

Abstract
The normal cellular function requires communication between mitochondria and the nucleus, termed mitochondria-to-nucleus retrograde signaling. Disruption of this mechanism has been implicated in the development of cancers. Many proteins are known modulators of retrograde signaling, but whether microRNAs (miRNAs) are also involved is unknown. We conducted an miRNA microarray analysis using RNA from a parental cell line, a Rho0 line lacking mitochondrial DNA (mtDNA) and a Rho0 line with restored mtDNA. We found that miR-663 was down-regulated in the mtDNA-depleted Rho0 line. mtDNA restoration reversed this miRNA to parental level, suggesting that miR-663 may be epigenetically regulated by retrograde signaling. By using methylation-specific PCR and bisulfite sequencing we demonstrate that miR-663 promoter is epigenetically regulated not only by genetic but also by pharmacological disruption of oxidative phosphorylation (OXPHOS). Restoration of OXPHOS Complex I inhibitor–induced miR-663 expression by N-acetylcysteine suggested that reactive oxygen species (ROS) play a key role in epigenetic regulation of miR-663. We determined that miR-663 regulates the expression of nuclear-encoded respiratory chain subunits involved in Complexes I, II, III, and IV. miR-663 also controlled the expression of the Complexes I (NDUFAF1), II (SDHAF2), III (UQCC2), and IV (SCO1) assembly factors and was required for stability of respiratory supercomplexes. Furthermore, using luciferase assays, we found that miR-663 directly regulates UQCC2. The anti–miR-663 reduced OXPHOS complex activity and increased in vitro cellular proliferation and promoted tumor development in vivo in mice. We also found that increased miR-663 expression in breast tumors consistently correlates with increased patient survival. We provide the first evidence for miRNA controlling retrograde signaling, demonstrating its epigenetic regulation and its role in breast tumorigenesis.
Keywords: breast cancer, DNA methylation, gene expression, microRNA (miRNA), mitochondria, miR-663, retrograde signaling, OXPHOS, tumorigenesis
Introduction
Mitochondria are principally known for their role in producing cellular energy, although they are integral in other processes such as cellular growth, death, and signaling. The mitochondrial proteome is nuclear encoded except for 13 proteins encoded by the mitochondrial genome. Therefore, the mitochondrion must import over 99% of its proteome. As a result, the coordinated communication of the nuclear and mitochondrial genomes is required for numerous cellular processes. The cell accomplishes this coordination through the anterograde and retrograde dispatch of signals in the nucleus-to-mitochondria and mitochondria-to- nucleus directions, respectively. Although anterograde communication has received much focus in the past given the nucleus' major role in regulating mitochondrial structure and function, retrograde communication has become an increasingly important area of research. In mammals, it has been linked to metabolic stress response by activation of AMP-activated protein kinase (AMPK) and subsequent stimulation of PGC1-α to promote mitochondrial biogenesis; it has also been shown to intersect with calcium signaling via several calcium-regulated kinases such as protein kinase C and p38 MAPK (1). The pathway has also been shown to regulate the p53 tumor suppressor pathway (2, 3). Importantly, dozens of genes from several functional categories including transcription, cell signaling, and cell architecture are known to be involved in retrograde communication in breast cancer (2, 4, 5). Moreover, mitochondrial dysfunction is a hallmark of many cancers, suggesting an important role for retrograde signaling as well (6–9). Cytoplasmic hybrid (cybrid) studies, in which nuclear backgrounds are kept constant but mitochondrial genetic profiles are modified, suggest that retrograde signaling may play an important role in tumorigenesis (10, 11).
microRNAs (miRNAs)2 are small, noncoding RNAs that impose genetic regulation by pairing with complementary sequences in a transcript's 3′-untranslated region (UTR). As a class of molecules, miRNAs are well-known to play important roles in a variety of cancers (12). The prospect of their utility as biomarkers for cancer is an increasingly important area of research (13, 14). miRNAs have been shown to regulate anterograde communication between the nucleus and mitochondria. For example, mitochondrial COX1 mRNA expression is increased by nuclear-encoded miR-181c (15), and miR-1 increases mitochondrial translation during muscle differentiation (16). Moreover, miR-214 modulates mitochondrial morphology by down-regulating MFN2 (17), and miR-7 regulates the permeability transition pore by targeting VDAC1 (18). It is clear that miRNAs play a role in mediating anterograde communication, but there are currently no reports of the involvement of miRNAs in retrograde communication. We show that the miR-663 is epigenetically regulated and suppresses tumor development. It regulates mitochondrial function and mediates the retrograde response.
