

Abstract
PD-L1 (programmed death ligand 1) and PD-L2 are cell-surface glycoproteins that interact with programmed death 1 (PD-1) on T cells to attenuate inflammation. PD-1 signaling has attracted intense interest for its role in a pathophysiological context: suppression of anti-tumor immunity. Similarly, vitamin D signaling has been increasingly investigated for its non-classical actions in stimulation of innate immunity and suppression of inflammatory responses. Here, we show that hormonal 1,25-dihydroxyvitamin D (1,25D) is a direct transcriptional inducer of the human genes encoding PD-L1 and PD-L2 through the vitamin D receptor, a ligand-regulated transcription factor. 1,25D stimulated transcription of the gene encoding PD-L1 in epithelial and myeloid cells, whereas the gene encoding the more tissue-restricted PD-L2 was regulated only in myeloid cells. We identified and characterized vitamin D response elements (VDREs) located in both genes and showed that 1,25D treatment induces cell-surface expression of PD-L1 in epithelial and myeloid cells. In co-culture experiments with primary human T cells, epithelial cells pretreated with 1,25D suppressed activation of CD4+ and CD8+ cells and inhibited inflammatory cytokine production in a manner that was abrogated by anti-PD-L1 blocking antibody. Consistent with previous observations of species-specific regulation of immunity by vitamin D, the VDREs are present in primate genes, but neither the VDREs nor the regulation by 1,25D is present in mice. These findings reinforce the physiological role of 1,25D in controlling inflammatory immune responses but may represent a double-edged sword, as they suggest that elevated vitamin D signaling in humans could suppress anti-tumor immunity.
Keywords: gene expression, immunology, inflammation, steroid hormone receptor, vitamin D, anti-tumor immunity, checkpoint inhibitors, inflammatory T cell responses, vitamin D signaling
Introduction
Programmed death ligand 1 (PD-L1, B7-H1, or CD274) and its homologue programmed death ligand 2 (PD-L2, B7-DC, or CD273) are surface glycoproteins essential for peripheral tolerance (1). Binding of programmed death ligands to their cognate receptor, programmed death 1 (PD-1), on T cells results in a blockade of downstream T cell receptor signaling, inducing anergy, exhaustion, and apoptosis in inflammatory effector T (Teff)5 cells (2), while stimulating de novo differentiation and existing pool expansion of regulatory T (Treg) cells (3, 4). This effectively decreases the ratio of inflammatory to anti-inflammatory cytokines (1, 3, 5, 6). PD-L1 also interferes with priming of naive T cells (6), with polarization of CD4+ T cells toward the TH1 subtype (3), with Teff cell proliferation (3, 6), or it simply acts to reduce the time of interaction between cytotoxic T lymphocytes (CTLs) and target cells, essentially acting as a shield to protect the latter against T cell–mediated immune responses (7). CD274 (which codes for PD-L1) displays a very wide pattern of tissue gene expression, but PD-L1 is only seen at the protein level in myeloid cells, airway and kidney tubular epithelium, heart, placenta, and intestinal colon epithelium of inflammatory bowel disease (IBD) patients (6). PD-L2 expression is restricted to professional antigen-presenting cells (APCs) and is generally present at much lower levels on the cell surface compared with PD-L1 (6).
PD-L/PD-1 signaling has come under intense scrutiny because its physiological pro-tolerogenic effects are exploited by a number of cancers (e.g. carcinomas of the lung, ovary, head and neck, bladder, colon, melanomas, and gliomas) to escape immune detection and clearance (2, 8, 9). Greater PD-L1 surface expression in tumors or tumor-associated macrophages has been linked to poor prognosis and increased proliferation, epithelial–mesenchymal transition, and metastasis despite adequate numbers of tumor-infiltrating lymphocytes (2, 10). In this context, antibody therapies targeting PD-L1 or its receptor, PD-1, have proven remarkably efficacious in clinical and preclinical settings for a number of cancers (2, 11), including recurrent/metastatic head and neck squamous cell carcinoma (HNSCC) (12). Recent meta-analyses have provided evidence that clinical response to PD-1–blocking therapy correlates positively with the level of expression of PD-L1 in tumors (13–16), underlining the importance of understanding the signaling pathways regulating PD-L1 expression.
The pro-tolerogenic actions of PD-L1 have also been linked to beneficial effects in a plethora of immune-related disorders (6), namely multiple sclerosis, IBD, systemic lupus erythematosus, and diabetes. For example, intestinal epithelial ablation of PD-L1 expression in mice leads to IBD (17). We noted that several of these conditions overlap with those linked to vitamin D (VD) deficiency. VD was discovered as the curative agent for nutritional rickets, a disease of bone growth, and is a critical regulator of calcium homeostasis (18). However, it is now recognized to have pleiotropic actions (18). It undergoes sequential hydroxylations to produce its hormonal form, 1,25-dihydroxyvitamin D (1,25D), which signals through the vitamin D receptor (VDR), a ligand-regulated transcription factor. The VDR is expressed throughout the immune system, and 1,25D has emerged as a key regulator of innate immunity via its actions in both myeloid and epithelial cells (19–22). The VDR regulates the transcription of several genes implicated in innate immune responses (e.g. 1,25D signaling lies upstream and downstream of pattern recognition receptor engagement and is a direct inducer of antimicrobial peptide gene transcription) (19–21). Notably, 1,25D directly and indirectly induces signaling through the NOD2–DEFB4 innate immune pathway (21), whose deficiency has been linked to Crohn's disease, a form of IBD. Remarkably, however, many of the mechanisms of 1,25D signaling identified appear to be human/primate-specific and are not conserved in mice (23, 24).
Whereas 1,25D generally enhances innate immune responses, it induces a more tolerogenic adaptive immunity associated with higher Treg/Teff cell and anti-inflammatory (IL-10) to inflammatory (IFN-γ, TNF-α, IL-17, and IL-21) cytokine (19, 25, 26) ratio. Apart from the above, little is known about the effects of VD signaling on cross-talk between target cells or cells of the innate and those of the adaptive arms of the immune system. Here, we show that 1,25D directly up-regulates the transcription of the genes encoding PD-L1 and PD-L2 in human epithelial and myeloid cells. We found that the VDR binds to enhancers located in the CD274 and CD273 (encoding PD-L2) genes, which are adjacent in the human genome. We also provide evidence that 1,25D-induced PD-L1 expression on epithelial or myeloid cells inhibits T cell cytokine production. However, similar to other immune-related actions of 1,25D (23), the observed regulatory events are not conserved in mice. The induction of PD-L1 and PD-L2 expression is a mechanism accounting for the effects of VD signaling in T cell tolerance and is in accord with other studies providing evidence that it is protective against IBD (21, 27, 28). Importantly, however, this may prove to be a double-edged sword in terms of physiological versus potential pathophysiological actions of VD signaling, as elevated 1,25D-induced PD-L1 expression may be detrimental to anti-tumor immunity.
Results
Tissue-specific 1,25D-regulated CD274 and CD273 expression in humans but not mice
Analysis of our previous profiling studies of 1,25D-regulated gene expression in human cells of epithelial or myeloid origin revealed CD274 and CD273 as potential VDR targets (20, 29). These data were validated by performing RT-qPCR on RNA extracted from human HNSCC cell lines SCC25 and SCC4 and human differentiated THP-1 macrophages treated with 1,25D for up to 24 h (Fig. 1A). SCC25 cells are well differentiated and sensitive to the antiproliferative effects of 1,25D, whereas SCC4 cells are poorly differentiated and 1,25D-resistant, although they retain 1,25D signaling (30). CD274 expression increased in all cell lines exposed to 1,25D relative to vehicle (Fig. 1A). Consistent with its tissue-specific expression pattern, CD273 was only up-regulated in differentiated THP-1 cells (Fig. 1A, right) and was unchanged in SCC25 and SCC4 cells (Fig. 1A, left and middle). 1,25D also induced CD274, but not CD273, expression in two cultures of primary human keratinocytes (Fig. 1B). Similarly, CD274 expression was stimulated (along with positive-control genes CYP24A1, AREG, and NOD2) by 1,25D in primary human nasal epithelial cells (supplemental Fig. S1, A and B). Consistent with results obtained in differentiated THP-1 cells, 1,25D enhanced the expression of both genes in primary human myeloid cells, namely macrophages (Mϕs) (Fig. 1C). Whereas the fold inductions of CD274 and CD273 in myeloid cells were comparable, CD273 was generally expressed more weakly than CD274 (supplemental Fig. S2). Interestingly, 1,25D and the pathogen-associated molecular pattern LPS, a known PD-L1/PD-L2 inducer (31), up-regulated CD274 cooperatively in THP-1 cells (supplemental Fig. S3A, left), but not in SCC25 cells, where LPS had no effect (supplemental Fig. S3A, right). A similar combined effect of 1,25D and LPS on CD273 expression was seen in THP-1 cells (supplemental Fig. S3B). These observations provide evidence for cooperative effects of Toll-like receptor 4 and vitamin D signaling pathways in regulation of CD274 and CD273 transcription.
