

Abstract
P-glycoprotein (Pgp) is an efflux pump important in multidrug resistance of cancer cells and in determining drug pharmacokinetics. Pgp is a prototype ATP-binding cassette transporter with two nucleotide-binding domains (NBDs) that bind and hydrolyze ATP. Conformational changes at the NBDs (the Pgp engines) lead to changes across Pgp transmembrane domains that result in substrate translocation. According to current alternating access models (substrate-binding pocket accessible only to one side of the membrane at a time), binding of ATP promotes NBD dimerization, resulting in external accessibility of the drug-binding site (outward-facing, closed NBD conformation), and ATP hydrolysis leads to dissociation of the NBDs with the subsequent return of the accessibility of the binding site to the cytoplasmic side (inward-facing, open NBD conformation). However, previous work has not investigated these events under near-physiological conditions in a lipid bilayer and in the presence of transport substrate. Here, we used luminescence resonance energy transfer (LRET) to measure the distances between the two Pgp NBDs. Pgp was labeled with LRET probes, reconstituted in lipid nanodiscs, and the distance between the NBDs was measured at 37 °C. In the presence of verapamil, a substrate that activates ATP hydrolysis, the NBDs of Pgp reconstituted in nanodiscs were never far apart during the hydrolysis cycle, and we never observed the NBD–NBD distances of tens of Å that have previously been reported. However, we found two main conformations that coexist in a dynamic equilibrium under all conditions studied. Our observations highlight the importance of performing studies of efflux pumps under near-physiological conditions, in a lipid bilayer, at 37 °C, and during substrate-stimulated hydrolysis.
Keywords: ABC transporter, fluorescence resonance energy transfer (FRET), membrane, membrane bilayer, multidrug transporter, spectroscopy, LRET, luminescence resonance energy transfer, nanodisc
Introduction
P-glycoprotein (Pgp,7 ABCB1, MDR1) exports hundreds of chemically unrelated, hydrophobic compounds out of cells, including many therapeutic drugs (1). Pgp has been studied for over 4 decades for its involvement in multidrug resistance of cancer cells, and more recently for its importance in determining the pharmacokinetics of drugs used for treatment of HIV/AIDS and neurodegenerative and cardiovascular disorders (2, 3). Because of its effects on pharmacokinetics, the United States Food and Drug Administration mandates documentation of Pgp–drug interactions for approval of any new drug (4, 5). An ongoing goal of the pharmaceutical industry has been the development of drugs that either selectively block Pgp or evade recognition by Pgp to achieve more favorable pharmacokinetics. Consequently, there is a great interest in understanding the mechanism by which drugs are transported by Pgp.
Pgp is a prototype ABC exporter that harnesses the energy from ATP binding and hydrolysis at the two nucleotide-binding domains (NBDs) to power conformational changes in the transmembrane domains that lead to substrate translocation across the cell membrane. Pgp has been crystallized in nucleotide-free conformations with the NBDs ∼30 Å apart, where a central cavity formed by the transmembrane helices is exposed to the inner leaflet of the membrane and the cytoplasm (Fig. 1, left) (6, 7). More recent crystal structures of Pgp revealed even larger separation of the NBDs (8–10). Comparison of X-ray crystal structures of nucleotide-free and nucleotide-bound ABC transporters has led to the proposal of an alternating-access model where the central cavity is only accessible to one side of the membrane at a time (Fig. 1) (11). Despite decades of research, the molecular mechanism of ABC transporters is still controversial. Data on different ABC transporters using diverse experimental methods such as Cys cross-linking (12, 13), fluorescence (or Förster) resonance energy transfer (FRET), and luminescence (or lanthanide-based) resonance energy transfer (LRET) (14–18), double electron-electron paramagnetic resonance (DEER) spectroscopy (15, 19–22), mass spectrometry (23), and electron microscopy (24–27) are not always in agreement. A number of models have been proposed as a result of experimental discrepancies, very limited number of structural studies under “physiological” conditions, and lack of high-resolution kinetic information. These models can be broadly divided into two categories, monomer/dimer models (28–32), where ATP hydrolysis is followed by the complete dissociation of the NBDs, with NBD–NBD separations of tens of Å, and constant-contact models (33–36), where the NBDs remain in contact throughout the hydrolysis cycle, or remain in close proximity, and the power stroke results from smaller conformational changes at the NBD–NBD interface. In one constant-contact model, ATP hydrolysis is proposed to alternate between the two ATP-binding sites, with the site that just performed the hydrolysis opening to allow ADP/ATP exchange in a partially opened NBD dimer (37, 38). Independently of the extent of the conformational changes on the NBD side, formation of a closed NBD dimer upon ATP binding seems to be coupled to rearrangements in the transmembrane helices that result in transition from an inward-facing conformation (Fig. 1, left, open conformation; dissociated NBDs, or loosely-associated NBDs; binding pocket open to the membrane/cytoplasm) to an outward-facing conformation (Fig. 1, right, closed NBD dimer; dimeric NBDs; binding pocket open to extracellular side) from which the drug can dissociate into the external medium. Finally, hydrolysis of ATP promotes NBD dissociation, or dimer opening, resetting the pump for the next transport cycle.
Figure 1.
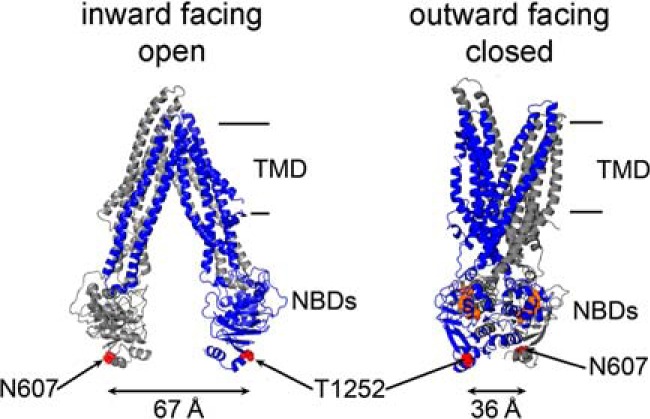
Alternating access model of Pgp with large NBD–NBD separation in the apo-state. Binding of ATP to the NBDs promotes the transition from the nucleotide-free inward-facing conformation (apo; PDB code 4Q9H) to the outward-facing conformation (nucleotide-bound model (46) based on the X-ray crystal structure of nucleotide-bound Sav1866; PDB code 2ONG). The first and second homologous halves are represented in gray and blue, respectively. In this study, Asn-607 and Thr-1252 (red spheres) were mutated to Cys to chemically attach LRET probes. The approximate position of the membrane is between the lines on the right. TMD, transmembrane domain; NBDs, nucleotide-binding domains.
Here, we used LRET to measure distance changes between the NBDs during the transport cycle of Pgp reconstituted in lipid nanodiscs (NDSCs). NDSCs are nanometer-scale discoidal structures that contain a phospholipid bilayer encased by membrane scaffold proteins (MSPs), which are soluble and stable (39). Although NDSCs differ from native membranes in physical properties such as curvature, and in the complexity of lipid composition, they are excellent lipid–bilayer platforms for spectroscopy and other applications. LRET is a highly sensitive spectroscopic technique that allows the study of working proteins in a native-like membrane environment and at physiological temperature (16, 40–43). LRET, like the traditional FRET, is based on energy transfer from a donor to an acceptor, but in LRET a lanthanide (Tb3+ or Eu3+) is used as donor. The use of luminescent lanthanides as donors has advantages for membrane protein studies over the traditional fluorescent donors used for FRET. Tb3+ displays sharp atomic-like emission peaks with dark regions between the peaks that allow for measurements of the sensitized acceptor emission (due to resonance energy transfer) without contamination from the donor emission. Also, the long emission lifetime of the Tb3+-excited state (milliseconds versus nanoseconds of traditional fluorophores) makes delayed (gated) acquisition possible (generally acquisition is delayed 60–200 μs from the excitation pulse). Gated acquisition minimizes the light-scattering effects of structures such as detergent micelles, liposomes, and NDSCs, and results in minimal background with high signal-to-noise ratio. Another major advantage is that the long lifetime-sensitized acceptor emission makes the calculation of donor/acceptor distances independent of labeling stoichiometry because long lifetime emission from acceptors with intrinsic lifetimes in the nanoseconds range can only arise from energy transfer (43). We have recently shown the usefulness of our approach to study the NBD dimerization/dissociation process during the ATP hydrolysis cycle of the bacterial Pgp homolog MsbA (16, 18). For the studies presented here, we introduced two Cys in a fully functional Cys-less Pgp (CL Pgp), at positions equivalent to those that we have used to characterize NBD movements in MsbA. The LRET probes (donor and acceptor) were chemically attached to these Cys, and alterations in the distance that separates the probes were assessed from the changes in the sensitized-emission lifetimes. We show conformational changes on the Pgp cytoplasmic side in response to binding of the transport substrate in the transmembrane domain and a degree of NBD dissociation much wider for Pgp in detergent than for Pgp reconstituted into lipid bilayers. Our data stresses the importance of performing structural studies of ABC exporters in a lipid bilayer and at physiological temperature.
Results
Characterization of the N606C/T1252C Pgp (NT Pgp)
Starting with a fully-functional CL Pgp, we introduced two Cys in positions equivalent to those that we used to study MsbA (16, 18). To generate NT Pgp, we started with our improved CL Pgp engineered by directed-evolution mutagenesis (44). In contrast to a previous all Cys-to-Ala variant (44, 45), this CL Pgp is fully capable of drug transport. In NT Pgp, residues Asn-607 and Thr-1252 of CL Pgp, near the carboxyl ends of each NBD, were substituted with Cys. These Cys are readily accessible to the aqueous environment for efficient labeling (6, 8, 10), and major changes in the distance between Cys-607 and Cys-1252 are expected between the inward-facing (open NBD dimer) (38, 46) and outward-facing (closed NBDs) conformations (at least 30 Å; Fig. 1) (6–10). The NT Pgp mutant displayed wild-type-like drug resistance in vivo, indicating that it was capable of exporting drugs across the plasma membrane (Fig. 2A). Purified WT Pgp, CL Pgp, and NT Pgp were >95% pure (supplemental Fig. 1) and displayed robust Ver-stimulated ATPase activity in detergent-lipid micelles: 3.9 ± 0.4, 3.5 ± 0.3, and 4.0 ± 0.5 μmol/min/mg, respectively (n = 3 or more different preparations each) (47). In two experiments, the Ver-stimulated ATPase activity of the catalytically-inactive mutant NT/AA Pgp (NT Pgp with substitution of the catalytic carboxylates Glu-552 and Glu-1197 with Ala) was undetectable. Also, labeling with the LRET probes did not affect the ATPase activity of NT Pgp in NDSCs. The activities in the presence of 30 μm Ver were 4.1 ± 0.24 and 4.1 ± 0.21 μmol/min/mg for the labeled (n = 3) and unlabeled (n = 5) NT Pgp, respectively.
Figure 2.