Results
miR-663 mediates mitochondria-to-nucleus retrograde signaling and is regulated by reactive oxygen species (ROS)
The mitochondria-to-nucleus retrograde response is a cascade of molecular events of which our understanding is incomplete (5, 19, 20). To identify miRNAs in this pathway, we conducted an miRNA microarray using RNA from a parental cell line, its Rho0 derivative devoid of mitochondrial DNA (mtDNA), and a cybrid line with repleted mtDNA from healthy platelets (Fig. 1A). The most significantly increased miRNAs in the Rho0 line were hsa-miR-186 and hsa-491–3p; the most significantly decreased miRNAs were hsa-miR-668, hsa-miR-625, hsa-miR-760, and hsa-miR-663. Restoration of mtDNA in cybrid cells restored expression of these miRNAs.
Figure 1.

miR-663 controls mitochondria-to-nucleus retrograde signaling. A, results of a microRNA microarray using RNA from 143B parental (WT) cells, parental cells devoid of mtDNA (Rho0), and Rho0 cells with restored mtDNA (Cybrid). The graph represents -fold change of the six most modulated microRNAs in the parental and cybrid lines relative to the Rho0 line. B, confirmation of the microarray results showing down-regulation of miR-663. Real-time PCR confirmation of down-regulated miR-663 at the mature transcript level in Rho0 cells compared with WT (parental) and cybrid cells. C, real-time PCR of the primary transcript and the mature miR-663 molecule shows down-regulation of both forms after inducing mitochondrial dysfunction at each complex in the electron transport chain. 143B cells were treated for 12 h with DMSO or inhibitors of mitochondrial oxidative phosphorylation (OXPHOS) complexes. Rot = rotenone (Complex I, 100 nm), mal = malonate (Complex II, 10 mm), AA = antimycin A (Complex III, 20 μm), KCN = potassium cyanide (Complex IV, 1 mm), and OM = oligomycin (Complex IV, 12.5 μm). # denotes p < 0.01 comparing primary transcript between DMSO and treatments, and * denotes p < 0.05 comparing mature transcript between DMSO and treatments. D, assessment of reactive oxygen species by detection of oxidized CM-H2DCFDA in 143B cells after treatment with rotenone or antimycin A. E, expression of primary miR-663 expression in 143B cells treated with H2O2. F, expression of primary miR-663 in 143B cells treated with rotenone alone or rotenone in combination with N-acetylcysteine. *, p < 0.05; **, p < 0.01 relative to DMSO. All real-time PCR data represent the average of three biological replicates. Error bars represent S.D.
Because its role in breast cancer is not fully characterized, we selected miR-663 for validation by real-time PCR. Mature miR-663 expression was reduced by depletion of mtDNA, and repletion restored its expression to parental levels (Fig. 1B). Because the miR-663 expression is dependent on mitochondrial function, we hypothesized that ROS produced by the impairment of oxidative phosphorylation (OXPHOS) may be the regulatory signal (21, 22). We treated parental cells with OXPHOS complex inhibitors and found down-regulation of primary and mature miR-663 (Fig. 1C). Detection of oxidized CM-H2DCFDA (5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester) by FACS showed that mitochondrial impairment significantly increases reactive oxygen species (Fig. 1D). H2O2 reduced primary miR-663 expression (Fig. 1E). Similarly, rotenone was able to down-regulate primary miR-663 expression, and the antioxidant N-acetylcysteine restored its expression (Fig. 1F). We conclude that mitochondria and mitochondrial ROS regulate miR-663.
Mitochondria regulate miR-663 promoter methylation
We showed previously that loss of mtDNA reversibly alters DNA methylation (19), suggesting mitochondrial control over nuclear gene expression. Our current study demonstrates the regulation of miR-663 expression by mitochondrial function, and its reversibility suggests an epigenetic mechanism. We used methylation-specific PCR (MSP) and genomic bisulfite sequencing (GBS) to show that mitochondrial dysfunction deregulates miR-663 through DNA methylation. The miR-663 promoter was hypermethylated in Rho0 (parental cells without mtDNA) cells compared with parental and cybrid cells, and the same was true of parental cells treated with rotenone or antimycin A (Fig. 2A). We confirmed this with genomic bisulfite sequencing (Fig. 2B). Furthermore, we found that hypermethylation and reduced expression of miR-663 by rotenone and antimycin A did not occur in the presence of 5-aza-2′-deoxycytidine, an inhibitor of DNA methylation (Fig. 2C). Importantly, treatment of cells with rotenone or antimycin A increased their methyltransferase activity (Fig. 2D).