Figure 1.
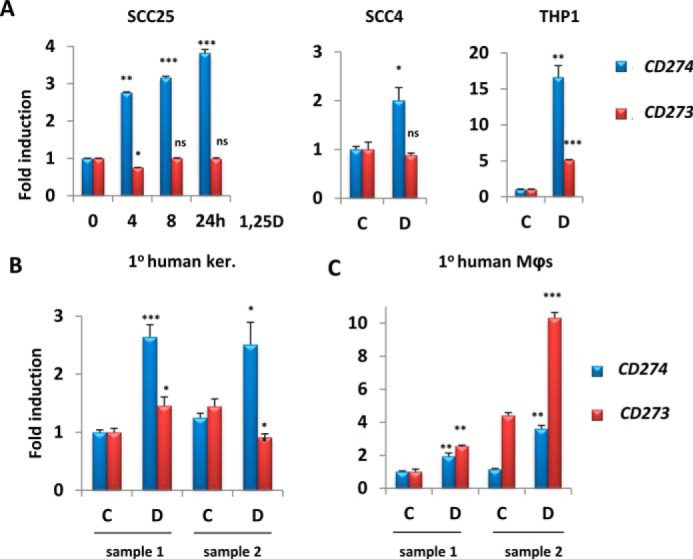
1,25D treatment increases mRNA levels of CD274 in epithelial cells and of CD274 and CD273 in myeloid cells. A–C, analysis by RT-qPCR of the regulation by 100 nm 1,25D of CD274 and CD273 gene expression in SCC25 and SCC4 cells (epithelial) and THP-1 cells (myeloid) (A), primary human keratinocytes (B), and primary human macrophages (C). -Fold change and p values are relative to control sample (C or 0 h) and are calculated separately for each gene (CD274 and CD273). C, vehicle (ethanol); D, 100 nm 1,25D (n = 3); ns > 0.05 ≥ * ≥ 0.01 ≥ ** ≥ 0.001 ≥ ***. Error bars, S.D.
Given that many aspects of innate immune regulation by 1,25D appear to be largely human/primate-specific (23, 24), we assessed the degree of conservation of the regulation by 1,25D of these genes in a model organism. We used the mouse HNSCC cell line AT84, which is essentially identical histologically and in terms of 1,25D responsiveness to human SCC25 cells. We also analyzed primary mouse Mϕs obtained from two mice and both non-activated and activated mouse dendritic cells (DCs). 1,25D treatment for 24 h had no effect on Cd274 and Cd273 expression in AT84 or in myeloid cells (supplemental Fig. S4, A and B). Note that Cd273 mRNA levels were below the detection limit in AT84 cells. The transcriptional stimulation of Cyp24a1, a target of VD in both mice and humans, was measured as a positive control for 1,25D genomic signaling in AT84 cells and in primary mouse myeloid cells, and as expected, 1,25D strongly up-regulated Cyp24a1 gene expression (supplemental Fig. S4, C and D).
To determine whether increased transcription of CD274 by 1,25D translates into elevated PD-L1 protein levels, we performed Western blotting in SCC25 and in SCC4 cells treated with vehicle or 1,25D for 24 h. 1,25D substantially up-regulated protein levels in both cell lines (Fig. 2A), consistent with its effects on CD274 gene expression. Note that the 1,25D-induced increase in PD-L1 protein levels (supplemental Fig. S5) paralleled CD274 mRNA stimulation (Fig. 1A, left) in a time-dependent experiment. This observation suggests that hormonal vitamin D increases PD-L1 abundance via stimulating its gene expression. PD-L2 expression was below the detection limit by Western blotting in human and mouse epithelial cells (data not shown).
Figure 2.

1,25D increases protein expression of PD-L1. A, analysis by Western blotting of PD-L1 expression in SCC25 and SCC4 cells following a 24-h exposure to 100 nm 1,25D (D) or ethanol (C). B, 1,25D-induced PD-L1 expression is stable in SCC25 cells after 1,25D withdrawal, as assessed by Western blotting. -Fold change relative to control and normalized to actin is indicated below each PD-L1 blot. C, wide-field microscopy for DAPI nuclear staining (i and ii) and PD-L1 expression (iii and iv) in differentiated THP-1 macrophages exposed for 24 h to vehicle (C) or to 100 nm 1,25D (D). v and vi, merge of the images. D, a compilation of z-stacks from several focal planes is presented from confocal microscopy for bronchiolar epithelium differentiation marker ZO-1 (i and ii), PD-L1 expression (iii and iv), and DAPI nuclear staining (v and vi) in primary bronchial epithelial cells treated for 48 h with vehicle (C) or with 100 nm 1,25D (D). vii and viii, merge for all images. ix, total percentage of PD-L1–positive cells across all seven fields. x, average number of PD-L1–positive cells from seven separate fields. ns > 0.05 ≥ 0.01 ≥ ** ≥ 0.001. Error bars, S.D.
1,25D-induced PD-L1 increase persisted for up to 48 h after 1,25D withdrawal in SCC25 cells (Fig. 2B). Note that we observed two bands, both up-regulated, for PD-L1 in SCC25 cells (Fig. 2A, left). These probably correspond to the smaller cytosolic and the larger cell-surface isoforms (1, 32, 33). We also analyzed the effect of 1,25D treatment on PD-L1 expression in primary human keratinocytes and human HT29 colon carcinoma cells and observed a similar up-regulation (supplemental Fig. S6, A and B). 1,25D-dependent changes in protein levels were reflected in cell-surface PD-L1 being robustly up-regulated by 1,25D in THP-1 cells, as measured by wide-field microscopy (Fig. 2C) or by flow cytometry (supplemental Fig. S6C). We also tested for 1,25D-dependent up-regulation of PD-L1 in primary human bronchial epithelial cells obtained from healthy donor explants and differentiated on air–liquid interface filters. Because of filter incongruities and varied cell height, we generated z-stacks from several focal planes to quantify accurately differences in staining. When epithelial marker zonula occludens-1 (ZO-1) was used to normalize for cell number (Fig. 2D, i and ii), these studies revealed that 1,25D treatment increased PD-L1 levels in bronchial epithelial cells relative to vehicle-treated cells both in terms of the percentage of PD-L1–positive cells in all fields and the average number of positive cells in all fields (Fig. 2D, ix and x). Elevated PD-L1 expression in 1,25D-treated cells is also evident from analysis of typical images from single focal planes (supplemental Fig. S7). Consistent with these results, we observed comparable effects of 1,25D on cell-surface expression of PD-L1 in SCC25 cells by wide-field microscopy (supplemental Fig. S6D). In contrast, Pd-l1 protein was unaffected by 1,25D treatment in mouse AT84 cells (supplemental Fig. S4E), consistent with the lack of gene regulation by 1,25D. Similarly, exposure to 1,25D had no effect on Pd-l1 protein expression in primary mouse DCs (supplemental Fig. S4F) or in primary mouse skin cells (supplemental Fig. S4G). Finally, we also tested in SCC25 cells for 1,25D-induced changes in the production of soluble PD-L1, which has been shown to retain immunosuppressive properties similar to those of the cell-surface molecule (34). However, if soluble PD-L1 was produced, it was below the detection limit in an ELISA performed on culture medium samples from 1,25D- or vehicle-treated SCC25 cells.
