

Abstract
Saccharomyces cerevisiae Mid1 is composed of 548 amino acids and a regulatory subunit of Cch1, a member of the eukaryotic pore-forming, four-domain cation channel family. The amino acid sequence and voltage insensitivity of Cch1 are more similar to those of Na+ leak channel non-selective (NALCN) than to the α1 subunit of voltage-gated Ca2+ channels (VGCCs). Despite a lack in overall primary sequence similarity, Mid1 resembles in some aspects VGCC α2/δ regulatory subunits and NALCN-associated proteins. Unlike animal α2/δ subunits, Mid1 and NALCN-associated proteins are essential for the function of the pore-forming subunit. We herein investigated the processing and membrane translocation of Mid1. Mid1 was found to have a 20-amino-acid-long N-terminal signal peptide and appeared to be entirely localized extracellularly. A signal peptide–deleted Mid1 protein, Mid1ΔN23, was N-glycosylated and retained Ca2+ influx activity through Cch1. Moreover, an N-terminal truncation analysis revealed that even truncated Mid1 lacking 209 N-terminal amino acid residues was N-glycosylated and maintained Ca2+ influx activity. A 219-amino-acid-truncated Mid1 protein lost this activity but was still N-glycosylated. In the sec71Δ and sec72Δ single mutants defective in the post-translational protein transport into the endoplasmic reticulum (ER), Mid1ΔN23 could not mediate Ca2+ influx and did not undergo N-glycosylation, whereas wild-type Mid1 exhibited normal Ca2+ influx activity and N-glycosylation in these mutants. Therefore, the signal peptide–lacking Mid1ΔN23 protein may be translocated to the ER exclusively through the post-translational protein translocation, which typically requires an N-terminal signal peptide. Mid1 may provide a tool for studying mechanisms of protein translocation into the ER.
Keywords: calcium channel, calcium transport, membrane protein, membrane transport, N-linked glycosylation, protein translocation, Saccharomyces cerevisiae, signal peptidase, yeast
Introduction
Among Ca2+-permeable ion channels, voltage-gated Ca2+ channels (VGCCs)3 have been the most intensively studied from basic to clinical viewpoints because they play crucial roles in synaptic transmission in neurons and contraction in skeletal muscle cells by mediating Ca2+ influx in response to changes in membrane potential in animal cells (1). VGCCs are composed of four subunits: the pore-forming α1 subunit and three auxiliary subunits, α2/δ, β, and γ. The α2/δ subunit is composed of four subtypes, each of which is the product of a single gene encoding an ∼140-kDa protein with a C-terminal potential transmembrane segment or glycosylphosphatidylinositol (GPI) anchor attachment site and a number of N-glycosylation sites (2). The single polypeptide is post-translationally cleaved into α2 and δ fragments, after which both are bound by disulfide bonding. In the mature form of the α2/δ protein, α2 is exposed entirely to the extracellular space; δ is anchored to the plasma membrane; and both are N-glycosylated, giving this protein a total molecular mass of ∼180 kDa. Although necessary for the modulation of VGCC properties, the α2/δ subunit does not affect single channel conductance, suggesting that it is not required for Ca2+ permeation of the α1 subunit (3–5).
The yeast Saccharomyces cerevisiae has a VGCC-related Ca2+ channel composed of essential Cch1 and Mid1 subunits. Cch1 was identified by scanning the yeast sequence database and is 24% identical to the α1 subunit of mammalian L-type VGCCs in overall amino acid sequences and is, thus, called an α1 homolog (6). Mid1 was identified by the screening of mutants showing the mating pheromone-induced death (mid) phenotype because of a defect in Ca2+ influx (7). Mid1 is composed of 548 amino acid residues and is localized in plasma and endoplasmic reticulum (ER) membranes (7–10). Although Mid1 has no homologous protein in animals or plants with respect to overall amino acid sequences, some features of this protein resemble those of animal α2/δ subunits, such as a potential N-terminal signal peptide, a number of potential N-glycosylation sites, and multiple cysteine residues (see Fig. 1) (7, 11). In addition, many of the fungal Mid1 orthologs have a GPI anchor attachment motif at the C-terminal region, similar to animal α2/δ proteins. Therefore, Mid1 may be called an α2/δ-like protein. In contrast to the α1 subunit of animal VGCCs, Cch1 is completely dependent on Mid1 to mediate Ca2+ influx (6, 12).
Figure 1.
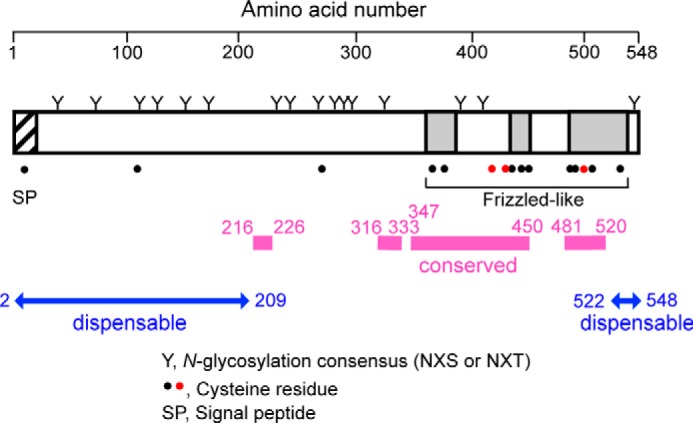
Schematic representation of the Mid1 protein. Twenty N-terminal amino acid residues constitute the signal peptide (hatched region). The N and C termini are present in the extracellular space and lumen of the endoplasmic reticulum. All of the 16 N-glycosylation consensus sites are N-glycosylated (designated by Y). Dots, Cys residues. Pink bars, regions highly conserved in the fungal Mid1 family. At least 209 N-terminal amino acid residues and the 27 C-terminal amino acid residues are non-essential for the function of Mid1 (blue double-headed arrows). There is a frizzled-like motif composed of ∼180 amino acid residues in the C-terminal region (three gray regions). This motif contains 10 Cys residues. Three red Cys residues (Cys417, Cys431, and Cys498) are reproducibly shown to be essential for the function of Mid1 (39) (K. Iida, unpublished data). Note that Cys417 and Cys431 are not included in the frizzled-like motif.
Although homologous to the α1 subunits of animal VGCCs, Cch1 is more homologous to a recently identified subfamily of animal VGCC α1 subunits, the Na+ leak channel non-selective (NALCN), as revealed by a phylogenetic analysis (13). Functionally, voltage insensitivity is similar between Cch1 and NALCN (14, 15), and this may be because both have fewer positive charges in the voltage-sensing S4 segment than the α1 subunit of VGCCs. In support of the close similarities between Cch1 and NALCN, their auxiliary subunits exhibit similar functional essentialities; as described above, Mid1 is essential for the function of Cch1. Likewise, the auxiliary subunits UNC80, UNC79, NLF-1, and CG33988 of animal NALCN are required for the function of NALCN; however, their localization and overall structure are divergent (16–18). The cysteine-rich domains of Mid1, NLF-1, CG33988, and FAM155A and -B are highly conserved and, thus, designated as the Mid1 domain, leading to the hypothesis that the relationship between the Mid1 domain and NALCN has persisted over one billion years of evolution (18). In this context, it has become more important to characterize the basic molecular nature of Mid1 as a model of essential subunits of the NALCN family.
There are two major protein translocation mechanisms that transfer secretory and plasma membrane proteins across the ER membrane (19, 20). One is co-translational translocation, which is enabled by a direct link between translation and translocation. Before the synthesis of these proteins is finished, their signal peptides are recognized and bound by the signal recognition particle (SRP) in the cytoplasm. The SRP mediates the binding of the complex to the SRP receptor at the ER membrane. The signal peptide is then inserted into the translocation channel or translocon composed of the Sec61 protein and others. S. cerevisiae cells with the sec61Δ mutation are inviable (21). The other mechanism is post-translational translocation. Whereas this mechanism also requires the translocon and the signal peptides of the proteins, it needs the Sec62/Sec63 complex, composed of Sec62, Sec63, Sec71 (also termed Sec66), and Sec72 (also termed Sec67) in place of SRP. Sec71 and Sec72 are fungi-specific subunits. In S. cerevisiae, the sec62Δ and sec63Δ single mutants are inviable (22), whereas the sec71Δ and sec72Δ single mutants are viable (23). The viability of the latter two mutants provides a useful tool for testing the dependence of protein translocation on the post-translational mechanism.
In the present study, we show that 20 N-terminal amino acid residues of Mid1 are a signal peptide. Although this signal peptide functions properly, the signal peptide–deleted forms of Mid1 are also N-glycosylated and become functional for mediating Ca2+ influx. One of these forms, Mid1ΔN23, is suggested to be delivered into the ER by the post-translational translocation mechanism in a Sec71- and Sec72-dependent fashion, whereas the complete Mid1 is delivered by the co-translational translocation mechanism. We suggest that Mid1 is delivered into the ER by at least two protein translocation mechanisms, and this is dependent on its molecular form. We also demonstrate that most of the 16 potential N-glycosylation sites of Mid1 are N-glycosylated and that the N and C termini are exposed to the extracellular space, suggesting that the entire sequence of Mid1 is present outside the cell.
Results
Identification of the N-terminal signal peptide
All of the public web software servers utilized, including PrediSi (24), Phobius (25), Signal-BLAST (26), and SignalP 4.1 (27), predicted that the Mid1 protein has a signal peptide with 20 amino acid residues at the N terminus. To confirm this prediction experimentally, we attempted to elucidate the N-terminal amino acid sequence of the mature Mid1 protein. We purified FLAG-tagged Mid1 from yeast cells using differential centrifugation and immunoprecipitation with an anti-FLAG antibody, followed by SDS-PAGE and Western blotting (Fig. 2). The purified protein was subjected to sequencing by Edman degradation, and the first five N-terminal amino acids were identified as LFQDF, which was identical to the Leu21-Phe22-Gln23-Asp24-Phe25 sequence deduced from the nucleotide sequence of the MID1 gene. Therefore, we concluded that the Mid1 protein has a signal peptide composed of 20 N-terminal amino acid residues (Fig. 2).
Figure 2.
