

Abstract
Lipins 1, 2, and 3 are Mg2+-dependent phosphatidic acid phosphatases and catalyze the penultimate step of triacylglycerol synthesis. We have previously investigated the biochemistry of lipins 1 and 2 and shown that di-anionic phosphatidic acid (PA) augments their activity and lipid binding and that lipin 1 activity is negatively regulated by phosphorylation. In the present study, we show that phosphorylation does not affect the catalytic activity of lipin 3 or its ability to associate with PA in vitro. The lipin proteins each contain a conserved polybasic domain (PBD) composed of nine lysine and arginine residues located between the conserved N- and C-terminal domains. In lipin 1, the PBD is the site of PA binding and sensing of the PA electrostatic charge. The specific arrangement and number of the lysines and arginines of the PBD vary among the lipins. We show that the different PBDs of lipins 1 and 3 are responsible for the presence of phosphoregulation on the former but not the latter enzyme. To do so, we generated lipin 1 that contained the PBD of lipin 3 and vice versa. The lipin 1 enzyme with the lipin 3 PBD lost its ability to be regulated by phosphorylation but remained downstream of phosphorylation by mammalian target of rapamycin. Conversely, the presence of the lipin 1 PBD in lipin 3 subjected the enzyme to negative intramolecular control by phosphorylation. These results indicate a mechanism for the observed differences in lipin phosphoregulation in vitro.
Keywords: mammalian target of rapamycin (mTOR), phosphatase, phosphatidic acid, phosphatidylethanolamine, phosphorylation, di-anionic, lipin, lpin1, lpin3
Introduction
In vertebrates, the lipin family of Mg2+-dependent phosphatidic acid (PA)3 phosphatases consists of three members (lipins 1–3), which form diacylglycerol from PA in neutral and phospholipid synthesis (1–6). Lipins are cytosolic enzymes that must associate with membranes to access substrate (1, 7–9). Their spatial regulation has been studied most extensively in the context of insulin signaling (10–13). More specifically, inhibition of mTOR and reduction in the phosphorylation of lipin 1 allow its translocation to the endoplasmic reticulum membrane and nucleus (10–14). We have shown that whereas lipin 1 is regulated by phosphorylation in vitro, the activity and localization of lipin 2 are not (15).
Lipins are PA effector proteins and are regulated by the lipid's intrinsic chemical properties (16, 17). The PA headgroup is a negatively charged phosphomonoester and contains two dissociable protons with pKa values of 3 and ∼7.9, pKa1 and pKa2, respectively (18, 19). At physiological pH, PA always carries a charge of at least −1. However, at least two phenomena can cause PA to become di-anionic: proximity to a hydrogen bond donor, which lowers the pKa2 below the physiological pH, or increase in the membrane pH above that of pKa2 (19). Positively charged, basic amino acids and phosphatidylethanolamine (PE), an abundant membrane lipid, are sources of hydrogen bond donors (18, 20).
Although there is no canonical lipid-binding site on PA effector proteins, many of them contain a cluster of lysines and/or arginines critical for binding to PA (16). Kooijman (21) et al. postulated that these residues are responsible for the initial attraction of PA effectors to the negatively charged membrane. The proteins sample the surface until they encounter a mono-anionic PA. They hydrogen-bond with the PA headgroup and cause a dissociation of the final proton. This causes a switch in the charge from mono- to di-anionic and locks the protein onto its lipid-ligand through strengthened electrostatic interactions. This model was termed the hydrogen bond switch mechanism and has been demonstrated experimentally by an induction in lipin activity in the presence of di-anionic PA (14, 15, 21).
Each lipin contains a cluster of nine lysines and arginines known as the polybasic domain (PBD). This region on lipin 1 was identified as the primary PA-binding site and a membrane/nuclear localization sequence (13, 22, 23). In addition, it is likely to also be the site of negative regulation of lipin 1 by phosphorylation (14). The PBD is conserved among PAP enzymes, but the specific arrangement and number of lysines and arginines vary between the mammalian lipins 1–3.
The present study is the first to investigate the in vitro activity of purified lipin 3 against PA-containing liposomes in the context of phosphorylation and the electrostatic charge of PA. Additionally, we present evidence that the specific PBDs of lipins 1 and 3 are responsible for the observed differences in their in vitro phosphoregulation.
Results
Purification and enzymatic activity of recombinant lipin 3
Adenovirus was used to overexpress Mus musculus FLAG-lipin 3 in HeLa cells. After 2 days, lipin 3 was affinity-purified, eluted with FLAG peptide, and dialyzed (Fig. 1A). The activity and biochemistry of purified lipin 3 were investigated using 0.5 mm PA solubilized in Triton X-100 micelles (24, 25). Lipin 3 activity was linear with time (Fig. 1B). Work by Carman et al. (24, 25) and our laboratory (15) demonstrated that lipins 1 and 2 adhere to surface dilution kinetics. The principles of this model can be demonstrated experimentally by testing the detergent concentration-dependent changes in enzyme activity while maintaining substrate at a constant bulk concentration (24). We show that lipin 3 activity decreases with increasing Triton X-100 concentration, suggesting that it also adheres to surface dilution kinetics (Fig. 1C). The optimal pH for lipin 3 function was determined, with the peak activity at pH 8.0 (Fig. 1D). Maximal lipin 3 PAP activity was reached at 0.5 mm Mg2+ (Fig. 1E). The addition of 0.01 mm Mn2+, another divalent cation, increased lipin 3 activity by only 35%, relative to the maximum activity of lipin 3 at 0.5 mm Mg2+ (Fig. 1F). The PAP enzymes are characterized by their sensitivity to an alkylating agent, N-ethylmaleimide (NEM) (7, 26). The addition of 2 mm NEM resulted in a nearly complete inhibition of lipin 3 PAP activity, suggesting that it contains NEM-sensitive cysteine residues (Fig. 1G, IC50 = 0.4 mm), as is the case for lipins 1 and 2 (10, 15, 25). The addition of reducing agents stimulates the activity of lipins 1 and 2 (15, 25). Indeed, 20 mm β-mercaptoethanol (β-ME) maximally stimulates lipin 3 activity (Fig. 1H).
Figure 1.

Purification and biochemical characterization of FLAG-lipin 3. A, HeLa cells were infected with adenovirus expressing FLAG-lipin 3. After 2 days, cells were homogenized in buffer A, and the lipin 3 protein was affinity-purified, eluted with FLAG peptide, and dialyzed. An aliquot of the purified FLAG-tagged lipin 3 was resolved by SDS-PAGE, and the gel was stained with Coomassie Blue. B, PAP enzymatic activity of 50 ng of purified lipin 3 was measured as a function of time in Triton X-100/PA mixed micelles. C, PAP activity of lipin 3 against PA at a constant concentration of 0.5 mm with an increasing concentration of Triton X-100. D, PAP activity of lipin 3 as a function of pH. E, PAP activity of lipin 3 as a function of MgCl2. F, PAP activity of lipin 3 against as a function of MnCl2. G, PAP activity of lipin 3 with increasing concentrations of NEM. H, PAP activity of lipin 3 with increasing concentrations of β-ME. Experiments shown in D–H were performed using Triton X-100/PA mixed micelles with a surface concentration and bulk concentration of PA at 10 mol % and 0.5 mm, respectively. Each data point is a mean of triplicate determinations ± S.E. (error bars).
Phosphorylation of lipin 3 and sensing of PA charge
Phosphorylation has been shown to play an important role in the regulation of lipin 1 PAP activity (10, 11, 14). To investigate the phosphorylation of lipin 3, the purified protein was trypsin-digested and subjected to LC-MS/MS. Phosphorylated residues were assigned a site localization probability describing the confidence that a particular site within the sequence region is phosphorylated. We found 15 novel phosphorylation sites with localization probabilities of 0.95 or greater throughout the length of the protein (Fig. 2A and Table 1). An important phosphorylation site for lipin 1 regulation is Ser106. This site was previously identified on lipins 1 and 2 (10, 15). Although LC-MS/MS analysis did not detect well-localized phosphorylation of this site on lipin 3, a confidently identified peptide with phosphorylation on Thr105 or Ser106 was observed in lipin 3 (see Table 5 (in red) and supplemental Table S1). An important lipin protein region, enriched in phosphorylation sites, is the serine-rich domain (SRD). The SRD contains several 14-3-3 consensus-binding motifs and is important for mediating lipin 1 subcellular localization (13). On lipin 3, the region corresponds to amino acids Ser185–Ser230, of which Ser198, Ser218, Ser220, and Ser230 are phosphorylated with a localization probability of 0.95 or greater and Ser215 falls just short of statistically significant localization probability of 0.93 (Table 1 and supplemental Table S1). Phosphorylation on residue Ser218 is also present in lipins 1 and 2 (Ser285 in lipin 1 and Ser243 in lipin 2), and Ser287 is also phosphorylated in lipin 1 (homologous to Ser220 in lipin 3), albeit with a localization probability of 0.77 (Table 1 and supplemental Table S1) (10, 15).
Figure 2.