Direct regulation of CD274 and CD273 gene expression by 1,25D via VDREs
To determine whether 1,25D signaling directly up-regulates transcription of CD274 and of CD273, we searched for potential VDREs in the two genes. Analysis of published ChIP followed by next-generation sequencing (ChIP-seq) data sets identified a VDR peak in an intronic region of CD273, located downstream of exon 5 and centered at 47,959 bp downstream of the transcription start site (TSS) (VDRECD273) (35) (Fig. 3A). This region contains a non-consensus VDRE-like sequence (Fig. 3A). Additionally, a putative near-consensus VDRE (VDRECD274) was identified at 829 bp upstream of the CD274 TSS in data generated by an in silico screen (29). Note that neither of these sites is conserved in mice (supplemental Fig. S8), consistent with the lack of regulation by 1,25D of gene or protein expression in this species. We employed ChIP assays followed by qPCR to monitor VDR binding to the VDREs described above. 1,25D treatment resulted in increased VDR association with VDRECD274 and with VDRECD273 in both SCC25 (Fig. 3B) and THP-1 cells (Fig. 3C) relative to vehicle, suggestive of potential enhancer activity. We probed further for changes in epigenetic markers denoting enhancer function. 1,25D up-regulated histone 3 lysine 4 monomethylation (H3K4me1) marks, indicative of active/poised enhancers, at VDRECD274 and VDRECD273 regions in both SCC25 (Fig. 3D) and THP-1 (Fig. 3E) cells, in a pattern similar to that of the VDR association with these elements. We also assessed the level of histone 3 lysine 27 acetylation (H3K27ac). In SCC25 cells, VDRECD274 displayed high levels of H3K27ac, which were not affected by 1,25D (Fig. 3F, left), whereas 1,25D increased H3K27ac at VDRECD273 (Fig. 3F, right). In THP-1 cells, 1,25D exposure was associated with increased H3K27ac marks at both enhancers (Fig. 3G). These results suggest that both VDREs function as active cis-acting enhancer elements. Moreover, 1,25D stimulated association of Pol II with both VDRECD274 and VDRECD273 in SCC25 (Fig. 4A) and in THP-1 cells (Fig. 4B). Notably, however, enhanced recruitment of Pol II was observed at the TSS of CD274, but not CD273, in SCC25 cells, whereas Pol II association with both TSSs was stimulated by 1,25D in THP-1 cells (Fig. 4, C and D). Essentially identical results were obtained when VDR recruitment to TSS was examined (Fig. 4, E and F). These observations demonstrate the direct regulation of CD274 and CD273 transcription by 1,25D and are consistent with their tissue specificity.
Figure 3.

CD274 and CD273 VDREs act as enhancer elements. A, tandem CD274 (5′-end only) and PDCD1LG1 genes and positions of VDREs. The dotted red arrow upstream of CD273 VDRE indicates eRNA. B–G, ChIP analysis in extracts of SCC25 (B, D, and F) or THP-1 cells (C, E, and G) of 1,25D-dependent binding of the VDR (B and C) to the VDREs in the CD274 and CD273 genes, along with effects of 1,25D on histone H3 Lys-4 monomethylation (D and E) and histone H3 Lys-27 acetylation (F and G). The -fold change is calculated relative to the nonspecific IgG IP performed with the control sample. C, vehicle (24 h); D, 100 nm 1,25D (24 h) (n = 3); ns > 0.05 ≥ * ≥ 0.01 ≥ ** ≥ 0.001 ≥ ***. Error bars, S.D.
Figure 4.

1,25D directly regulates CD274 and CD273 gene expression via both VDREs. A–D, ChIP analysis of the effects of 1,25D on recruitment of the large subunit of RNA polymerase II (Pol 2) to the VDREs (A and B) and TSSs (C and D) in SCC25 cells and THP-1 cells, as indicated. E and F, effects of 1,25D on recruitment of the VDR to the TSS of CD274 or CD273 genes in SCC25 (E) and THP-1 cells (F), as indicated. The -fold change is calculated relative to the nonspecific IgG IP performed with the control sample. G, 1,25D treatment for the indicated times stimulates eRNA synthesis upstream of VDRECD273 + 47,959. Directed RT-qPCR was employed to show 1,25D-dependent production of eRNA 5′ of VDRECD273 + 47,959 and centered at 47,810 bp downstream of the CD273 TSS in THP-1 cells. C, vehicle (24 h); D, 100 nm 1,25D (24 h). A–F, n = 3; G, n = 5; ns > 0.05 ≥ * ≥ 0.01 ≥ ** ≥ 0.001 ≥ ***. Error bars, S.D.
As described above, we observed 1,25D-dependent changes in VDR and Pol II recruitment and levels of epigenetic markers in SCC25 cells at the intronic CD273 VDRE despite lack of regulation of the adjacent gene. Pol II recruited to enhancer elements often undergoes a round of transcription at these sites, producing small non-coding so-called enhancer RNAs (eRNAs), whose expression correlates strongly with enhancer function and may contribute to target gene expression (36, 37). Therefore, as a further test for VDRECD273 function in SCC25 and THP-1 cells, we screened for production of eRNAs at various distances upstream of the VDREs using strand-specific directed RT-qPCR, which avoids detection of spliced intronic RNA species (see “Experimental procedures”). We did not detect any expression for myoblast-specific hMUNC eRNA (38), serving as a negative control. In contrast, 1,25D strongly induced the production of eRNAs (Fig. 4G) in THP-1 cells centered at 224 bp upstream of VDRECD273 and complementary to the 47,810–47,660 bp region downstream of CD273 TSS (see Fig. 3A). However, we did not find any eRNAs produced from the same, or any other, site in SCC25 cells, which highlights the tissue-specific effects of 1,25D action and strongly suggests that the intronic enhancer in the CD273 gene is fully functional in THP-1 cells and not in SCC25 cells.
Finally, to verify that the response elements in the CD274 and CD273 gene were capable of mediating 1,25D-dependent transactivation, we cloned them upstream of luciferase genes. Two fragments of the CD274 promoter were cloned upstream of a promoterless luciferase gene, both containing sequence from −840 to +23 relative to the CD274 TSS, with one containing and one lacking the VDRE (CD274+VDRE and CD274−VDRE, respectively). The results of reporter gene expression assays show that the VDRE was required to generate 1,25D-dependent induction of luciferase activity (supplemental Fig. S9A). Because of its far downstream location, the CD273 VDRE was cloned as an oligonucleotide upstream of minimal promoter-driven luciferase. Reporter gene assays showed that it, too, was capable of mediating 1,25D-dependent induction of luciferase activity (supplemental Fig. S9B).
1,25D-regulated epithelial PD-L1 expression inhibits T-cell function
Ablation of PD-L1 expression in epithelial cells in mouse intestine leads to an inflammatory phenotype (17), and other studies have provided evidence that epithelial PD-L1 can control T cell behavior (5, 6). To assess the impact of 1,25D-stimulated epithelial PD-L1 expression on T cell function, we set up a co-culture system consisting of primary human whole T cells in direct contact with SCC25 or THP-1 cells, which had been pretreated for 24 h with vehicle or 1,25D. T cells (both CD3+CD4+ and CD3+CD8+) were isolated by negative selection from peripheral blood mononuclear cell (PBMC) blood fractions of three healthy donors (supplemental Fig. S10, A–C) and were tested for purity (supplemental Fig. S10, D–G) (see “Experimental procedures”). The T cells obtained had no APCs (DCs and monocyte) (supplemental Fig. S10, D and E), natural killer (supplemental Fig. S10F), or B cells (supplemental Fig. S10G) contaminants.
Pretreated SCC25 and THP-1 cells and primary human whole T cells were co-cultured for 24 h in 1,25D-free medium in the presence of control IgG or PD-L1–specific blocking antibody (MIH1). The release of TNF-α and IFN-γ into the medium by co-cultured T cells was measured by ELISA. Production of IFN-γ or TNF-α was inhibited by co-culturing with cells pretreated with 1,25D (Fig. 5, A and B, left). Importantly, the addition of anti-PD-L1 blocking antibody completely reversed the inhibitory effect of 1,25D pretreatment on TNF-α release and partially abrogated the effect on IFN-γ (Fig. 5, A and B, right). In separate experiments, we also measured by ELISA the production of IL-2, constitutively secreted by activated Jurkat cells, co-cultured with SCC25 cells, as a readout for T-cell function (supplemental Fig. S11). 1,25D pretreatment of SCC25 cells resulted in a 2-fold reduction of IL-2 release into the medium, an effect reversed completely by blocking PD-L1.