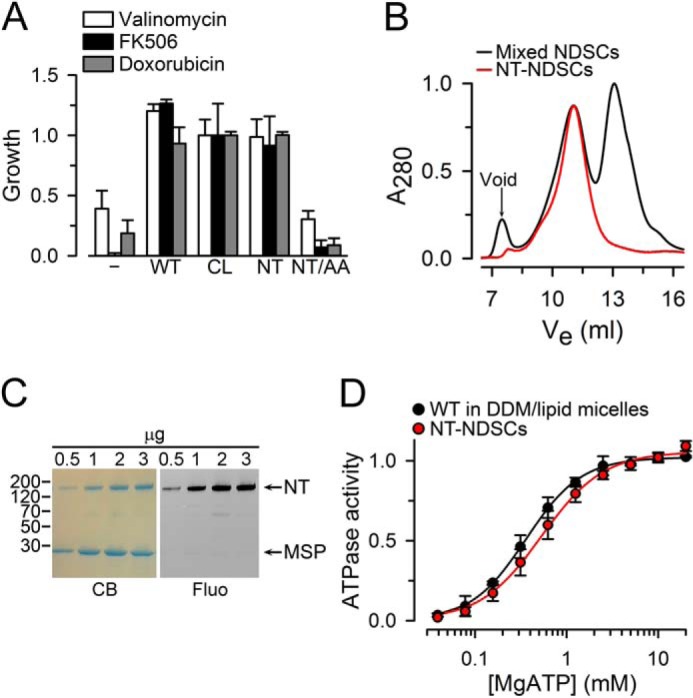
Characterization of NT Pgp. A, drug resistance assay in S. cerevisiae. The relative growth of cells transformed with the pVT vector control (−) and cells expressing wild-type Pgp (WT), Cys-less Pgp (CL), the N607C/T1252C (NT), or E552A/E1197A (NT/AA) mutants was monitored in the presence of 100 μm valinomycin, 62 μm FK506, or 40 μm doxorubicin for 25–30 h at 28 °C. Data were normalized to the CL Pgp average and are presented as means ± S.D. of three independent experiments performed in duplicate. B, size-exclusion chromatography elution profile of NT Pgp purified and reconstituted in lipid nanodiscs. The two main peaks of the black trace correspond to NDSCs containing Pgp (Ve ∼11 ml; Ve, elution volume) and empty NDSCs (Ve ∼13 ml). The red trace corresponds to NT Pgp in NDSCs (NT-NDSCs) purified by affinity chromatography (anti-FLAG M2 affinity gel) from the mixed NDSCs. C, NT Pgp in NDSCs. Coomassie-stained SDS-polyacrylamide gel (CB) and the corresponding fluorescent image (Fluo) of the fraction enriched in NDSCs containing NT Pgp labeled with Bodipy FL. Arrows point to the NT Pgp (NT) and MSP bands. Molecular masses of selected markers, in kDa, are shown on the left. D, ATPase activity measured in the presence of 30 μm verapamil. Data are averages ± S.E. of at least three independent experiments. Lines represent fits to the Hill equation, with Hills coefficients of 2 and 1.8 for WT Pgp and NT Pgp, respectively. Vmax and Km values were not significantly different between WT Pgp (Vmax = 3.9 ± 0.4 μmol/min/mg; Km = 0.31 ± 0.03 mm) and NT Pgp (Vmax = 4.1 ± 0.5 μmol/min/mg; Km = 0.27 ± 0.02 mm).
A typical size-exclusion chromatography elution profile of NT Pgp reconstituted in NDSCs is shown in Fig. 2B (black trace). The two main peaks correspond to NT Pgp in nanodiscs (Fig. 2B, left) and empty NDSCs (right). For most LRET experiments, we used the fraction enriched in NT Pgp-NDSCs, but we also used affinity-purified NT Pgp in NDSCs (Fig. 2B, red trace); the results obtained without and with this additional extra purification step were essentially identical. Fig. 2C shows a Coomassie Blue-stained SDS-PAGE of the fraction enriched in NT Pgp NDSC and a fluorescence image of the gel that confirms the specific labeling of NT Pgp by Bodipy FL. Fig. 2D shows that the dependence on [MgATP] of Ver-stimulated ATP hydrolysis of NT Pgp-NDSCs and WT-Pgp in detergent-lipid micelles was undistinguishable. However, the affinity for Ver was higher in the NDSCs (supplemental Fig. 2). The ATPase activity of WT Pgp and NT Pgp in detergent-lipid micelles or NDSCs was stable for several hours at 37 °C, whereas Pgp ATPase activity at 37 °C in detergent without lipids had a half-time (t½) of <5 min (supplemental Fig. 3). NT Pgp-NDSCs could be stored at 4 °C for up to a week with little loss of activity. Overall, these results demonstrate that NT Pgp is active in vivo and in vitro.
Conformational changes of the NBDs during the ATP hydrolysis cycle of Pgp in nanodiscs
We chose the Tb3+-chelate/Bodipy FL donor/acceptor pair (Forster distance of 41 Å) to label the two Cys of NT Pgp to maximize sensitivity of the conformational changes predicted. This LRET donor/acceptor pair is highly-sensitive to distance changes in the ∼30–55-Å range (supplemental Fig. 4). NDSCs containing 0.5–1.0 μm labeled NT Pgp were analyzed in a 3-mm pathlength cuvette at 37 °C. NT Pgp-NDSCs labeled only with donor (Fig. 3A, black dashed trace, Tb only) showed typical Tb3+ emission peaks centered about 490 and 545 nm. NT Pgp-NDSCs labeled with donor and acceptor showed an additional peak around 515 nm that corresponds to the sensitized Bodipy FL emission that arises from LRET from Tb3+ (Fig. 3A, solid lines). The intensity of this energy transfer is strongly dependent on the donor/acceptor distance and therefore, it is a sensitive indicator of conformational changes. Addition of 5 mm NaATP produced a large increase of the LRET intensity (Fig. 3A, blue) compared with the intensity in the apo-state (Fig. 3A, black, nucleotide- and drug-free). The EC50 for ATP estimated from similar LRET experiments was 0.26 ± 0.06 mm (n = 5); thus, 5 mm ATP is a saturating concentration. Subsequent addition of MgSO4 to start ATP hydrolysis caused a partial reversion (Fig. 3A, orange) toward the LRET intensity in the apo-state (Fig. 3A, black). Subsequent addition of sodium orthovanadate (Vi) (Fig. 3A, green), an inhibitor of Pgp ATPase activity that promotes the formation of a stable post-hydrolysis transition state (48), produced the strongest LRET intensity. A similar experiment performed with the catalytically inactive NT/AA Pgp (Fig. 3B) showed increased LRET intensity with the transition from the apo-state to the ATP-bound state (black to blue), whereas MgSO4 did not decrease LRET (orange) as in the active Pgp. This observation strongly suggests that the transition from the ATP-bound state to the MgATP state in Fig. 3A (blue to orange) was dependent on ATP hydrolysis. Together, these results show that LRET can be used to follow the conformational changes of Pgp in NDSCs during the ATP-hydrolysis cycle.
Figure 3.
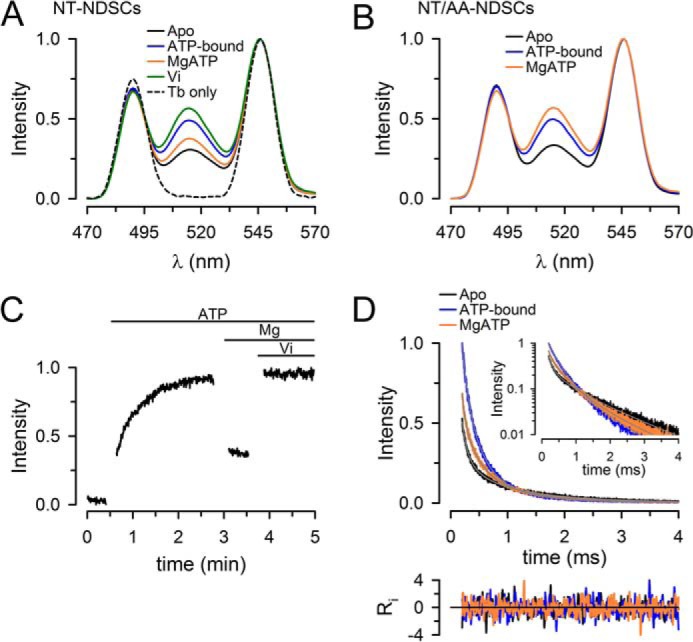
Conformational changes during the ATP hydrolysis cycle of NT Pgp in NDSCs at 37 °C. A, emission spectra from NDSCs containing NT Pgp labeled with donor only (Tb3+ chelate) or donor and acceptor (Tb3+ chelate and Bodipy FL). Traces were normalized to the Tb3+ emission at 546 nm. B, emission spectra of the catalytically-inactive mutant NT/AA Pgp in NDSCs. C, time course of the changes in Bodipy FL-sensitized emission, recorded at 520 nm, in response to sequential additions of NaATP, MgSO4, and Vi. The gaps during the recording correspond to time periods where additions and manual mixing were performed. The intensity was normalized to the maximum obtained after addition of Vi. D, Bodipy FL sensitized emission decays, recorded at 520 nm, from NT Pgp in NDSCs. The inset displays the same curves in a semi-log scale. Ri represents the weighted residuals of the multiexponential fits (gray lines in the main graph and inset). Decays were normalized to the emission of the ATP-bound protein at 200 μs. All the traces in the figures are representative of at least seven independent experiments and were obtained at 37 °C. Emission was recorded after a 200-μs delay from the 337-nm excitation pulse. Apo, nucleotide- and drug-free buffer with 1 mm EDTA; ATP-bound, + 5 mm NaATP; MgATP, +10 mm MgSO4.
A time course of the changes in LRET intensity is shown in Fig. 3C. The increase in intensity in response to the addition of NaATP (transition from apo-state to ATP-bound state) was slow (t½ ∼20 s). This is likely due to the fact that MgATP, and not NaATP, is the true substrate for binding to the NBDs. We have presented evidence of a much faster rate of dimerization of isolated NBDs in response to MgATP (versus NaATP) that might result from more favorable electrostatics for MgATP at the dimer interface (41). As expected, the changes in LRET intensities elicited by transitions to the MgATP and Vi states were much faster, and their kinetics could not be resolved in these manual mixing experiments.
The changes in LRET intensities described above can result from the following: 1) changes in donor/acceptor distances (higher LRET intensity results from shorter distances, e.g. dimerizing NBDs); 2) changes in the proportion of molecules with dimeric versus dissociated NBDs; or 3) a combination of both. To discriminate between these possibilities, we assessed donor/acceptor distances and the distribution of donor/acceptor pair distances in different states. Fig. 3D shows typical LRET decays from NT Pgp-NDSCs under different experimental conditions. As expected from the results in Fig. 3A, the transition from the apo-state to the ATP-bound state increased the Bodipy FL-sensitized emission intensity (see early part of the Fig. 3D, blue versus black) and accelerated the overall rate of decay. The latter is clearly evident in the semi-log plot (Fig. 3D, inset). During continuous ATP hydrolysis (MgATP state; Fig. 3D, orange) the initial intensity and the decay rate were intermediate between those in the apo- and ATP-bound states. In the semi-log plot inset (Fig. 3D), it is clear that the decays do not follow a single-exponential function, in which case they would appear as straight lines. The decays can be well fitted using a three-exponential decay function. As mentioned under “Experimental procedures,” the fastest component is very small and is largely the result of the instrument response time. Therefore, there were only two dominant donor/acceptor distances under all the experimental conditions studied. These distances and the proportion of donor/acceptor pairs displaying each of the distances are summarized in Table 1. The data revealed the coexistence of these two dominant conformations during the ATP hydrolysis cycle: 1) a short distance (d1) where the LRET probes are separated by ∼33 Å (d1 is likely to correspond to the closely-associated NBD dimer conformation; Fig. 1, right); and 2) a longer distance (d2) where the probes are separated by ∼47 Å (Δd2-d1 ∼15 Å; d2 is likely to correspond to a conformation with dissociated NBDs). These two distances remained essentially unchanged in the apo-, ATP-bound, and MgATP states. The data in Table 1 indicated that the observed changes in the LRET intensities and decays illustrated in Fig. 3 were mainly the result of changes in the proportion of Pgp molecules in each of the two conformations. This behavior was similar to that previously described for MsbA (16, 18). From the apo-state to the ATP-bound state, the % of molecules that adopted the closed NBD conformation d1 increased from only ∼10 to ∼40%. Transition from the ATP-bound state to the MgATP state decreased that number to ∼30%, as expected for a dynamic equilibrium between associating and dissociating NBDs during steady-state ATP hydrolysis (41, 49). These results suggest that the open conformation of the Pgp NBDs is favored even under maximal ATPase activity. This is in agreement with electron microscopy studies (27, 50). In summary, our data suggest the following: 1) the NBDs of NT Pgp in NDSCs are more frequently dissociated during basal ATP hydrolysis, and 2) even though the NBD–NBD distance is much shorter than that expected from the Pgp crystal structures in detergent, the NBDs dissociate, supporting a monomer–dimer mode of operation.