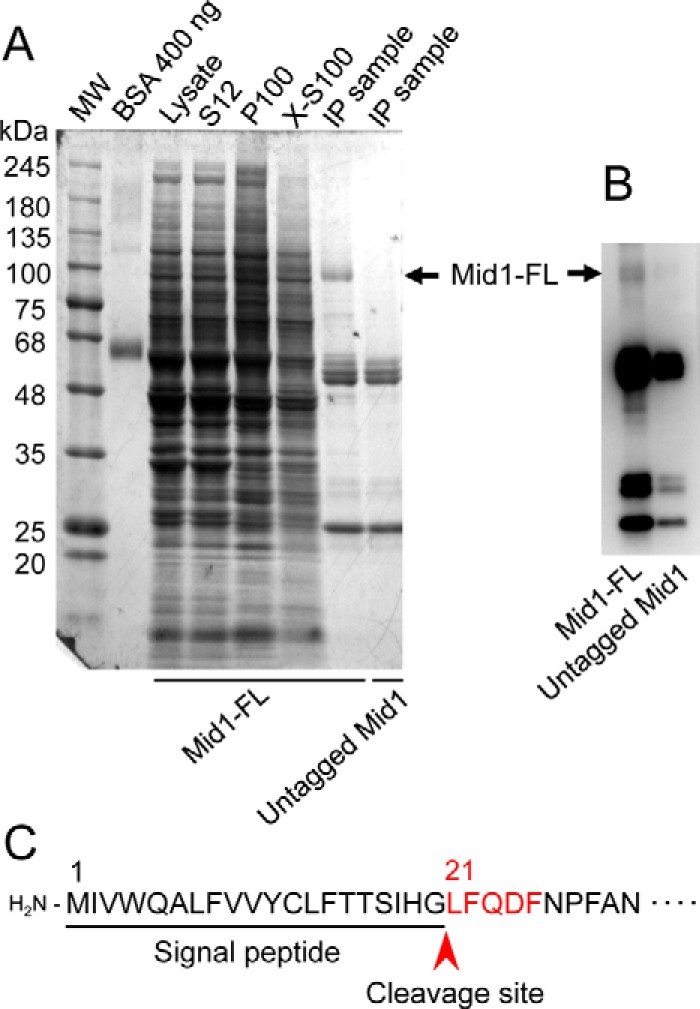
Purification of the Mid1 protein and the presentation of its signal peptide. A, purification steps of the Mid1 protein tagged C-terminally with the FLAG epitope (Mid1-FL). Mid1-FL was purified by differential centrifugation, followed by immunoprecipitation with an anti-FLAG antibody. Proteins in an SDS-polyacrylamide gel were visualized with Coomassie Brilliant Blue. S12, P100, and X-S100 represent the supernatant of 12,000 × g centrifugation, the pellet of 100,000 × g centrifugation, and the supernatant of 100,000 × g centrifugation of a solubilized P100 preparation with Triton X-100. IP, immunoprecipitation. The arrow represents the position of Mid1-FL. B, Western blotting of immunoprecipitated samples with an anti-FLAG antibody. C, a signal peptide of Mid1 represented with an underline. The amino acid sequence LFQDF shown in red is that elucidated by amino acid sequencing as the N terminus of the purified, mature Mid1 protein. The arrowhead indicates the cleavage site between Gly20 and Leu21.
N-terminally truncated forms of the Mid1 protein lacking the signal peptide are active
To establish whether the N-terminal signal peptide is important for Mid1 to become active through proper processing and modifications, we constructed a truncated form of this protein lacking 23 N-terminal amino acid residues (ΔN23), expressed it from the MID1 promoter in the mid1 mutant, and assayed its ability to rescue the mid1 phenotypes, such as weak Ca2+ uptake activity and the loss of viability in the presence of the mating pheromone α-factor. Fig. 3 (A and B) shows that the ΔN23 protein rescued both phenotypes; however, this rescuing activity was slightly weaker than that of the wild-type Mid1 protein. These results indicate that the signal peptide is not essential for Mid1 to acquire activity but is required for full activity. These results also suggest that the ΔN23 protein is targeted properly to the plasma membrane to cooperate with Cch1.
Figure 3.
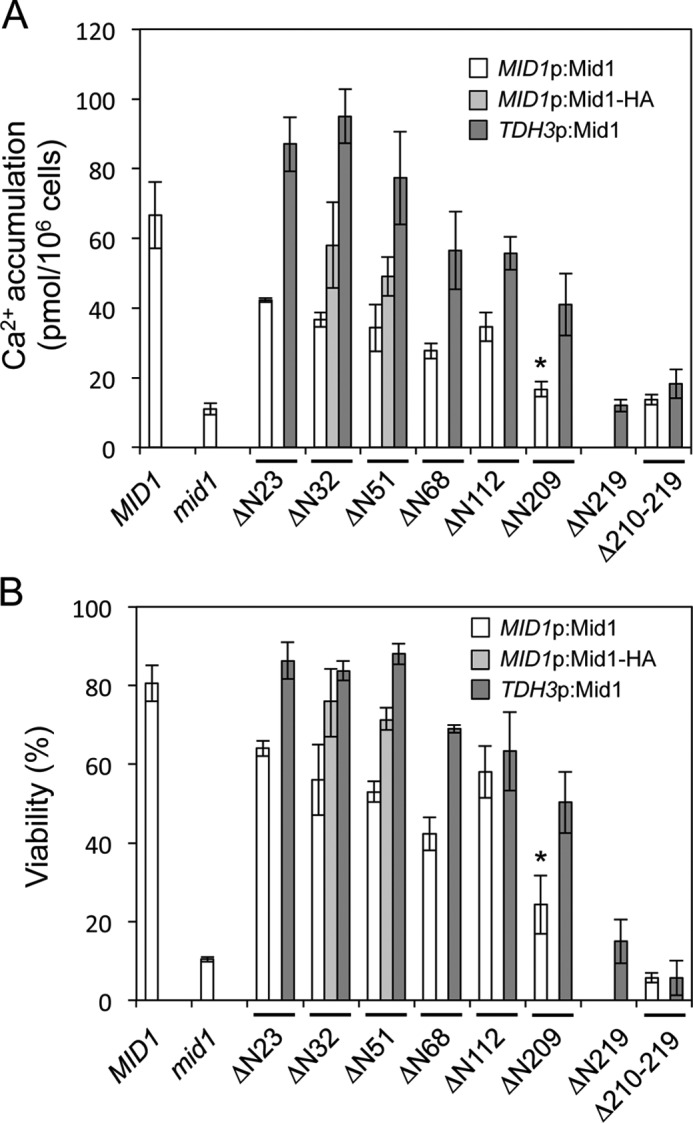
Activities of N-terminally truncated Mid1 proteins and an internally deleted Mid1 protein. Shown are Ca2+ accumulation (A) and the viability (B) of the mid1Δ mutant transformed with the plasmid that expresses either Mid1ΔN23, Mid1ΔN51, Mid1ΔN68, Mid1ΔN112, Mid1ΔN209, Mid1ΔN219, or Mid1Δ210–219 or their corresponding C-terminally HA-tagged derivatives. Cells of the transformants were exposed to 6 μm α-factor in SD.Ca100 medium for 2 h for the assessment of Ca2+ accumulation and for 8 h for the evaluation of cell viability. MID1p:Mid1, the Mid1 protein expressed from the MID1 promoter; MID1p:Mid1-HA, the Mid1-HA protein expressed from the MID1 promoter; TDH3p:Mid1, the Mid1 protein expressed from the transcriptionally strong TDH3 promoter. The means ± S.D. (error bars) from at least three independent experiments are given. *, p < 0.03 (mid1 versus ΔN209).
The above results with the ΔN23 protein prompted us to investigate how much of the N-terminal region may be truncated while maintaining the activity of Mid1. We constructed a series of N-terminally truncated forms of Mid1, such as ΔN32, ΔN51, ΔN68, ΔN112, ΔN209, and ΔN219, the numbers of which represent the deleted number of amino acid residues from the N terminus. We then expressed them from the MID1 promoter or a transcriptionally strong TDH3 promoter (28) in the mid1 mutant and measured their activities, similar to ΔN23. As shown in Fig. 3, all of the truncated Mid1 proteins except for ΔN219 retained Ca2+ influx activities and viability maintenance; however, their activities were one-half to one-third wild-type levels when these constructs were expressed from the promoter of MID1 (see white bars). The ΔN209 protein retained weak Ca2+ accumulation activity and viability (mid1 versus ΔN209 in white bars, p < 0.03). However, when these constructs were expressed from the TDH3 promoter, even the ΔN209 protein exhibited activity that was more than one-half that of wild-type levels, whereas the ΔN219 protein did not (see dark gray bars). These results indicate that 209 N-terminal amino acid residues are non-essential for the function of Mid1.
We also found that HA-tagged deletion mutant proteins (ΔN32-HA and ΔN51-HA), which were expressed from the MID1 promoter, had slightly stronger activities than the untagged counterparts (Fig. 3), suggesting that the HA tag does not impair the activity of the truncated forms of Mid1.
N-terminally truncated forms of Mid1 are N-glycosylated
Intact Mid1 is an N-glycosylated protein (7). Therefore, functionally active, N-terminally truncated forms of Mid1, such as ΔN23, ΔN32, ΔN51, ΔN68, ΔN112, and ΔN209, were expected to be N-glycosylated. To examine this hypothesis, we performed a Western blot analysis of the wild-type and ΔN112 proteins before and after the endoglycosidase H (Endo H) digestion of whole-cell extracts. Fig. 4A shows that the higher-molecular weight population of the two proteins was Endo H-sensitive and, thus, was N-glycosylated. Similar to ΔN112, all of the other truncated proteins were N-glycosylated; however, the amounts of the proteins were reduced to approximately one-half to one-third those of wild-type Mid1 (Fig. 4, A and B). Furthermore, even the non-active form of Mid1, ΔN219, was N-glycosylated. These results indicate that the truncation of at least 219 amino acid residues from the N terminus did not affect N-glycosylation and also imply that yeast cells possess a mechanism by which proteins that have lost the N-terminal signal peptide may still be incorporated into the lumen of the ER and then N-glycosylated.
Figure 4.
