNA and DNA in polyacrylamide gels. Electrophoresis 8: 93–99. [Google Scholar]
- Chaker-Margot M, Hunziker M, Barandun J, Dill BD, Klinge S. 2015. Stage-specific assembly events of the 6-MDa small-subunit processome initiate eukaryotic ribosome biogenesis. Nat Struct Mol Biol 22: 920–923. [DOI] [PubMed] [Google Scholar]
- Chaker-Margot M, Barandun J, Hunziker M, Klinge S. 2017. Architecture of the yeast small subunit processome. Science 355: eaal1880. [DOI] [PubMed] [Google Scholar]
- Charette JM, Baserga SJ. 2010. The DEAD-box RNA helicase-like Utp25 is an SSU processome component. RNA 16: 2156–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilova N, Gazda HT. 2015. Ribosomopathies: how a common root can cause a tree of pathologies. Dis Model Mech 8: 1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Folter S, Immink RG. 2011. Yeast protein–protein interaction assays and screens. Methods Mol Biol 754: 145–165. [DOI] [PubMed] [Google Scholar]
- Dragon F, Gallagher JE, Compagnone-Post PA, Mitchell BM, Porwancher KA, Wehner KA, Wormsley S, Settlage RE, Shabanowitz J, Osheim Y, et al. 2002. A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature 417: 967–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutca LM, Gallagher JE, Baserga SJ. 2011. The initial U3 snoRNA:pre-rRNA base pairing interaction required for pre-18S rRNA folding revealed by in vivo chemical probing. Nucleic Acids Res 39: 5164–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley KI, Surovtseva Y, Merkel J, Baserga SJ. 2015. Determinants of mammalian nucleolar architecture. Chromosoma 124: 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Pevida A, Kressler D, de la Cruz J. 2015. Processing of preribosomal RNA in Saccharomyces cerevisiae. Wiley Interdiscip Rev RNA 6: 191–209. [DOI] [PubMed] [Google Scholar]
- Funakoshi M, Hochstrasser M. 2009. Small epitope-linker modules for PCR-based C-terminal tagging in Saccharomyces cerevisiae. Yeast 26: 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher JE, Dunbar DA, Granneman S, Mitchell BM, Osheim Y, Beyer AL, Baserga SJ. 2004. RNA polymerase I transcription and pre-rRNA processing are linked by specific SSU processome components. Genes Dev 18: 2506–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelbart ME, Rechsteiner T, Richmond TJ, Tsukiyama T. 2001. Interactions of Isw2 chromatin remodeling complex with nucleosomal arrays: analyses using recombinant yeast histones and immobilized templates. Mol Cell Biol 21: 2098–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelperin DM, White MA, Wilkinson ML, Kon Y, Kung LA, Wise KJ, Lopez-Hoyo N, Jiang L, Piccirillo S, Yu H, et al. 2005. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev 19: 2816–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandi P, Rybin V, Bassler J, Petfalski E, Strauss D, Marzioch M, Schäfer T, Kuster B, Tschochner H, Tollervey D, et al. 2002. 90S pre-ribosomes include the 35S pre-rRNA, the U3 snoRNP, and 40S subunit processing factors but predominantly lack 60S synthesis factors. Mol Cell 10: 105–115. [DOI] [PubMed] [Google Scholar]
- Hamperl S, Wittner M, Babl V, Perez-Fernández J, Tschochner H, Griesenbeck J. 2013. Chromatin states at ribosomal DNA loci. Biochim Biophys Acta 1829: 405–417. [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. 1993. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75: 805–816. [DOI] [PubMed] [Google Scholar]
- Hart GT, Lee I, Marcotte ER. 2007. A high-accuracy consensus map of yeast protein complexes reveals modular nature of gene essentiality. BMC Bioinformatics 8: 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Luhrmann R, Stelzl U. 2012. Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell 45: 567–580. [DOI] [PubMed] [Google Scholar]
- Hughes JM. 1996. Functional base-pairing interaction between highly conserved elements of U3 small nucleolar RNA and the small ribosomal subunit RNA. J Mol Biol 259: 645–654. [DOI] [PubMed] [Google Scholar]