Table 1.
Distances between the Tb3+ and Bodipy FL probes during the catalytic cycle of NT Pgp
The measurements in the absence of Ver were obtained first for the apo-state and then after sequential addition of 5 mm NaATP (ATP-bound state), 10 mm MgSO4 (MgATP state), and 0.25 mm Vi (Vi state). In the presence of Ver, measurements were obtained first for the apo-state and then after sequential addition of Ver (Ver-bound state), 5 mm NaATP (Ver/ATP-bound state), 10 mm MgSO4 (Ver/MgATP state), and 0.25 mm Vi (Ver/Vi state). The two dominant exponential components of the sensitized emission decays are presented as distances d1 and d2. Lifetimes of Bodipy FL-sensitized emission (τDA), the calculated donor-acceptor distances (R), and the percentage of NT Pgp molecules displaying each distance are shown. The latter were calculated from multiexponential fits as the fractional intensity contribution of each exponential component divided by the rate of energy transfer (k = 1/τDA − 1/τD). Data are presented as means ± S.E., and n is the number of independent experiments. All the LRET decays used for these calculations were obtained from NT Pgp in NDSCs at 37 °C.
| State | d1 |
d2 |
||||
|---|---|---|---|---|---|---|
| τDA | R1 | Molecules | τDA | R2 | Molecules | |
| μs | Å | % | μs | Å | % | |
| NT Pgp in NDSCs in the absence of transport substrate (n = 7) | ||||||
| Apo | 378 ± 12 | 32.7 ± 0.2 | 12 ± 1 | 1286 ± 18 | 47.1 ± 0.4 | 88 ± 1 |
| ATP-bound | 369 ± 11 | 32.5 ± 0.2 | 40 ± 4 | 1212 ± 11 | 45.6 ± 0.2 | 60 ± 4 |
| MgATP | 414 ± 13 | 33.3 ± 0.2 | 29 ± 2 | 1282 ± 19 | 47.0 ± 0.4 | 71 ± 2 |
| Vi | 439 ± 16 | 33.7 ± 0.3 | 49 ± 4 | 1088 ± 34 | 43.6 ± 0.5 | 51 ± 4 |
| NT Pgp in NDSCs in the presence of 30 μm verapamil (Ver; n = 5) | ||||||
| Apo | 373 ± 11 | 32.6 ± 0.2 | 12 ± 1 | 1265 ± 37 | 46.7 ± 0.8 | 88 ± 1 |
| Ver-bound | 389 ± 16 | 32.9 ± 0.3 | 17 ± 1 | 1136 ± 13 | 44.3 ± 0.2 | 83 ± 1 |
| Ver/ATP-bound | 349 ± 20 | 32.1 ± 0.4 | 24 ± 3 | 1150 ± 11 | 44.5 ± 0.2 | 76 ± 3 |
| Ver/MgATP | 394 ± 17 | 33.0 ± 0.3 | 39 ± 4 | 1164 ± 33 | 44.8 ± 0.6 | 61 ± 4 |
| Ver/Vi | 430 ± 28 | 33.6 ± 0.5 | 46 ± 6 | 1119 ± 32 | 44.0 ± 2.0 | 54 ± 6 |
It is important to note that the shorter separation between the NBDs in our studies is not the result of confining the protein in the NDSCs (∼110-Å diameter), because it was also observed in MsbA reconstituted in unilamellar liposomes of ∼1,000-Å diameter (18). Therefore, an important finding of this study is that reconstitution of Pgp in a lipid bilayer constrains its conformations and results in a more compact protein, in agreement with previous findings on MsbA (18). It is also important to stress that the standard deviations below 1 Å (Table 1) do not represent the error in the calculated versus “real” distances, but they are the result of the very high experimental reproducibility of LRET with atomic resolution. There is an error when the donor/acceptor distance is assumed to correspond to the distance between the reacted thiols in the Cys side chains (unavoidable uncertainty about the position of probes due to their length). However, measurements by others and us have shown donor/acceptor LRET distances very close to those expected from crystal structures (16, 41–43). Even though the size of the optical probes can introduce errors in the donor/acceptor versus the Cys side-chain absolute distances, LRET, as FRET, is an excellent method to determine distance changes. Considering the free mobility of the optical probes in LRET and that we positioned them outside the active NBD interface, in a region that does not experience conformational changes during the nucleotide binding/hydrolysis cycle, LRET seems more than adequate to address domain movements in Pgp, and it seems a good indicator of the changes in distance under different conditions. In any case, the important point is that the maximal difference between states can be as small as ∼10 Å (see below).
Conformational changes in the NBDs during Pgp activation by verapamil
The ATPase activity of Pgp in detergent/lipid micelles or NDSCs is stimulated 5–10-fold by Ver (supplemental Fig. 2). It has been proposed that the ATPase activity is stimulated because binding of transport substrate in the transmembrane region causes conformational changes that are transmitted to the NBDs, promoting their dimerization (51, 52). Here, we tested this possibility with studies of the NBD–NBD interactions in NT Pgp reconstituted in NDSCs. Fig. 4A shows typical emission spectra of donor/acceptor-labeled NT Pgp in NDSCs under experimental conditions similar to those in Fig. 3A but in the Ver-bound state (30 μm Ver was added to the NT Pgp in the apo-state). Transition from the apo-state (Fig. 4A, black) to the Ver-bound state (Fig. 4A, red) increased the LRET intensity, but subsequent addition of NaATP had little effect (Fig. 4A, blue, Ver/ATP-bound). This is in contrast to the clear increase observed in the transition from the apo-state to the ATP-bound state in the absence of Ver (Fig. 3A, black versus blue). Transition to the MgATP state in the presence of Ver increased LRET intensity (Fig. 4A, orange, Ver/MgATP), in marked contrast to the magnesium-induced decrease in LRET intensity in the absence of Ver (Fig. 3A, orange versus blue). The effect of Ver was also observed in experiments performed in reversed order, where addition of Ver to NT Pgp-NDSCs actively hydrolyzing MgATP increased the LRET intensity to a value similar to that in the Ver/MgATP state in Fig. 4A (supplemental Fig. 5). The effect of Ver on Pgp was fast, as can be seen in the time course of the LRET intensity changes in Fig. 4B. Transition from the Ver-bound state to the Ver/ATP-bound state produced little change in intensity, whereas transition to the Ver/MgATP state after addition of MgSO4 produced a rapid increase in intensity, to a level close to the maximum reached after inhibition of ATP hydrolysis by Vi (Vi state). Again, the increase in LRET with the transition from the Ver/ATP-bound state to the state during continuous ATP hydrolysis (Ver/MgATP) is in clear contrast with the decrease in LRET intensity when hydrolysis starts in the absence of Ver (Fig. 3C).
Figure 4.
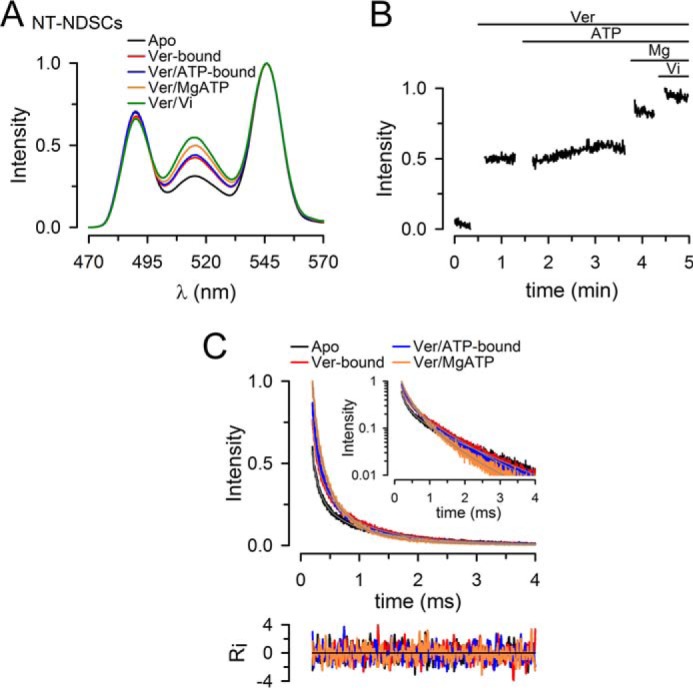
Conformational changes during the ATP hydrolysis cycle of NT Pgp in NDSCs at 37 °C in the presence of verapamil. A, emission spectra from NDSCs containing NT Pgp labeled with donor (Tb3+ chelate) and acceptor (Tb3+ chelate and Bodipy FL). B, time course of the changes in Bodipy FL-sensitized emission in response to sequential additions of Ver, NaATP, MgSO4, and Vi. Intensity was normalized to the maximum obtained after addition of Vi. C, Bodipy FL sensitized emission decays, recorded at 520 nm, from NT Pgp in NDSCs. Decays were normalized to the emission of the Ver/Vi state at 200 μs. All the traces shown in these figures are representative of at least five independent experiments and were obtained at 37 °C. Apo, nucleotide- and drug-free buffer with 1 mm EDTA; Ver, +30 μm verapamil; Ver/ATP-bound, +5 mm NaATP; Ver/MgATP, +10 mm MgSO4; Ver/Vi, +0.25 mm Vi. See legend to Fig. 3 for details.
The LRET decays showed higher intensities and faster decays for the Ver-bound (Fig. 4C, red) and Ver/ATP-bound (Fig. 4C, blue) NT Pgp-NDSCs compared with the apo-state (Fig. 4C, black), and the intensity and speed of decay increased further in the Ver/MgATP state (Fig. 4C, orange). The analysis of the data (Table 1) indicates that the increase in LRET intensity in the presence of Ver was the result of a combination of two changes: shortening of the longer distance d2 by 2.2 ± 0.3 Å (p < 0.003) and increase in the % molecules displaying closed NBD dimers (d1) by 5 ± 1% (p < 0.003). The distance d2 in the Ver-bound state (∼44 Å) was maintained in the Ver/ATP, Ver/MgATP, and Ver/Vi states and might represent a semi-open conformation similar to that in the Vi state in the absence of Ver (Table 1).
Unexpectedly, transition from the Ver-bound state to the Ver/ATP-bound state had only a small effect on % of molecules with closed NBD dimers (Table 1 and Fig. 5A). However, the % of molecules displaying the distance d1 did increase in the Ver/MgATP state to ∼40% (Table 1 and Fig. 5A). This result is in marked contrast with the results in the absence of Ver, where the transition from the ATP-bound to the MgATP state was accompanied by a decrease in the % of molecules with donor/acceptor pairs showing distance d1 (Table 1 and Fig. 5A). Hence, ATP hydrolysis induced by addition of Mg2+ results in different conformational changes in the ATP-bound state (absence of transport substrate) and Ver/ATP-bound state (basal versus Ver-stimulated hydrolysis). In summary, under steady-state ATP hydrolysis Ver promoted the formation of the closed NBD dimer conformation (∼40 versus ∼30% of the molecules) and reduced the separation between the NBDs in the open conformation (Table 1 and Fig. 5A).