Figure 5.
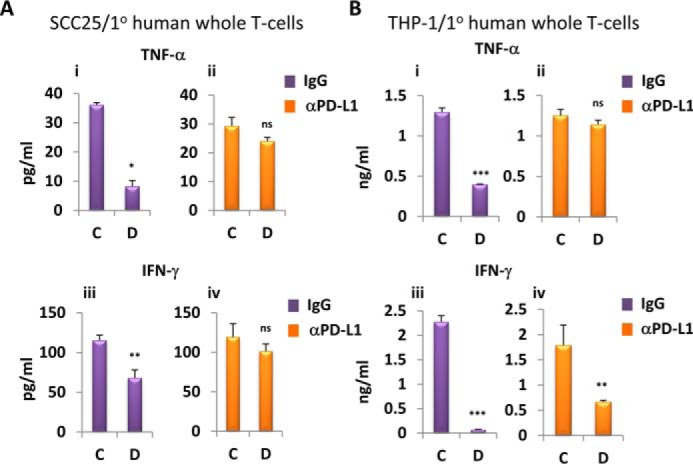
1,25D-dependent PD-L1 increase in epithelial and myeloid cells results in diminished cytokine production by co-cultured T cells. A and B, effects of 1,25D pretreatment of SCC25 (A) and THP-1 (B) cells followed by co-culture for 24 h with primary human whole T cells in the presence of nonspecific IgG (i and iii) or anti-PD-L1 (iii and iv) blocking antibody on secretion of TNF-α (i and ii) and IFN-γ (iii and iv), assessed by ELISA. C, vehicle pretreatment for 24 h; D, 100 nm 1,25D pretreatment for 24 h (n = 3); ns > 0.05 ≥ * ≥ 0.01 ≥ ** ≥ 0.001 ≥ ***. Error bars, S.D.
PD-L1 engagement by T cells has been linked to inhibition of activation and a resulting decrease in inflammatory cytokine production. We therefore used the experimental co-culture system described above to assess the effect of 1,25D-dependent PD-L1 expression in SCC25 cells on the activation status of co-cultured pan-T cells obtained from three healthy donors (one male and two female). Notably, 1,25D pretreatment significantly reduced early (CD69), mid-early (CD71), and intermediate (CD25) activation markers on CD4+ T cells obtained from all three donors and co-cultured in the presence of normal nonspecific IgG (Fig. 6, D–F). A similar trend was observed in CD8+ (Fig. 6, A–C) cells, where changes in CD25+ populations did not reach statistical significance (Fig. 6A) but conformed to the pattern observed in the corresponding CD4+ T cells (Fig. 6D). Blocking of PD-L1 signaling by αPD-L1 antibody partially or completely rescued this effect (Fig. 6). The observed PD-L1–dependent effects of 1,25D pretreatment on T cell activation become even more obvious upon examination of density plots of CD25 (supplemental Figs. S12 and S15), CD69 (supplemental Figs. S13 and S16), and CD71 (supplemental Figs. S14 and S17) activation markers in CD4+ (supplemental Figs. S12–S14) and CD8+ subpopulations (supplemental Figs. S15–S17) for each patient and experimental condition. Note that there was no significant effect of 1,25D pretreatment in the presence of nonspecific IgG or αPD-L1 blocking antibody on T cell apoptosis (supplemental Fig. S18). Therefore, we conclude that 1,25D-induced surface expression of PD-L1 inhibits activation of effector T cells, which translates in reduced inflammatory cytokine production. These results highlight the importance of induced epithelial PD-L1 expression in regulation of T cell function by 1,25D.
Figure 6.

1,25D-dependent PD-L1 increase in epithelial cells results in reduction of activation of co-cultured CD4+ and CD8+ T cell populations. Flow cytometry of T cells co-cultured with 1,25D-pretreated SCC25 cells and stained for CD8 (A–C), CD4 (D–F), and the activation markers CD25 (A and D), CD71 (B and E), and CD69 (C and F). p values were calculated for treated (vitamin D, 1,25D) versus control (vehicle) in IgG and αPD-L1 groups separately and for control (IgG) versus control (PD-L1). Shown is vitamin D pretreatment for 24 h with 100 nm 1,25D (n = 3); ns > 0.05 ≥ *. Error bars, S.D.
Discussion
A role for VD signaling in suppression of inflammatory responses has been well established (25, 26). However, the molecular-genetic events underlying this regulation have been poorly characterized. The results presented here reveal that 1,25D acting through the VDR directly induces the transcription of the genes encoding PD-L1 and PD-L2 in human cell lines and primary cultures. These findings complement previous observations showing that 1,25D treatment induces a stable semi-mature DC phenotype capable of stimulating Treg and IL-10 production (39). Induction of PD-L1 expression was observed in epithelial and myeloid cells, whereas 1,25D-regulated expression of PD-L2 was myeloid-specific, consistent with the expression patterns of the two genes.
We identified VDREs in both genes, which appeared to function as poised or active enhancers in both epithelial and myeloid cells, given the 1,25D-dependent recruitment of Pol II and regulation of enhancer marks at these sites. However, we only detected 1,25D-dependent Pol II recruitment to the CD273 TSS in myeloid cells, conditions under which the gene is regulated. We also detected the 1,25D-dependent production of eRNAs from the CD273 VDRE only in myeloid cells. Consistent with other findings (36), it appears likely, therefore, that the eRNA produced following exposure to 1,25D in THP-1 cells may act to stimulate CD273 transcription. The absence of this eRNA species in epithelial SCC25 cells is consistent with lack of transcriptional control of CD273 by 1,25D in these cells. Note that we performed chromosome conformation capture assays to detect the formation of a loop between the CD273 VDRE and the TSS of the CD274 gene but failed to detect any interaction. It thus appears that whereas 1,25D-dependent recruitment of the VDR and cofactors to the CD273 VDRE occurs in both epithelial and myeloid cells, it is only fully functional as an enhancer in myeloid cells, consistent with the expression pattern of CD273. In this regard, numerous ChIP-seq studies of the VDR, other nuclear receptors, and other classes of transcription factors have generally identified far greater numbers of binding sites than regulated genes, indicating that many bona fide binding sites do not correspond to fully functional enhancers under the conditions of the ChIP-seq experiment (35, 41).
We further demonstrated cell-surface up-regulation of PD-L1 and a PD-L1–dependent suppression of T cell cytokine production in the presence of 1,25D. Neither the regulatory events nor the VDR-binding sites characterized in the two human genes were conserved in mice. This lack of conservation was not unexpected, as many of the previously identified innate immune responses driven by VD signaling in human cells appear to be (largely) human/primate-specific. This includes the 1,25D-induced expression of antimicrobial peptide genes CAMP and HBD2/DEFB4, the gene encoding the pattern recognition receptor NOD2 (21), and the IL1B gene (20). Notably, the VDRE in the promoter-proximal region encoding the CAMP gene is embedded in a human/primate-specific Alu repeat that appears to have been inserted at the dawn of the primate lineage (24). Therefore, our observations of species-specific regulation of PD-L1 and PD-L2 expression reinforce the notion that many aspects of VD-regulated innate immunity appear to have evolved with the primate lineage.
1,25D-regulated expression of PD-L1 and PD-L2 is of considerable physiological and clinical significance, given their critical role in controlling T cell activation and suppression of inflammatory immune responses. Notably, intestinal epithelial ablation of Pd-l1 expression in mice led to intestinal inflammation through defects in innate immune signaling (17). The maintenance of intestinal PD-L1 and PD-L2 expression through 1,25D signaling is thus entirely consistent with an emerging picture of a role for VD in maintenance of intestinal innate immune homeostasis. Previous studies showed that the hormone-bound VDR directly stimulates the transcription of the NOD2 and HBD2/DEFB4 genes, which lie at either end of an innate immune pathway that is defective or attenuated in a subset of patients with Crohn's disease (CD) (21, 42). These results suggested that VD deficiency may contribute to the pathogenesis of CD, a notion that is reinforced by the results of intervention trials that strongly support a role for VD supplementation in suppression of symptoms and enhancing the quality of life in CD patients (42–44). 1,25D-induced expression of PD-L1 and PD-L2 thus provides another mechanism supporting a central role for VD signaling in controlling intestinal inflammation.