Figure 2.

Mitochondrial dysfunction induces hypermethylation of miR-663 promoter. A, methylation-specific PCR assessing methylation of the miR-663 promoter in Rho0 cells compared with WT or Cybrid cells and after treatment of WT cells with rotenone or antimycin A. M denotes methylated DNA amplified by primers specific for methylated DNA. U denotes unmethylated DNA amplified by primers specific for unmethylated DNA. B, bisulfite sequencing depicts 21 CpG dinucleotides present in the miR-663 promoter. Each row represents the sequencing results of a single clone. Each black circle represents a methylated CpG dinucleotide, and each white circle represents an unmethylated CpG dinucleotide. C, the bar graphs on top show real-time PCR of primary miR-663 in 143B cells after treatment with rotenone or antimycin A in the presence or absence of 5-aza-2′-deoxycytidine. The lower panels show MSP with rotenone or antimycin A in the presence or absence of 5-aza-2′-deoxycytidine. D, cellular methyltransferase activity after inhibition of Complex I (rotenone) or III (antimycin). *, p < 0.05; **, p < 0.01. Error bars represent S.D. Real-time PCR data represent the average of three biological replicates. MSP and bisulfite sequencing results are representative of two biological replicates. Methyltransferase activity data represent two biological replicates run in triplicate.
We extended our study to breast cancer cells demonstrating that mitochondrial dysfunction, induced by depletion of mtDNA by a POLG-dominant negative mutant or chemically by rotenone, results in the down-regulation of miR-663 (Fig. 3, A–C). We also observed increased methylation of the miR-663 promoter after treatment with rotenone as shown by combined bisulfite restriction analysis (COBRA) and bisulfite sequencing (Fig. 3, B and C). We analyzed the methylation status of miR-663 in primary breast tumors from The Cancer Genome Atlas (TCGA) database. Consistent with our studies, we discovered hypermethylation of the miR-663 promoter in tumors compared with normal samples (Fig. 3D). These data suggest that miR-663 is down-regulated by mitochondrial dysfunction through an epigenetic mechanism involving DNA hypermethylation and that miR-663 may play a role in cancer.
Figure 3.

miR-663 is regulated by mitochondrial dysfunction and DNA methylation. A, top panel shows quantification of mtDNA content after depletion of mtDNA in MCF7 cells. Depletion was achieved by activating a doxycycline-inducible dominant negative POLG mutant, D1135A. Bottom panel shows real-time PCR of the primary miR-663 transcript in MCF7 cells showing down-regulation of miR-663 after depletion of mtDNA. B and C, MCF7 cells (B) and MDA-MB-231 (C) treated with rotenone also had reduced expression of miR-663. Middle panels show COBRA analyses in which bisulfite converted DNA was digested with BstUI. Lower molecular weight bands indicate CGCG sequences that were methylated. Bottom panels show bisulfite sequencing results in breast cancer cells treated with DMSO or rotenone. D, median beta values from 533 TCGA breast tumors analyzed on the Infinium HumanMethylation450 BeadChip platform show the methylation status at nine CpG loci across the miR-663 promoter region. Each locus on the bead chip array is denoted by its distance from the beginning of the miR-663 precursor transcript. *, p < 0.05; ***, p < 0.001. Real-time PCR data represent the average of three biological replicates. Error bars represent S.D. Error bars for tumor methylation beta values are S.E.