Figure 5.
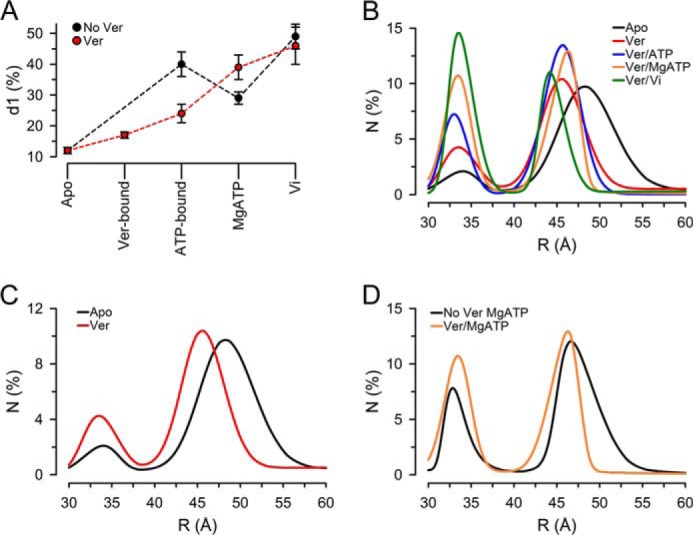
Distance distributions of NT Pgp in NDSCs at 37 °C in different states during the hydrolysis cycle. A, percentage of NT Pgp molecules displaying the shorter distance (d1, ∼33 Å). Means ± S.E. in the absence (No Ver; n = 7) and presence of Ver (n = 5) were calculated from the multiexponential fits results (Table 1) as the fractional intensity contribution of each exponential component divided by the rate of energy transfer (k = 1/τDA − 1/τD). B, distance distributions calculated from LRET-sensitized emission intensity decays analyzed by an exponential series method. N, % molecules. The percentage of molecules in each conformation, calculated as described in the legend to A, was adjusted based on the cumulative number of molecules versus donor/acceptor pair distance obtained from the exponential series method analysis. See “Experimental procedures” and supplemental Fig. 8 for details. Solid lines correspond to the best fits obtained using Peak Fit (Systat Software Inc., San Jose, CA). Apo, nucleotide- and drug-free buffer with 1 mm EDTA; Ver, +30 μm Ver; Ver/ATP-bound, +5 mm NaATP; Ver/MgATP, +10 mm MgSO4; Ver/Vi, + 0.25 mm Vi. C, comparison of distance distributions in the apo-state (No Ver) and in the presence of Ver (Ver). D, comparison of distance distributions during hydrolysis in the absence (MgATP) and presence of Ver (Ver/MgATP). Data from NT Pgp in NDSCs at 37 °C were used for all calculations. The curves in B--D are averages from five independent experiments.
Distribution of Pgp conformations during the hydrolysis cycle
Fig. 5B shows the average distribution of distances calculated from the LRET-sensitized emission decays of NT Pgp NDSC in the presence of Ver using an exponential series method (see “Experimental procedures”). Equivalent data in the absence of Ver can be found in supplemental Fig. 6. The exponential series method analysis can recover lifetime distributions (which can be converted to distance distributions) without assumptions about the distributions shapes, and it can therefore be used to discriminate between discrete and continuous distributions (18, 53, 54). Clear peaks centered at distances that approximate d1 and d2 obtained from the multiexponential fits (Table 1) confirmed the presence of two discrete conformations in NT Pgp-NDSCs under all experimental conditions studied (Fig. 5B): the first peak corresponding to the “closed NBD” conformation d1, and the second peak to the “open NBD” conformation d2. The analysis suggests the predominance of well-defined conformations of Pgp as opposed to a broad distribution of conformations.
Fig. 5B also illustrates the shift of the d2 conformation peak toward shorter distances in the presence of Ver (Fig. 5C), as suggested by the discrete multiexponential fittings discussed above. Particularly interesting is the observation that NT Pgp in NDSCs is more compact during hydrolysis in the presence of Ver (compare black and orange traces in Fig. 5D); 90% of the d2 Ver/MgATP molecules were within ∼7 Å (42–49 Å), and only 65% were in the absence of Ver.
Effects of temperature
The ATPase activity of Pgp is highly sensitive to temperature (55). Here, we focused on the conformational changes of NT Pgp in NDSCs at 20 °C. Fig. 6A shows representative emission spectra of NT Pgp-NDSCs where it is clear that the LRET intensity at 515 nm was lower than at 37 °C (compare with Figs. 3A and 4A). The calculated distances d1 and d2 in the apo-state at 20 °C were similar to those determined at 37 °C (32.6 ± 0.8 and 48.7 ± 3.1 Å, respectively; n = 3). Very different from NT Pgp in NDSCs at 37 °C (Fig. 3A), the responses to addition of ATP and MgSO4 at 20 °C were absent, although incubation with Vi for ∼10 min produced a significant increase in the LRET intensity (Fig. 6A, green). Because Vi trapping of Pgp requires NBD dimerization and MgATP hydrolysis, the slow increase in LRET intensity coincides with the lower basal ATPase rate of Pgp at 20 °C versus 37 °C (55). These results indicate that even in a lipid bilayer, at lower temperatures in the absence of substrate Pgp does not experience significant conformational changes during the catalytic cycle. The emission spectra in Fig. 6B illustrate the effect of Ver on NT Pgp-NDSCs at 20 °C. Similar to the effect at 37 °C (Fig. 4A), there was an increase in LRET intensity with the transition from the apo-state (Fig. 6B, black) to the Ver-bound state (Fig. 6B, red). Also similar to the responses at 37 °C, the LRET intensity remained essentially unchanged after addition of ATP (Fig. 6B, blue; Ver/ATP-bound state), but it increased significantly in the Ver/MgATP state (Fig. 6B, orange), and even more in the Vi state (Fig. 6B, green). The steady-state increase in LRET intensity was reached rapidly, consistent with the relatively high Ver-stimulated ATPase activity of NT Pgp-NDSCs at 25 °C (1.6 ± 0.1 μmol/min/mg). The changes observed for the Ver-bound NT Pgp-NDSCs at 20 and 37 °C were similar, although the average % of molecules displaying the shorter distance d1 was consistently lower at 20 °C (apo-state, 10 ± 2%; Ver-bound state, 12 ± 1%; Ver/ATP-bound state, 12 ± 1%; Ver/MgATP state, 27 ± 3%; Ver/Vi state, 37 ± 5%; n = 3; compare with Table 1). The similar effect of Ver at 20 and 37 °C is consistent with the high degree of ATPase stimulation observed at both temperatures (5- and 7-fold, respectively). These results suggest that although less efficiently than at physiological temperature, at 20 °C Ver activates the ATPase activity of Pgp in NDSCs and also shifts the dynamic equilibrium toward the closed NBD dimer conformation.
Figure 6.
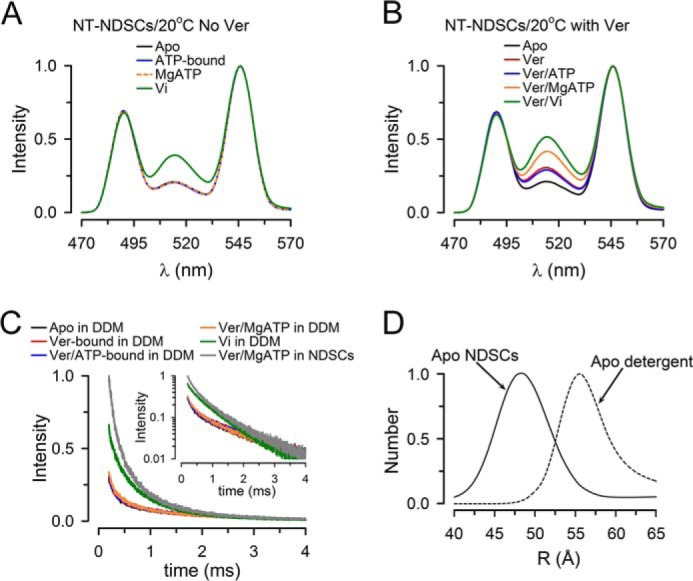
Conformational changes during the ATP hydrolysis cycle of NT Pgp in NDSCs at 20 °C. A, emission spectra from NDSCs containing NT Pgp measured in the absence of Ver. Apo, nucleotide- and drug-free buffer with 1 mm EDTA; ATP-bound, +5 mm NaATP; MgATP, +10 mm MgSO4; Vi, +0.25 mm Vi. B, emission spectra from NDSCs containing NT Pgp measured in the presence of Ver. Apo, nucleotide- and drug-free buffer with 1 mm EDTA; Ver, +30 μm Ver; Ver/ATP-bound, +5 mm NaATP; Ver/MgATP, +10 mm MgSO4; Ver/Vi, +0.25 mm Vi. C, Bodipy FL sensitized emission decays from NT Pgp in detergent (0.1% DDM) or NDSCs. The inset displays the same curves in a semi-log scale. Decays were normalized to the emission at 200 μs of NT Pgp in NDSCs in the Ver/MgATP state. D, distribution of Pgp molecules displaying the d2 distance (longer distance) determined at 20 °C in the apo-state of NT Pgp in DDM and in NDSCs. The y axis represents the relative number of molecules displaying the d2 distance. All the traces shown in these figures are representative of at least three independent experiments and were obtained at 20 °C. Emission was recorded after a 200-μs delay from the 337-nm excitation pulse. See legend to Fig. 3 for details.
Conformational changes of Pgp in nanodiscs versus detergent
Here, we studied the conformational changes of Pgp in detergent at 20 °C. Although Ver was present, the LRET decay curves in the apo, Ver-bound, Ver/ATP-bound, and Ver/MgATP states were indistinguishable (Fig. 6C, black, red, blue, and orange, superimposed lower records), and they only changed after incubation with Vi for several minutes (Fig. 6C, green; recorded after a 20-min incubation with Vi). For comparison, the LRET intensity at 20 °C in the Ver/MgATP state of NT Pgp-NDSCs was much larger (Fig. 6C, gray). These results indicate that the effect of Ver on Pgp conformation requires the presence of lipids, as has been previously suggested (56). Analysis of the LRET decays of NT Pgp in detergent at 20 °C pointed to two coexisting conformations with a short distance of ∼33 Å and a long distance of ∼56 Å. The first distance is similar to those for NT Pgp-NDSCs at 20 and 37 °C; this is not surprising because it corresponds to the closed NBD dimer. The longer distance, however, was significantly longer than that calculated for NT Pgp in NDSCs at the same temperature. This is in agreement with our previous findings of a more compact conformation for MsbA in a lipid bilayer than in detergent (18). The longer average distance for NT Pgp in detergent is likely to underestimate the actual value because the calculated distance of ∼56 is ∼15 Å away from the Tb3+/Bodipy FL Förster distance, with E ∼0.15 (see supplemental Fig. 4). The difference between NT Pgp in detergent and nanodiscs is clearly illustrated by the d2 distance distributions in Fig. 6D.