Phosphorylation, enzymatic activity, and PA binding of lipin 3. A, affinity-purified lipin 3 was digested into peptides and subjected to LC-MS/MS to identify phosphorylated residues. Shown is a schematic of the identified phosphorylation sites. B, lipin 3 was affinity-purified from HeLa cells with and without prior incubation with 2000 units of λ protein phosphatase. Phosphorylated (−λ) and dephosphorylated (+λ) lipin 3 proteins were separated on an SDS-polyacrylamide gel along with BSA standards and stained with Coomassie Blue dye. C, to verify that +λ lipin 3 residues were deficient in phosphorylation, HeLa cells overexpressing FLAG-lipin 3 were radiolabeled. Cells were harvested, homogenized, and incubated with anti-FLAG beads for 4 h. After immunoprecipitation, the radiolabeled lipin 3 was incubated with 200 units of λ phosphatase. The protein was separated on an SDS-polyacrylamide gel, and incorporation of 32P was visualized using autoradiography (top inset, [32P]lipin 3) and then immunoblotted for FLAG (bottom inset, FLAG-lipin 3). The scatter plot displays the amount of 32P in phosphorylated and dephosphorylated lipin 3 normalized to the amount of total lipin 3. Shown are the mean and points from analysis of three independent experiments ± S.E. (error bars). Student's t test was used to analyze statistical significance. D, PAP activity of purified lipin 3 using PC/PA (90 mol % PC, 10 mol % PA) and PC/PE/PA (30 mol % PE) liposomes. −λ and +λ lipin 3 activities were assayed at pH 7.5. E, −λ and +λ lipin 3 PAP activity was measured using Triton X-100/PA mixed micelles. Each data point is a mean of triplicate determinations ± S.E. (error bars). Phosphorylated (F) and dephosphorylated (G) lipin 3 were subjected to liposome floatation assays using PC/PA (20 mol % PA) and PC/PE/PA (30 mol % PE, 20 mol % PA) liposomes. Shown is a scatter plot with the mean of triplicate determinations from one representative experiment ± S.E. Student's t test was used to analyze statistical significance. *, p < 0.0009; **, p < 0.0005.
Table 1.
Lipin 3 phosphorylation analysis by LC-MS/MS
The first column indicates the phosphopeptide sequence identified by mass spectrometry, containing a phosphorylated Ser/Thr, annotated as (ph). The second column indicates the identity and the position of the phosphorylated residue. The final column indicates localization probability: between 0.95 and 1. Only the sites for which we had a high sequence position confidence (≥0.95) and a high peptide identity confidence (PEP score of ≤0.05) are listed.
| Modified sequence lipin 3 | Residue | Localization probability |
|---|---|---|
| LGVLRS(ph)REK | S60 | 1 |
| LGDSGEAFFVQELDS(ph)DEEDVPPR | S94 | 1 |
| PTPES(ph)PSAQEAEEPSSQPK | S178 | 0.96 |
| PTPESPS(ph)AQEAEEPSSQPK | S180 | 0.99 |
| DIHPYS(ph)DGECTPQANLSSGDLMSPK | S198 | 1 |
| S(ph)DSELELR | S218 | 0.96 |
| SDS(ph)ELELR | S220 | 0.96 |
| LRSLEPS(ph)PLRAE | S230 | 1 |
| TQNS(ph)RGAGHPPATK | S332 | 1 |
| SWS(ph)WTTPESHTPSGHPQVSR | S345 | 0.99 |
| RWS(ph)EPSNQK | S408 | 1 |
| LLES(ph)PNPEHIAECTLDSVDK | S418 | 0.99 |
| FTQHMVS(ph)YEDLTK | S460 | 0.99 |
| TEVLSSDDDVPDS(ph)PVILEVPPLPSSTPGYVPTYKK | S567 | 0.99 |
| LLFPPVVRGPSTDLAS(ph)PE | S826 | 1 |
Table 5.
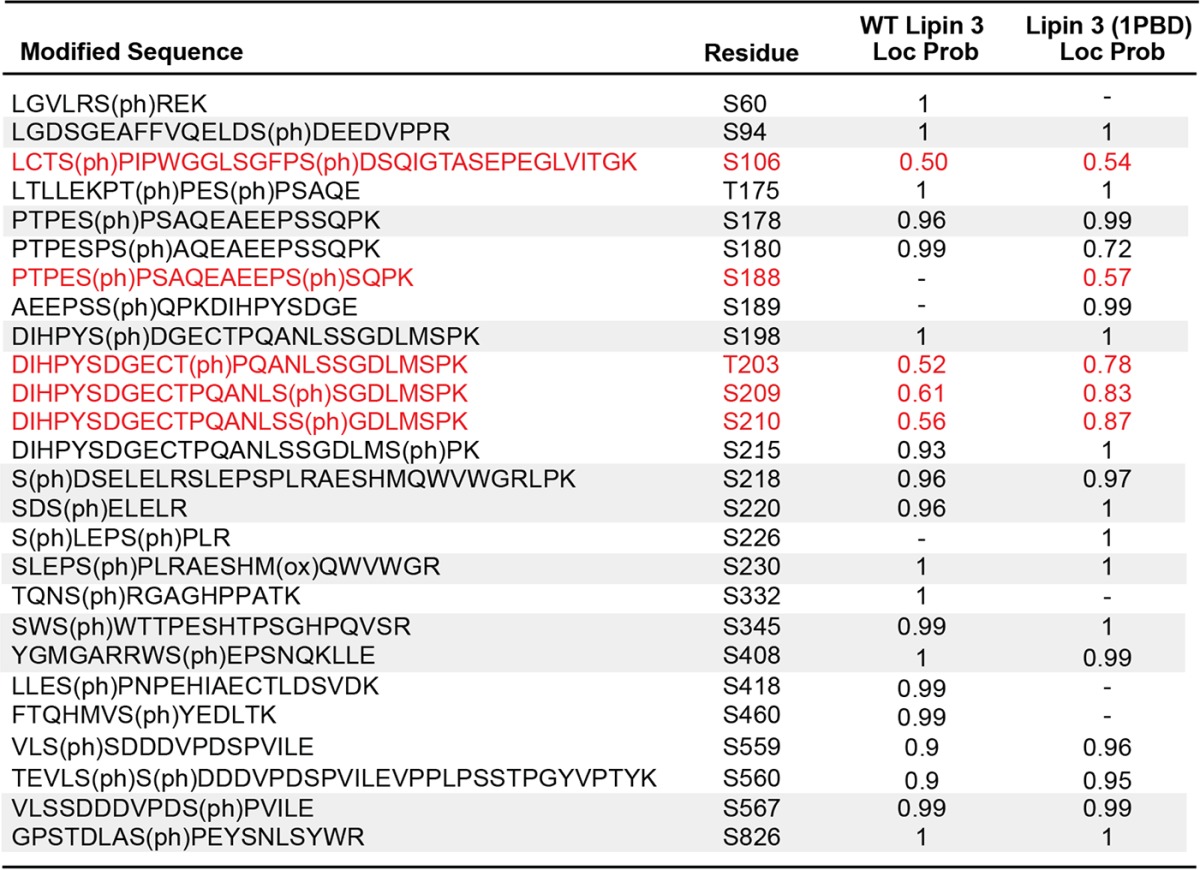
WT lipin 3 and lipin 3 (1PBD) phosphorylation analysis by LC-MS/MS
Rows highlighted in grey indicate phosphorylated residues with localization probabilities of ≥ 0.95 in both proteins. Localization probabilities are left blank if no peptide was detected or if the localization probability was < 0.5. With the exception of the sites in red, sites are only reported if they have localization probabilities ≥ 0.95 in at least one of WT lipin 3 or lipin 3 (1PBD). Sites in red are those believed to be important in lipin activity and regulation but were not detected with a localization probability of ≥ 0.95 in either protein. However, we have greater than 99% confidence that there is a phosphorylation site in these peptide regions. Peptide sequences come from the WT protein unless the WT localization probability is < 0.95, in which case the peptide corresponding to the highest site localization probability is shown. Only sites with high peptide identity confidence (PEP ≤ 0.05) are listed.
To determine whether phosphorylation affects lipin 3 enzymatic activity, FLAG-lipin 3 was purified using affinity chromatography as described above. Before protein elution, the protein was incubated in phosphatase buffer with and without protein λ phosphatase, eluted, and dialyzed. This yielded phosphorylated (−λ) and dephosphorylated (+λ) lipin 3 (Fig. 2B). We verified that all of the phosphates were removed by overexpressing and isolating lipin 3 from 32P-radiolabeled HeLa cells expressing FLAG-lipin 3. A 30-min treatment with λ protein phosphatase in vitro removed almost all phosphates (+λ; Fig. 2C). Next, purified protein and either phosphatidylcholine (PC)/PA liposomes or Triton X-100/PA mixed micelles were used to determine whether the removal of phosphates from lipin 3 affects its basal PAP activity or affinity for substrate (Fig. 2 (D and E) and Table 2). The Kmapp values of phosphorylated (−λ) and dephosphorylated (+λ) lipin 3 for PA in liposomes were around 24.6 ± 4 and 22.5 ± 5 μm, respectively (Table 2).
Table 2.