Whereas our findings are entirely consistent with the previously established roles of VD in regulating immune system function, they represent something of a double-edged sword, given the implication of elevated signaling through PD-1 in suppression of anti-tumor immunity. They also represent a conundrum, given the extensive evidence that maintenance of VD sufficiency reduces the incidence of several cancers. The cancer-preventive activities of 1,25D signaling are supported by epidemiological data (45), experiments in animal models, and several mechanistic studies (46). 1,25D signaling can block cancer cell proliferation in some in vitro models and induce differentiation (46). Moreover, it can suppress oncogenic pathways driven by Wnt signaling (46, 47), c-MYC (48), and others and can promote the activity of tumor suppressors, such as FoxO proteins (49). However, whereas the activated VDR may be effective at blocking aberrant signaling at early stages of the oncogenic process and may suppress the growth of some tumors (at least in animal models), there is ample evidence for acquisition of resistance to 1,25D signaling during tumorigenesis. Several cell lines derived from malignancies of various origins are partially or wholly resistant to the anti-proliferative effects of 1,25D although VDR expression and 1,25D-dependent transactivation are maintained (21). These observations are consistent with the failure of 1,25D and several of its analogues as cancer therapeutics because of tumor resistance.
Our results provide another potential mechanism of tumor resistance to 1,25D therapy through maintenance of elevated PD-L1 and PD-L2 signaling in the tumor microenvironment, thereby suppressing T cell-mediated anti-tumor immunity. These findings may also provide a potential explanation for the observations in some studies of a reverse J-shaped curve in the relationship between cancer incidence and levels of the major circulating VD metabolite 25-hydroxyvitamin D (50) (i.e. a correlation between increased incidence of some malignancies and superphysiological circulating 25-hydroxyvitamin D levels), an observation for which there was previously no mechanistic basis. Based on our findings, it can also be argued that it would be important to take the VD status of patients into account in settings of tumor immunotherapy. It is perhaps paradoxical that, whereas elevated PD-L1 expression may suppress anti-tumor immunity, its level of expression in tumors also correlates positively with clinical responses to anti-PD-L1/PD-1 therapy (13–16). In conclusion, we have shown that 1,25D stimulates the expression of the genes encoding PD-L1 and PD-L2, an observation that strengthens the role of VD signaling in immune system regulation, although it may represent a risk factor because of its potential to contribute to suppression of anti-tumor immunity.
Experimental procedures
Cell isolation and tissue culture
All cell lines were cultured under conditions recommended by the American Type Culture Collection (ATCC). SCC25, SCC4, and AT84 cell lines were obtained from the ATCC and were passaged in DMEM/F-12 (Wisent, 319-085-CL) containing 10% FBS (Wisent, 080150), 10% DMEM/F-12. HEK293 cells were cultured in 10% DMEM (Wisent, 319-005). Primary human normal epidermal keratinocytes (catalog no. 2110) with the appropriate culture medium (catalog no. 2101) supplemented with keratinocyte growth supplement (catalog no. 2152), and antibiotics (penicillin/streptomycin; catalog no. 0503) were purchased from ScienCell. THP-1 and Jurkat (ATCC) cell lines were cultured in 10% RPMI 1640 (Wisent, 350-005-CL). Primary mouse DCs and Mϕs were obtained by flushing C57BL6 tibia and femur, followed by lysing erythrocytes using BD Pharm Lyse buffer (BD Biosciences, 555,899) and culturing for 4 h. The non-adherent cells were replated in fresh culture medium. Mouse DCs were differentiated in 10% RPMI 1640 (Wisent, 350-005-CL) containing 20 ng/ml GM-CSF for 8 days. Where applicable, DCs were activated with LPS (Sigma-Aldrich, L3012-5MG). Mouse macrophages were cultured in 10% DMEM containing 30% conditioned medium from L929 cells (containing M-CSF). Human primary cells were obtained from healthy subjects following informed consent according to the McGill policy on ethical conduct of research involving human subjects and approved by the McGill Ethics Committee (A06-M64-14A and 14-234-BMB). Human primary monocytes were purified as described (20) from the PBMC fractions of two donors, using Ficoll–Paque Premium (GE Healthcare, 17-5442-02) and differentiated using GM-CSF (Life Technologies, Inc., PHC2011). Primary bronchial epithelial cells were obtained from healthy donors and were cultured and differentiated as described previously (51). Primary human pan-T cells were obtained from PBMC fraction of three donors by negative selection using EasySep kit (StemCell, 17951) and were cultured in 10% Iscove's (Wisent, 319-105-CL) medium. T cell purity was assessed through flow cytometry (supplemental Fig. S10) by quantifying the markers of various cell populations found in PBMCs, namely CD3 (T cells) (supplemental Fig. S10B) CD11c (DCs) (supplemental Fig. S10D), CD14 (monocytes) (supplemental Fig. S10E), CD56 (NK cells) (supplemental Fig. S10F), and CD19 (B cells) (supplemental Fig. S10G). In addition, the CD3+ cells (T cells) were subdivided into CD4+ and CD8+ (CD3+/CD4−) populations (supplemental Fig. S10C). All treatments were done using 100 nm 1,25D or vehicle (DMSO). THP-1 cells were first differentiated with 10 nm phorbol 12-myristate 13-acetate overnight and washed three times with complete medium before exposure to 1,25D/vehicle. 1 μg/ml LPS was used where applicable.
Co-culture experiments
The co-culture procedure was inspired by mixed lymphocyte reaction and was performed essentially as described (52). Briefly, 28,000 pretreated SCC25 or differentiated THP-1 cells were preblocked with 20 μg/ml anti-PD-L1 antibody (eBioscience, 14-5983-82) or isotype normal IgG (eBioscience, 14-4714-85) and FcR blocking solution (BioLegend, 422302) in the case of THP-1 for 2 h. Primary human T cells were resuspended in 10% Iscove's medium containing blocking antibodies at the concentrations indicated above and added to the target cells. For Jurkat cells, 50 ng/ml phorbol 12-myristate 13-acetate and 1 μg/ml PHA, required for activation, were also added. T/Jurkat (2.8 × 105) cells were in a 10:1 ratio with SCC25/THP-1 cells and were co-cultured for 24 h.
RNA extraction, reverse transcription, and qPCR
RNA was extracted using TRIzol reagent (Invitrogen, 15596-018) as per the manufacturer's instructions. The iScript cDNA synthesis kit (Bio-Rad, 170-8891) and 1 μg of RNA template was used to generate cDNA, which was diluted 5 times and used in qPCR with SsoFast EvaGreen Supermix (Bio-Rad, catalog no. 172-5201) in a Roche Applied Science LightCycler 96 machine. eRNA production was tested essentially as described (40). Briefly, RT was performed using specific stem–loop oligonucleotides for detection of directed strand-specific RNA production. supplemental Table 1 contains a full list of primers used.
Western blotting
A standard Western blotting protocol (20) was employed. Rabbit polyclonal anti-PD-L1 (reactive to human, mouse, and rat) (H-130, sc-50298), goat anti-β-actin (C-11, sc-1615), and donkey HRP-conjugated anti-goat (sc-2020) antibodies were purchased from Santa Cruz Biotechnology, Inc. Goat anti-rabbit-HRP (catalog no. 7074) was obtained from Cell Signaling. Goat anti-mouse Pd-l1 was purchased from R&D Systems (AF1019). Changes in protein levels were quantified relative to control using ImageJ after normalization to actin; the -fold change is displayed below each Western blot figure. Representative images of at least three biological trials are presented.
VDRE screens
Peaks from VDR ChIP-seq studies were aligned with the human genome (build hg19) using the USCS genome browser. The VDRE upstream of the CD274 TSS was identified by an in silico screen for consensus human VDREs taken from JASPAR database; both positive and negative strands of the human genome (build hg19) encompassing the CD274 gene locus were used as a template.