The importance of lipids to make Pgp more compact was also apparent in experiments where we added Escherichia coli lipids to NT Pgp in detergent at 20 °C in the apo-state. In these experiments the low intensity, slow decay measured in NT Pgp in detergent (Fig. 6C, black) was gradually replaced with faster decays as the amount of lipids increased (supplemental Fig. 7). Analysis of the LRET-sensitized emission decays suggested that the effect of lipid addition was mostly the result of a decrease in the longer donor/acceptor distance. This suggests that the lipids help stabilize and activate Pgp by promoting NBD association even in the presence of detergent. Significant light scattering prevented detailed studies of the conformational changes under different conditions, which points to the limitation of mixed detergent/lipid micelles and highlights one of the advantages of the use of soluble NDSCs.
Discussion
Translocation of substrate by ABC exporters is believed to occur when the proteins switch from an inward- to an outward-facing conformation (57–59). However, it is unclear whether the alternated accessibility of the binding pocket is the result of large conformational changes that include association/dissociation of the NBDs, where the NBDs can be tens of Å apart (monomer/dimer models) (30–33, 60, 61), or smaller conformational changes at the NBD–NBD interface, with the NBDs remaining associated during the transport cycle (constant-contact models) or in close proximity (34–38, 60, 62). The main evidence supporting a large distance between the NBDs of Pgp in the apo-state comes from X-ray crystal structures in the inward-facing conformation (6–10, 50), which may not be physiologically relevant (17, 18, 25, 37, 60, 63, 64). Therefore, we decided to investigate the degree of separation of the NBDs during the ATP-hydrolysis cycle of Pgp using LRET under near-physiological conditions: using a fully-active Pgp mutant reconstituted in a lipid bilayer and studied at 37 °C in the absence and presence of a transport substrate, including studies during hydrolysis.
Under all conditions studied, in the absence or presence of transport substrate, in detergent or reconstituted in NDSCs, at 20 or 37 °C, Pgp adopted two dominant conformations. One of the Pgp conformations displays a donor/acceptor distance of ∼33 Å (d1), and the other conformation displays a donor/acceptor distance that was longer than d1 (d2) by an average of ∼10 to ∼25 Å, depending on the experimental conditions. It seems reasonable to assume that d1 corresponds to the closed NBD dimer because it is close to the distance expected between the probes in the nucleotide-bound Pgp model, where the distance between the α carbons of Asn-607 and Thr-1252 is ∼36 Å (Fig. 1) (46). This close correspondence between Cys α carbons and calculated LRET distance (within 3 Å) is a common finding for LRET-based measurements, and it is likely the result of the position of the probes on the outside of the NBD structure, away from other areas of the Pgp, and the unpolarized long lifetime of the LRET-sensitized emission that allows the probes to sample all orientations, centered close to the α carbon (16, 40, 42, 43).
The longer d2 (Δd2-d1 ∼25 Å) was calculated for Pgp in detergent, at 20 °C, in the absence of Ver. The NBD–NBD separation in detergent and in the absence of transport substrate is consistent with that in crystal structures (6, 7). The shorter d2 (Δd2-d1 ∼10 Å) was calculated for Pgp in NDSCs in the presence of Ver and at 37 °C, and it is compatible with NBDs loosely associated or barely dissociated. These data are in agreement with an electron microscopy study of Pgp in two-dimensional lipid crystals showing NBDs close to each other even in the absence of nucleotide (25). A close proximity between the “dissociated” NBDs was also found for MsbA in NDSCs versus detergent (18). Together, the data suggest that the large separation of the NBDs in the crystal structures is the result of studying the proteins under non-physiological conditions and that in a lipid bilayer Pgp adopts a partially-open conformation with NBDs that are never far apart.
Although the inward-facing open conformation with widely separated NBDs has been observed in structures such as those of MsbA and Pgp (6, 7, 65), its physiological relevance is unclear (18, 64). A recent study of Pgp using DEER spectroscopy agrees with the view of a large separation between the NBDs in the apo-state (22). In this study, the protein was studied “locked” in different states during the hydrolysis cycle. Although we do not know the origin of the differences with our work, there are several potential reasons besides the methodologies employed. One is the study of Pgp in NDSCs versus detergent/lipid micelles. We and others have shown that Pgp in detergent/lipid micelles behaves closer to Pgp in a lipid bilayer than the protein in detergent, but it still shows significantly lower affinity for substrates (supplemental Fig. 2) (66). Another difference is the CL Pgp version used. The CL Pgp used in the DEER spectroscopy work is an older version that we no longer use because of its reduced stability and profound alterations in drug transport ability (44, 45). In this work, we used our newer CL Pgp, generated by directed evolution, which has wild-type-like properties (44). Also, the drug-stimulated activity reported in the DEER study was low (22), varying between ∼3 and 20% of the values we report here. One of the most striking differences, however, is the absence of significant conformational changes in all the conditions studied, except for the Vi state (22). This is in sharp contrast with our studies, where we found differences between all states with Pgp in NDSC at 37 °C; the results in the DEER study are reminiscent of our finding of Pgp in detergent or in NDSCs at low temperature in the absence of substrate (see Fig. 6).
Recently, we found that the degree of the NBD–NBD separation in the bacterial Pgp homolog MsbA was significantly smaller than that predicted from the X-ray crystal structures (16, 18). More importantly, we found significant differences between MsbA reconstituted in a lipid bilayer and MsbA in detergent (16, 18). The NBDs of MsbA in NDSCs barely dissociate if they do at all, and ∼50% of the molecules displayed closed NBD dimers in the apo-state (18). We suggested that the structural differences between the crystal structures of MsbA and MsbA in a lipid bilayer were the result of the presence of the membrane (18). Similarities between the related exporters Pgp and MsbA are expected; however, mechanistic differences are also possible. For example, MsbA has a high basal ATPase activity that is only marginally stimulated by the transport substrate lipid A (67–69), a behavior very different from that of Pgp (70), making extrapolation of MsbA data to Pgp uncertain. The fact that Pgp and MsbA reconstituted in a lipid bilayer share a similar separation of the NBDs in the open conformation (d2) is not surprising considering that they are homologous ABC exporters that can transport similar substrates (71). However, there were significant differences between the two proteins in the relative proportion of molecules that adopt the closed NBD dimer conformation versus the open NBD conformation. In the ATP-bound state ∼70% of MsbA molecules display closed NBD dimers (18), whereas under identical experimental conditions only ∼40% of Pgp molecules adopted that conformation. The difference was even more pronounced in the apo-state, where the % of MsbA and Pgp molecules displaying closed NBD dimers were ∼50 and ∼10%, respectively. These observations are consistent with the lower probability of finding Pgp in the outward-facing conformation in an electron microscope study of MsbA and Pgp stabilized with amphiphiles (27). Mass spectrometry data also suggest that Pgp exists predominantly in the inward-facing conformation (23). It has been proposed that the preference of Pgp for the inward-facing conformation when compared with Sav1866 (34) is the result of a relatively larger hydrophobicity of the drug-binding pocket and an increased charge density of the NBD interface (46).
One unique aspect of our studies is that we could observe conformational changes of Pgp reconstituted in a bilayer while it is hydrolyzing ATP at 37 °C and in the absence or presence of the transport substrate Ver. During ATP hydrolysis in the presence of Ver, Pgp adopted a more compact NBD conformation than in the absence of Ver, and it also displayed a higher % of molecules with closed NBD dimers (d1 ∼40%), the closest to that of the Vi state. Because the Vi state could correspond to a semi-open asymmetric NBDs dimer, with MgADP·Vi trapped in one closed site and the other site open (57), given the similarity between the Ver/MgATP and Vi states, it seems possible that a similar conformation is predominant in the Ver/MgATP state during Ver-stimulated hydrolysis. An alternative explanation is that the results arise from a mixture consisting of Pgp molecules with closed NBD dimers and Pgp molecules with partially-dissociated NBDs. However, this last possibility will be harder to reconcile with the ability of Pgp to trap nucleotides with a stoichiometry of one per Pgp in the Vi state with nearly complete inhibition of ATPase activity (69). Unfortunately, there is no available structural information for Pgp or other ABC exporters in an asymmetric dimer conformation with nucleotide trapped in only one site. Such structural information would allow us to determine whether the short d2 measured in the Ver/MgATP and Vi states could correspond to an asymmetric dimer.
In general, our results agree with Cys cross-linking studies that showed that the C-terminal ends of the two Pgp NBDs do not need to separate significantly during the drug-stimulated catalytic cycle (51) and that Ver activates ATP hydrolysis by bringing the NBDs together (52). Our data also agree with a FRET study of Pgp in liposomes that included single-molecule recordings (17), where it was proposed that the Ver-stimulated catalytic cycle proceeds via a series of relatively small steps. Our results also suggest that the mechanisms of basal and drug-stimulated ATPase activities are different, consistent with biochemical studies (57), and that drugs stimulate Pgp ATPase activity by a combination of reducing the NBD–NBD distance and increasing the % of the molecules with closed NBD dimers. It seems likely that during Ver-stimulated ATP hydrolysis the NBDs do not dissociate completely (or barely dissociate), as observed during ATP hydrolysis in the absence of drug.
The existence of effects of Ver binding on the NBDs is in agreement with prior reports (52, 72–74). The conformational effects resulting from Ver binding to Pgp were observed at 20 and 37 °C but required reconstitution in a lipid bilayer. Transition from the apo-state to the Ver-bound state did not produce noticeable effects when Pgp was studied in detergent micelles. This observation agrees with data showing that Pgp responds to vinblastine and Ver when it is reconstituted in liposomes but not when it is in detergent (56). Possible interpretations include masking of the substrate-binding pocket by detergent and structural alterations that prevent the conformational changes needed for substrate-induced activation. Recent results showed a major effect of the Pgp substrate-binding pocket environment on the response to ligand binding (66). In that study, Pgp in n-dodecyl-β-d-maltopyranoside (DDM) displayed reduced affinity for Ver compared with the protein in liposomes, and it even turned high-affinity inhibitors such as tariquidar into low-affinity activators (66). In our hands, the EC50 value for stimulation of Pgp ATPase activity by Ver in the NDSCs was low (1.5 ± 0.1 μm; n = 3; see supplemental Fig. 2) and similar to that reported in membranes and proteoliposomes (66). It has also been suggested that detergent partially unfolds Pgp, reducing the coupling between the drug-binding sites and the catalytic domains (75). Addition of phospholipids increases the thermal stability of Pgp (70) and appears to assist Pgp refolding into a more native conformation in which coupling between the drug-binding site and the NBD is restored (73). A recent cross-linking study in Pgp has suggested that the presence of lipids is important for the communication between the NBDs and transmembrane domains required for activation of the ATPase activity (76). Our results suggest that addition of lipids to the detergent-solubilized Pgp promoted a conformation with closer NBDs. The lack of conformational effects in response to drug binding without lipids could explain the absence of structural changes in the crystal structures of drug-free versus drug-bound Pgp (6).
In summary, our results reconcile different models that have been proposed to explain the mechanism of Pgp and present new structural information that contributes to our understanding of the molecular mechanism of stimulation of the Pgp ATPase activity by transport substrates. We found that in the absence of transport substrate Pgp operates in a monomer/dimer mode. However, the separation between the NBDs is smaller than that found in crystal structures (6–10). Comparison of data from Pgp in NDSCs versus detergent suggests that the inward-facing conformation in the crystal structures does not correspond to a physiological conformation and is, at least in part, the result of the absence of the membrane. However, at this point we cannot rule out larger NBD–NBD separations under conditions such as binding of large substrates (e.g. β amyloid). In the presence of substrate, Pgp seems to switch to a mode where the NBDs remain either associated (asymmetric dimer) or in close proximity during the hydrolysis cycle, which could account for the drug-stimulated ATPase activity. It is tempting to speculate that the monomer/dimer mechanism that would take place when Pgp displays basal ATPase activity switches in the presence of Ver to a constant-contact mode of operation or a mode where the NBDs separate less. In this mode, the NBDs would never dissociate completely, or would barely dissociate, which could account for the drug-stimulated ATPase activity.