Steady-state kinetic data for phosphorylated (−λ) and dephosphorylated (+λ) lipin 3
The data were generated from assays using PC/PA or PC/PE/PA liposomes containing 10 mol % PA, the indicated mol % PE, and additional PC to bring the combined concentration of phospholipids to 100 mol %. The specific activity was measured as Vmax/mg protein. Student's t test was used to analyze statistical significance. *, p < 0.05.
| PE | Kmapp | Kcat | Kcat/Kmapp | |
|---|---|---|---|---|
| mol % | μm | s−1 | μm−1 s−1 | |
| −λ | 0 | 24.6 ± 4 | 0.50 ± 0.02 | 0.020 |
| −λ | 30 | 37.3 ± 3 | 0.78 ± 0.12 | 0.023 |
| +λ | 0 | 22.5 ± 3 | 0.51 ± 0.01 | 0.023 |
| +λ | 30 | 31.9 ± 9 | 0.91 ± 0.05 | 0.028 |
Under these conditions, the majority of PA is mono-anionic (18, 21). Phosphatidylethanolamine causes the pKa2 of PA to decrease to 6.9 and switches the charge of PA to −2 at physiological pH (18). The incorporation of 30 mol % PE in the PC/PA liposomes resulted in a 2-fold augmentation of phosphorylated and dephosphorylated lipin 3 PAP activity and an increase in kcat (Fig. 2D and Table 2). These data suggest that lipin 3 has greater activity toward di-anionic than mono-anionic PA independent of phosphorylation. Proximity to a hydrogen bond donor is not the only mechanism whereby PA can regulate association with to its own effector proteins. The charge of the PA headgroup can also be altered by changes in pH (14, 21). For example, lowering pH below the pKa2 of PA will result in the protonation of the headgroup due to an increase in the concentration of hydrogen ions. Conversely, an increase in pH would result in stabilization of di-anionic PA. Indeed, the activity of lipin 3 against Triton X-100/PA mixed micelles was more than 2-fold greater at pH 8.0 than at pH 7.5 (Fig. 2E).
As a more direct measure of association with PA, the ability of phosphorylated and dephosphorylated Venus-tagged lipin 3 to bind PC/PA and PC/PE/PA liposomes was investigated using liposome flotation on a sucrose gradient. Venus is a yellow fluorescent protein and is easily detectable by spectrometry. It was found that in the absence of PE, the binding of lipin 3 to PC/PA liposomes was unaffected by dephosphorylation. In the presence of 30 mol % PE, the binding of lipin 3 was enhanced by 50%, regardless of its phosphorylation state (Fig. 2, F and G). These data further support phosphorylation-independent augmentation of lipin 3 PAP activity under these conditions.
Phosphorylation of lipin 3 in 3T3-L1 cells
Lipin 1 is reported to be phosphorylated and negatively regulated upon acute insulin treatment, in a rapamycin- and Torin 1-sensitive manner (10, 11, 13, 14). We sought to determine whether lipin 3 is similarly affected by phosphorylation. Lipins 1 and 3 were expressed in differentiated 3T3-L1 cells by adenoviral transduction. Two days post-infection, the cells were serum-starved in low-phosphate buffer for 2 h, incubated with 32P orthophosphate, and treated with vehicle or a pan-mTOR inhibitor, Torin 1, for 45 min plus 10 milliunits/ml insulin for the last 15 min or insulin alone, and phosphate incorporation was assessed (Fig. 3, A and B). Concurrent with previous studies, we show that insulin treatment stimulated the phosphorylation of lipin 1 in a Torin 1-sensitive manner (10, 12). However, insulin and Torin 1 treatment had no effect on the levels of phosphate incorporation into lipin 3.
Figure 3.

Phosphorylation of lipin 3 in 3T3-L1 cells. A, 3T3-L1 cells infected with adenovirus expressing FLAG-lipin 3 were serum-starved in low-phosphate buffer for 2 h, incubated with 0.2 mCi/ml orthophosphate, and treated as indicated. The cells were harvested, incubated with anti-FLAG beads for 4 h, displaced from beads, and separated on an SDS-polyacrylamide gel and transferred to PVDF. Top, a phosphor image of 32P incorporation into lipin 3 (top; 32P) and an immunoblot image of total lipin 3 protein (bottom; lipin 3). Bottom, quantitation of three independent experiments. B, the experiment was performed as in A, but with FLAG-lipin 1. Shown are scatter plots with mean of the quantitation of three independent experiments ± S.E. (error bars). One-way ANOVA was used to analyze statistical significance followed by Sidak post hoc analysis. *, p < 0.05; N.S., no statistical significance.
The PAP activity of PBD mutants with liposomes
All three lipin family members contain homologous PBDs immediately C-terminal to the N-terminal domain at residues 153–161 in lipin 1 and 136–144 in lipin 3 (Fig. 4A) (23, 25). In lipin 1, deletion of this site excluded the enzyme from the nucleus (22). Mutation of the lipin 1 PBD to alanines confirmed that it is a nuclear localization sequence and further that it is critical for PA binding but not for PAP activity (13, 14, 22). Our laboratory has shown that PBD is important for sensing of the PA electrostatic charge, and phosphorylation sites within lipin 1 may hinder PA association with the PBD (14). Our data thus far contrast with those for lipin 1 and indicate that under the present conditions, lipin 3 phosphorylation does not prevent its recognition and binding to di-anionic PA (Fig. 2).
Figure 4.

Enzymatic activity of lipin PBD exchange mutants in liposomes. A, schematic of lipin PBD exchange mutants. B, PBD mutants were expressed, affinity-purified, incubated in phosphatase buffer with (+λ) or without (−λ) λ phosphatase, and eluted as described before. Proteins were separated on an SDS-polyacrylamide gel along with BSA standards and stained with Coomassie Blue dye. The PAP activities of purified phosphorylated (−λ) and dephosphorylated (+λ) lipin 1 (3PBD) were measured with PC/PA (C) and PC/PE/PA (D) liposomes. Shown are the PAP activities of −λ and +λ purified lipin 3 (1PBD) with PC/PA (E) and PC/PE/PA (F) liposome. Activities were assayed at pH 7.5. Each data point is a mean of triplicate determinations ± S.E. (error bars).
We wondered about the molecular basis for such differences in the phosphoregulation of lipin enzymes. One possibility is that the distinct PBDs of lipins 1 and 3 are at least partially responsible for conferring sensitivity to phosphoregulation. To test this hypothesis, constructs exchanging the PBD of WT lipin 1 with that of WT lipin 3 (lipin 1 (3PBD)) and the PBD of WT lipin 3 with that of WT lipin 1 (lipin 3 (1PBD)) were expressed. The PBD mutant constructs (Fig. 4A) were purified from HeLa cells to yield phosphorylated (−λ) and dephosphorylated (+λ) proteins (Fig. 4B). The PAP activities of the PBD exchange mutants were tested in the context of PC/PA and PC/PE/PA liposomes. Insertion of the lipin 3 PBD in lipin 1 eliminated the ability of phosphorylation to hinder the recognition of the electrostatic state of PA, and PE significantly augmented the specific activity of both phosphorylated and dephosphorylated enzyme (Fig. 4, C and D). As seen in Table 3, the Kmapp did not significantly decrease with 30 mol % PE, but the catalytic efficiency and turnover number of the lipin 1 (3PBD) mutant increased independent of phosphorylation.
Table 3.
Steady-state kinetic data for phosphorylated (−λ) and dephosphorylated (+λ) lipin PBD mutants
The data were generated from assays using PC/PA or PC/PE/PA liposomes containing 10 mol % PA, the indicated mol % PE, and additional PC to bring the combined concentration of phospholipids to 100 mol %. The specific activity was measured as Vmax/mg protein. Student's t test was used to analyze statistical significance.
| PE | Kmapp | Kcat | Kcat/Kmapp | |
|---|---|---|---|---|
| mol % | μm | s−1 | μm−1 s−1 | |
| Lipin 1 (3PBD) | ||||
| −λ | 0 | 211 ± 170 | 0.76 ± 0.23 | 0.003 |
| −λ | 30 | 191 ± 88 | 1.66 ± 0.17 | 0.008 |
| +λ | 0 | 170 ± 99 | 0.64 ± 0.06 | 0.004 |
| +λ | 30 | 146 ± 54 | 1.64 ± 0.34 | 0.011 |
| Lipin 3 (1PBD) | ||||
| −λ | 0 | 120 ± 69 | 0.67 ± 0.09 | 0.005 |
| −λ | 30 | 117 ± 31 | 0.92 ± 0.05 | 0.007 |
| +λ | 0 | 107 ± 40 | 0.78 ± 0.19 | 0.007 |
| +λ | 30 | 155 ± 38 | 2.02 ± 0.26 | 0.014 |
In contrast, only dephosphorylated lipin 3 (1PBD) activity was significantly augmented in the presence of 30 mol %, PE but the activity of phosphorylated lipin 3 (1PBD) was not (Fig. 4, E and F). The kcat of dephosphorylated lipin 3 (1PBD) increased by about 2.5-fold, compared with 1.3-fold for the phosphorylated enzyme (Table 3). These data suggest that the phosphoregulation of lipin 1 occurs, at least in part, because of its specific PBD.
The PAP activity of PBD mutants within micelles
In the context of Triton X-100/PA mixed micelles, the activity of WT lipin 1 is significantly higher than its specific activity in liposomes (14). In contrast, lipin 3 activity is closely matched between these two modes of substrate presentation. It is possible that lipin 3 enzymatic activity is sensitive to inhibition by detergent. To test whether the variation in lipin 1 and 3 PAP activity in micelles is due to the PBD, we measured the activity of the lipin PBD mutants under the described conditions. The lipin 1 (3PBD) displayed specific activity comparable with that of WT lipin 1 (Fig. 5A) (14). Lipin 3 (1PBD) activity was also similar to that of WT lipin 3 (Figs. 2E and 5B). Similar results were obtained using Tween 20 (data not shown) to solubilize PA, suggesting that the effect is not a result of detergent acting on the enzymatic activity but may be a consequence of micellar presentation of substrate. Therefore, the functional portion of the lipin proteins that defines the high PAP activity is not within the PBD.
Figure 5.

Enzymatic activity of lipin PBD exchange mutants in Triton X-100/PA micelles. Phosphorylated (−λ) and dephosphorylated (+λ) lipin 1 (3PBD) (A) and lipin 3 (1PBD) (B) activities were assayed using Triton X-100/PA mixed micelles at pH 7.5. The surface concentration of PA was 10 mol %. Each data point is a mean of triplicate determinations ± S.E. (error bars).