miR-663 regulates nuclear-encoded OXPHOS subunits
Because miR-663 responds to mitochondrial dysfunction, we hypothesized a role for the miRNA in regulating mitochondrial function. To investigate the role of miR-663 in regulating the expression of nuclear-encoded mitochondrial genes, we transfected a control vector, an miR-663 expression vector or an anti–miR-663–expressing vector in MCF7 and MDA-MB-231 breast cancer cells. We achieved 3- to 3.5-fold up-regulation of miR-663 as assessed by real-time PCR (supplemental Fig. S2). Because the anti–miR-663 only sequesters miR-663 rather than degrading it, we determined miR-663 inhibition by analyzing the expression of known target genes. Significant down-regulation of miR-663 (supplemental Fig. 2) was demonstrated by increased expression of the well-established target genes, TGF-β1 (23–26) and JunB (27, 28). Western blotting of mitochondrial extracts with an antibody mixture against OXPHOS subunits for each complex showed that the anti–miR-663 reduced the expression of subunits in both MCF7 and MDA-MB-231 cells (Fig. 4A). We expanded our analysis to assess OXPHOS gene expression in MCF7 cells and found that modulation of miR-663 resulted in broad transcriptional reprogramming of Complexes I–IV respiratory subunits (Fig. 4B). The anti–miR-663 reduced the expression of most subunits assayed; in particular NDUFB8 (Complex I), SDHB and SDHC (Complex II), UQCRFS1 and UQCRC2 (Complex III), and COX4L1 and COX7C (Complex IV). Ectopic expression of miR-663 resulted in increased expression of several of those genes. We conclude that miR-663 regulates nuclear-encoded respiratory chain subunits involved in Complexes I–IV.
Figure 4.
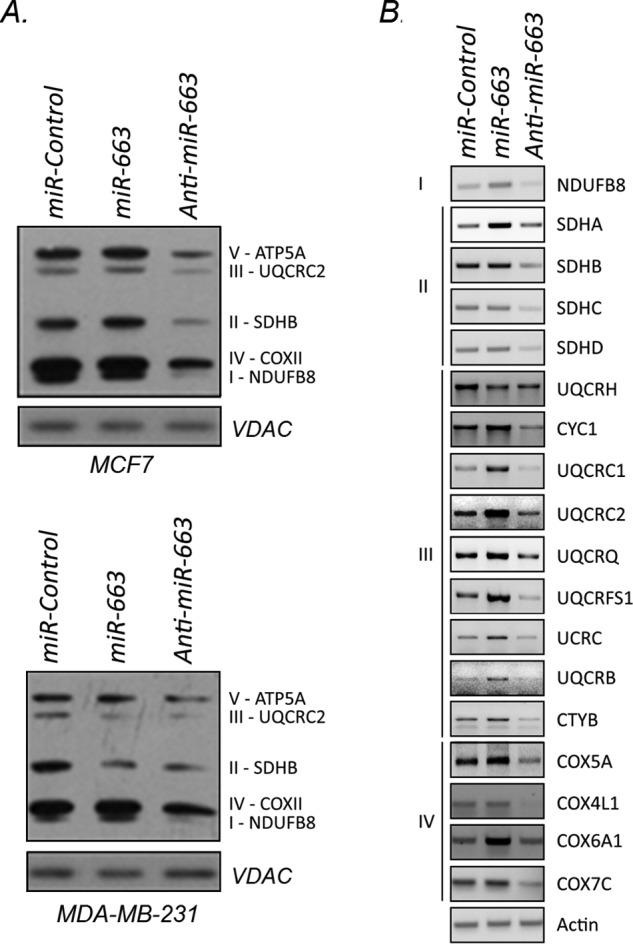
Inhibition of miR-663 reprograms OXPHOS gene expression. A, protein expression of subunits for each OXPHOS complex in mitochondria from miR-663–modulated MCF7 cells (top blot) and MDA-MB-231 cells (bottom blot). B, semi-quantitative PCRs of respiratory complex subunits using mRNA isolated from miR-663–modulated MCF7 cells. All PCRs were run at the same time as their corresponding actin controls and represent two biological replicates.
miR-663 regulates expression of OXPHOS assembly factors and stability of supercomplexes
In addition to relying on the expression of OXPHOS subunits, the mitochondrial function requires assembly factors for the construction of OXPHOS complexes (29, 30). We tested the role of miR-663 in regulating key assembly factors. We found that some assembly factors for Complexes I–IV were reprogrammed at the transcriptional level after modulation of miR-663 (Fig. 5A). The anti–miR-663 reduced the expression of nearly all assembly factors assayed, including NDUFAF1 (Complex I), SDHAF2 (Complex II), UQCC2 (Complex III), and SCO1 (Complex IV). Ectopic expression of miR-663 increased expression of several of those genes and others (Fig. 5A). Next, we used luciferase assay to test if miR-663 directly targeted any of the dysregulated respiratory genes. Using the miRNA target prediction software miRanda (http://www.microrna.org/microrna/home.do)3, we identified SDHAF2 and UQCC2 as potential targets of miR-663. We tested these two predicted targets and one unpredicted Complex I subunit, NDUFB8 (regulated by miR-663). We found that miR-663 directly interacts with the 3′-UTR of UQCC2 (Fig. 5B). These data demonstrate that miR-663 regulates the respiratory chain directly by modulating the expression of a Complex III assembly factor; importantly we found no evidence that miR-663 localizes to mitochondria (supplemental Fig. S3).