Experimental procedures
Construction of Pgp mutants
For expression in Saccharomyces cerevisiae, codon-optimized mouse Pgp (abcb1a, mdr1a, GenBankTM JF834158) in the pVT expression vector (pVT-opti-mdr3) was used as a template to generate a fully-functional CL Pgp (44, 70). A tobacco etch virus (TEV) protease site (ENLYFQ), a FLAG epitope (DYKDDDDK), and a His6 tag were added to the Pgp C terminus immediately downstream of the mdr1a ORF. The TEV, FLAG, and His tag sequences underlined in the 5′ to 3′ sense strand, in order, were 5′-CTCGAGGAAAACTTGTACTTCCAGGGTGGTGGAGGTTCTGGTGGTTCTGACTACAAGGATGACGATGACAAGGGCGCCTCTGGTGGTTCTCACCACCATCACCACCATTGA-3′. A wild-type Pgp (WT Pgp) plasmid containing the same TEV, FLAG, and His tag sequences was also constructed. The new CL Pgp construct was used as a template to generate two mutants by directed recombination-assisted mutagenesis (77), N607C/T1252C Pgp (NT Pgp) and NT/E1197A. The former is a catalytically-active double-Cys mutant, and the latter is an NBD2 catalytic carboxylate mutant (E1197A) based on NT Pgp. Using the last mutant as template, PCR-based mutagenesis was used to mutate the NBD1 catalytic carboxylate (Glu-552) to generate the catalytically-inactive NT/E552A/E1197A mutant (NT/AA Pgp). Plasmid sequences were confirmed by DNA sequencing. For expression in Pichia pastoris, DNA sequences coding for CL Pgp, NT Pgp or NT/AA Pgp were subcloned into the pLIC-CL-opti-mdr3-His6 vector as described (70), and the FLAG epitope was replaced by a double Strep II tag (WSHPQFEK) that is more cost-effective for large-scale purifications.
Drug resistance assay
Plasmids were transformed into S. cerevisiae strain JP201, and drug resistance assays were performed as described previously (70, 78).
Pgp expression and purification
For purification from S. cerevisiae, plasmids were transformed into the BY4743 strain (79). Fourteen-liter cultures were grown in a New Brunswick BioFlow IV fermentor to a maximal A600 of 4.0, which produce 100–120 g of cells. Microsomal membrane preparation, Pgp solubilization in 0.6% DDM and purification based on the affinity of the Pgp His6 tag for Ni-NTA were performed as described (44, 70). Pgp was eluted from the Ni-NTA resin with buffer A (250 mm NaCl, 10% glycerol, 0.1% DDM, 1 mm β-mercaptoethanol, 0.1 mm tris(2-carboxyethyl) phosphine (TCEP), and 50 mm Tris/HCl, pH 7.4) containing 200 mm imidazole. A second purification step was performed by binding the imidazole eluate containing Pgp to an anti-FLAG M2 affinity gel (A2220, Sigma), with elution by competition in buffer A containing 100 μg/ml FLAG peptide; the reducing agent TCEP was omitted from all anti-FLAG gel buffers. Pgp concentrated to ∼1.5 mg/ml using 100-kDa cutoff centrifugal filter units (Amicon, EMD Millipore, Billerica, MA) was stored in aliquots at −80 °C until used. Protein concentration was calculated from the absorbance at 280 nm using a Pgp molar extinction coefficient of 109,750 m−1 cm−1.
For larger-scale expression we used the P. pastoris strain KM71H. Cells transformed with the corresponding expression plasmids were grown in 6–8-liter fermentor cultures and were induced with methanol (44, 70, 80). The first purification step was based on Pgp affinity for the Ni-NTA resin (see above). Pgp was subjected to an additional purification step consisting of binding to a streptactin superflow resin (Qiagen, Valencia, CA) in buffer A with 1 mm DTT, with elution by competition with 2.5 mm desthiobiotin. We did not detect biochemical differences between Pgp expressed and purified from S. cerevisiae and P. pastoris expression/purification tag systems.
Labeling of Pgp and reconstitution into nanodiscs
Purified CL Pgp, NT Pgp, and NT/AA Pgp mutant proteins were first reduced with 1 mm DTT for 15 min on ice, followed by removal of most DTT using 1-ml Sephadex G-50 spin columns (81) or Zeba columns (Thermo Fisher Scientific, Bellafonte, PA) pre-equilibrated in buffer A without TCEP. Pgp was then labeled with a 2-fold molar excess (2:1 dye/Cys) of both donor (Tb3+–chelate diethylenetriaminepentaacetate–cs124–ϵ-maleimidocaproic acid hydrazide; thiol-reactive Lanthascreen, Life Technologies, Inc.) (82) and acceptor (Bodipy FL maleimide, Life Technologies, Inc.) for 1 h at room temperature. For some experiments, NT Pgp was labeled only with donor or acceptor. Bodipy FL/Pgp stoichiometry, estimated from the absorbances of Pgp (280 nm) and Bodipy FL (504 nm; using a Bodipy FL molar extinction coefficient of 84,000 m−1 cm−1) was ∼0.6 mol/mol when NT Pgp was labeled with donor and acceptor and or ∼1.2 when it was labeled only with Bodipy FL. Compared with NT Pgp and NT/AA Pgp, CL Pgp showed essentially no labeling under the conditions described above, consistent with our previous studies (44). For LRET experiments of Pgp in detergent, the unreacted free labels were removed by size-exclusion chromatography on a Superdex 200 column (3.2/30 mm, GE Healthcare) equilibrated with buffer A. Alternatively, labeled Pgp was reconstituted in NDSCs following previously described procedures (18, 39). Pgp in detergent micelles has very low ATPase activity at 20–25 °C, and its activity at 37 °C decreases rapidly (t½ ∼5 min). Supplementation of the detergent-solubilized protein with lipids increases the thermostability of Pgp (supplemental Fig. 3) (70), but light scattering hinders the detection of fluorescence changes. Therefore, we decided to study Pgp in lipid NDSCs using LRET, an approach that we used successfully to study MsbA (16, 18). For reconstitution in NDSCs, labeled Pgp, E. coli polar lipids (Avanti Polar Lipids, Alabaster, AL), and the membrane scaffold protein MSP1E3D1 were mixed (at a molar ratio of 1:6 Pgp/MSP and 1:110 MSP/lipids). The mixture was incubated for 1 h at 4 °C, and then the detergent was removed by overnight incubation with Bio-Beads SM-2 (Bio-Rad). The NDSC sample was run on a Superdex 200 column (10/300 mm) equilibrated with detergent-free NDSC buffer (150 mm NaCl, 0.1 mm TCEP, 50 mm Tris/HCl, pH 7.4) to remove free unreacted labels and to isolate a fraction enriched in Pgp-loaded NDSCs (Fig. 2B, black). For some experiments, Pgp-NDs were further separated from empty NDSCs by anti-FLAG affinity chromatography as described above, except that detergent-free NDSC buffer was used for binding, washing, and elution (Fig. 2B, red). Pgp concentration in the NDSC preparation was determined in SDS-PAGE gels stained with Coomassie Blue, using known amounts of detergent-solubilized Pgp as standard. As expected from the absence of Cys in MSP1E3D1 (Fig. 2C), the results using NT Pgp labeled with the LRET probes after reconstitution in NDSCs were identical to those obtained using NT Pgp labeled in detergent and then reconstituted in NDSCs.
LRET studies
For LRET studies, ∼1 μm CL Pgp, NT Pgp, or NT/AA Pgp subjected to the labeling protocol, in either detergent or NDSCs, was analyzed in 3-mm pathlength quartz cuvettes, essentially as described (16, 18, 40). The samples were first analyzed in nucleotide- and drug-free state (apo-state) using NDSC buffer with 1 mm EDTA to chelate trace divalent cations and to prevent ATP hydrolysis. Then the samples were analyzed after successive additions of NaATP (5 mm NaATP; ATP-bound state), MgSO4 (10 mm MgSO4; MgATP; during continuous ATP hydrolysis), and sodium orthovanadate (0.25 mm Vi; Vi state; post-hydrolysis intermediate state). We used 30 μm Ver to determine the effect of binding of transport substrate. Emission spectra (QM3SS spectrometer; Photon Technology International, London, Canada), LRET decays, and intensity time courses (EasyLife L phosphorescence lifetime photometer, Optical Building Blocks, NJ) were recorded as described (18, 40, 49, 83). Emission was acquired with a 200-μs delay from the beginning of the ∼1-μs excitation pulse from a xenon flash lamp (gated mode). This 200-μs delay allows for the selective recording of long-lifetime processes (donor emission or sensitized emission from the acceptor) after the short-lifetime processes (e.g. acceptor emission resulting from direct excitation and scattering of the excitation pulse) have largely disappeared. Excitation was set to 335 nm, and emission was collected through bandpass filters (Tb3+, 490/10 nm; Bodipy FL, 520/10 nm, Omega Optical, Brattleboro, VT) or an emission monochromator. As expected, control experiments mixing equal proportions of NT Pgp labeled with Tb3+-only and Bodipy FL-only showed essentially no intermolecular LRET. LRET experiments were performed at 37 °C unless stated otherwise.
LRET decay analysis
Donor/acceptor distances were calculated according to the following: E = 1 − τDA/τD, and R = R0 (E−1 − 1)1/6, where E is the efficiency of energy transfer; τD is the lifetime of the donor in the absence of acceptor; τDA is the sensitized emission lifetime (lifetime of the acceptor that arises from LRET); R is the donor/acceptor distance; and R0 is the Förster distance (the distance at which E = 0.5). The R0 determined for the Tb3+/Bopidy Fl pair was 41 Å, making this LRET pair very sensitive to distance changes in the ∼30–55 Å range (supplemental Fig. 4).
The donor-only decay from NT Pgp produced in S. cerevisiae, without the strep tag, or in P. pastoris, studied after removal of the tag with TEV protease, was indistinguishable. The Tb3+ emission decays recorded at 490 ± 10 nm and at 37 °C were well fitted by a two-exponential function with lifetimes of 722 ± 5 μs (τD1, 5% of the signal) and 1,850 ± 10 μs (τD2, 95% of the signal) (n = 6). This longest Tb-only lifetime (τD2) was used as the donor-only τD. The donor-only τD2 from NT Pgp with the strep-tag was shorter (1,517 ± 11 μs; n = 3) but returned to the normal value after removal of the tag with TEV protease. Therefore, for the LRET experiments the strep tag from NT Pgp expressed in P. pastoris was removed with TEV protease as described (84). At 20 °C τD2 lengthened to 2,204 ± 37 μs (n = 3).
The sensitized emission decays from Bodipy FL, recorded at 520 ± 10 nm, were well fitted by a three-exponential decay function. This function was chosen by the improving of the fitting from the two- to the three-exponential function, without significant additional improvement in the goodness of fit by fitting a four-exponential function to the data. The goodness of fit with the increase from two to four exponential components was assessed by the decreases in χ2 and the value of weighted residuals, a random distribution of weighted residuals, and a rapid decay of the autocorrelation of weighted residuals. Once χ2 did not change appreciably by increasing the number of parameters (fitting from three to four exponentials), we performed a one-sided F-test of sample variance (s2) with null (H0: s23-exp = s24-exp) and alternative (Ha: s23-exp > s24-exp) hypotheses. With s23-exp/s24-exp typically <1.005, Ha was rejected (p < 0.001). The fastest component of the three-exponential fits (τDA 100–200 μs) accounted for ≤5% of the signal and was disregarded for the calculations because it did not change appreciably under different experimental conditions, and it is largely the consequence of the instrument response time (16, 18, 40). The presence of only two relevant exponential decay components was also apparent from the analysis of lifetime distributions using an exponential series method designed to recover lifetime distributions without a priori assumptions about their shapes (see below) (53).