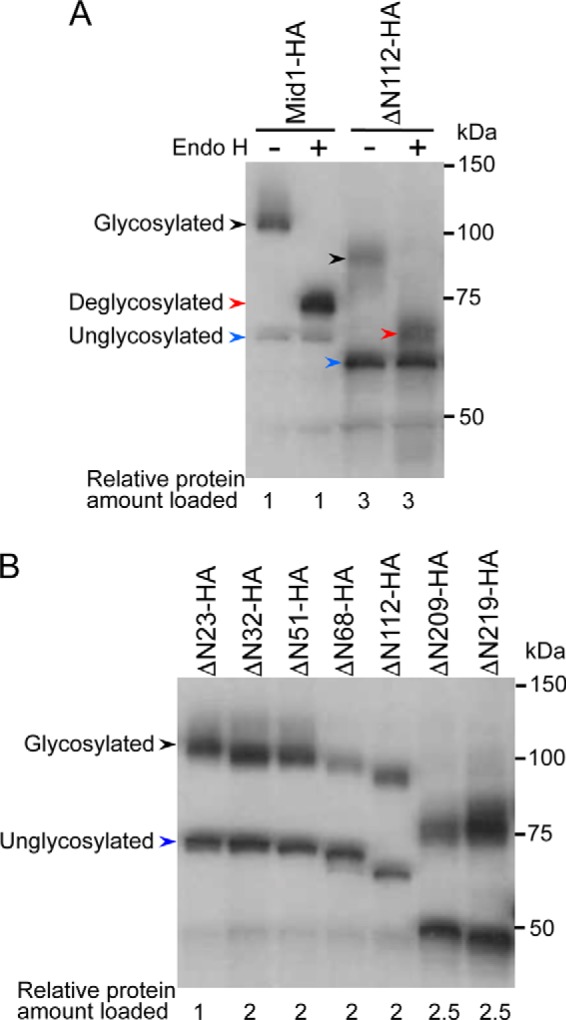
N-glycosylation of N-terminally truncated Mid1-HA proteins as revealed by Western blotting. The yeast strains used were those expressing the N-terminally truncated forms of Mid1 expressed from the MID1 promoter, described in the legend to Fig. 3. A, identification of N-glycosylated proteins with the Endo H treatment. Crude extracts were split into two aliquots, one of which was treated with Endo H and the other with Milli-Q water. Both were then subjected to SDS-PAGE followed by Western blotting with an anti-HA antibody. The Endo H treatment converted the ∼110 kDa band to the ∼75 kDa band in Mid1-HA and the ∼95 kDa band to the ∼66 kDa band in ΔN112-HA, indicating that the ∼110 and ∼95 kDa bands are N-glycosylated forms. B, all of the N-terminally truncated forms of Mid1-HA examined are N-glycosylated. Crude extracts were directly subjected to SDS-PAGE followed by Western blotting. In the bottom of both panels, the relative protein amount loaded to the gel was described. Black arrowhead, N-glycosylated protein; red arrowhead, deglycosylated protein; blue arrowhead, unglycosylated protein.
The Pro210–Met219 region is essential
Because the above results suggest that the region spanning from Pro210 to Met219 is essential for the function of Mid1 (Fig. 3, A and B), we deleted this region and examined the activity of the deletion mutant (designated Δ210–219). The Δ210–219 protein completely lost its activity (see the first and second bars from the right ends in Fig. 3, A and B). We then compared the amino acid sequence of this region of the S. cerevisiae Mid1 protein with those of 96 orthologs and found that Asp216 and Asp218 were highly conserved during evolution (Fig. 5A). Therefore, we performed an amino acid replacement analysis for the two acidic residues. Asp216 and Asp218 were replaced by the small, non-polar amino acid, Ala, or the polar, uncharged amino acid, Asn, the side-chain volume of which was similar to that of Asp, resulting in the D216A, D216N, D218A, and D218N mutant proteins. Fig. 5 (B and C) shows that D216A and D216N exhibited Ca2+ influx activities and viability maintenance similar to those of the wild-type Mid1, suggesting that the acidity and side-chain volume of Asp216 are not important for the activity of Mid1.
Figure 5.
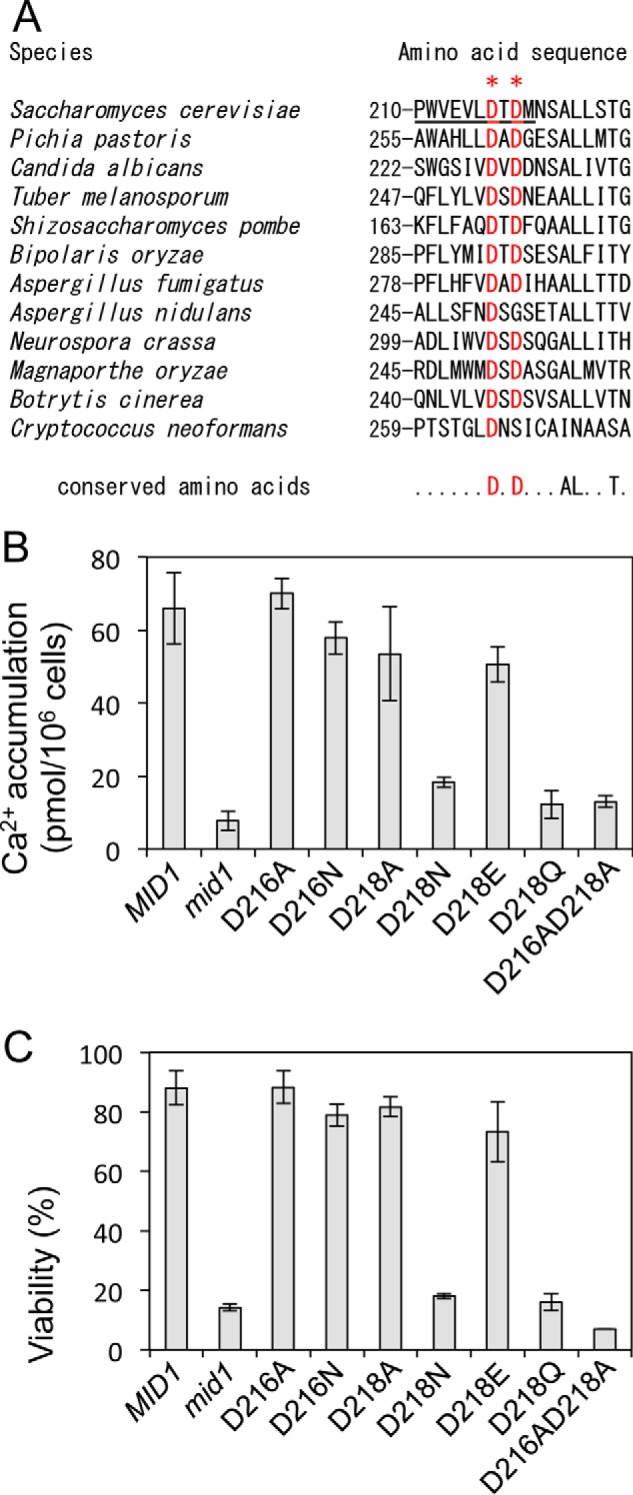
Asp216 and Asp218 are important for the function of Mid1. A, amino acid sequence alignment of the 210–227 region of Mid1 and its corresponding regions of fungal orthologs. Highly conserved Asp216 and Asp218 are shown in red and marked with asterisks. B, Ca2+ accumulation. C, the viability of cells. Cells expressing an amino acid substitution mutant Mid1 protein were incubated and subjected to Ca2+ accumulation and viability assays as described in the legend to Fig. 3. The means ± S.D. (error bars) of at least three independent experiments are given.
On the other hand, D218N lost both activities, whereas D218A did not. Furthermore, D218Q, but not D218E, lost both activities. In terms of the physicochemical properties of amino acids, the hydrophilicities of Asp, Asn, Glu, and Gln are known to be similar (29), whereas Ala possesses a small side chain. Therefore, we speculated that an amino acid residue at the position of Asp218 may be acidic or small. Both factors may be important for Mid1. We also found that the D216A/D218A double mutant lost both activities (Fig. 5, B and C), suggesting that at least one Asp residue is essential for the function of Mid1 in this region.
N-terminal signal peptide–deleted Mid1 is delivered into the ER post-translationally
To elucidate how the N-terminal signal peptide–deleted forms of Mid1 are delivered into the ER, we focused on yeast mutants lacking Sec proteins, which are present in the ER membrane. Secretory and plasma membrane proteins are translocated into the ER through two major protein translocation mechanisms. One is co-translational and the other post-translational (19, 20). These co-translational and post-translational protein translocation mechanisms both utilize the common translocation channel composed of the Sec61 protein. However, the former depends on the SRP for the targeting of the ribosome-nascent polypeptide complex to the Sec61 complex, whereas the latter depends on the Sec62/63/71/72 complex for the targeting of fully translated proteins to the Sec61 complex in S. cerevisiae. In the present study, we employed the sec71Δ and sec72Δ mutations to investigate the mechanisms by which the N-terminal signal peptide–deleted forms of Mid1 were delivered into the ER and became functional. We also employed the Mid1ΔN23 protein as a model of the N-terminal signal peptide–deleted forms of Mid1. We did not employ the sec61Δ, sec62Δ, or sec63Δ mutation because these are lethal mutations (21, 22) and also because Sec61 and Sec63 are involved in co-translational and post-translational protein translocation (19, 20, 30).
Mid1ΔN23 and a control protein, wild-type Mid1, were expressed individually from the MID1 promoter in either mid1Δ, mid1Δ sec71Δ, or mid1Δ sec72Δ mutants, and the Ca2+ accumulation activity and viability of each mutant expressing one of the two proteins were examined after cells were exposed to α-factor. Fig. 6A shows that in the mid1Δ mutant, Mid1ΔN23 exhibited approximately half the activity of wild-type Mid1, suggesting that yeast cells possess a mechanism that properly translocates N-terminal signal peptide–deleted proteins. This was also implied from the results shown in Fig. 3A (see the bar of MID1 and ΔN23).
Figure 6.