The binding of lipin 1 and 3 PBD mutants to PC/PA liposomes
The lipin PBDs are the main site of substrate interaction (14, 22). It is likely that changes in activity caused by the PBD mutants are due to altered association of the mutant enzymes with PA. Using liposome flotation assays, we investigated the ability of phosphorylated and dephosphorylated Venus-lipin 1 (3PBD) and Venus-lipin 3 (1PBD) purified enzymes to associate with PA-containing liposomes. The binding of Lipin 1 (3PBD) increased by about 25–30% in the presence of 30 mol % PE regardless of phosphorylation state (Fig. 6, A and B). The binding of phosphorylated lipin 3 (1PBD) increased slightly from 0 to 30 mol % PE, whereas the dephosphorylated enzyme binding increased by ∼40% from PC/PA to PC/PE/PA liposomes (Fig. 6, C and D). It should be pointed out that the PBD exchange mutants were catalytically active but displayed significantly reduced Kmapp for PA, compared with WT lipins 1 and 3 (Fig. 4 (C–F) and Table 3) (14), suggesting that the PBD exchange did not affect affinity to bulk substrate. However, Kmapp takes into account both bulk and surface concentration (24).
Figure 6.
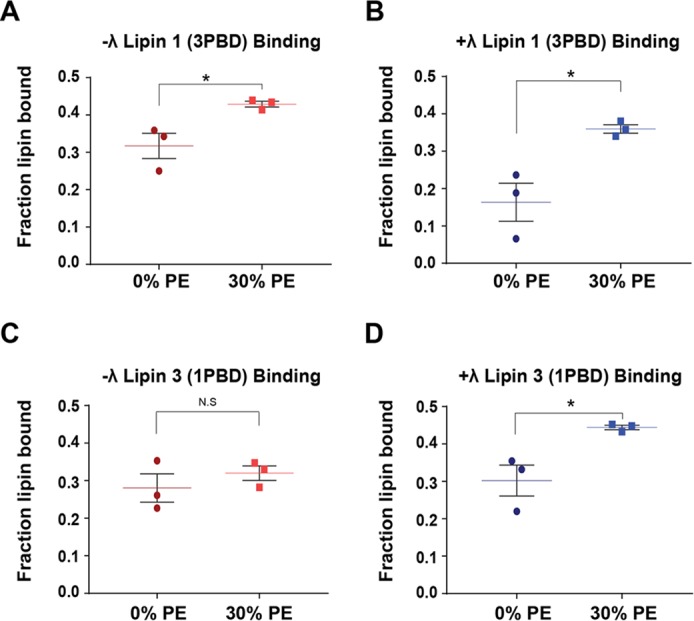
Binding of Venus-lipin PBD exchange mutants with PA containing liposomes. Phosphorylated (−λ) Venus-tagged lipin 1 (3PBD) (A), dephosphorylated (+λ) Venus-tagged lipin 1 (3PBD) (B), −λ Venus-tagged lipin 3 (1PBD) (C), and +λ Venus-tagged lipin 3 (1PBD) (D) were subjected to liposome floatation assays using PC/PA and PC/PE/PA (30 mol % PE) liposomes, containing 20 mol % PA and 0.1 mol % PC-pyrene. The final concentration of PA in all assays was 2 mm. Shown is a scatter plot with a mean of triplicate determinations from one representative experiment ± S.E. (error bars). Student's t test was used to analyze statistical analysis. *, p < 0.05.
Phosphorylation of Ser106 on lipin 1 (3PBD)
There are two possibilities by which exchanging the PBDs alters the ability of phosphorylation to regulate enzymatic activity. Either the phosphorylation sites themselves are altered by the generation of the PBD mutants or the existing phosphorylation sites can regulate the PBD of lipin 1 but not lipin 3. Serine 106 is a conserved site among the lipins and in lipin 1 is phosphorylated in response to insulin, downstream of mTOR (10, 12). Ser106 on lipin 2 is not responsive to insulin stimulation or Torin 1 treatment (15). We attempted to investigate the phosphorylation of this site on lipin 3 and the PBD mutants but could not detect phospho-Ser106 on lipin 3 or lipin 3 (1PBD) using Western blot analysis. Lipin amino acid alignments revealed that the region surrounding Ser106 that was used to generate phospho-specific antibodies is conserved within lipins 1 and 2 but not in lipin 3. The LC-MS/MS analysis of lipin 3 revealed that either Thr105 or Ser106 was phosphorylated, but the precise localization could not be resolved with statistical significance (supplemental Table S1). Therefore, we investigated the regulation of Ser106 phosphorylation in lipin 1 (3PBD). Wild-type lipin 1 and lipin 1 (3PBD) proteins were overexpressed in HeLa cells and treated with vehicle or 250 nm Torin 1. Changes in the phosphorylation of Ser106 were detected using the phospho-Ser106 antibody. Under basal conditions, the residue Ser106 on lipin 1 (3PBD) was phosphorylated (Fig. 7A). Treatment with Torin 1 decreased phosphorylation of Ser106 on WT lipin 1, as reported previously (Fig. 7, A and B) (12). Exchanging the lipin 1 PBD with the PBD of lipin 3 had no effect on the ability of Torin 1 to inhibit phosphorylation of this site (Fig. 7, A and B). In addition, gel mobility shifts between phosphorylated and dephosphorylated PBD mutants were similar to what has been seen with the WT enzymes (Fig. 7C). Together, immunoblotting data suggest that mutation of the lipin 1 PBD does not alter the phosphorylation of Ser106 and does not eliminate the lipin 1 (3PBD) as a downstream substrate of mTOR.
Figure 7.

Phosphorylation of lipin 1 (3PBD) at Ser106. HeLa cells infected with adenovirus expressing either FLAG-lipin 1 or FLAG-lipin 1 (3PBD) were treated with vehicle or Torin 1, harvested, and incubated with anti-FLAG beads. The enzymes were displaced, separated on SDS-polyacrylamide gel, and probed for phospho-Ser106 or total lipin protein. A, image of phosphorylated Ser106 and total enzyme. B, quantitation of three independent experiments. C (left), Coomassie image of purified phosphorylated lipin 1 and lipin 1 (3PBD); right, Coomassie image of purified phosphorylated lipin 3 and lipin 3 (1PBD). Shown is a scatter plot with mean of the quantitation of three independent experiments. One-way ANOVA was used to analyze statistical significance followed by Sidak post hoc analysis. *, p < 0.05. Error bars, S.E.
To more thoroughly investigate whether the mutation of the lipin PBD domains altered the global phosphorylation of the rest of the protein, we analyzed phosphorylation sites of lipin 1 (3PBD) and lipin 3 (1PBD) using LC-MS/MS analysis and compared them with those of WT lipins 1 and 3. Previous work investigating lipin 1 phosphorylation was performed on HA-tagged lipin 1 purified from 3T3-L1 cells after acute insulin stimulation (10). To maintain consistency in protein acquisition, purification, and LC-MS/MS analysis, we purified FLAG-tagged WT lipin 1 from HeLa cells in an identical manner and analyzed phosphorylation sites for all four proteins.
Twenty-two phosphorylation sites were identified on WT lipin 1 and 16 on lipin 1 (3PBD) and localized with a probability of 0.95 or greater (Table 4). Many of the sites found on WT lipin 1 agreed with previous reports (10). Of the 16 phosphosites on lipin 1 (3PBD), 11 were in common with WT lipin 1, including Ser106 and sites within the SRD (Table 4 and Fig. 8). Fifteen phosphorylation sites were identified on lipin 3 and 16 on lipin 3 (1PBD) with a localization probability of 0.95 or greater (Fig. 2 and Tables 1 and 5). Of these, 10 were shared with WT lipin 3, including residues within the conserved SRD (Table 5 and Fig. 8). Four additional sites in the SRD with localization probabilities between 0.52 and 0.88 were also shared between WT lipin 3 and lipin 3 (1PBD) (supplemental Table S1). These data suggest that the switching of the PBDs between lipins 1 and 3 does not significantly alter the overall phosphorylation of the proteins.
Table 4.
WT lipin 1 and lipin 1 (3PBD) phosphorylation analysis by LC-MS/MS
Rows highlighted in grey indicate phosphorylated residues with localization probabilities of ≥ 0.95 in both proteins. Localization probabilities are left blank if no peptide was detected or if the localization probability was < 0.5. With the exception of the sites in red, sites are only reported if they have localization probabilities ≥ 0.95 in at least one of WT lipin 1 or lipin 1 (3PBD). Sites in red are those believed to be important in lipin activity and regulation but were not detected with a localization probability of ≥ 0.95 in either protein. However, we have > 99% confidence that there is a phosphorylation site in these peptide regions. Peptide sequences come from the WT protein unless the WT localization probability is < 0.95, in which case the peptide corresponding to the highest site localization probability is shown. Only sites with high peptide identity confidence (PEP ≤ 0.05) are listed.