Figure 5.

Inhibition of miR-663 destabilizes supercomplexes. A, semi-quantitative PCRs of respiratory complex assembly factors using mRNA isolated from miR-663–modulated MCF7 cells. All PCRs were done at the same time as their corresponding actin control. B, luciferase assays assessing direct interactions between miR-663 and the 3′-UTRs of predicted miR-663 targets. C, BN-PAGE of OXPHOS supercomplexes in MCF7 cells (left blot) and MDA-MB-231 cells (right blot) stably expressing either a microRNA control vector, an miR-663 expression vector, or an anti–miR-663 vector. Mitochondria were solubilized in 1% digitonin. Blots were probed with an OXPHOS antibody mixture (MitoSciences) including antibodies against NDUFB8 (Complex I), SDHB (Complex II), UQCRC2 (Complex III), COXII (Complex IV), and ATP5A (Complex V). D, Blue Native blots showing supercomplex stability after CRISPR disruption of subunits or assembly factors for Complexes I–IV in MCF7 cells. Mitochondria were solubilized in 1% digitonin or 1% Triton as noted. Antibodies are indicated below each blot and dilutions of 1:1000 were used for all. E, primary miR-663 transcript expression by real-time PCR using cDNA prepared from CRISPR transfected MCF7 cells. *, p < 0.05; **, p < 0.01. BN-PAGE and semi-quantitative PCR data are representative of two biological replicates. Real-time PCR data represent three biological replicates. Luciferase assays represent two biological replicates run in quadruplicate. Error bars depict S.D.
Large macromolecular OXPHOS complexes, or supercomplexes, are assembled from smaller complexes and are important for electron transfer between complexes (31, 32). These supercomplexes are thus an important factor in mitochondrial function (33). We used Blue Native (BN)-PAGE to assess the role of miR-663 in supercomplex stability. In accord with the subunit and assembly factor expression changes, we observed a robust destabilization of OXPHOS supercomplex ComplexI/ComplexIII2/ComplexIV, ComplexIII2/IV, ComplexIII2, and ComplexII in MCF7 cells by the anti–miR-663 (Fig. 5C). In MDA-MB-231 cells, we observed destabilization of ComplexIII2 and ComplexII (Fig. 5C). Ectopic expression of miR-663 increased the expression of many subunits and assembly factors without affecting the stability of supercomplexes. Unexpectedly, ectopic expression of miR-663 reduced the stability of the ComplexIII2 in both MCF7 and MDA-MB-231 lines.
To determine the impact of subunit and assembly factor reprogramming on supercomplex assembly, we used CRISPRs to disrupt many miR-663–target subunits and assembly factors and assessed the effects by BN-PAGE. Genetic disruptions were confirmed by heteroduplex mobility assay (HMA) or Western blotting (supplemental Fig. S4). Disruption of each subunit or assembly factor resulted in complex-specific disruption of supercomplexes (Fig. 5D). Disruption of the Complex I subunit NDUFB8 destabilized ComplexI/ComplexIII2/ComplexIV and ComplexI/ComplexIII2. Disruption of the Complex II assembly factor SDHAF2 destabilized Complex II. Disruption of the Complex III assembly factor UQCC1 destabilized ComplexIII2/IV and ComplexIII2. Disruption of the Complex IV assembly factor COX10 destabilized ComplexIII2/ComplexIV and increased ComplexIII2 and ComplexIV. Furthermore, except for SDHAF2, disruption of each of these OXPHOS genes reduced miR-663 expression by a feedback mechanism (Fig. 5E).
miR-663 regulation of OXPHOS gene expression modulates complex activity
The expression of OXPHOS subunits, their assembly factor–mediated construction into larger complexes, and the stability of those complexes determine, in part, the enzymatic activity of each complex (34). Except for Complex IV, the anti–miR-663 reduced the activity of all OXPHOS complexes (Fig. 6). In most cases, ectopic expression of miR-663 did not alter the enzyme activity, except Complex II. Complex III activity was reduced by ectopic expression of miR-663, which may be a result of the observed reduction in UQCRH expression and the destabilization of ComplexIII2 after expressing miR-663. These results indicate that miR-663 regulates OXPHOS complex activity by mediating OXPHOS gene expression and complex assembly.