Mid1ΔN23 is translocated to the ER post-translationally. Shown are Ca2+ accumulation (A) and the viability (B) of the mid1Δ, mid1Δ sec71Δ, and mid1Δ sec72Δ mutants transformed with the plasmid YCpS-MID1 or YCpS-MID1ΔN23. Cells of the transformants were treated with α-factor and subjected to the measurement of Ca2+ accumulation and cell viability as described in the legend to Fig. 3. C, identification of N-glycosylation of the MID1ΔN23-FL52 protein with the Endo H treatment. The mid1Δ, mid1Δ sec71Δ, and mid1Δ sec72Δ mutants were transformed with the plasmid YCpS-MID1-FL52 or YCpS-MID1ΔN23-FL52. Crude extracts prepared from the transformants were subjected to the Endo H treatment, followed by SDS-PAGE and Western blotting, as described in the legend to Fig. 4, except that an anti-FLAG antibody was used to detect the proteins to be identified. The amount of whole proteins in the crude extracts loaded to lanes 7, 8, 11, and 12 was 5 times as high as that loaded to other lanes. Black arrowhead, N-glycosylated protein; red arrowhead, deglycosylated protein; blue arrowhead, unglycosylated protein. For simplicity, arrowheads are shown only in lanes 9–12. Error bars, S.D.
In the sec71Δ background, Mid1ΔN23-expressing cells lost Ca2+ accumulation activity to an almost negative control level (compare mid1Δ sec71Δ/MID1ΔN23 with mid1Δ/vector), whereas wild-type Mid1-expressing cells did not lose this activity (Fig. 6A), indicating that Mid1ΔN23 depends almost completely on Sec71 to become functional.
In the sec72Δ background, Mid1ΔN23-expressing cells lost Ca2+ accumulation activity completely (Fig. 6A). In addition, the activity of wild-type Mid1-expressing cells was decreased to approximately two-thirds that of wild-type Mid1-expressing cells in the mid1Δ mutant (compare mid1Δ sec72Δ/MID1 with mid1Δ/MID1). These results suggest that Mid1ΔN23 is completely dependent on Sec72 to become functional. The results obtained with the sec71Δ and sec72Δ mutations suggest that Mid1ΔN23 is translocated into the ER post-translationally. The difference in the activity of wild-type Mid1 between the sec71Δ and sec72Δ backgrounds was not unexpected because individual proteins with the N-terminal signal peptide are known to be differentially sensitive to the mutations. For example, the Sec63-invertase fusion protein (an ER membrane protein) was translocated more efficiently into the ER in the sec72Δ mutant than in the sec71Δ mutant, whereas pro-α-factor (a secretory protein) was translocated more efficiently into the ER in the sec71Δ mutant than in the sec72Δ mutant (23).
Viability assays led to essentially the same conclusion (Fig. 6B). In the mid1Δ mutant, cells expressing Mid1ΔN23 slightly lost viability 8 h after exposure to α-factor. In the sec71Δ and sec72Δ backgrounds, cells expressing Mid1ΔN23 lost viability completely, whereas those expressing wild-type Mid1 did not.
To clarify whether the absence of Ca2+ accumulation activity and lack of viability of Mid1ΔN23-expressing cells in the sec71Δ and sec72Δ backgrounds is due to a failure in the translocation of the Mid1ΔN23 protein across the ER membrane, we performed a Western blot analysis of the wild-type Mid1 and Mid1ΔN23 proteins tagged with the epitope FLAG between the N-terminal 51st and 52nd amino acid residues (Mid1-FL52 and Mid1ΔN23-FL52, respectively). Mid1-FL52 exhibited the full activity of untagged Mid1 for Ca2+ accumulation and viability maintenance (data not shown). Whole-cell extracts were prepared from the mid1Δ, mid1Δ sec71Δ, and mid1Δ sec72Δ mutants expressing either Mid1-FL52 or Mid1ΔN23-FL52, incubated with or without Endo H, and then subjected to SDS-PAGE followed by Western blotting (Fig. 6C). In the mid1Δ mutant, Mid1-FL52 and Mid1ΔN23-FL52 were both found to be N-glycosylated to produce a ∼110-kDa protein, and the N-glycosylated forms of the two proteins were equally sensitive to Endo H to produce ∼75-Da proteins (lanes 1–4). we concluded that Mid1-FL52 and Mid1ΔN23-FL52 were both equally N-glycosylated in the SEC71+ and SEC72+ backgrounds. On the other hand, Mid1ΔN23-FL52 was not N-glycosylated in the sec71Δ or sec72Δ background and stayed as a ∼72-kDa protein (lanes 7, 8, 11, and 12), whereas full-length Mid1-FL52 underwent N-glycosylation in the same backgrounds (lanes 5, 6, 9, and 10). These results suggest that Mid1ΔN23-FL52 is translocated to the ER, depending upon the function of Sec71 and Sec72 post-translationally, whereas full-length Mid1-FL52 is translocated to the ER co-translationally. We also noted that the amount of the Mid1ΔN23-FL52 protein was markedly less than that of the Mid1-FL52 protein in sec71Δ and sec72Δ cells. We speculate that the Mid1ΔN23-FL52 protein that failed to be translocated to the ER may be susceptible to proteolytic degradation in the cytoplasm. Taken together, the results shown in Fig. 6 (A–C) are consistent with the N-terminal signal peptide–deleted forms of Mid1 entering the ER by the post-translational protein translocation mechanism and then being N-glycosylated.
Prediction of membrane topology by assessing N-glycosylation sites
To elucidate the role of Mid1 in vivo, it is important to assess the membrane topology of Mid1. This protein has four hydrophobic regions (7), one of which was found to be the signal peptide described above, whereas the others may be a transmembrane segment. We attempted to predict its topology using several web-based prediction servers, including MEMSAT2 (31), Phobius (25), TMHMM version 2.0 (32), and TMpred (33); however, these servers gave divergent results. Thus, we predicted membrane topology experimentally. To achieve this, we focused on putative N-glycosylation sites (NXS or NXT) because Mid1 has 16 putative N-glycosylation sites that are distributed in most parts of the Mid1 sequence (Fig. 1), and these sites must be located at the extracellular space or in the lumen of the ER.
Because Mid1 is a highly N-glycosylated protein (7), difficulties were associated with unambiguously identifying N-glycosylation sites using single amino acid substitutions followed by mobility shift assays on SDS-polyacrylamide gels (data not shown). In an attempt to overcome this, we constructed a mutant Mid1 protein, named Mid1(11Q), in which 11 Asn residues of 16 NX(S/T) sites were replaced by Gln and 5 Asn residues remained unchanged (Fig. 7A); therefore, a single change in N-glycosylation became detectable using mobility shift assays. We found that the Mid1(11Q) protein maintained weak but significant activity to complement the mid1 mutation, whereas the Mid1(16Q) protein with 16 Asn to Gln substitutions did not (data not shown). Therefore, the Mid1(11Q) protein was suitable for subsequent experiments. The 11 replaced sites covered the positions before and after the four hydrophobic regions named H1–H4, which may be potential transmembrane segments (7), except for the position between H3 and H4. To compensate for this exception, we replaced Val363 with Asn to introduce a novel N-glycosylation motif (Asn363-Cys364-Ser365) in the Mid1(11Q) protein (see V363N in Fig. 7A).
Figure 7.

All putative N-glycosylation sites examined are N-glycosylated. A, schematic diagram of the Mid1(11Q) protein, the 11 putative N-glycosylation sites (Asn) of which were replaced with Gln. Open circle, intact N-glycosylation site; closed circle, mutated N-glycosylation site with the Asn → Gln substitution; *, a newly substituted Asn residue, which produced the Q146N or V363N substitution. Note that the latter generated a new N-glycosylation site, whereas the former did not. SP, signal peptide. H1–H4, the top four hydrophobic regions in Mid1 (7). Note that among the 16 putative N-glycosylation sites, only Asp282 (one of the open circles) was not tested for N-glycosylation because this residue was expected to receive the same modification as the 6 Asn residues near it. In addition, the reason for constructing Q285N/Q291N was that the two positions were so close to each other that it was unnecessary to construct the corresponding single substitution mutants because the purpose of this experiment was to assess the membrane topology of Mid1. B, a putative N-glycosylation site added to the Mid1(11Q) protein decreases its electrophoretic mobility, indicating that it is N-glycosylated. *, newly substituted Asn residue; arrow, the position of Mid1(11Q) on the Western blot. C, the Asn → Gln substitution at a putative N-glycosylation site in the Mid1(11Q) protein increases its electrophoretic mobility, indicating that the substituted site is originally an N-glycosylation site. In B and C, crude extracts prepared from cells expressing each substitution mutant protein were subjected to SDS-PAGE and a Western blot analysis using affinity-purified rabbit polyclonal antibodies against 20 C-terminal amino acid residues of Mid1 (10).
When single Gln residues were returned individually to Asn at Gln32, Gln70, Gln228, Gln238, Gln265, Gln285 + Gln291, Gln324, Gln389, Gln407, and Gln536 of the Mid1(11Q) protein to restore a single N-glycosylation motif (NX(S/T)), all proteins shifted to an upper position from the Mid1(11Q) protein on an SDS-polyacrylamide gel (Fig. 7B), suggesting that the restored sites were really N-glycosylated. In addition, the V363N protein shifted to the upper position. The result suggests that the V363N-containing area between H3 and H4 is not localized intracellularly but extracellularly. As a control of the Gln to Asn replacement experiment, we replaced the intrinsic Gln146 residue with Asn to give Q146N with the amino acid sequence of Asn146-Ala147-Leu148, which was not an N-glycosylation motif, and examined its mobility on the SDS-polyacrylamide gel. As expected, the Q146N protein did not shift on the gel (Fig. 7B).
To investigate whether the unreplaced Asn residues in the N-glycosylation motif, Asn112, Asn125, Asn159, and Asn175, were N-glycosylated in the Mid1(11Q) protein, we individually replaced these Asn residues with Gln and examined their mobility on an SDS-polyacrylamide gel. Fig. 7C shows that the N112Q, N125Q, N159Q, and N175Q proteins shifted to a lower position from the Mid1(11Q) protein, suggesting that these four Asn residues are real N-glycosylation sites.