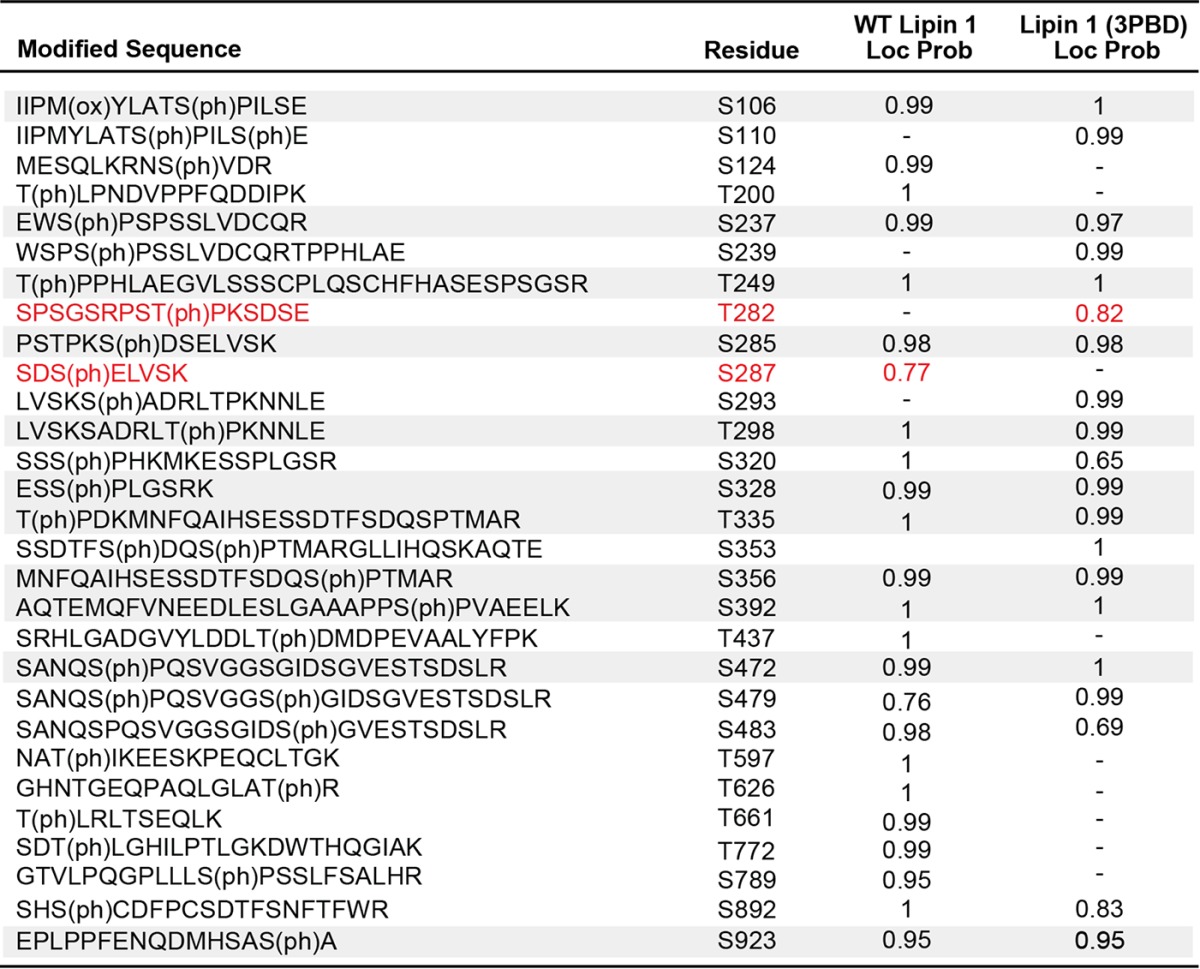
Figure 8.
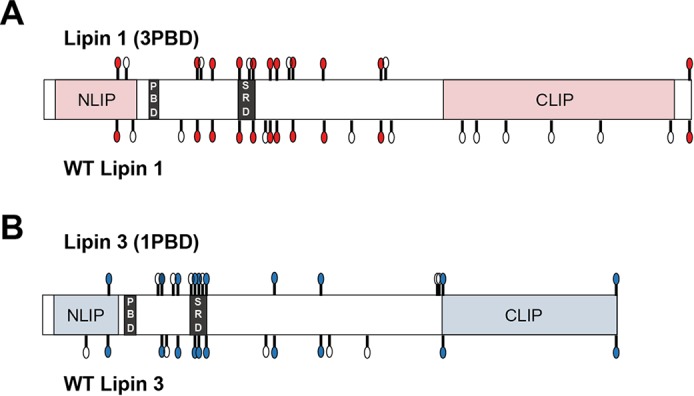
Shared phosphorylated sites among WT lipins 1 and 3 and the PBD mutants. Shown is a schematic of the phosphorylation sites found in the four indicated proteins. The white circles indicate the position of the phosphorylation sites unique to the given protein. The colored circles indicate the position of the phosphorylation sites shared between WT lipin 1 and lipin 1 (3PBD) (A) and WT lipin 3 and lipin 3 (1PBD) (B). CLIP and NLIP, C-terminal and N-terminal domain, respectively.
Discussion
The lipins are enzymes with complex and multifunctional roles in cellular lipid metabolism. Whereas lipins serve the same catalytic function to dephosphorylate PA, their roles on the physiological level are distinct. Deleterious mutations in Lpin1 gene result in hepatic and adipose irregularities in mice, whereas in humans they cause muscle rhabdomyolysis (23, 27, 28). Mice lacking lipin 2 appear to have age-dependent neuronal dysfunction, and human LPIN2 mutations have been linked to the rare immunological disorder Majeed syndrome (29). Lipin 3-deficient rodents do not show an obvious phenotype but may be critical for adipogenesis (30). Such findings are perhaps a reflection of the intricate and distinct means by which these enzymes are regulated on the molecular level. The in vitro control of lipin 1 localization and activity by phosphorylation and the electrostatic charge of PA is well understood, but in these contexts, lipin 3 is relatively understudied.
In the present work, we investigated lipin 3 activity, identified phosphorylated residues, showed that phosphorylation does not negatively regulate its ability to recognize the electrostatic charge of PA, and demonstrated that lipin 3 phosphorylation is not acutely controlled by insulin or mTOR inhibition in vitro. These findings are similar to what was observed for lipin 2 but contrast with what is known of lipin 1 phosphoregulation (14, 15). The molecular basis for such differences among enzymes, which otherwise catalyze the same reaction, is unclear.
One possibility is that the protein-wide phosphorylation of lipins 1 and 3 is intrinsically unique. Both proteins are highly phosphorylated, as shown previously for lipin 1 and demonstrated here by LC-MS/MS analysis and the incorporation of 32P (Figs. 2 and Fig. 3 and Table 1) (10). However, subtle differences in phosphorylation may alter enzyme activity and regulation. In lipin 1, the SRD is a region enriched in serines and involved in the nucleocytoplasmic trafficking of this protein (10, 13). Lipin 3 contains a homologous domain that contains nine serines and one threonine. Of these, we found that four serines are phosphorylated (Fig. 2A and Table 1). One serine in this region, Ser226, is phosphorylated in lipin 1 (as determined by Harris et al. (10)) but not in lipin 3. Although this may play a role in the regulatory differences between lipins 1 and 3, it is probably not the only factor.
PA effector proteins, including lipins, do not contain a recognized lipid association motif, but their recruitment and ability to interact with membranes is important for enzymatic activity (16, 17). Kooijman et al. (18) showed that small peptides composed of lysines and arginines can alter the electrostatic charge of PA by lowering pKa2 below the physiological pH and causing proton dissociation. Basic residues in native proteins demonstrate these principles. For example, the FRBP12 rapamycin-binding domain of mTOR contains a single arginine, crucial for the interaction of the FRBP12 rapamycin-binding domain with PA (31). Other examples include Raf-1, which binds PA at a non-conserved sequence of 35 amino acids that includes a cluster of four positively charged residues responsible for the initial electrostatic attraction between this enzyme and lipid (32).
In yeast, the best-described example of protein-PA association is with Opi1, a gene repressor (33–35). The activity of Opi1 is regulated by its sequestration to the endoplasmic reticulum membrane, mediated by binding to PA (36). Young et al. (37) showed that a basic domain in Opi1 is responsible for direct interaction with PA, and this interaction was diminished when cells were acidified. Further, they identified two lysines and one arginine as being critical for the pH-dependent electrostatic interactions with this lipid. A methyl-PA lacking the pKa2, displayed weak and pH-independent binding to Opi1 (37, 38). These studies eloquently demonstrate the dynamic nature of PA ionization state and the importance of the interactions between basic residues and PA for proper protein function.
Due to the significance of basic residues in protein-PA association, we hypothesized that the unique PBD of lipin 1 plays an important role in the ability of phosphorylation to regulate its activity. In the present work, we show that replacing the lipin 1 PBD with that of lipin 3 eliminates the ability of phosphorylation to negatively regulate lipin 1 activity (Fig. 4, C and D). Conversely, the specific activity of lipin 3 (1PBD) increased significantly only after treatment with protein phosphatase and only in the presence of PE, indicating that phosphorylation hinders its recognition of di-anionic PA (Fig. 4, E and F). In accordance with this finding, the kcat of dephosphorylated lipin 3 (1PBD) increased to a much greater extent, from 0 to 30% PE, than its phosphorylated counterpart (Table 3).