The percentage of donor/acceptor pairs with a discrete average distance (percentage Pgp molecules in each conformation) was estimated from the relative intensity of each lifetime component and the rate of energy transfer (k = 1/τDA − 1/τD) as described (16, 18, 85). Analysis of the distribution of lifetimes and distances was performed by an exponential series method (Felix 32 analysis software, Photon Technology International) designed to recover lifetime distributions without a priori assumptions about their shapes (53). We analyzed the decays using a series of 200 exponentials with fixed, logarithmically-spaced lifetimes and variable pre-exponentials. The use of logarithmically-spaced lifetimes assigns more points to shorter lifetimes, which can be visually misleading regarding the relative proportion of molecules in different conformations. To minimize this effect, areas under the peaks were normalized to the cumulative percentage of molecules, also calculated by the exponential series method. Average data without normalization, in the presence of Ver, and the corresponding percentage cumulative distances data are presented in supplemental Fig. 8.
ATPase activity assays
Purified Pgp (0.5–1.0 μg) in detergent was activated by incubation with 10 mm DTT and 1% (w/v) E. coli polar lipids for 15 min at room temperature. For Pgp in NDSCs, the protein (0.5–1.0 μg) was directly diluted into the assay mixture without DTT or lipid. The rate of ATP hydrolysis was measured at 37 °C, except as stated otherwise, by an ATPase-linked enzyme assay, as described (70), in the absence and presence of 30 μm Ver.
Author contributions
M. E. Z., L. M., I. L. U., and G. A. A. conceived and designed the study and analyzed and interpreted the data. I. L. U., D. J. S., L. M., and A. S. developed the expression, purification, and labeling protocols of highly active CL and NT Pgp and analyzed temperature-sensitive ATPase activity of detergent-soluble and reconstituted Pgp. G. A. F. made the NT/AA mutant. G. A. A., M. E. Z., and L. M. performed most LRET experiments and analyzed LRET data. M. E. Z., I. L. U., and G. A. A. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank the members of the Center for Membrane Protein Research (School of Medicine, Texas Tech University Health Sciences Center) and Dr. Qinghai Zhang (The Scripps Research Institute, La Jolla, CA) for critical discussions.
This work was supported in part by the Cancer Prevention and Research Institute of Texas Grant RP101073, National Institute of Health Grants RGM102928 and R01GM118594, and the South Plains Foundation from Lubbock, TX. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S8.
- Pgp
- P-glycoprotein
- ABC
- ATP-binding cassette
- CL
- Cys-less
- DDM
- n-dodecyl-β-d-maltopyranoside
- DEER
- double electron-electron resonance
- LRET
- luminescence resonance energy transfer
- MSP
- membrane scaffold protein
- NBD
- nucleotide-binding domain
- NDSC
- nanodisc
- NT
- N607C/T1256C Pgp mutant
- TCEP
- tris(2-carboxyethyl) phosphine hydrochloride
- Ver
- verapamil
- Vi
- sodium orthovanadate
- Bodipy FL
- N-(2-aminoethyl)maleimide
- PDB
- Protein Data Bank
- TEV
- tobacco etch virus
- Ni-NTA
- nickel-nitrilotriacetic acid.
References
- 1. Sharom F. J. (2011) The P-glycoprotein multidrug transporter. Essays Biochem. 50, 161–178 [DOI] [PubMed] [Google Scholar]
- 2. Gottesman M. M., and Ling V. (2006) The molecular basis of multidrug resistance in cancer: the early years of P-glycoprotein research. FEBS Lett. 580, 998–1009 [DOI] [PubMed] [Google Scholar]
- 3. Eckford P. D., and Sharom F. J. (2009) ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 109, 2989–3011 [DOI] [PubMed] [Google Scholar]
- 4. International Transporter Consortium, Giacomini K. M., Huang S. M., Tweedie D. J., Benet L. Z., Brouwer K. L., Chu X., Dahlin A., Evers R., Fischer V., Hillgren K. M., Hoffmaster K. A., Ishikawa T., Keppler D., Kim R. B., et al. (2010) Membrane transporters in drug development. Nat. Rev. Drug Discov. 9, 215–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee S. C., Arya V., Yang X., Volpe D. A., and Zhang L. (2017) Evaluation of transporters in drug development: current status and contemporary issues. Adv. Drug Deliv. Rev. 116, 100–118 [DOI] [PubMed] [Google Scholar]
- 6. Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., and Chang G. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li J., Jaimes K. F., and Aller S. G. (2014) Refined structures of mouse P-glycoprotein. Protein Sci. 23, 34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ward A. B., Szewczyk P., Grimard V., Lee C. W., Martinez L., Doshi R., Caya A., Villaluz M., Pardon E., Cregger C., Swartz D. J., Falson P. G., Urbatsch I. L., Govaerts C., Steyaert J., and Chang G. (2013) Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. U.S.A. 110, 13386–13391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jin M. S., Oldham M. L., Zhang Q., and Chen J. (2012) Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 490, 566–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Szewczyk P., Tao H., McGrath A. P., Villaluz M., Rees S. D., Lee S. C., Doshi R., Urbatsch I. L., Zhang Q., and Chang G. (2015) Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr. D Biol. Crystallogr. 71, 732–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hollenstein K., Dawson R. J., and Locher K. P. (2007) Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 17, 412–418 [DOI] [PubMed] [Google Scholar]
- 12. Loo T. W., Bartlett M. C., and Clarke D. M. (2010) Human P-glycoprotein is active when the two halves are clamped together in the closed conformation. Biochem. Biophys. Res. Commun. 395, 436–440 [DOI] [PubMed] [Google Scholar]
- 13. Loo T. W., and Clarke D. M. (2014) Identification of the distance between the homologous halves of P-glycoprotein that triggers the high/low ATPase activity switch. J. Biol. Chem. 289, 8484–8492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qu Q., and Sharom F. J. (2001) FRET analysis indicates that the two ATPase active sites of the P-glycoprotein multidrug transporter are closely associated. Biochemistry 40, 1413–1422 [DOI] [PubMed] [Google Scholar]
- 15. Borbat P. P., Surendhran K., Bortolus M., Zou P., Freed J. H., and Mchaourab H. S. (2007) Conformational motion of the ABC transporter MsbA induced by ATP hydrolysis. PLoS Biol. 5, e271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cooper R. S., and Altenberg G. A. (2013) Association/dissociation of the nucleotide-binding domains of the ATP-binding cassette protein MsbA measured during continuous hydrolysis. J. Biol. Chem. 288, 20785–20796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Verhalen B., Ernst S., Börsch M., and Wilkens S. (2012) Dynamic ligand-induced conformational rearrangements in P-glycoprotein as probed by fluorescence resonance energy transfer spectroscopy. J. Biol. Chem. 287, 1112–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zoghbi M. E., Cooper R. S., and Altenberg G. A. (2016) The lipid bilayer modulates the structure and function of an ATP-binding cassette exporter. J. Biol. Chem. 291, 4453–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zou P., Bortolus M., and McHaourab H. S. (2009) Conformational cycle of the ABC transporter MsbA in liposomes: detailed analysis using double electron-electron resonance spectroscopy. J. Mol. Biol. 393, 586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wen P. C., Verhalen B., Wilkens S., Mchaourab H. S., and Tajkhorshid E. (2013) On the origin of large flexibility of P-glycoprotein in the inward-facing state. J. Biol. Chem. 288, 19211–19220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Wonderen J. H., McMahon R. M., O'Mara M. L., McDevitt C. A., Thomson A. J., Kerr I. D., MacMillan F., and Callaghan R. (2014) The central cavity of ABCB1 undergoes alternating access during ATP hydrolysis. FEBS J. 281, 2190–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Verhalen B., Dastvan R., Thangapandian S., Peskova Y., Koteiche H. A., Nakamoto R. K., Tajkhorshid E., and Mchaourab H. S. (2017) Energy transduction and alternating access of the mammalian ABC transporter P-glycoprotein. Nature 543, 738–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marcoux J., Wang S. C., Politis A., Reading E., Ma J., Biggin P. C., Zhou M., Tao H., Zhang Q., Chang G., Morgner N., and Robinson C. V. (2013) Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc. Natl. Acad. Sci. U.S.A. 110, 9704–9709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee J. Y., Urbatsch I. L., Senior A. E., and Wilkens S. (2002) Projection structure of P-glycoprotein by electron microscopy. Evidence for a closed conformation of the nucleotide-binding domains. J. Biol. Chem. 277, 40125–40131 [DOI] [PubMed] [Google Scholar]
- 25. Lee J. Y., Urbatsch I. L., Senior A. E., and Wilkens S. (2008) Nucleotide-induced structural changes in P-glycoprotein observed by electron microscopy. J. Biol. Chem. 283, 5769–5779 [DOI] [PubMed] [Google Scholar]
- 26. Fribourg P. F., Chami M., Sorzano C. O., Gubellini F., Marabini R., Marco S., Jault J. M., and Lévy D. (2014) 3D cryo-electron reconstruction of BmrA, a bacterial multidrug ABC transporter in an inward-facing conformation and in a lipidic environment. J. Mol. Biol. 426, 2059–2069 [DOI] [PubMed] [Google Scholar]
- 27. Moeller A., Lee S. C., Tao H., Speir J. A., Chang G., Urbatsch I. L., Potter C. S., Carragher B., and Zhang Q. (2015) Distinct conformational spectrum of homologous multidrug ABC transporters. Structure 23, 450–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smith P. C., Karpowich N., Millen L., Moody J. E., Rosen J., Thomas P. J., and Hunt J. F. (2002) ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell 10, 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hopfner K. P., Karcher A., Shin D. S., Craig L., Arthur L. M., Carney J. P., and Tainer J. A. (2000) Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell 101, 789–800 [DOI] [PubMed] [Google Scholar]
- 30. Higgins C. F. (2007) Multiple molecular mechanisms for multidrug resistance transporters. Nature 446, 749–757 [DOI] [PubMed] [Google Scholar]
- 31. Janas E., Hofacker M., Chen M., Gompf S., van der Does C., and Tampé R. (2003) The ATP hydrolysis cycle of the nucleotide-binding domain of the mitochondrial ATP-binding cassette transporter Mdl1p. J. Biol. Chem. 278, 26862–26869 [DOI] [PubMed] [Google Scholar]
- 32. Vergani P., Lockless S. W., Nairn A. C., and Gadsby D. C. (2005) CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433, 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oswald C., Holland I. B., and Schmitt L. (2006) The motor domains of ABC-transporters. What can structures tell us? Naunyn Schmiedebergs Arch. Pharmacol. 372, 385–399 [DOI] [PubMed] [Google Scholar]
- 34. Dawson R. J., and Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 35. Jones P. M., and George A. M. (2007) Nucleotide-dependent allostery within the ABC transporter ATP-binding cassette: a computational study of the MJ0796 dimer. J. Biol. Chem. 282, 22793–22803 [DOI] [PubMed] [Google Scholar]
- 36. Jones P. M., and George A. M. (2011) Molecular-dynamics simulations of the ATP/apo-state of a multidrug ATP-binding cassette transporter provide a structural and mechanistic basis for the asymmetric occluded state. Biophys. J. 100, 3025–3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jones P. M., and George A. M. (2009) Opening of the ADP-bound active site in the ABC transporter ATPase dimer: evidence for a constant contact, alternating sites model for the catalytic cycle. Proteins 75, 387–396 [DOI] [PubMed] [Google Scholar]