Collectively, these results suggest that most of the putative N-glycosylation sites were N-glycosylated, leading to the hypothesis that the whole Mid1 protein is present outside the cell and in the lumen of the ER with no transmembrane segment.
N and C termini of Mid1 are present extracellularly
If the above hypothesis is correct, the N and C termini of Mid1 must both be present outside the cell and in the lumen of the ER. To address this issue, we used the Suc2C reporter fused to the N or C terminus of the Mid1 protein to assess N-glycosylation. Suc2C is a fragment (60 amino acid residues) near the C terminus of invertase Suc2 and has four potential N-glycosylation sites (34). As a control, the Asn residues in the four sites were replaced with Gln to make Suc2CQ. The reporters were fused to the N terminus of Mid1 to produce Suc2C-Mid1 and Suc2CQ-Mid1, which were found to exhibit mid1-complementing activity (data not shown). As shown in Fig. 8A, the molecular weight of Suc2C-Mid1 was greater than that of Suc2CQ-Mid1 before the Endo H treatment but shifted to smaller-sized proteins with the same molecular weights after the Endo H treatment. These results suggest that Suc2C, but not Suc2CQ, fused to the N terminus of Mid1, is N-glycosylated, and, thus, the N terminus is located outside the cell and in the lumen of the ER. These results also suggest that N-terminally fused Suc2C and Suc2CQ interfere with the removal of the signal peptide but do not inhibit the function of Mid1.
Figure 8.
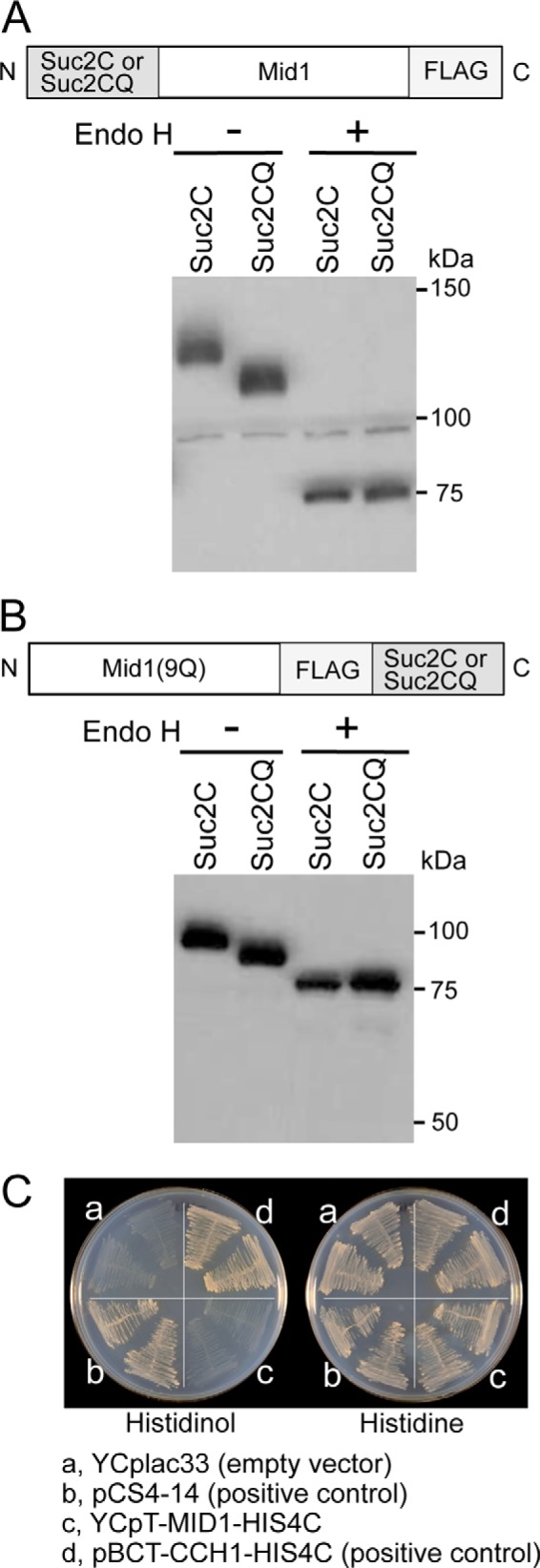
N and C termini of Mid1 are present outside the cell and in the ER lumen. A, subcellular localization of the N terminus of Mid1. The Mid1 protein tagged N-terminally with the N-glycosylation reporter Suc2C or its negative control Suc2CQ was treated with Endo H or mock-treated and then subjected to Western blotting with an anti-FALG antibody. B, subcellular localization of the C terminus of Mid1. The Mid1(9Q) protein tagged C-terminally with Suc2C or Suc2CQ was subjected to the same treatment and analysis as described above. C, histidinol plate assays. The yeast strain FC2a transformed with each of the following four plasmids was streaked onto a histidinol plate (left) and histidine plate (right), incubated at 30 °C for 3 days, and then photographed. a, YCplac33 (empty vector; negative control); b, pCS4–14 (positive control); c, YCpT-MID1-HIS4C; d, pBCT-CCH1-HIS4C (positive control).
When both reporters were fused to the C terminus of Mid1, the resulting Mid1-Suc2C and Mid1-Suc2CQ were hard to distinguish from each other based on apparent molecular weights on SDS-polyacrylamide gels (data not shown), and this may have been due to their tertiary structures with multiple N-glycosylated sites. Therefore, we constructed the Mid1(9Q) protein tagged C-terminally with Suc2C and Suc2CQ, in which nine Asn residues were replaced by Gln to reduce the number of N-glycosylation sites in Mid1 (see the legend to Table 1). The resulting Mid1(9Q)-Suc2C and Mid1(9Q)-Suc2CQ exhibited mid1-complementing activity and were subjected to the same experiments as Suc2C-Mid1 and Suc2CQ-Mid1. Fig. 8B showed that a molecular weight difference was clearly present between Mid1(9Q)-Suc2C and Mid1(9Q)-Suc2CQ, with the former being greater than the latter before the Endo H treatment. After this treatment, the two fused proteins shifted to smaller-sized proteins with the same molecular weights. These results suggest that the C terminus of Mid1 is also present outside the cell and in the lumen of the ER.
Table 1.
Plasmids used in this study
| Plasmid name | Genotype |
|---|---|
| YCpT-MID1-FL | LEU2 CEN4 ARS1 TDH3p:MID1–5xFLAG ampr |
| YCpS-MID1 | LEU2 CEN4 ARS1 MID1p:MID1 ampr |
| YCpS-MID1ΔN23a | LEU2 CEN4 ARS1 MID1p:MID1 (Δ2–23) ampr |
| YCpS-MID1ΔN23-HAa | LEU2 CEN4 ARS1 MID1p:MID1 (Δ2–23)-HAx4 ampr |
| YCpT-MID1ΔN23a | LEU2 CEN4 ARS1 TDH3p:MID1 (Δ2–23) ampr |
| YCpS-MID1Δ210–219 | LEU2 CEN4 ARS1 MID1p:MID1 (Δ210–219) ampr |
| YCpT-MID1Δ210–219 | LEU2 CEN4 ARS1 TDH3p:MID1 (Δ209–219) ampr |
| YCpS-MID1-FL52 | LEU2 CEN4 ARS1 MID1p:MID1-5xFLAG52ampr |
| YCpS-MID1ΔN23-FL52 | LEU2 CEN4 ARS1 MID1p:MID1 (Δ2–23)-5xFLAG52ampr |
| YCpT-MID1(11Q) | LEU2 CEN4 ARS1 TDH3p:MID1 (11Q)bampr |
| YCpS-SUC2C-MID1-FL | LEU2 CEN4 ARS1 MID1p:SUC2C-MID1–5xFLAG ampr |
| YCpS-SUC2CQ-MID1-FL | LEU2 CEN4 ARS1 MID1p:SUC2CQ-MID1–5xFLAG ampr |
| YCpS-MID1(9Q)-FL-SUC2C | LEU2 CEN4 ARS1 MID1p:MID1 (9Q)c-5xFLAG-SUC2C ampr |
| YCpS-MID1(9Q)-FL-SUC2CQ | LEU2 CEN4 ARS1 MID1p:MID1 (9Q)c-5xFLAG-SUC2CQ ampr |
| YCpT-MID1-HIS4C | URA3 CEN4 ARS1 TDH3p:MID1-HIS4C ampr |
| pBCT-CCH1-HIS4C | URA3 CEN4 ARS1 TDH3p:CCH1-HIS4C ampr |
| pCS4-14d | URA3 2 μm-ori HMG1-SUC2-HIS4C ampr |
a Plasmids that express a series of N-terminally truncated forms of the Mid1 and Mid1-HA proteins, including Mid1ΔN32, Mid1ΔN51, Mid1ΔN68, Mid1ΔN112, Mid1ΔN209, and Mid1ΔN219 and their corresponding C-terminally HA-tagged derivatives were also constructed. However, to avoid redundancy, only the three listed plasmids were described as representatives.
b 11Q, a series of MID1 mutants with 11 Asn → Gln substitutions: N32Q, N70Q, N228Q, N238Q, N265Q, N285Q, N291Q, N324Q, N389Q, N407Q, and N536Q.
c 9Q, MID1 mutants with nine Asn → Gln substitutions: N32Q, N70Q, N228Q, N238Q, N275Q, N324Q, N389Q, N407Q, and N536Q.
d A gift from Sengstag (35).
To confirm this, we employed a growth assay on histidinol plates (35). His4C, a histidinol dehydrogenase that catalyzes the conversion of histidinol to histidine, was fused to the C terminus of Mid1 to produce Mid1-His4C. If the C terminus of Mid1 is located in the cytoplasm, fused His4C allows the growth of the his4-101 mutant (strain FC2a) on SD agar plates containing histidinol but lacking histidine. However, if the C terminus is located outside the cell or in the lumen of the ER, fused His4C cannot allow the growth of the mutant. As shown in Fig. 8C, the his4-101 mutant expressing Mid1-His4C did not grow on the histidinol plate. In contrast, the mutant expressing Cch1-His4C, in which His4C was fused to the cytoplasmic C terminus of Cch1, or the positive control protein Hmg1-Suc2-His4C (35) was allowed to grow on the histidinol plate. This result supports the conclusion that the C terminus of Mid1 is located outside the cell and in the lumen of the ER. Collectively, these results suggest that the entire molecule of Mid1 is located in the extracellular space and lumen of the ER.