- Hunziker M, Barandun J, Petfalski E, Tan D, Delan-Forino C, Molloy KR, Kim KH, Dunn-Davies H, Shi Y, Chaker-Margot M, et al. 2016. UtpA and UtpB chaperone nascent pre-ribosomal RNA and U3 snoRNA to initiate eukaryotic ribosome assembly. Nat Commun 7: 12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James P, Halladay J, Craig EA. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144: 1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornprobst M, Turk M, Kellner N, Cheng J, Flemming D, Kos-Braun I, Kos M, Thoms M, Berninghausen O, Beckmann R, et al. 2016. Architecture of the 90S pre-ribosome: a structural view on the birth of the eukaryotic ribosome. Cell 166: 380–393. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Peng WT, Cagney G, Robinson MD, Haw R, Zhong G, Guo X, Zhang X, Canadien V, Richards DP, et al. 2004. High-definition macromolecular composition of yeast RNA-processing complexes. Mol Cell 13: 225–239. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, et al. 2006. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440: 637–643. [DOI] [PubMed] [Google Scholar]
- Lim YH, Charette JM, Baserga SJ. 2011. Assembling a protein-protein interaction map of the SSU processome from existing datasets. PLoS One 6: e17701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan H, Gailiín MÓ, McStay B. 2017. Integrating the genomic architecture of human nucleolar organizer regions with the biophysical properties of nucleoli. FEBS J. 10.1111/febs.14108. [DOI] [PubMed] [Google Scholar]
- Marmier-Gourrier N, Clery A, Schlotter F, Senty-Segault V, Branlant C. 2011. A second base pair interaction between U3 small nucleolar RNA and the 5′-ETS region is required for early cleavage of the yeast pre-ribosomal RNA. Nucleic Acids Res 39: 9731–9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann KL, Baserga SJ. 2013. Genetics. Mysterious ribosomopathies. Science 341: 849–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann KL, Charette JM, Vincent NG, Baserga SJ. 2015. A protein interaction map of the LSU processome. Genes Dev 29: 862–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McStay B. 2016. Nucleolar organizer regions: genomic ‘dark matter’ requiring illumination. Genes Dev 30: 1598–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JH, Apeltsin L, Newman AM, Baumbach J, Wittkop T, Su G, Bader GD, Ferrin TE. 2011. clusterMaker: a multi-algorithm clustering plugin for Cytoscape. BMC Bioinformatics 12: 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M. 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156: 119–122. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Kikuno R, Ohara O. 2002. Protein-protein interactions between large proteins: two-hybrid screening using a functionally classified library composed of long cDNAs. Genome Res 12: 1773–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhoul H, Ke J, Zhou X, Liao W, Zeng SX, Lu H. 2014. Ribosomopathies: mechanisms of disease. Clin Med Insights Blood Disord 7: 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nastou KC, Tsaousis GN, Kremizas KE, Litou ZI, Hamodrakas SJ. 2014. The human plasma membrane peripherome: visualization and analysis of interactions. Biomed Res Int 2014: 397145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogi Y, Yano R, Dodd J, Carles C, Nomura M. 1993. Gene RRN4 in Saccharomyces cerevisiae encodes the A12.2 subunit of RNA polymerase I and is essential only at high temperatures. Mol Cell Biol 13: 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeffinger M, Wei KE, Rogers R, DeGrasse JA, Chait BT, Aitchison JD, Rout MP. 2007. Comprehensive analysis of diverse ribonucleoprotein complexes. Nat Methods 4: 951–956. [DOI] [PubMed] [Google Scholar]
- Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, Campbell NH, Chavali G, Chen C, del-Toro N, et al. 2014. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res 42: D358–D363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osheim YN, French SL, Keck KM, Champion EA, Spasov K, Dragon F, Baserga SJ, Beyer AL. 2004. Pre-18S ribosomal RNA is structurally compacted into the SSU processome prior to being cleaved from nascent transcripts in Saccharomyces cerevisiae. Mol Cell 16: 943–954. [DOI] [PubMed] [Google Scholar]
- Pereira-Leal JB, Enright AJ, Ouzounis CA. 2004. Detection of functional modules from protein interaction networks. Proteins 54: 49–57. [DOI] [PubMed] [Google Scholar]