Chromatin immunoprecipitation assays
ChIP was performed as described previously (21) with minor modifications. For histones, cell membrane was first lysed (10 mm Tris, pH 7.5, 10 mm NaCl, 0.2% Nonidet P-40), and nuclei were washed three times with MNase buffer (New England Biolabs, 7007BC), followed by digestion with MNase (New England Biolabs, M0247S) for 30 min at 37 °C rotating. Nuclei were pelleted and resuspended in ChIP lysis buffer. Mild sonication was applied to break the nuclear membrane and extract the DNA. For transcription factors, cells were directly lysed in ChIP lysis buffer and sonicated to shear the DNA to a fragment of length 200–600 bp. 4 μg of antibodies for VDR (D-6; Santa Cruz Biotechnology, sc-13133), Pol 2 (Abcam, ab5131), H3K4me1 (Abcam, ab8895), and H3K27ac (Abcam, ab4729) were used for IP in 500 μl of dilution buffer. DNA was purified using a FavorGen PCR/gel DNA purification kit (FAGCK001-1), and qPCR was performed with primers specific for each region (supplemental Table S1).
Molecular cloning
The promoter region of CD274 starting at −840 bp to +23 bp (chr9:5449663–5450525) relative to the CD274 TSS and either containing or lacking VDRECD274 was synthesized in vitro (IDT-gBlock) with EcoRI (GAATTC) and XhoI (CTCGAG) added to the 5′- and 3′-ends of the fragment, respectively (see supplemental Fig. S9A). To ensure proper restriction enzyme digestion, a 6-bp random oligonucleotide (TAAGCA) was also added to both ends of the fragment. After double digestion with EcoRI and XhoI of the pGLuc (New England Biolabs, N8082S) plasmid and both CD274 promoter fragments overnight, ligation was performed between the pGLuc vector and CD274 promoter containing the VDRE or CD274 promoter lacking the VDRE. Sanger sequencing of the resulting constructs was done at the McGill University and Genome Quebec Innovation Centre and confirmed the proper integration and orientation of the CD274 promoter region. We inserted a 17-bp oligonucleotide encompassing the CD273 VDRE upstream of the pGL4.24 (Promega, E8421) minimal promoter by PCR and the Q5 site-directed mutagenesis kit (New England Biolabs, E0554S) using a forward primer complementary to a sequence upstream of the minimal promoter and containing the VDRE and a reverse primer complementary to a sequence near the pGL4.24 minimal promoter (supplemental Table S1). The presence of the VDRE was confirmed through sequencing.
Luciferase assay
HEK293 cells grown in 6-well plates were transfected with 1 μg of plasmid using CalFectin (SignaGen, SL100478) following the manufacturer's protocol. 16 h post-transfection, culture medium was changed with a fresh one, and cells were treated with vehicle or 100 nm 1,25D for 12 h. In the case of pGL4.24 vector, cells were lysed, and firefly luciferase activity was assessed using the Dual-Luciferase Reporter Assay kit (Promega, E1910). For the pGLuc-basic 2 vector, the culture medium was used directly in conjunction with the BioLux Gaussia Luciferase Assay kit (New England Biolabs, E3300S) to quantify changes in luciferase activity. Luciferase readings from non-transfected cells (background) were subtracted from transfected HEK293 cells, and changes in luciferase activity were normalized to the relative amount of pGLuc or pGL4.24 luciferase gene, as assessed by qPCR.
Flow cytometry and imaging
Adherent cells were detached using trypsin-EDTA (Wisent, 325-542-EL). Adherent and suspension cells were centrifuged at 500 rcf for 5 min, and supernatant was removed. Cells were resuspended in FACS buffer (0.5–1% BSA in PBS) at a concentration of 1 × 106 cells/ml. 2 μg of anti-mouse (BioLegend, 124308) or anti-human PD-L1-PE (eBioscience, 12-5983-426), FITC-CD3 (eBioscience, 11-0038-80), APC-CD56 (Invitrogen, 17-0566-41), PE/Cy7-CD19 (eBioscience, 25-0198-41), PerCP/Cy5.5-CD11c (BioLegend, 301623), PE-CD14 (Invitrogen, 12-0149-41), and APC/Cy7-CD4 (eBioscience, 56-0048-41) antibodies were added and incubated for 30 min at room temperature in the dark. Cells were washed three times with ice-cold FACS buffer and were run immediately on FACSCalibur (BD Biosciences) instruments. At least 20,000 cells/sample were monitored. Results were analyzed using FlowJo version 10.6.
SCC25 and THP-1 were fixed for 10 min with 4% paraformaldehyde and then washed with PBS twice. After a 5-min permeabilization with 0.1% Triton X-100, cells were incubated with anti-PD-L1-PE antibody (eBiosciences, 12-5983-42) in PBS containing 0.2% BSA at room temperature for 1 h in a dark, humidified chamber. Following three washes, slides were mounted in Prolong Gold containing DAPI (Life Technologies, Inc., P36935) and observed with a Zeiss Axiovert X100 bright field microscope. Images were acquired using Zen software (processing and analysis was performed at the McGill University Life Sciences Complex Advanced BioImaging Facility).
Primary bronchial epithelium was stained as described previously. ZO-1 staining was included as a mark for differentiation. Confocal images were taken on an LSM750 microscope (Zeiss; ×63 oil immersion objective), using Zen blue. Image stacks were processed with Zen Black and ImageJ and are shown as representations with maximum intensity projection. Quantification was performed with MetaExpress and analyzed using multiwavelength scoring. All of the images (n = 7) in each condition (DMSO and 1,25D) were processed and quantified using the same settings. We quantified the image by obtaining the average of the number of PD-L1+ cells (normalized to cell numbers assessed via ZO-1) across all seven fields and by calculating the total percentage of PD-L1+ cells (PD-L1+ cells/total cells). Image processing and analysis was performed at the McGill University Life Sciences Complex Advanced BioImaging Facility.
T cells were collected by centrifugation at 350 rcf for 10 min at room temperature and washed twice with ice-cold PBS. They were then blocked with human FcR binding inhibitor (eBioscience, 14-9161-73) and stained with the following antibodies: PE-Cy7-CD71 (eBioscience, 25-0719-41), PE-CD69 (eBioscience, 12-0699-41), PerCP-Cy5.5-CD44 (eBioscience, 45-0441-80), APC-CD25 (eBioscience, 17-0259-41), AlexaFluor-700-CD4 (eBioscience, 56-0048-41), and APC-eFluor-780-CD8 (eBioscience, 47-0088-41). Cells were washed and cross-linked in 2% paraformaldehyde. Flow cytometry was performed using a BD-LSRFortessa analyzer.
ELISA
Supernatants of SCC25/THP-1 cells co-cultured with Jurkat/T cells were centrifuged at 4 °C for 10 min at 500 rcf to pellet cells and debris. Supernatant was filtered through a 0.22-μm sterile filter. Samples were frozen in liquid nitrogen and shipped on dry ice for analysis to the University of Maryland Cytokine Core Laboratory for IL-2, TNF-α, and IFN-γ ELISA.
Statistics
Student's t test or one-way analysis of variance followed by Tukey's HSD post-hoc test were performed to assess significance in the case of two or more than two samples, respectively. A p value of less than or equal to 0.05 was considered significant. Symbols used to denote p value are as follows: ns > 0.05 ≥ * ≥ 0.01 ≥ ** ≥ 0.001 ≥ ***. Statistical analysis was performed using R (version 3.2.3).
Author contributions
V. D. conceived and performed experiments and wrote most of the manuscript. M. B. performed the microscopy studies. R. S.-T. and B. M. helped with gene expression studies and qPCR. G. B. isolated primary mouse myeloid cells in the C. M. K. laboratory and helped with flow cytometry, and primary T cells were obtained by C. M. K. and cultured by B. H. in the C. M. K. laboratory. R. A. in the G. L. L. laboratory isolated and cultured primary bronchial epithelial cells and provided help with imaging and quantification. G. L. and C. M. K. provided support and critical reading of the manuscript. J. H. W. conceived experiments, wrote part of the manuscript, and provided editing.
Supplementary Material
This work was supported by grants from the Canadian Institutes of Health Research (MOP 106439) and Genome Quebec (to J. H. W.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Table S1 and Figs. S1–S18.