Discussion
In the present study, we characterized the basic molecular features of Mid1, an S. cerevisiae α2/δ-like protein. We found that Mid1 had an N-terminal signal peptide composed of 20 amino acid residues that was cleaved off between Gly20 and Leu21. This result is consistent with the prediction made by several signal peptide prediction software servers. Mid1 has 16 potential N-glycosylation sites almost throughout its entire sequence (7), and most of them were found to be N-glycosylated in the present study. We also found that the N and C termini of Mid1 were exposed to the extracellular space and lumen of the ER. Based on these results and previous findings showing that Mid1 is present at the ER and plasma membranes, we propose that the whole body of Mid1 is present in the lumen side of the ER membrane and outside the plasma membrane. This proposal is supported by the previous finding that Mid1 is cleaved by Zymolyase applied to intact yeast cells (7). Zymolyase is a commercial lytic enzyme preparation that exhibits endoglucanase and protease activities (36).
GPI-anchored or not
The extracellular localization of the entire Mid1 protein is consistent with the features of the α2/δ subunits of animal VGCCs. With a conventional view, the α2/δ subunit is completely exposed to the extracellular space, except for a potential transmembrane segment present in the C terminus of the δ subunit. However, a recent study reported that the α2/δ subunit is GPI-anchored at the C terminus of the δ subunit (37). In either case, it is clear that most of the animal α2/δ subunits are present outside the cell. Our online search for a GPI anchor attachment site of Mid1 orthologs with PredGPI (38) predicted that S. cerevisiae Mid1 is not a GPI-anchored protein (Fig. 9). This prediction does not contradict the following findings from previous studies: Mid1 does not have the C-terminal hydrophobic sequence required by GPI-anchored proteins (7); C-terminal truncation up to 27 amino acid residues does not affect the function of Mid1 (39); and wild-type Mid1 proteins tagged C-terminally with HA or GFP are localized to the ER and plasma membranes and are capable of complementing the mid1 mutation (7, 10, 40).
Figure 9.
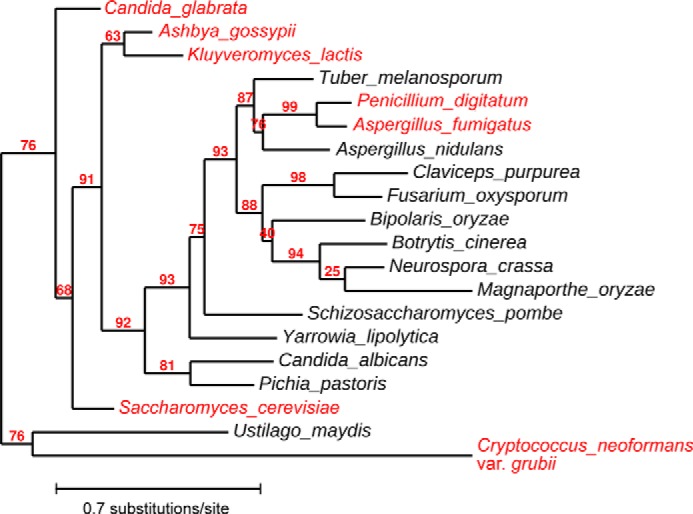
Maximum-likelihood phylogeny of Mid1 orthologs in representative fungi. The amino acid sequences of Mid1 orthologs from 20 fungi were analyzed with the online program Phylogeny.fr (50) to construct a phylogenetic tree. The value at each branch is bootstrap support (%). Amino acid sequences were also examined with the GPI anchor predictor PredGPI (38) to predict whether each Mid1 ortholog is a GPI-anchored protein. Species with a non-GPI-anchored Mid1 ortholog are shown in red.
The GPI anchor search also provided an interesting prediction. Among Mid1 orthologs from 20 representative fungi, those from six species, including Candida glabrata and Cryptococcus neoformans as well as S. cerevisiae, were predicted to not be GPI-anchored, whereas the remainder, including Schizosaccharomyces pombe, Candida albicans, Neurospora crassa, and Aspergillus nidulans, were expected to be GPI-anchored (Fig. 9). Based on the phylogenetic tree of the Mid1 orthologs, the Mid1 protein of some species such as S. pombe and C. albicans appears to have acquired a GPI anchor attachment site during evolution. This speculation raises the question of what molecule attaches the non-GPI-anchored Mid1 protein to the ER and plasma membranes. A previous study demonstrated that the Mid1 protein was not released from the membrane fraction with NaCl, Na2CO3, or urea, but was released with Triton X-100, leading to the hypothesis that Mid1 is an integral membrane protein (7). Therefore, Mid1 may be attached to the ER and plasma membranes by an as yet unidentified integral membrane molecule. We suggest that Cch1 is not a partner of membrane trafficking because Mid1 is localized properly in cch1Δ cells.4
Sec71- and Sec72-dependent protein translocation to the ER and plasma membrane
An unexpected result of the present study was that the signal peptide–deleted derivatives of Mid1 were able to be N-glycosylated and became functional for Ca2+ influx. Up to 209 N-terminal amino acid residues were found to be non-essential for Mid1 to become N-glycosylated and maintain Ca2+ influx activity, although the amount and activity of the protein gradually decreased as the extent of truncation increased. In eukaryotic cells, plasma membrane proteins and secreted proteins are translocated to the ER by one of two translocation mechanisms, co-translational translocation and post-translational translocation, after which N-glycosylation occurs appropriately (41).
The present study demonstrated an interesting feature of Mid1 in protein translocation. When the Mid1ΔN23 protein was examined as a model of the N-terminal signal peptide–deleted derivatives of Mid1, the translocation of this protein was found to be totally dependent on Sec71 and Sec72, which are required for post-translational protein translocation in yeast. Namely, Mid1ΔN23 possessed Ca2+ accumulation and cell viability maintenance activities in SEC71+ and SEC72+ cells, but lost these activities in sec71Δ or sec72Δ cells. Furthermore, the loss of these activities correlated with a lack of N-glycosylation and a decrease in the amount of Mid1ΔN23 in these mutant cells. Therefore, it is obvious that Mid1ΔN23 is translocated to the ER by the post-translational protein translocation mechanism. A typical model depicts that the post-translational protein translocation mechanism as well as the co-translational protein translocation mechanism require an N-terminal signal peptide to translocate substrate proteins to the ER (19, 20). However, it is possible that an internal signal peptide or a signal patch, both of which are located downstream of the N-terminal 219-amino acid residue, may be responsible for the translocation of the signal peptide–deleted derivatives of Mid1. In this context, it is possible to speculate that the H3 to H4 region, as two closely spaced, hydrophobic segments separated by a short turn, forms a helical hairpin that acts as an internal signal peptide and holds the Mid1 protein to the membrane (42); however, this speculation seems to be unlikely, because the V363N-containing area, which could be the turn of the hairpin produced by the H3 and H4 segments, was found to be localized extracellularly (Fig. 7, A and B). Based on the present finding that Mid1 is localized in the lumenal side of the ER membrane and outside the plasma membrane, it is also possible that hydrophobic H3 and/or H4 segments may individually serve as a hairpin loop tethering the Mid1 protein to the exoplasmic leaflet of the membrane. Another possibility is that Mid1 has a partner molecule that helps it to be translocated to the ER. These possibilities warrant further study.
We noted that the results of our truncation experiments were partly inconsistent with previous findings showing that the Mid1 derivative composed of only 22 N-terminal amino acid residues tagged with GFP exhibited weak Ca2+ influx–mediating activity (40). In light of the present finding that 20 N-terminal amino acid residues constitute a signal peptide, it is unlikely that the 22-amino acid fragment exhibits this activity. We also found that Ca2+ influx–mediating activity associated with two C-terminally truncated forms of Mid1 tagged with GFP, Mid1(1–360)-GFP and Mid1(1–400)-GFP (40), was unlikely because even C-terminally truncated forms with longer sequences, such as Mid1(1–501) and Mid1(1–509), did not maintain Ca2+ influx activity (39). Therefore, the GFP-tagged Mid1 proteins need to be re-examined.
In conclusion, Mid1 has an N-terminal signal peptide and is N-glycosylated and localized to the extracellular space. N-terminal signal peptide–deleted derivatives of Mid1 are translocated to the ER by the post-translational protein translocation mechanism in a Sec71- and Sec72-dependent manner. Up to at least 209 N-terminal amino acid residues may be deleted without losing the acceptability of N-glycosylation and the activity of mediating Ca2+ influx, whereas 219 N-terminal amino acid residues cannot be deleted to maintain the activity. We have shown that the region spanning amino acid residues 210–219 is essential for the activity. Mid1 may serve not only as a model of the regulatory subunit of the Cch1/VGCC/NALCN cation channel family but also as a tool for studying mechanisms of protein translocation into the ER.
Experimental procedures
Yeast strains
The parental strain H207 (MATa his3-Δ1 leu2-3,112 trp1-289 ura3-52 sst1-2) and its derivative mutant strain H311 (MATa mid1-Δ5::HIS3 his3-Δ1 leu2-3,112 trp1-289 ura3-52 sst1-2) were described previously (8). The isogenic mutant strains TC71 (sec71Δ), TC71m (mid1Δ sec71Δ), TC72 (sec72Δ), and TC72m (mid1Δ sec72Δ) were constructed by a one-step gene replacement procedure (43), in which a PCR product carrying the URA3 cassette flanked with the 5′- and 3′-ends of the SEC71 or SEC72 gene was integrated into a respective gene of the strains H207 and H311. The strain FC2a (MATa his4-101 leu2-3,112 tryp1-1 ura3-52 HOL1-1) was a gift from Dr. C. Sengstag and was used in histidinol plate assays (35).