What remains unclear is how minor changes in the amino acid arrangement of the lipin PBD can drastically alter the ability of phosphorylation to control lipin activity. Kooijman at el. (21) demonstrated that the increase in the PA charge induced by lysine residues is greater than the increase induced by arginines. It is possible that lysines more readily hydrogen-bond with the phosphorylated amino acids within the protein, which weakens their interaction with PA. Interestingly, lipin 1 contains lysines on three sites within its PBD where lipin 3 contains arginines (Fig. 4A). Perhaps this underlies the mechanism whereby lipin 1 but not lipin 3 is negatively regulated by its own phosphorylation. In addition, it may be that not the number of lysines but a very precise arrangement of basic residues, both lysines and arginines, is necessary for phosphorylation to hinder recognition of PA charge and substrate association. Unfortunately, there are no structural data available for lipins, and the region between the C-terminal and N-terminal domains does not conform to any known protein fold (Fig. 2A) (5). As such, the precise mechanisms whereby the lipin 1 PBD allows for phosphoregulation are speculative. The elimination of phosphoregulation of lipin 1 (3PBD) could alternatively be explained by inherent changes in the overall phosphorylation of the mutant enzyme. However, the observed changes in gel mobility upon λ phosphatase treatment of the purified PBD mutants suggest that this is not the case (Fig. 7C). Additionally, Ser106 of lipin 1 (3PBD) is phosphorylated under basal conditions and remains downstream of mTOR in vitro, as demonstrated by reduction of phospho-Ser106 with Torin 1 treatment (Fig. 7B). Furthermore, LC-MS/MS analysis of lipin PBD mutants revealed that they shared many of the same phosphorylation sites as their WT counterparts (Tables 4 and 5 and Fig. 8). Based on these data, it appears that the exchange of the PBDs between lipins 1 and 3 does not significantly affect their phosphorylation. However, it is possible that the phosphorylated residues not found in the PBD mutants play crucial role in the regulation of PAP activity.
It is of interest whether the lipin PBD was always subject to this intermolecular regulatory mechanism rather than gaining this function later, during the emergence of more complex organisms. The PBD does not exist in yeast, and the recruitment of the yeast Pah1 is dependent on an N-terminal amphipathic helix rather than a PBD-like domain (39). The PBD domain appeared before the evolution of vertebrate species and the three distinct lipins. Whether the single lipin PBD was regulated by phosphorylation is unknown. However, although the lipin 1 domain notably changed from its first appearance to mammals, it is highly conserved, hinting at its important role in lipin function. Conversely, the lipin 3 PBD sequence is more variable in its evolution, at least from reptiles to humans. These observations suggest that perhaps lipin 3 PBD evolved to function under different cellular conditions and that its recognition of substrate charge was fine-tuned to be unresponsive to control by the enzyme's phosphorylation sites.
Another feature of the lipin PBDs is their unusual length. The regions implicated in PA association in most PA effector proteins are composed of no more than 2–5 basic residues, and often just one lysine or arginine is critical for binding (16). Lipins, however, have a nine-residue PBD, and the differences between lipin 1 and 3 PBDs are in the first five residues. Interestingly, mutation of the first four amino acids in this domain (KKRR) perturbed the binding of lipin 1 to PA. Mutations of the remaining five amino acids did not negatively affect binding to any further extent but abolished nuclear localization (22). Lipins are known to be multifunctional; for example, they are PA phosphatases but also transcriptional co-regulators (1, 3–5, 40). In the context of the literature, our data suggest that whereas the PBDs are critical for substrate recognition and association, they are probably involved in other lipin functions. These and maybe yet unknown roles for lipins are perhaps reasons for the evolution of a relatively longer PBD. The nuances surrounding the differences in the molecular regulation of lipins may be indicative of the specialization of their activities or perhaps speak to their tissue-specific functions and necessity for either more lax or alternative regulatory mechanisms (6, 30, 41). Our findings elucidate a novel intramolecular mode of lipin 1 regulation, implicating the PBD as the determinant factor in the ability of lipin activity to be controlled by phosphorylation.
Experimental procedures
Materials
[32P]ATP was from PerkinElmer Life Sciences. The 1,2-dioleoyl-sn-glycerol, 1,2-dioleoyl-sn-glycero-3-phosphate, 1,2-sn-glycero-3-phosphocholine, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, and all materials for the preparation of liposomes were from Avanti Polar Lipids (Alabaster, AL). The Escherichia coli DGK, FLAG beads, FLAG peptide, primary FLAG antibody, and alkaline-conjugated secondary anti-rabbit and anti-mouse antibodies were from Sigma-Aldrich. The generation of anti-rabbit polyclonal antibody against phosphoserine 106 of lipin 1 was described by Harris et al. (10). All other commonly used reagents were also from Sigma-Aldrich unless otherwise indicated. All cell lines were obtained from ATCC (Manassas, VA).
Cell culture
Human cervical cancer cells (HeLa) were cultured in DMEM supplemented with 5% FBS (VMR Life Science Seradigm, Radnor, PA) and 1% penicillin/streptomycin (Life Technologies, Inc.). Fibroblasts were cultured in DMEM with 10% FCS (Thermo Fisher Scientific, Waltham, MA), 1% FBS, and 1% penicillin/streptomycin. The fibroblasts were grown to confluence and differentiated to 3T3-L1 cells by treatment with 100 units/ml regular human insulin (Humulin R) (Lilly), 0.5 mm isobutylmethylxanthine, and 0.25 μm dexamethasone. Following differentiation, cells were cultured in DMEM containing 10% FBS and antibiotics.
Recombinant expression plasmids
The M. musculus lipin 3 cDNA was subcloned in frame with FLAG tag in the pCMV-TAG2B vector. The PBD exchange mutants (lipin 1 (3PBD) and lipin 3 (1PBD)) were generated by PCR mutagenesis. Venus-tagged lipin 3, lipin 1 (3PBD), and lipin 3 (1PBD) were generated by subcloning the Venus cDNA in frame with the N terminus of lipin 3 downstream from the FLAG epitope tag. Adenoviruses were generated using the pAdEASY system (42). All amino acid numbering conforms to M. musculus phosphatidic acid phosphatase LPIN3 (accession number NP_075021.1) or, when appropriate, to LPIN1 isoform b (accession number NP_056578).
Purification of recombinant lipin proteins
Cells from 40 15-cm plates were cultured in DMEM with 5% FBS and infected with an adenoviral vector expressing the indicated FLAG-lipin constructs for 48 h. The cells were fed daily during this time, harvested, and homogenized using a 22-gauge needle in buffer A (20 mm HEPES, 150 mm NaCl, 0.1% Brij, pH 7.2). The cell homogenates were centrifuged at 16,000 × g for 10 min, and the cleared lysates were incubated with 15 μl/plate of FLAG (M2) beads for 4 h at 4 °C. Following the incubation, beads were separated into two fractions and incubated in phosphatase buffer (50 mm HEPES, 100 mm NaCl, 1 mm MnCl2, 2 mm DTT, pH 7.0) with (+λ) and without (−λ) 2000 units of λ protein phosphatase for 30 min at 30 °C. The beads were then loaded on an affinity column and washed in 1 ml of buffer A 10 times. The lipin proteins were eluted with five successive additions of an equal volume of 0.5 mg/ml FLAG peptide and dialyzed against buffer A without detergent. The purified lipins were loaded on an 8.57% acrylamide SDS-polyacrylamide gel along with BSA standards and stained with Coomassie Blue dye for quantification of protein yield.
Preparation of [32P]PA
1,2-Dioleoyl-sn-glycerol was phosphorylated by E. coli diacylglycerol kinase using [γ-32P]ATP. The labeled PA was purified by thin-layer chromatography as described by Han and Carman (43).
Substrate preparation
Briefly, 90 mol % PC and 10 mol % PA or 60 mol % PC, 30 mol % PE and 10 mol % PA were dissolved in chloroform and combined. To this was added [32P]PA (10,000 cpm/nmol). The lipids were dried in vacuo. The dried lipids were hydrated in buffer B (50 mm Tris-HCl, 0.5 mm MgCl2, and 10 mm β-ME, pH 7.4) to 10 mm and subjected to five freeze-thaw cycles, followed by extrusion 11 times with a mini-extruder through a 100-nm polycarbonate filter (44). The preparation of Triton X-100/PA mixed micelles followed a protocol described previously (14). Briefly, PA and [32P]PA were combined in chloroform and dried in vacuo. Triton X-100 was added to solubilize the lipids to the indicated concentrations. Micelles were generated with 10 mol % PA and were calculated using the formula, mol % = 100 × [lipid (m)]/([lipid (m)] [Triton X-100 (m)]).
Measurement of PAP activity
The basic principles for measuring PAP activities were derived from previous work by Han and Carman (43) and further elaborated by Eaton et al. (14). Briefly, purified lipin proteins, radioactive liposomes, and buffer B were combined to a final volume of 100 μl. The final concentrations were as follows: 50 mm Tris-HCl, 0.5 mm MgCl2, 10 mm β-ME, and the indicated concentrations of PA. The reactions were allowed to proceed for 20 min at 30 °C with gentle agitation and were terminated with the addition of 500 μl of acidic methanol (methanol + 0.1 n HCl). To this was added 1 ml of chloroform followed by 1 ml of 1 m NaCl. The organic extraction was vortexed, and 500 μl of the aqueous phase was placed in scintillation vials to measure the removal of 32P from PA by a scintillation counter. The activity from assays containing lipins was normalized to activity in assays without enzyme.