- Teff
- effector T
- Treg
- regulatory T
- CTL
- cytotoxic T lymphocyte
- IBD
- inflammatory bowel disease
- APC
- antigen-presenting cell
- HNSCC
- head and neck squamous cell carcinoma
- VD
- vitamin D
- VDR
- vitamin D receptor
- VDRE
- vitamin D response element
- 1,25D
- 1,25-dihydroxyvitamin D
- qPCR
- quantitative PCR
- Mϕs
- macrophages
- DC
- dendritic cell
- ChIP-seq
- ChIP followed by next-generation sequencing
- TSS
- transcription start site
- H3K4me1
- histone 3 lysine 4 monomethylation
- H3K27ac
- histone 3 lysine 27 acetylation
- Pol II
- polymerase II
- eRNA
- enhancer RNA
- PBMC
- peripheral blood mononuclear cell
- CD
- Crohn's disease
- IP
- immunoprecipitation
- rcf
- relative centrifugal force
- ZO-1
- zonula occludens-1.
References
- 1. Dong H., Zhu G., Tamada K., and Chen L. (1999) B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 5, 1365–1369 [DOI] [PubMed] [Google Scholar]
- 2. Homet Moreno B., and Ribas A. (2015) Anti-programmed cell death protein-1/ligand-1 therapy in different cancers. Br. J. Cancer 112, 1421–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gupta N., Hegde P., Lecerf M., Nain M., Kaur M., Kalia M., Vrati S., Bayry J., Lacroix-Desmazes S., and Kaveri S. V. (2014) Japanese encephalitis virus expands regulatory T cells by increasing the expression of PD-L1 on dendritic cells. Eur. J. Immunol. 44, 1363–1374 [DOI] [PubMed] [Google Scholar]
- 4. Fukaya T., Takagi H., Sato Y., Sato K., Eizumi K., Taya H., Shin T., Chen L., Dong C., Azuma M., Yagita H., Malissen B., and Sato K. (2010) Crucial roles of B7-H1 and B7-DC expressed on mesenteric lymph node dendritic cells in the generation of antigen-specific CD4+Foxp3+ regulatory T cells in the establishment of oral tolerance. Blood 116, 2266–2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Okiyama N., and Katz S. I. (2014) Programmed cell death 1 (PD-1) regulates the effector function of CD8 T cells via PD-L1 expressed on target keratinocytes. J. Autoimmun. 53, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gianchecchi E., Delfino D. V., and Fierabracci A. (2013) Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmun. Rev. 12, 1091–1100 [DOI] [PubMed] [Google Scholar]
- 7. Hirano F., Kaneko K., Tamura H., Dong H., Wang S., Ichikawa M., Rietz C., Flies D. B., Lau J. S., Zhu G., Tamada K., and Chen L. (2005) Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 65, 1089–1096 [PubMed] [Google Scholar]
- 8. Dong H., Strome S. E., Salomao D. R., Tamura H., Hirano F., Flies D. B., Roche P. C., Lu J., Zhu G., Tamada K., Lennon V. A., Celis E., and Chen L. (2002) Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 8, 793–800 [DOI] [PubMed] [Google Scholar]
- 9. Parsa A. T., Waldron J. S., Panner A., Crane C. A., Parney I. F., Barry J. J., Cachola K. E., Murray J. C., Tihan T., Jensen M. C., Mischel P. S., Stokoe D., and Pieper R. O. (2007) Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 13, 84–88 [DOI] [PubMed] [Google Scholar]
- 10. Curran M. A., Montalvo W., Yagita H., and Allison J. P. (2010) PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. U.S.A. 107, 4275–4280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ansell S. M., Lesokhin A. M., Borrello I., Halwani A., Scott E. C., Gutierrez M., Schuster S. J., Millenson M. M., Cattry D., Freeman G. J., Rodig S. J., Chapuy B., Ligon A. H., Zhu L., Grosso J. F., et al. (2015) PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N. Engl. J. Med. 372, 311–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bell R. B., Leidner R., Feng Z., Crittenden M. R., Gough M. J., and Fox B. A. (2015) Developing an immunotherapy strategy for the effective treatment of oral, head and neck squamous cell carcinoma. J. Oral Maxillofac. Surg. 73, S107–S115 [DOI] [PubMed] [Google Scholar]
- 13. Aguiar P. N. Jr., Santoro I. L., Tadokoro H., de Lima Lopes G., Filardi B. A., Oliveira P., Castelo-Branco P., Mountzios G., and de Mello R. A. (2016) A pooled analysis of nivolumab for the treatment of advanced non-small-cell lung cancer and the role of PD-L1 as a predictive biomarker. Immunotherapy 8, 1011–1019 [DOI] [PubMed] [Google Scholar]
- 14. Gandini S., Massi D., and Mandalà M. (2016) PD-L1 expression in cancer patients receiving anti PD-1/PD-L1 antibodies: a systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 100, 88–98 [DOI] [PubMed] [Google Scholar]
- 15. Yang Y. F., Pang Z., Ding N., Dong W., Ma W., Li Y., Du J., and Liu Q. (2016) The efficacy and potential predictive factors of PD-1/PD-L1 blockades in epithelial carcinoma patients: a systematic review and meta analysis. Oncotarget 7, 74350–74361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang T., Xie J., Arai S., Wang L., Shi X., Shi N., Ma F., Chen S., Huang L., Yang L., Ma W., Zhang B., Han W., Xia J., Chen H., and Zhang Y. (2016) The efficacy and safety of anti-PD-1/PD-L1 antibodies for treatment of advanced or refractory cancers: a meta-analysis. Oncotarget 7, 73068–73079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scandiuzzi L., Ghosh K., Hofmeyer K. A., Abadi Y. M., Lázár-Molnar E., Lin E. Y., Liu Q., Jeon H., Almo S. C., Chen L., Nathenson S. G., and Zang X. (2014) Tissue-expressed B7-H1 critically controls intestinal inflammation. Cell Rep. 6, 625–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin R., and White J. H. (2004) The pleiotropic actions of vitamin D. Bioessays 26, 21–28 [DOI] [PubMed] [Google Scholar]
- 19. White J. H. (2012) Vitamin D metabolism and signaling in the immune system. Rev. Endocr. Metab. Disord. 13, 21–29 [DOI] [PubMed] [Google Scholar]
- 20. Verway M., Bouttier M., Wang T. T., Carrier M., Calderon M., An B. S., Devemy E., McIntosh F., Divangahi M., Behr M. A., and White J. H. (2013) Vitamin D induces interleukin-1β expression: paracrine macrophage epithelial signaling controls M. tuberculosis infection. PLoS Pathog. 9, e1003407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang T. T., Dabbas B., Laperriere D., Bitton A. J., Soualhine H., Tavera-Mendoza L. E., Dionne S., Servant M. J., Bitton A., Seidman E. G., Mader S., Behr M. A., and White J. H. (2010) Direct and indirect induction by 1,25-dihydroxyvitamin D3 of the NOD2/CARD15-defensin β2 innate immune pathway defective in Crohn disease. J. Biol. Chem. 285, 2227–2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang T. T., Nestel F. P., Bourdeau V., Nagai Y., Wang Q., Liao J., Tavera-Mendoza L., Lin R., Hanrahan J. W., Mader S., White J. H., and Hanrahan J. H. (2004) Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J. Immunol. 173, 2909–2912 [DOI] [PubMed] [Google Scholar]
- 23. Dimitrov V., and White J. H. (2016) Species-specific regulation of innate immunity by vitamin D signaling. J. Steroid Biochem. Mol. Biol. 164, 246–253 [DOI] [PubMed] [Google Scholar]
- 24. Gombart A. F., Saito T., and Koeffler H. P. (2009) Exaptation of an ancient Alu short interspersed element provides a highly conserved vitamin D-mediated innate immune response in humans and primates. BMC Genomics 10, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jeffery L. E., Burke F., Mura M., Zheng Y., Qureshi O. S., Hewison M., Walker L. S., Lammas D. A., Raza K., and Sansom D. M. (2009) 1,25-Dihydroxyvitamin D3 and IL-2 combine to inhibit T cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J. Immunol. 183, 5458–5467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mahon B. D., Wittke A., Weaver V., and Cantorna M. T. (2003) The targets of vitamin D depend on the differentiation and activation status of CD4 positive T cells. J. Cell. Biochem. 89, 922–932 [DOI] [PubMed] [Google Scholar]