Media
Synthetic media SD.Ca100 and SD were prepared as described previously (7). SD.Ca100 and SD media contained 100 and 681 μm CaCl2, respectively. In histidinol plate assays, 6 mm histidinol was added to SD agar plates instead of histidine. Synthetic complete medium, SC.Ca100, contained 100 μm CaCl2, and supplemented with 0.01 mg/ml adenine, 0.05 mg/ml l-arginine, 0.08 mg/ml l-aspartic acid, 0.05 mg/ml l-isoleucine, 0.05 mg/ml l-lysine, 0.02 mg/ml l-methionine, 0.05 mg/ml l-phenylalanine, 0.10 mg/ml l-threonine, 0.05 mg/ml l-tyrosine, and 0.14 mg/ml valine. Where necessary, 0.02 mg/ml l-histidine, 0.10 mg/ml l-leucine, 0.05 mg/ml l-tryptophan, and 0.02 mg/ml uracil were supplemented to synthetic media.
Plasmids
The plasmids used in this study are listed in Table 1. Standard methods were used to manipulate genetic materials (44). All of the plasmids carrying the MID1 gene and its derivatives were constructed from YCpS-MID1 (45), YCpT-MID1 (46), and YCpT-MID1-HA4 (47). The plasmids that express the Mid1-His4C and Cch1-His4C proteins, YCpT-MID1-HIS4C and pBCT-CCH1-HIS4C, respectively, were constructed by fusing the HIS4C ORF on pCS4-14 (35) to the 3′-end of the MID1 ORF on YCpT-MID1 and that of CCH1 ORF on pBCT-CCH1H (48). The plasmids that express a series of N-terminally truncated forms of the Mid1 and Mid1-HA proteins as well as an internal deletion mutant of Mid1 were constructed from YCpS-MID1, YCpT-MID1-HA, and YCpT-MID1 by introducing appropriate restriction enzyme sites in the MID1 ORF by PCR. These include Mid1ΔN23, Mid1ΔN51, Mid1ΔN68, Mid1ΔN112, Mid1ΔN209, Mid1ΔN219, and Mid1Δ210–219 and their corresponding C-terminally HA-tagged derivatives. The symbol ΔN23 indicates that the 23 N-terminal amino acid residues of the Mid1 protein have been truncated. To avoid redundancy, only plasmids expressing the Mid1ΔN23 and Mid1ΔN23-HA proteins and those expressing the internal deletion mutant are listed in Table 1. The plasmids expressing a series of the Mid1 proteins with Asn → Gln substitutions or Gln → Asn substitutions were constructed by introducing a corresponding substitution mutation with the KOD-Plus Mutagenesis Kit (Toyobo Co., Ltd., Osaka, Japan). The same methodology and kit were applied to construct the plasmids expressing the Mid1 proteins with point mutations at Asp216 and/or Asp218. The fusion of the Suc2C or Suc2CQ tag to the Mid1 protein with five FLAG epitopes was performed according to the method described previously (34). Suc2C is a fragment of Suc2 invertase and has four potential N-glycosylation sites (34), and Suc2CQ is an N-glycosylation-less derivative of Suc2C with four Asn to Gln substitutions. All of the plasmids constructed were confirmed by DNA sequencing.
Purification of Mid1-FLAG
The preparation of the microsomal fraction and immunoprecipitation were performed according to the method described by Locke et al. (49). Cells of strain H311 (mid1-Δ5) carrying YCpT-MID1-FL were grown in SC.Ca100 medium (2 liters) to the late log phase (OD600 = 1.6–1.8), harvested, washed once with Milli-Q water by centrifugation at 800 × g for 15 min, and then suspended in 10 ml of BB buffer (300 mm sorbitol, 100 mm NaCl, 5 mm MgCl2, and 10 mm Tris-HCl (pH 7.5)). Cells were disrupted using a French Press (Thermo Electron Corp., FA-078A, Madison, WI) at 200,000 p.s.i. to obtain the cell lysate. Cell debris was removed by centrifugation (at 3,000 × g and 4 °C for 15 min), and the supernatant (Lysate in Fig. 2A) was centrifuged at 12,000 × g and 4 °C for 10 min to obtain a high-speed supernatant (S12), which was then centrifuged at 100,000 × g and 4 °C for 30 min. The pellet (P100) was solubilized in 2 ml of IP buffer containing Triton X-100 (50 mm Tris-HCl (pH 8.0), 1.0% Triton X-100, 150 mm NaCl, 2 mm EDTA, 1 mm PMSF, and protease inhibitor mixture (04-693-159-001, Roche Diagnostics GmbH, Mannheim, Germany)) and then centrifuged again at 100,000 × g and 4 °C for 30 min. The supernatant (X-S100) was used for immunoprecipitation as follows. Five micrograms of a mouse monoclonal anti-FLAG antibody (012-22384, Wako, Osaka, Japan) was bound to 2.5 μg of Dynabeads Protein A (10002D, Thermo Fisher Scientific, Waltham, MA) in 200 μl of IP buffer. X-S100 (1 ml) was then mixed with the antibody-Dynabead complex and incubated at 4 °C for 2 h with rotation. The Mid1-FLAG–antibody–Dynabead complex was washed three times with 500 μl of IP buffer and once with 100 μl of wash buffer (50 mm Tris-HCl (pH 8.0)) and solubilized in 24 μl of SDS-sample buffer without reducing reagents (62.5 mm Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, and 0.001% bromphenol blue). The solution was briefly centrifuged to recover the Mid1-FLAG protein from the ternary complex.
N-terminal amino acid sequencing of Mid1-FLAG
The N-terminal amino acid sequence of Mid1-FLAG was elucidated by Edman degradation at the Protein Engineering Office of Nippi, Inc. (Tokyo, Japan).
Endo H treatment
The method used was essentially the same as that described by Kamano et al. (34). Briefly, ∼1.2 × 107 cells were harvested, washed once with Milli-Q water, and resuspended in TE buffer (10 mm Tris-HCl (pH 8.0) and 1 mm EDTA) containing 1 mm PMSF and protease inhibitor mixture. Cells in the suspension were disrupted with glass beads as described previously (7). The crude extracts received one-tenth volume of 10× denaturation buffer (5% SDS, 0.4 m DTT) and were heated at 70 °C for 5 min. The heated sample was cooled on ice, one-tenth volume of the 10× reaction buffer (0.5 m sodium acetate buffer (pH 5.2)) was added, and the mixture was then divided into two equal aliquots, one of which received Endo H (a final concentration of 0.17 units/ml) (catalog no. 11088726001, Roche Diagnostics GmbH); the other received an equal volume of the solvent Milli-Q water. Both were incubated at 37 °C for 1 h and subjected to SDS-PAGE followed by Western blotting.
SDS-PAGE and Western blotting
Previously described methods were generally followed (7). To detect the Mid1 protein tagged with the HA or FLAG epitope, a mouse monoclonal anti-HA antibody (12CA5, BabCo, Richmond, CA) or mouse monoclonal anti-FLAG antibody (Wako), respectively, was used. To detect amino acid–substituted Mid1 proteins with no tag, affinity-purified rabbit polyclonal antibodies against the C-terminal peptide (20 amino acid residues) of Mid1 (10) were used.
Assessment of Ca2+ accumulation and cell viability
Cells were grown in SD.Ca100 medium to the mid-log phase and incubated with 6 μm α-factor for 2 h to assess Ca2+ accumulation and for 8 h to evaluate cell viability, according to the methods described previously (7).
Histidinol plate assays
The strain FC2a carrying an appropriate plasmid was streaked onto SD agar plates containing 6 mm histidinol or 0.13 mm histidine, incubated at 30 °C for 3 days, and then photographed.
Statistical analysis
The significance of differences was evaluated using an unpaired Student's t test, with a maximum p value of <0.05 being required for significance.
Author contributions
K. I. performed plasmid construction, N-glycosylation, and histidinol plate assays and wrote the manuscript. J. T. performed Ca2+ accumulation and viability assays. T. C. performed protein purification, signal peptide assessments, and protein translocation assays. S. Y.-K. performed N-glycosylation and histidinol plate assays. K. I., J. T., T. C., S. Y.-K., and H. I. contributed to the experimental design and data analysis. H. I. conceived the project, secured funding, and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Drs. Akira Hosomi and Kohji Kasahara for valuable discussions, Dr. Christian Sengstag for the yeast strain FC2a and plasmid pCS4-14, and Yumiko Higashi for secretarial assistance.
This work was supported by Grant-in-Aid for Scientific Research B 26291026 (to H. I.) from the Japan Society for the Promotion of Science. The authors declare that they have no conflicts of interest with the contents of this article.
H. Kobayashi and H. Iida, unpublished data.
- VGCC
- voltage-gated Ca2+ channel
- GPI
- glycosylphosphatidylinositol
- ER
- endoplasmic reticulum
- NALCN
- Na+ leak channel non-selective
- SRP
- signal recognition particle
- Endo H
- endoglycosidase H.