Figure 6.

Inhibition of miR-663 alters OXPHOS enzyme activity. Isolated mitochondria were used to conduct enzymatic activity assays of OXHPOS Complexes I–V in MCF7 cells expressing a microRNA control vector, an miR-663 expression vector, or an anti–miR-663 vector. Activities were normalized to total protein. *, p < 0.05; **, p < 0.01. Assays represent two biological replicates run in triplicate. Error bars depict S.D.
miR-663 regulates tumorigenic properties in vitro and tumor growth in vivo
Mitochondrial dysfunction is a hallmark of cancer (7, 9, 35). Because miR-663 regulates mitochondrial function, we tested the impact of miR-663 on in vitro tumorigenic properties and in vivo tumorigenesis. Ectopic expression of miR-663 decreased cellular invasion (Fig. 7A) in both MCF7 and MDA-MB-231 cells in vitro but did not affect in vitro proliferation (Fig. 7B). Consistent with in vitro studies, we demonstrated increased tumor growth rate in anti–miR-663–expressing xenografts and decreased growth rate in miR-663–expressing xenografts compared with the control (Fig. 7C). Tumor weight demonstrated the same relationship; the anti-miR-663 and ectopic expression increased and decreased tumor weight, respectively (Fig. 7D). Based on these studies, we conclude that miR-663 suppresses breast tumorigenesis.
Figure 7.

miR-663 regulates cellular growth, invasion, and tumor progression. A and B, after stable transfection of MCF7 and MDA-MB-231 breast cancer cell lines with the microRNA control, miR-663 expression vector, or an anti–miR-663 vector, tumorigenic assays were carried out to assess the role of miR-663 in cellular invasion (A) and cellular proliferation (B). C and D, tumor growth (C) and tumor weight (D) were assessed in mouse subcutaneous xenografts stably expressing a microRNA control, miR-663 expression vector, or an anti–miR-663 vector. E, analysis of TCGA tumors (bar graph) showing mature miR-663 expression by tumor stage I–IV (tumor sample n = 82, 121, 390, 150, and 13 for normal, stage I, stage II, stage III, and stage IV, respectively). Kaplan-Meier curves (right plot) derived from TCGA data showing 10-year survival of patients with group stage II breast tumors, stratified by high (n = 15) or low (n = 18) mature miR-663 expression. F, analysis of TCGA tumors (bar graph) showing mature miR-663 expression by tumor metastatic stage (tumor sample n = 82, 690, and 15 for normal, M0, and M1, respectively). Survival of patients (right plot) with metastatic stage 0 breast tumors stratified by high (n = 11) and low (n = 22) mature miR-663 expression. Patients were stratified by high (expression values >0) and low (expression values of 0) mature miR-663 expression. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Cellular invasion and proliferation assays present at least two biological replicates. Proliferation assays were run in quadruplicate. In vitro assay error bars represent S.D. Xenograft and primary tumor error bars denote S.E.
We expanded our analysis into the TCGA database and found that miR-663 expression correlates with stage-specific survival in breast tumors. miR-663 was elevated in stage II tumors. Stage II tumors are characterized by being smaller than 5 cm with lymph node involvement, or larger than 5 cm with no node involvement. Interestingly, miR-663 expression imparted a positive survival effect (Fig. 7E). We also found miR-663 to be elevated at metastatic stage M0 (no detectible metastases) where it also imparted a positive survival effect (Fig. 7F). These data suggest that miR-663 plays an important role in early stage breast tumor biology.