- 27. Sadeghian M., Saneei P., Siassi F., and Esmaillzadeh A. (2016) Vitamin D status in relation to Crohn's disease: meta-analysis of observational studies. Nutrition 32, 505–514 [DOI] [PubMed] [Google Scholar]
- 28. Kabbani T. A., Koutroubakis I. E., Schoen R. E., Ramos-Rivers C., Shah N., Swoger J., Regueiro M., Barrie A., Schwartz M., Hashash J. G., Baidoo L., Dunn M. A., and Binion D. G. (2016) Association of vitamin D level with clinical status in inflammatory bowel disease: a 5-year longitudinal study. Am. J. Gastroenterol. 111, 712–719 [DOI] [PubMed] [Google Scholar]
- 29. Wang T. T., Tavera-Mendoza L. E., Laperriere D., Libby E., MacLeod N. B., Nagai Y., Bourdeau V., Konstorum A., Lallemant B., Zhang R., Mader S., and White J. H. (2005) Large-scale in silico and microarray-based identification of direct 1,25-dihydroxyvitamin D3 target genes. Mol. Endocrinol. 19, 2685–2695 [DOI] [PubMed] [Google Scholar]
- 30. Akutsu N., Lin R., Bastien Y., Bestawros A., Enepekides D. J., Black M. J., and White J. H. (2001) Regulation of gene expression by 1α,25-dihydroxyvitamin D3 and its analog EB1089 under growth-inhibitory conditions in squamous carcinoma cells. Mol. Endocrinol. 15, 1127–1139 [DOI] [PubMed] [Google Scholar]
- 31. Loke P., and Allison J. P. (2003) PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc. Natl. Acad. Sci. U.S.A. 100, 5336–5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cho Y. A., Yoon H. J., Lee J. I., Hong S. P., and Hong S. D. (2011) Relationship between the expressions of PD-L1 and tumor-infiltrating lymphocytes in oral squamous cell carcinoma. Oral Oncol. 47, 1148–1153 [DOI] [PubMed] [Google Scholar]
- 33. He X. H., Xu L. H., and Liu Y. (2005) Identification of a novel splice variant of human PD-L1 mRNA encoding an isoform-lacking Igv-like domain. Acta Pharmacol. Sin. 26, 462–468 [DOI] [PubMed] [Google Scholar]
- 34. Chen Y., Wang Q., Shi B., Xu P., Hu Z., Bai L., and Zhang X. (2011) Development of a sandwich ELISA for evaluating soluble PD-L1 (CD274) in human sera of different ages as well as supernatants of PD-L1+ cell lines. Cytokine 56, 231–238 [DOI] [PubMed] [Google Scholar]
- 35. Heikkinen S., Väisänen S., Pehkonen P., Seuter S., Benes V., and Carlberg C. (2011) Nuclear hormone 1α,25-dihydroxyvitamin D3 elicits a genome-wide shift in the locations of VDR chromatin occupancy. Nucleic Acids Res. 39, 9181–9193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lam M. T., Li W., Rosenfeld M. G., and Glass C. K. (2014) Enhancer RNAs and regulated transcriptional programs. Trends Biochem. Sci. 39, 170–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rahman S., Zorca C. E., Traboulsi T., Noutahi E., Krause M. R., Mader S., and Zenklusen D. (2017) Single-cell profiling reveals that eRNA accumulation at enhancer-promoter loops is not required to sustain transcription. Nucleic Acids Res. 45, 3017–3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mueller A. C., Cichewicz M. A., Dey B. K., Layer R., Reon B. J., Gagan J. R., and Dutta A. (2015) MUNC, a long noncoding RNA that facilitates the function of MyoD in skeletal myogenesis. Mol. Cell. Biol. 35, 498–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Unger W. W., Laban S., Kleijwegt F. S., van der Slik A. R., and Roep B. O. (2009) Induction of Treg by monocyte-derived DC modulated by vitamin D3 or dexamethasone: differential role for PD-L1. Eur. J. Immunol. 39, 3147–3159 [DOI] [PubMed] [Google Scholar]
- 40. Chen C., Ridzon D. A., Broomer A. J., Zhou Z., Lee D. H., Nguyen J. T., Barbisin M., Xu N. L., Mahuvakar V. R., Andersen M. R., Lao K. Q., Livak K. J., and Guegler K. J. (2005) Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 33, e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Welboren W. J., van Driel M. A., Janssen-Megens E. M., van Heeringen S. J., Sweep F. C., Span P. N., and Stunnenberg H. G. (2009) ChIP-Seq of ERα and RNA polymerase II defines genes differentially responding to ligands. EMBO J. 28, 1418–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Raftery T., Martineau A. R., Greiller C. L., Ghosh S., McNamara D., Bennett K., Meddings J., and O'Sullivan M. (2015) Effects of vitamin D supplementation on intestinal permeability, cathelicidin and disease markers in Crohn's disease: results from a randomised double-blind placebo-controlled study. United European Gastroenterol. J. 3, 294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jørgensen S. P., Agnholt J., Glerup H., Lyhne S., Villadsen G. E., Hvas C. L., Bartels L. E., Kelsen J., Christensen L. A., and Dahlerup J. F. (2010) Clinical trial: vitamin D3 treatment in Crohn's disease: a randomized double-blind placebo-controlled study. Aliment. Pharmacol. Ther. 32, 377–383 [DOI] [PubMed] [Google Scholar]
- 44. Samson C. M., Morgan P., Williams E., Beck L., Addie-Carson R., McIntire S., Booth A., Mendez E., Luzader C., Tomer G., Saeed S., Donovan E., Bucuvalas J., and Denson L. A. (2012) Improved outcomes with quality improvement interventions in pediatric inflammatory bowel disease. J. Pediatr. Gastroenterol. Nutr. 55, 679–688 [DOI] [PubMed] [Google Scholar]
- 45. Deeb K. K., Trump D. L., and Johnson C. S. (2007) Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat. Rev. Cancer 7, 684–700 [DOI] [PubMed] [Google Scholar]
- 46. Chiang K. C., and Chen T. C. (2013) The anti-cancer actions of vitamin D. Anticancer Agents Med. Chem. 13, 126–139 [PubMed] [Google Scholar]
- 47. Shah S., Islam M. N., Dakshanamurthy S., Rizvi I., Rao M., Herrell R., Zinser G., Valrance M., Aranda A., Moras D., Norman A., Welsh J., and Byers S. W. (2006) The molecular basis of vitamin D receptor and β-catenin crossregulation. Mol. Cell 21, 799–809 [DOI] [PubMed] [Google Scholar]
- 48. Salehi-Tabar R., Nguyen-Yamamoto L., Tavera-Mendoza L. E., Quail T., Dimitrov V., An B. S., Glass L., Goltzman D., and White J. H. (2012) Vitamin D receptor as a master regulator of the c-MYC/MXD1 network. Proc. Natl. Acad. Sci. U.S.A. 109, 18827–18832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. An B. S., Tavera-Mendoza L. E., Dimitrov V., Wang X., Calderon M. R., Wang H. J., and White J. H. (2010) Stimulation of Sirt1-regulated FoxO protein function by the ligand-bound vitamin D receptor. Mol. Cell. Biol. 30, 4890–4900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mayne S. T., Ferrucci L. M., and Cartmel B. (2012) Lessons learned from randomized clinical trials of micronutrient supplementation for cancer prevention. Annu. Rev. Nutr. 32, 369–390 [DOI] [PubMed] [Google Scholar]
- 51. Veit G., Bossard F., Goepp J., Verkman A. S., Galietta L. J., Hanrahan J. W., and Lukacs G. L. (2012) Proinflammatory cytokine secretion is suppressed by TMEM16A or CFTR channel activity in human cystic fibrosis bronchial epithelia. Mol. Biol. Cell 23, 4188–4202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barsoum I. B., Smallwood C. A., Siemens D. R., and Graham C. H. (2014) A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 74, 665–674 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.