Measurement of PA binding
Binding of lipin to liposome vesicles was measured following a slightly modified version of Hofer et al. (45). In short, PC/PA (80 mol % PC, 20 mol % PA) and PC/PE/PA (50 mol % PC, 30 mol % PE, 20 mol % PA) were hydrated to 10 mm in buffer C (50 mm Tris-HCl, 1 mm EDTA, pH 7.2). Liposomes were prepared as described above. The reaction volume was 100 μl and contained Venus-FLAG-lipins, 2 mm PA, and buffer C. The reactions were allowed to proceed for 30 min at 30 °C with gentle agitation and were terminated by the addition of an equal volume of 80% sucrose (w/v). This mixture was layered over 270 μl of 80% sucrose (w/v) in 5 × 41-mm Beckman centrifuge tubes, followed by 150 μl of 20% sucrose (w/v) and then 100 μl of buffer C. The sucrose gradients were centrifuged in an SW 55Ti swinging bucket rotor containing nylon inserts at 240,000 × g for 1 h. Following the centrifugation, the top 100 μl of the gradients were collected, and PC-pyrene and Venus absorption were measured. The Venus absorption from samples was normalized to input Venus readout from protein and buffer only. Similarly, PC-pyrene from samples was normalized to input PC-pyrene measurement from liposomes and buffer only. To determine the fraction of lipins bound to liposomes, Venus absorption values were normalized to that of PC-pyrene. The inability of lipin to associate with PE as well as PC has been reported previously (14, 22).
Mass spectrometry analysis of purified lipin 1, lipin 3, lipin 1 (3PBD), and lipin 3 (1PBD)
Protein was precipitated from affinity-purified lipin aliquots in 20% TCA; solubilized in 8 m urea, 75 mm NaCl, 50 mm Tris, pH 8.2; reduced using 5 mm dithiothreitol at 55 °C for 30 min; alkylated with 10 mm iodoacetamide at room temperature, diluted in 4 equal volumes of 50 mm Tris, pH 8.2; and digested overnight using trypsin (Promega Corp., Madison, WI), Lys-C (Wako Chemicals, Tokyo, Japan), or Glu-C (Worthington). The resulting peptides were desalted and analyzed in duplicate by LC-MS/MS on both Thermo LTQ-Velos-Orbitrap and Q-Exactive mass spectrometers using a data-dependent acquisition strategy. Raw mass spectra were searched using Maxquant (version 1.5.5.1) (46) against the UniProt human protein sequence database concatenated to sequences of the WT recombinant mouse lipins including the lipin 1 and 3 PBD-switch mutants, allowing for phosphorylated serine, threonine, or tyrosine. The “match between runs” feature of Maxquant was disabled, and peptide-spectral match false discovery rate was filtered to <1%. Phosphorylation sites were mapped if the corresponding peptide had a posterior error probability of ≤0.05 and if the site localization probability was 0.95 or greater. The posterior error probability score is a measure of confidence in the identity of the peptide. The site localization probability is a measure of confidence in the precise localization of the phosphorylation to a residue within the peptide.
Analysis of phosphate removal by λ protein phosphatase
HeLa cells were cultured on 4 15-cm plates and infected with adenovirus overexpressing FLAG-lipin 3 for 48 h. Following the infection period, cells were incubated in low-phosphate buffer (145 mm NaCl, 5.4 mm KCl, 1.4 mm CaCl2, 1.4 mm MgSO4, 25 mm NaHCO3, 0.2 mm sodium phosphate, 5 mm glucose, 10 mm HEPES, 0.1% insulin-free BSA, pH 7.4), radiolabeled with 0.04 mCi/ml [32P]ATP, and incubated at 30 °C for 2 h. The radiochemical specific activities were always between 4000 and 6000 Ci/mmol. Following the incubation, the cells were harvested, homogenized as described above, and then incubated with 15 μl/plate of anti-FLAG beads. Following the incubation, the beads were separated into two factions and were treated with (+λ) or without (−λ) 400 units of λ protein phosphatase. The lipin 3 was detached from the beads by the addition of 1× Laemmli buffer containing β-ME. The proteins were separated by an 8.75% SDS-PAGE gel under reducing conditions and transferred onto a PVDF membrane (Immobilon, Darmstadt, Germany). The radiolabeled protein and removal of phosphate were visualized using autoradiography.
Phosphorylation of lipin 3 in 3T3-L1 cells
7–10 days post-differentiation, 3T3-L1 cells (2 10-cm plates/condition) were infected with an adenoviral vector expressing FLAG-lipin 3 or FLAG-lipin 1 for 48 h. In the last 2 h of the infection, cells were serum-starved in low-phosphate buffer supplemented with 0.2 mCi/ml [32P]orthophosphate. During this time, the cells were treated with vehicle or Torin 1 (Tocris Bioscience, Bristol, UK) for the last 45 min, followed by 10 milliunits/ml of insulin for the last 15 min. The cells were harvested and homogenized in buffer A with protease inhibitors and cleared by centrifugation at 16,000 x g for 10 min. The supernatant was incubated with 15 μl/plate of anti-FLAG (M2) beads for 4 h at 4 °C. Proteins were displaced from the beads with the addition of Laemmli buffer containing β-ME and were separated on an 8.75% SDS-polyacrylamide gel. Phosphate incorporation into lipins 1 and 3 was visualized using autoradiography, and total protein expression was detected as described using Western blot analysis. The incorporation of 32P was normalized to total protein.
Western immunoblot analysis
To verify the expression of the indicated proteins and phosphorylation sites, the PVDF membrane containing proteins was blocked by incubation in Tris-buffered saline with detergent Tween 20 (TBST) containing 10% (w/v) dried milk for 1 h at 25 °C. The TBST contained the following: 50 mm Tris, 150 mm NaCl, and 0.05% (w/v) Tween 20, pH 7.4. After the block, the membrane was incubated with the indicated antibodies (1:1000) in TBST at 25 °C for 1 h with gentle agitation. The membrane was then incubated with alkaline phosphatase–conjugated mouse or rabbit secondary antibody (1:10,000) diluted in TBST with 2% (w/v) dried milk for 1 h at 25 °C. After three 15-min washes with TBST, the membrane was briefly incubated in chemiluminescent alkaline phosphatase substrate (Applied Biosystems, Foster City, CA). The immunoreactivity was detected using a Fuji LAS 4000.
Statistical analysis
The statistical analysis and determination of all kinetic constants were done using GraphPad Prism software. For calculation of Kmapp values, non-linear regression of the Michaelis–Menten plots was used. The kcat values were calculated using the equation, kcat = Vmax/Et, where Et is the amount of enzyme catalytic sites in nmol and Vmax is the maximal rate of the reaction in nmol/min. Statistical significance between two groups was determined using Student's t test. Statistical significance between more than two groups was determined using one-way analysis of variance (ANOVA) followed by the indicated post hoc analysis. The statistical tests used for each figure are indicated. All values are expressed as means ± S.E. Unless otherwise noted, data show one of at least three independent experiments. When appropriate, all experiments were repeated with at least two separate protein preparations. Significance was set to p < 0.05.
Author contributions
S. B. and T. E. H. conceived, coordinated, and designed the study. T. E. H. also significantly contributed to the editing of the manuscript. S. B. designed, performed, and analyzed the experiments shown in Figs. 1 (C, D, and H), 2 (B–E), 3, 4, 5, 6, and 7 and Tables 2 and 3 and wrote the manuscript. S. T. designed, performed, and analyzed the experiments shown in Fig. 2F, and R. T. L., S. W. E., and J. V. designed, performed, and analyzed the experiments shown in Figs. 2A and 8 and Tables 1, 4, and 5. S. B., T. E. H., and M. E. G. analyzed and displayed the data seen in Figs. 2A and 8 and Tables 1 and 4. M. E. G. also assisted in the experiments performed in Fig. 7A. J. M. P. performed and analyzed the experiments shown in Fig. 1 (A, B, E, and F). J. M. E. designed, performed, and analyzed the experiments shown in Fig. 1A. G. R. M. assisted in the experiments performed in Fig. 2B and made significant contributions to the editing of the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Carl Creutz, Katelyn Ahern, and Mitchell Granade for help with the editing of the manuscript.
This work was supported by the National Institutes of Health Grants R01 DK101946 (to T. E. H.) and R35 GM119536 (to J. V). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Table S1.
- PA
- phosphatidic acid
- mTOR
- mammalian target of rapamycin
- PE
- phosphatidylethanolamine
- PBD
- polybasic domain
- NEM
- N-ethylmaleimide
- βME
- β-mercaptoethanol
- SRD
- serine-rich domain
- PC
- phosphatidylcholine.