- Pérez-Fernández J, Roman A, De Las Rivas J, Bustelo XR, Dosil M. 2007. The 90S preribosome is a multimodular structure that is assembled through a hierarchical mechanism. Mol Cell Biol 27: 5414–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Fernández J, Martin-Marcos P, Dosil M. 2011. Elucidation of the assembly events required for the recruitment of Utp20, Imp4 and Bms1 onto nascent pre-ribosomes. Nucleic Acids Res 39: 8105–8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipps KR, Charette J, Baserga SJ. 2011. The small subunit processome in ribosome biogenesis-progress and prospects. Wiley Interdiscip Rev RNA 2: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poll G, Li S, Ohmayer U, Hierlmeier T, Milkereit P, Perez-Fernández J. 2014. In vitro reconstitution of yeast tUTP/UTP A and UTP B subcomplexes provides new insights into their modular architecture. PLoS One 9: e114898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu S, Vlasblom J, Emili A, Greenblatt J, Wodak SJ. 2007. Identifying functional modules in the physical interactome of Saccharomyces cerevisiae. Proteomics 7: 944–960. [DOI] [PubMed] [Google Scholar]
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. 2001. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24: 218–229. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Galán O, Garcia-Gomez JJ, de la Cruz J. 2013. Yeast and human RNA helicases involved in ribosome biogenesis: current status and perspectives. Biochim Biophys Acta 1829: 775–790. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Tollervey D. 1999. Base pairing between U3 small nucleolar RNA and the 5′ end of 18S rRNA is required for pre-rRNA processing. Mol Cell Biol 19: 6012–6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan KE, Bohnsack MT, Schneider C, Watkins NJ. 2014. The roles of SSU processome components and surveillance factors in the initial processing of human ribosomal RNA. RNA 20: 540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondalle SB, Baserga SJ. 2014. Human diseases of the SSU processome. Biochim Biophys Acta 1842: 758–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Zhu X, Qi J, An W, Lan P, Tan D, Chen R, Wang B, Zheng S, Zhang C, et al. 2017. Molecular architecture of the 90S small subunit pre-ribosome. eLife 6: e22086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter B, Fetchko MJ, Imhof R, Graham CI, Stoffel-Studer I, Zbinden C, Raghavan M, Lopez L, Beneti L, Hort J, et al. 2007. Examining protein–protein interactions using endogenously tagged yeast arrays: the Cross-and-Capture system. Genome Res 17: 1774–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turowski TW, Tollervey D. 2015. Cotranscriptional events in eukaryotic ribosome synthesis. Wiley Interdiscip Rev RNA 6: 129–139. [DOI] [PubMed] [Google Scholar]
- Wang J, Huo K, Ma L, Tang L, Li D, Huang X, Yuan Y, Li C, Wang W, Guan W, et al. 2011. Toward an understanding of the protein interaction network of the human liver. Mol Syst Biol 7: 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner JR. 1999. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci 24: 437–440. [DOI] [PubMed] [Google Scholar]
- Watkins NJ, Segault V, Charpentier B, Nottrott S, Fabrizio P, Bachi A, Wilm M, Rosbash M, Branlant C, Luhrmann R. 2000. A common core RNP structure shared between the small nucleoar box C/D RNPs and the spliceosomal U4 snRNP. Cell 103: 457–466. [DOI] [PubMed] [Google Scholar]
- Wehner KA, Gallagher JE, Baserga SJ. 2002. Components of an interdependent unit within the SSU processome regulate and mediate its activity. Mol Cell Biol 22: 7258–7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolford JL Jr, Baserga SJ. 2013. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 195: 643–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelick PC, Trainor PA. 2015. Ribosomopathies: global process, tissue specific defects. Rare Dis 3: e1025185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Lin J, Liu W, Chen X, Chen R, Ye K. 2014. Structure of Utp21 tandem WD domain provides insight into the organization of the UTPB complex involved in ribosome synthesis. PLoS One 9: e86540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Sun Q, Chen R, Chen X, Lin J, Ye K. 2016a. Integrative structural analysis of the UTPB complex, an early assembly factor for eukaryotic small ribosomal subunits. Nucleic Acids Res 44: 7475–7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wu C, Cai G, Chen S, Ye K. 2016b. Stepwise and dynamic assembly of the earliest precursors of small ribosomal subunits in yeast. Genes Dev 30: 718–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.