Discussion
Mitochondria play a regulatory role in tumorigenesis, evidenced by their connection with tumor suppressor function (36) and apoptosis (37). Furthermore, mitochondrial dysfunction induces the Warburg phenomenon and metabolic reprogramming (37, 38). Although it is well-appreciated that mitochondrial dysfunction plays an important role in tumorigenesis, the mechanisms underlying this are not fully understood. Retrograde signaling is a response to mitochondrial dysfunction in which nuclear gene transcription is reprogrammed through signaling cascades mediated by second messengers such as ROS and cytosolic calcium. Our knowledge of the mechanisms by which these alterations occur is currently incomplete; our understanding of the role of miRNA in retrograde signaling is particularly lacking. Among other processes, miRNAs have been shown to regulate mitochondrial function. miR-30 targets the p53-Drp1 axis, whereas miR-181c translocates into mitochondria to directly modulate gene expression (39, 40). Current research describes an increasingly complex interplay of nuclear-encoded miRNAs that localize to mitochondria, perhaps joining a pool of miRNAs that are transcribed by mtDNA itself (41–43). miRNAs, therefore, appear poised to modulate mitochondria-to-nucleus communication, which could play an important role in cancer. In this study, we identified miR-663 as an important regulator of the mitochondrial retrograde response and tumorigenesis.
The miR-663 gene family is a primate-specific group of miRNAs of which only miR-663a (or miR-663) and miR-663b have been identified in humans. The role of miR-663 in tumorigenesis appears to be tissue specific; in papillary thyroid carcinoma (25), glioblastoma (44, 45), and pancreatic cancer (46), miR-663 suppresses tumorigenic characteristics, whereas in nasopharyngeal carcinoma (47), lung cancer (48), and prostate cancer (49), miR-663 induces tumorigenic characteristics. Furthermore, miR-663 expression is decreased in pediatric acute myeloid leukemia patients (50), whereas its overexpression was associated with chemoresistance in breast tumors (51). The complexity of miR-663's role in tumorigenesis is at least partly because of the complex nature of its target genes. On the one hand, miR-663 targets oncogenes like H-Ras (52), PIK3CD (44), and CXCR4 (45). H-Ras is highly mutated in bladder cancer (53) and is a driving factor in breast cancer (54, 55). PIK3CD is ubiquitously expressed in follicular lymphoma and is overexpressed in many solid tumors (56, 57); it regulates cell chemotaxis in breast cancer (58). On the other hand, miR-663 targets the tumor suppressor APC (59, 60) which is known to be frequently modulated epigenetically in breast cancer and regulates the Wnt/β-catenin pathway (61, 62). Furthermore, several miR-663 targets are known to switch between their tumor suppressive and oncogenic functions depending on the stage of cancer (e.g. TGF-β1) (24, 26, 48, 63) or their genetic context (e.g. p21 (47) and JunD (27)). It is therefore important to characterize the role of miR-663 in other cellular and tumorigenic contexts. From an miRNA microarray, we identified miR-663 and a number of other miRNAs that were dysregulated in Rho0 cells and restored to parental expression levels after restoring mtDNA, presenting us with reversible, candidate mediators of retrograde communication. We chose miR-663 for further investigation for several reasons. Firstly, its role in breast cancer remains unclear. Secondly, the miR-663 promoter has been shown to be labile to DNA methylation. This, in conjunction with our previous results showing that mitochondrial dysfunction alters nuclear DNA methylation, highlights miR-663 as a potentially important factor in understanding this mechanism in breast cancer. We validated the dysregulation of miR-663 in Rho0 cells by real-time PCR (Fig. 1B) and showed that it is dysregulated by mitochondrial dysfunction (Fig. 1C). We demonstrated that miR-663 is regulated by ROS (Fig. 1, D and F). This is supported by previous reports showing that oxidative stress alters miR-663 expression (27, 64, 65).
Our previous studies demonstrated a link between mitochondrial function and nuclear DNA methylation (19, 66, 67). Here we expanded our studies to elucidate this mechanism and identify its targets. The miRNAs that were dysregulated in our screen returned to parental levels upon restoration of mtDNA (Fig. 1A). We observed that mitochondrial dysfunction resulted in the reduction of both the primary and mature miR-663 molecules (Fig. 1C). Our previous study showing the reversal of epigenetic changes by replenishing mtDNA indicated that these miRNA forms were epigenetically regulated. We demonstrated that mitochondrial dysfunction (pharmacological inhibition of OXPHOS complexes or removal of mtDNA) hypermethylated the miR-663 promoter and down-regulated its expression (Fig. 2, A and B). Hypermethylation and down-regulation of miR-663 was not observed in the presence of 5-aza-2′-deoxycytidine (Fig. 2C), confirming the methylation mechanism. Total DNA methyltransferase activity was elevated in cells treated with complex inhibitors (Fig. 2D), indicating that mitochondrial dysfunction alters methylation of miR-663 and other miRNAs identified in this study by modulating the activity of DNA methyltransferases. These results are consistent with a previous report showing that depletion of mtDNA increases expression of DNMT1 and methylation of the promoters of MGMT, EDNRB, and CHD1 (68).