References
- 1. Coleman R. A., and Lee D. P. (2004) Enzymes of triacylglycerol synthesis and their regulation. Prog. Lipid Res. 43, 134–176 [DOI] [PubMed] [Google Scholar]
- 2. Carman G. M., and Han G. S. (2009) Phosphatidic acid phosphatase, a key enzyme in the regulation of lipid synthesis. J. Biol. Chem. 284, 2593–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carman G. M., and Han G. S. (2006) Roles of phosphatidate phosphatase enzymes in lipid metabolism. Trends Biochem. Sci. 31, 694–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reue K., and Zhang P. (2008) The lipin protein family: dual roles in lipid biosynthesis and gene expression. FEBS Lett. 582, 90–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harris T. E., and Finck B. N. (2011) Dual function lipin proteins and glycerolipid metabolism. Trends Endocrinol. Metab. 22, 226–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reue K., and Dwyer J. R. (2009) Lipin proteins and metabolic homeostasis. J. Lipid Res. 50, S109–S114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brindley D. N. (1984) Intracellular translocation of phosphatidate phosphohydrolase and its possible role in the control of glycerolipid synthesis. Prog. Lipid Res. 23, 115–133 [DOI] [PubMed] [Google Scholar]
- 8. Gomez-Muñoz A., Hatch G. M., Martin A., Jamal Z., Vance D. E., and Brindley D. N. (1992) Effects of okadaic acid on the activities of two distinct phosphatidate phosphohydrolases in rat hepatocytes. FEBS Lett. 301, 103–106 [DOI] [PubMed] [Google Scholar]
- 9. Grimsey N., Han G. S., O'Hara L., Rochford J. J., Carman G. M., and Siniossoglou S. (2008) Temporal and spatial regulation of the phosphatidate phosphatases lipin 1 and 2. J. Biol. Chem. 283, 29166–29174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harris T. E., Huffman T. A., Chi A., Shabanowitz J., Hunt D. F., Kumar A., and Lawrence J. C. Jr. (2007) Insulin controls subcellular localization and multisite phosphorylation of the phosphatidic acid phosphatase, lipin 1. J. Biol. Chem. 282, 277–286 [DOI] [PubMed] [Google Scholar]
- 11. Huffman T. A., Mothe-Satney I., and Lawrence J. C. Jr. (2002) Insulin-stimulated phosphorylation of lipin mediated by the mammalian target of rapamycin. Proc. Natl. Acad. Sci. U.S.A. 99, 1047–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peterson T. R., Sengupta S. S., Harris T. E., Carmack A. E., Kang S. A., Balderas E., Guertin D. A., Madden K. L., Carpenter A. E., Finck B. N., and Sabatini D. M. (2011) mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Péterfy M., Harris T. E., Fujita N., and Reue K. (2010) Insulin-stimulated interaction with 14-3-3 promotes cytoplasmic localization of lipin-1 in adipocytes. J. Biol. Chem. 285, 3857–3864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eaton J. M., Mullins G. R., Brindley D. N., and Harris T. E. (2013) Phosphorylation of lipin 1 and charge on the phosphatidic acid head group control its phosphatidic acid phosphatase activity and membrane association. J. Biol. Chem. 288, 9933–9945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eaton J. M., Takkellapati S., Lawrence R. T., McQueeney K. E., Boroda S., Mullins G. R., Sherwood S. G., Finck B. N., Villén J., and Harris T. E. (2014) Lipin 2 binds phosphatidic acid by the electrostatic hydrogen bond switch mechanism independent of phosphorylation. J. Biol. Chem. 289, 18055–18066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stace C. L., and Ktistakis N. T. (2006) Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim. Biophys. Acta 1761, 913–926 [DOI] [PubMed] [Google Scholar]
- 17. Shin J. J., and Loewen C. J. (2011) Putting the pH into phosphatidic acid signaling. BMC Biol. 9, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kooijman E. E., Carter K. M., van Laar E. G., Chupin V., Burger K. N., and de Kruijff B. (2005) What makes the bioactive lipids phosphatidic acid and lysophosphatidic acid so special? Biochemistry 44, 17007–17015 [DOI] [PubMed] [Google Scholar]
- 19. Kooijman E. E., and Burger K. N. (2009) Biophysics and function of phosphatidic acid: a molecular perspective. Biochim. Biophys. Acta 1791, 881–888 [DOI] [PubMed] [Google Scholar]
- 20. van Meer G., Voelker D. R., and Feigenson G. W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kooijman E. E., Tieleman D. P., Testerink C., Munnik T., Rijkers D. T., Burger K. N., and de Kruijff B. (2007) An electrostatic/hydrogen bond switch as the basis for the specific interaction of phosphatidic acid with proteins. J. Biol. Chem. 282, 11356–11364 [DOI] [PubMed] [Google Scholar]
- 22. Ren H., Federico L., Huang H., Sunkara M., Drennan T., Frohman M. A., Smyth S. S., and Morris A. J. (2010) A phosphatidic acid binding/nuclear localization motif determines lipin1 function in lipid metabolism and adipogenesis. Mol. Biol. Cell 21, 3171–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Péterfy M., Phan J., Xu P., and Reue K. (2001) Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat. Genet. 27, 121–124 [DOI] [PubMed] [Google Scholar]
- 24. Carman G. M., Deems R. A., and Dennis E. A. (1995) Lipid signaling enzymes and surface dilution kinetics. J. Biol. Chem. 270, 18711–18714 [DOI] [PubMed] [Google Scholar]
- 25. Han G. S., and Carman G. M. (2010) Characterization of the human LPIN1-encoded phosphatidate phosphatase isoforms. J. Biol. Chem. 285, 14628–14638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jamal Z., Martin A., Gomez-Muñoz A., and Brindley D. N. (1991) Plasma membrane fractions from rat liver contain a phosphatidate phosphohydrolase distinct from that in the endoplasmic reticulum and cytosol. J. Biol. Chem. 266, 2988–2996 [PubMed] [Google Scholar]
- 27. Phan J., and Reue K. (2005) Lipin, a lipodystrophy and obesity gene. Cell Metab. 1, 73–83 [DOI] [PubMed] [Google Scholar]
- 28. Schweitzer G. G., Collier S. L., Chen Z., Eaton J. M., Connolly A. M., Bucelli R. C., Pestronk A., Harris T. E., and Finck B. N. (2015) Rhabdomyolysis-associated mutations in human LPIN1 lead to loss of phosphatidic acid phosphohydrolase activity. JIMD Rep. 23, 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Majeed H. A., Al-Tarawna M., El-Shanti H., Kamel B., and Al-Khalaileh F. (2001) The syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia: report of a new family and a review. Eur. J. Pediatr. 160, 705–710 [DOI] [PubMed] [Google Scholar]
- 30. Csaki L. S., Dwyer J. R., Li X., Nguyen M. H., Dewald J., Brindley D. N., Lusis A. J., Yoshinaga Y., de Jong P., Fong L., Young S. G., and Reue K. (2014) Lipin-1 and lipin-3 together determine adiposity in vivo. Mol. Metab. 3, 145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Veverka V., Crabbe T., Bird I., Lennie G., Muskett F. W., Taylor R. J., and Carr M. D. (2008) Structural characterization of the interaction of mTOR with phosphatidic acid and a novel class of inhibitor: compelling evidence for a central role of the FRB domain in small molecule-mediated regulation of mTOR. Oncogene 27, 585–595 [DOI] [PubMed] [Google Scholar]
- 32. Ghosh S., Strum J. C., Sciorra V. A., Daniel L., and Bell R. M. (1996) Raf-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf-1 in 12-O-tetradecanoylphorbol-13-acetate-stimulated Madin-Darby canine kidney cells. J. Biol. Chem. 271, 8472–8480 [DOI] [PubMed] [Google Scholar]
- 33. Carman G. M., and Han G. S. (2009) Regulation of phospholipid synthesis in yeast. J. Lipid Res. 50, S69–S73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wagner C., Dietz M., Wittmann J., Albrecht A., and Schüller H. J. (2001) The negative regulator Opi1 of phospholipid biosynthesis in yeast contacts the pleiotropic repressor Sin3 and the transcriptional activator Ino2. Mol. Microbiol. 41, 155–166 [DOI] [PubMed] [Google Scholar]
- 35. Henry S. A., Kohlwein S. D., and Carman G. M. (2012) Metabolism and regulation of glycerolipids in the yeast Saccharomyces cerevisiae. Genetics 190, 317–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Salas-Santiago B., and Lopes J. M. (2014) Saccharomyces cerevisiae essential genes with an Opi− phenotype. G3 4, 761–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Young B. P., Shin J. J., Orij R., Chao J. T., Li S. C., Guan X. L., Khong A., Jan E., Wenk M. R., Prinz W. A., Smits G. J., and Loewen C. J. (2010) Phosphatidic acid is a pH biosensor that links membrane biogenesis to metabolism. Science 329, 1085–1088 [DOI] [PubMed] [Google Scholar]
- 38. Loewen C. J., Gaspar M. L., Jesch S. A., Delon C., Ktistakis N. T., Henry S. A., and Levine T. P. (2004) Phospholipid metabolism regulated by a transcription factor sensing phosphatidic acid. Science 304, 1644–1647 [DOI] [PubMed] [Google Scholar]
- 39. Karanasios E., Han G. S., Xu Z., Carman G. M., and Siniossoglou S. (2010) A phosphorylation-regulated amphipathic helix controls the membrane translocation and function of the yeast phosphatidate phosphatase. Proc. Natl. Acad. Sci. U.S.A. 107, 17539–17544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Finck B. N., Gropler M. C., Chen Z., Leone T. C., Croce M. A., Harris T. E., Lawrence J. C. Jr., Kelly D. P. (2006) Lipin 1 is an inducible amplifier of the hepatic PGC-1α/PPARα regulatory pathway. Cell Metab. 4, 199–210 [DOI] [PubMed] [Google Scholar]
- 41. Donkor J., Sariahmetoglu M., Dewald J., Brindley D. N., and Reue K. (2007) Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J. Biol. Chem. 282, 3450–3457 [DOI] [PubMed] [Google Scholar]
- 42. He T. C., Zhou S., da Costa L. T., Yu J., Kinzler K. W., and Vogelstein B. (1998) A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. U.S.A. 95, 2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Han G. S., and Carman G. M. (2004) Assaying lipid phosphate phosphatase activities. Methods Mol. Biol. 284, 209–216 [DOI] [PubMed] [Google Scholar]
- 44. MacDonald R. C., MacDonald R. I., Menco B. P., Takeshita K., Subbarao N. K., and Hu L. R. (1991) Small-volume extrusion apparatus for preparation of large, unilamellar vesicles. Biochim. Biophys. Acta 1061, 297–303 [DOI] [PubMed] [Google Scholar]
- 45. Höfer C. T., Herrmann A., and Müller P. (2010) Use of liposomes for studying interactions of soluble proteins with cellular membranes. Methods Mol. Biol. 606, 69–82 [DOI] [PubMed] [Google Scholar]
- 46. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.