References
- 1. Catterall W. A. (2011) Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 3, a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dolphin A. C. (2013) The α2/δ subunits of voltage-gated calcium channels. Biochim. Biophys. Acta 1828, 1541–1549 [DOI] [PubMed] [Google Scholar]
- 3. Wakamori M., Mikala G., and Mori Y. (1999) Auxiliary subunits operate as a molecular switch in determining gating behaviour of the unitary N-type Ca2+ channel current in Xenopus oocytes. J. Physiol. 517, 659–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barclay J., Balaguero N., Mione M., Ackerman S. L., Letts V. A., Brodbeck J., Canti C., Meir A., Page K. M., Kusumi K., Perez-Reyes E., Lander E. S., Frankel W. N., Gardiner R. M., Dolphin A. C., and Rees M. (2001) Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J. Neurosci. 21, 6095–6104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brodbeck J., Davies A., Courtney J. M., Meir A., Balaguero N., Canti C., Moss F. J., Page K. M., Pratt W. S., Hunt S. P., Barclay J., Rees M., and Dolphin A. C. (2002) The ducky mutation in Cacna2d2 results in altered Purkinje cell morphology and is associated with the expression of a truncated α2/δ-2 protein with abnormal function. J. Biol. Chem. 277, 7684–7693 [DOI] [PubMed] [Google Scholar]
- 6. Paidhungat M., and Garrett S. (1997) A homolog of mammalian, voltage-gated calcium channels mediates yeast pheromone-stimulated Ca2+ uptake and exacerbates the cdc1(Ts) growth defect. Mol. Cell. Biol. 17, 6339–6347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Iida H., Nakamura H., Ono T., Okumura M. S., and Anraku Y. (1994) MID1, a novel Saccharomyces cerevisiae gene encoding a plasma membrane protein, is required for Ca2+ influx and mating. Mol. Cell. Biol. 14, 8259–8271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tada T., Ohmori M., and Iida H. (2003) Molecular dissection of the hydrophobic segments H3 and H4 of the yeast Ca2+ channel component Mid1. J. Biol. Chem. 278, 9647–9654 [DOI] [PubMed] [Google Scholar]
- 9. Iida K., Tada T., and Iida H. (2004) Molecular cloning in yeast by in vivo homologous recombination of the yeast putative α1 subunit of the voltage-gated calcium channel. FEBS Lett. 576, 291–296 [DOI] [PubMed] [Google Scholar]
- 10. Yoshimura H., Tada T., and Iida H. (2004) Subcellular localization and oligomeric structure of the yeast putative stretch-activated Ca2+ channel component Mid1. Exp. Cell Res. 293, 185–195 [DOI] [PubMed] [Google Scholar]
- 11. Martin D. C., Kim H., Mackin N. A., Maldonado-Báez L., Evangelista C. C. Jr., Beaudry V. G., Dudgeon D. D., Naiman D. Q., Erdman S. E., and Cunningham K. W. (2011) New regulators of a high affinity Ca2+ influx system revealed through a genome-wide screen in yeast. J. Biol. Chem. 286, 10744–10754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fischer M., Schnell N., Chattaway J., Davies P., Dixon G., and Sanders D. (1997) The Saccharomyces cerevisiae CCH1 gene is involved in calcium influx and mating. FEBS Lett. 419, 259–262 [DOI] [PubMed] [Google Scholar]
- 13. Liebeskind B. J., Hillis D. M., and Zakon H. H. (2012) Phylogeny unites animal sodium leak channels with fungal calcium channels in an ancient, voltage-insensitive clade. Mol. Biol. Evol. 29, 3613–3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu B., Su Y., Das S., Liu J., Xia J., and Ren D. (2007) The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell 129, 371–383 [DOI] [PubMed] [Google Scholar]
- 15. Hong M.-P., Vu K., Bautos J., and Gelli A. (2010) Cch1 restores intracellular Ca2+ in fungal cells during endoplasmic reticulum stress. J. Biol. Chem. 285, 10951–10958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu B., Zhang Q., Wang H., Wang Y., Nakayama M., and Ren D. (2010) Extracellular calcium controls background current and neuronal excitability via an UNC79-UNC80-NALCN cation channel complex. Neuron 68, 488–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xie L., Gao S., Alcaire S. M., Aoyagi K., Wang Y., Griffin J. K., Stagljar I., Nagamatsu S., and Zhen M. (2013) NLF-1 delivers a sodium leak channel to regulate neuronal excitability and modulate rhythmic locomotion. Neuron 77, 1069–1082 [DOI] [PubMed] [Google Scholar]
- 18. Ghezzi A., Liebeskind B. J., Thompson A., Atkinson N. S., and Zakon H. H. (2014) Ancient association between cation leak channels and Mid1 proteins is conserved in fungi and animals. Front. Mol. Neurosci. 7, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zimmermann R., Eyrisch S., Ahmad M., and Helms V. (2011) Protein translocation across the ER membrane. Biochim. Biophys. Acta 1808, 912–924 [DOI] [PubMed] [Google Scholar]
- 20. Delic M., Valli M., Graf A. B., Pfeffer M., Mattanovich D., and Gasser B. (2013) The secretory pathway: exploring yeast diversity. FEMS Microbiol. Rev. 37, 872–914 [DOI] [PubMed] [Google Scholar]
- 21. Deshaies R. J., and Schekman R. (1987) A yeast mutant defective at an early stage in import of secretory protein precursors into the endoplasmic reticulum. J. Cell Biol. 105, 633–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rothblatt J. A., Deshaies R. J., Sanders S. L., Daum G., and Schekman R. (1989) Multiple genes are required for proper insertion of secretory proteins into the endoplasmic reticulum in yeast. J. Cell Biol. 109, 2641–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Green N., Fang H., and Walter P. (1992) Mutations in three novel complementation groups inhibit membrane protein insertion into and soluble protein translocation across the endoplasmic reticulum membrane of Saccharomyces cerevisiae. J. Cell Biol. 116, 597–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hiller K., Grote A., Scheer M., Münch R., and Jahn D. (2004) PrediSi: prediction of signal peptides and their cleavage positions. Nucleic Acids Res. 32, W375–W379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Käll L., Krogh A., and Sonnhammer E. L. L. (2004) A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 26. Frank K., and Sippl M. J. (2008) High performance signal peptide prediction based on sequence alignment techniques. Bioinformatics 24, 2172–2176 [DOI] [PubMed] [Google Scholar]
- 27. Petersen T. N., Brunak S., von Heijne G., and Nielsen H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 [DOI] [PubMed] [Google Scholar]
- 28. Nacken V., Achstetter T., and Degryse E. (1996) Probing the limits of expression levels by varying promoter strength and plasmid copy number in Saccharomyces cerevisiae. Gene 175, 253–260 [DOI] [PubMed] [Google Scholar]
- 29. Kyte J., and Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 30. Brodsky J. L., Goeckeler J., and Schekman R. (1995) Bip and Sec63p are required for both co- and posttranslational protein ttranslocation into the yeast endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 92, 9643–9646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jones D. T., Taylor W. R., and Thornton J. M. (1994) A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry 33, 3038–3049 [DOI] [PubMed] [Google Scholar]
- 32. Sonnhammer E. L. L., von Heijne G., and Krogh A. (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. In Proceedings of the Sixth International Conference on Intelligent Systems for Molecular Biology (Glasgow J., Littlejohn T., Major F., Lathrop R., Sankoff D., and Sensen C., eds) pp. 175–182, AAAI Press, Menlo Park, CA: [PubMed] [Google Scholar]
- 33. Hofmann K. K., and Stoffel W. (1993) TMbase: a database of membrane spanning proteins segments. Biol. Chem. Hoppe-Seyler 374, 166 [Google Scholar]
- 34. Kamano S., Kume S., Iida K., Lei K.-J., Nakano M., Nakayama Y., and Iida H. (2015) Transmembrane topologies of Ca2+-permeable mechanosensitive channels MCA1 and MCA2 in Arabidopsis thaliana. J. Biol. Chem. 290, 30901–30909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sengstag C. (2000) Using SUC2-HIS4C reporter domain to study topology of membrane proteins in Saccharomyces cerevisiae. Methods Enzymol. 327, 175–190 [DOI] [PubMed] [Google Scholar]
- 36. Scott J. H., and Schekman R. (1980) Lyticase: endoglucanase and protease activities that act together in yeast cell lysis. J. Bacteriol. 142, 414–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davies A., Kadurin I., Alvarez-Laviada A., Douglas L., Nieto-Rostro M., Bauer C. S., Pratt W. S., and Dolphin A. C. (2010) The α2/δ subunits of voltage-gated calcium channels from GPI-anchored proteins, a posttranlational modification essential for function. Proc. Natl. Acad. Sci. U.S.A. 107, 1654–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pierleoni A., Martelli P. L., and Casadio R. (2008) PredGPI: a GPI-anchor predictor. BMC Bioinformatics 9, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maruoka T., Nagasoe Y., Inoue S., Mori Y., Goto J., Ikeda M., and Iida H. (2002) Essential hydrophilic carboxyl-terminal regions including cysteine residues of the yeast stretch-activated calcium-permeable channel Mid1. J. Biol. Chem. 277, 11645–11652 [DOI] [PubMed] [Google Scholar]
- 40. Ozeki-Miyawaki C., Moriya Y., Tatsumi H., Iida H., and Sokabe M. (2005) Identification of functional domains of Mid1, a stretch-activated channel component, necessary for localization to the plasma membrane and Ca2+ permeation. Exp. Cell Res. 311, 84–95 [DOI] [PubMed] [Google Scholar]
- 41. Mandon E. C., Trueman S. F., and Gilmore R. (2013) Protein translocation across the rough endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 5, a013342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Engelman D. M., and Steitz T. A. (1981) The spontaneous insertion of proteins into and across membranes: the helical hairpin hypothesis. Cell 23, 411–422 [DOI] [PubMed] [Google Scholar]
- 43. Rothstein R. (1991) Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 194, 281–301 [DOI] [PubMed] [Google Scholar]
- 44. Sambrook J., and Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 45. Teng J., Iida K., Imai A., Nakano M., Tada T., and Iida H. (2013) Hyperactive and hypoactive mutations in Cch1, a yeast homologue of the voltage-gated calcium-channel pore-forming subunit. Microbiology 159, 970–979 [DOI] [PubMed] [Google Scholar]
- 46. Teng J., Goto R., Iida K., Kojima I., and Iida H. (2008) Ion-channel blocker sensitivity of voltage-gated calcium-channel homologue Cch1 in Saccharomyces cerevisiae. Microbiology 154, 3775–3781 [DOI] [PubMed] [Google Scholar]
- 47. Cho T., Ishii-Kato A., Fukata Y., Nakayama Y., Iida K., Fukata M., and Iida H. (2017) Coupling of a voltage-gated Ca2+ channel homologue with a plasma membrane H+-ATPase in yeast. Genes Cells 22, 94–104 [DOI] [PubMed] [Google Scholar]
- 48. Iida K., Teng J., Tada T., Saka A., Tamai M., Izumi-Nakaseko H., Adachi-Akahane S., and Iida H. (2007) Essential, completely conserved glycine residue in the domain III S2-S3 linker of voltage-gated calcium channel α1 subunits in yeast and mammals. J. Biol. Chem. 282, 25659–25667 [DOI] [PubMed] [Google Scholar]
- 49. Locke E. G., Bonilla M., Liang L., Takita Y., and Cunningham K. W. (2000) A homolog of voltage-gated Ca2+ channels stimulated by depletion of secretory Ca2+ in yeast. Mol. Cell. Biol. 20, 6686–6694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J. F., Guindon S., Lefort V., Lescot M., Claverie J. M., and Gascuel O. (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469 [DOI] [PMC free article] [PubMed] [Google Scholar]