- 38. Sauna Z. E., Kim I. W., Nandigama K., Kopp S., Chiba P., and Ambudkar S. V. (2007) Catalytic cycle of ATP hydrolysis by P-glycoprotein: evidence for formation of the E.S reaction intermediate with ATP-γ-S, a nonhydrolyzable analogue of ATP. Biochemistry 46, 13787–13799 [DOI] [PubMed] [Google Scholar]
- 39. Ritchie T. K., Grinkova Y. V., Bayburt T. H., Denisov I. G., Zolnerciks J. K., Atkins W. M., and Sligar S. G. (2009) Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zoghbi M. E., Krishnan S., and Altenberg G. A. (2012) Dissociation of ATP-binding cassette nucleotide-binding domain dimers into monomers during the hydrolysis cycle. J. Biol. Chem. 287, 14994–15000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zoghbi M. E., Fuson K. L., Sutton R. B., and Altenberg G. A. (2012) Kinetics of the association/dissociation cycle of an ATP-binding cassette nucleotide-binding domain. J. Biol. Chem. 287, 4157–4164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Posson D. J., Ge P., Miller C., Bezanilla F., and Selvin P. R. (2005) Small vertical movement of a K+ channel voltage sensor measured with luminescence energy transfer. Nature 436, 848–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Selvin P. R. (2002) Principles and biophysical applications of lanthanide-based probes. Annu. Rev. Biophys. Biomol. Struct. 31, 275–302 [DOI] [PubMed] [Google Scholar]
- 44. Swartz D. J., Mok L., Botta S. K., Singh A., Altenberg G. A., and Urbatsch I. L. (2014) Directed evolution of P-glycoprotein cysteines reveals site-specific, non-conservative substitutions that preserve multidrug resistance. Biosci. Rep. 34, e00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tombline G., Urbatsch I. L., Virk N., Muharemagic A., White L. B., and Senior A. E. (2006) Expression, purification, and characterization of cysteine-free mouse P-glycoprotein. Arch. Biochem. Biophys. 445, 124–128 [DOI] [PubMed] [Google Scholar]
- 46. Pan L., and Aller S. G. (2015) Equilibrated atomic models of outward-facing P-glycoprotein and effect of ATP binding on structural dynamics. Sci. Rep. 5, 7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tombline G., Bartholomew L. A., Urbatsch I. L., and Senior A. E. (2004) Combined mutation of catalytic glutamate residues in the two nucleotide-binding domains of P-glycoprotein generates a conformation that binds ATP and ADP tightly. J. Biol. Chem. 279, 31212–31220 [DOI] [PubMed] [Google Scholar]
- 48. Urbatsch I. L., Tyndall G. A., Tombline G., and Senior A. E. (2003) P-glycoprotein catalytic mechanism: studies of the ADP-vanadate inhibited state. J. Biol. Chem. 278, 23171–23179 [DOI] [PubMed] [Google Scholar]
- 49. Zoghbi M. E., and Altenberg G. A. (2013) Hydrolysis at one of the two nucleotide-binding sites drives the dissociation of ATP-binding cassette nucleotide-binding domain dimers. J. Biol. Chem. 288, 34259–34265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Frank G. A., Shukla S., Rao P., Borgnia M. J., Bartesaghi A., Merk A., Mobin A., Esser L., Earl L. A., Gottesman M. M., Xia D., Ambudkar S. V., and Subramaniam S. (2016) Cryo-EM analysis of the conformational landscape of human P-glycoprotein (ABCB1) during its catalytic cycle. Mol. Pharmacol. 90, 35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Verhalen B., and Wilkens S. (2011) P-glycoprotein retains drug-stimulated ATPase activity upon covalent linkage of the two nucleotide-binding domains at their C-terminal ends. J. Biol. Chem. 286, 10476–10482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Loo T. W., Bartlett M. C., and Clarke D. M. (2003) Drug binding in human P-glycoprotein causes conformational changes in both nucleotide-binding domains. J. Biol. Chem. 278, 1575–1578 [DOI] [PubMed] [Google Scholar]
- 53. James D. R., and Ware W. R. (1986) Recovery of underlying distributions of lifetimes from fluorescence decay data. Chem. Phys. Lett. 126, 7–11 [Google Scholar]
- 54. Siemiarczuk A., Wagner B. D., and Ware W. R. (1990) Comparison of the maximum entropy and exponential series methods for the recovery of distributions of lifetimes from fluorescence lifetime data. J. Phys. Chem. 94, 1661–1666 [Google Scholar]
- 55. Al-Shawi M. K., Polar M. K., Omote H., and Figler R. A. (2003) Transition state analysis of the coupling of drug transport to ATP hydrolysis by P-glycoprotein. J. Biol. Chem. 278, 52629–52640 [DOI] [PubMed] [Google Scholar]
- 56. Ambudkar S. V., Lelong I. H., Zhang J., Cardarelli C. O., Gottesman M. M., and Pastan I. (1992) Partial purification and reconstitution of the human multidrug-resistance pump: characterization of the drug-stimulatable ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 89, 8472–8476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Al-Shawi M. K. (2011) Catalytic and transport cycles of ABC exporters. Essays Biochem. 50, 63–83 [DOI] [PubMed] [Google Scholar]
- 58. Bouige P., Laurent D., Piloyan L., and Dassa E. (2002) Phylogenetic and functional classification of ATP-binding cassette (ABC) systems. Curr. Protein Pept. Sci. 3, 541–559 [DOI] [PubMed] [Google Scholar]
- 59. Sharom F. J. (2008) ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 9, 105–127 [DOI] [PubMed] [Google Scholar]
- 60. Jones P. M., O'Mara M. L., and George A. M. (2009) ABC transporters: a riddle wrapped in a mystery inside an enigma. Trends Biochem. Sci. 34, 520–531 [DOI] [PubMed] [Google Scholar]
- 61. Moody J. E., Millen L., Binns D., Hunt J. F., and Thomas P. J. (2002) Cooperative, ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-binding cassette transporters. J. Biol. Chem. 277, 21111–21114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jones P. M., and George A. M. (2013) Mechanism of the ABC transporter ATPase domains: catalytic models and the biochemical and biophysical record. Crit. Rev. Biochem. Mol. Biol. 48, 39–50 [DOI] [PubMed] [Google Scholar]
- 63. George A. M., and Jones P. M. (2013) An asymmetric post-hydrolysis state of the ABC transporter ATPase dimer. PLoS ONE 8, e59854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Locher K. P. (2016) Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 23, 487–493 [DOI] [PubMed] [Google Scholar]
- 65. Ward A., Reyes C. L., Yu J., Roth C. B., and Chang G. (2007) Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. U.S.A. 104, 19005–19010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shukla S., Abel B., Chufan E. E., and Ambudkar S. V. (2017) Effects of a detergent micelle environment on P-glycoprotein (ABCB1)-ligand interactions. J. Biol. Chem. 292, 7066–7076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Eckford P. D., and Sharom F. J. (2008) Functional characterization of Escherichia coli MsbA: interaction with nucleotides and substrates. J. Biol. Chem. 283, 12840–12850 [DOI] [PubMed] [Google Scholar]
- 68. Bretscher L. E., Buchaklian A. H., and Klug C. S. (2008) Spin-labeled lipid A. Anal. Biochem. 382, 129–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Doshi R., and van Veen H. W. (2013) Substrate binding stabilizes a pre-translocation intermediate in the ATP-binding cassette transport protein MsbA. J. Biol. Chem. 288, 21638–21647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bai J., Swartz D. J., Protasevich I. I., Brouillette C. G., Harrell P. M., Hildebrandt E., Gasser B., Mattanovich D., Ward A., Chang G., and Urbatsch I. L. (2011) A gene optimization strategy that enhances production of fully functional P-glycoprotein in Pichia pastoris. PLoS ONE 6, e22577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Reuter G., Janvilisri T., Venter H., Shahi S., Balakrishnan L., and van Veen H. W. (2003) The ATP binding cassette multidrug transporter LmrA and lipid transporter MsbA have overlapping substrate specificities. J. Biol. Chem. 278, 35193–35198 [DOI] [PubMed] [Google Scholar]
- 72. Loo T. W., Bartlett M. C., and Clarke D. M. (2003) Permanent activation of the human P-glycoprotein by covalent modification of a residue in the drug-binding site. J. Biol. Chem. 278, 20449–20452 [DOI] [PubMed] [Google Scholar]
- 73. Liu R., and Sharom F. J. (1996) Site-directed fluorescence labeling of P-glycoprotein on cysteine residues in the nucleotide-binding domains. Biochemistry 35, 11865–11873 [DOI] [PubMed] [Google Scholar]
- 74. Wang G., Pincheira R., Zhang M., and Zhang J. T. (1997) Conformational changes of P-glycoprotein by nucleotide binding. Biochem. J. 328, 897–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sharom F. J., Yu X., Chu J. W., and Doige C. A. (1995) Characterization of the ATPase activity of P-glycoprotein from multidrug-resistant Chinese hamster ovary cells. Biochem. J. 308, 381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Loo T. W., and Clarke D. M. (2016) P-glycoprotein ATPase activity requires lipids to activate a switch at the first transmission interface. Biochem. Biophys. Res. Commun. 472, 379–383 [DOI] [PubMed] [Google Scholar]
- 77. Stindt J., Ellinger P., Stross C., Keitel V., Häussinger D., Smits S. H., Kubitz R., and Schmitt L. (2011) Heterologous overexpression and mutagenesis of the human bile salt export pump (ABCB11) using DREAM (Directed REcombination-Assisted Mutagenesis). PLoS ONE 6, e20562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Beaudet L., and Gros P. (1995) Functional dissection of P-glycoprotein nucleotide-binding domains in chimeric and mutant proteins. Modulation of drug resistance profiles. J. Biol. Chem. 270, 17159–17170 [DOI] [PubMed] [Google Scholar]
- 79. Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., and Boeke J. D. (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 [DOI] [PubMed] [Google Scholar]
- 80. Lerner-Marmarosh N., Gimi K., Urbatsch I. L., Gros P., and Senior A. E. (1999) Large scale purification of detergent-soluble P-glycoprotein from Pichia pastoris cells and characterization of nucleotide binding properties of wild-type, Walker A, and Walker B mutant proteins. J. Biol. Chem. 274, 34711–34718 [DOI] [PubMed] [Google Scholar]
- 81. Urbatsch I. L., Gimi K., Wilke-Mounts S., and Senior A. E. (2000) Conserved Walker A Ser residues in the catalytic sites of P-glycoprotein are critical for catalysis and involved primarily at the transition state step. J. Biol. Chem. 275, 25031–25038 [DOI] [PubMed] [Google Scholar]
- 82. Chen J., and Selvin P. R. (1999) Thiol-reactive luminescent chelates of terbium and europium. Bioconjug. Chem. 10, 311–315 [DOI] [PubMed] [Google Scholar]
- 83. Zoghbi M. E., and Altenberg G. A. (2014) ATP binding to two sites is necessary for dimerization of nucleotide-binding domains of ABC proteins. Biochem. Biophys. Res. Commun. 443, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fiori M. C., Krishnan S., Cortes D. M., Retamal M. A., Reuss L., Altenberg G. A., and Cuello L. G. (2015) Functional hemichannels formed by human connexin 26 expressed in bacteria. Biosci. Rep. 35, e00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Heyduk T., and Heyduk E. (2001) Luminescence energy transfer with lanthanide chelates: interpretation of sensitized acceptor decay amplitudes. Anal. Biochem. 289, 60–67 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.