Mitochondrial dysfunction also hypermethylated the promoter and reduced the expression of miR-663 in breast cancer cell lines (Fig. 3, C and D). Using the TCGA database, we observed hypermethylation of the miR-663 promoter in breast tumors (Fig. 3D), as also reported previously (69). Expression analysis of DNA methylation modifying enzymes in the TCGA database showed that the tumors with hypermethylated miRNA-663 also demonstrate elevated expression of the DNA methylases DNMT1, DNMT3A, DNMT3B, and DNMT3L and reduced expression of the DNA demethylases ALKBH3 and FTO (supplemental Fig. S1). This supports our in vitro work suggesting a shift in the methylation machinery favoring hypermethylation. Considering that low expression of miR-663 correlated with reduced patient survival in the same tumors (Fig. 7, E and F), DNA methylation of miR-663 appears to be a significant event in breast cancer. Our previous study demonstrated that DNA methylation is a broad mechanism of mitochondrial regulation of nuclear transcription (19), and our current study defines miR-663 as the first miRNA regulated by this mechanism.
In this study, we report that miR-663 positively regulates expression of OXPHOS subunit genes and genes involved in the assembly of OXPHOS complexes. We demonstrated the direct regulation of UQCC2 by miR-663 using luciferase assays. Although an indirect correlation between an miRNA and its target genes is most common, several alternative regulatory mechanisms have been recently observed in which an miRNA can stabilize its target genes or activate their promoters thereby increasing target transcript abundance (70). Interestingly, we found that miR-663 ectopic expression stabilized the UQCC2 transcript and the anti–miR-663 destabilized it (Fig. 5A). Mutations in UQCC2 destabilize both ComplexI/III2 and ComplexIII2/IV (71). They also decrease Complexes I, III, and IV enzymatic activity. The anti–miR-663 in our study induced all of these defects in MCF7 cells, suggesting that UQCC2 alone may explain many of the mitochondrial defects that we observed after inhibiting miR-663.
We show that up-regulation of mitochondria-dependent miR-663 in breast cancer cell lines decreased Matrigel invasion; antisense inhibition of miR-663 increased cellular proliferation (Fig. 7, A and B). We demonstrate the same role for miR-663 in vivo. We show that xenografts of MCF7 cells stably transfected with the anti–miR-663 form larger tumors than xenografts of control transfected cells (Fig. 7, C and D). Conversely, miR-663 ectopic expression reduced tumor volume. In agreement, TCGA survival analyses show that breast cancer patients with stage II or M0 tumors with high miR-663 expression had increased survival compared with those with low miR-663–expressing tumors (Fig. 7, E and F). Collectively our data indicate that miR-663 down-regulation promotes breast tumorigenesis, whereas up-regulation inhibits it.
In conclusion, we established the role of miR-663 in controlling mitochondria-to-nuclear cross talk that plays a crucial role in regulating mitochondrial function (see Fig. 8 for a summary). Briefly, cells containing normal mitochondria and normal miR-663 expression show proper expression of OXPHOS subunits, assembly factors, stable OXPHOS supercomplexes, and normal OXPHOS enzyme activities. However, dysfunctional mitochondria induce oxidative stress, increase methyltransferase activity leading to a hypermethylated miR-663 promoter and reduced miR-663 expression. Inhibition of miR-663 expression reduces OXPHOS gene expression, destabilizes the supercomplexes, and reduces OXPHOS enzymatic activities which together contribute to increased tumorigenesis. Restoration of mitochondrial function reverses these changes, thereby inhibiting tumor progression. Taken together our studies highlight miR-663 as a central player in controlling mitochondria-to-nucleus retrograde communication and development of cancer.
Figure 8.

miR-663 retrograde regulation of OXPHOS and its role in tumorigenesis. A summary figure explains the regulatory mechanism underlying retrograde regulation of miR-663 and its role in controlling mitochondrial function and tumorigenesis.
Experimental procedures
Cell culture
Cell lines were purchased from American Type Culture Collection (ATCC) (Manassas, VA), except for the 143B Rho0 line which was a gift from Dr. James R. Smiley. The Rho0 cells were generated genetically by transfecting parental cells with a mitochondrially-targeted nuclease, UL12.5 (72). The cybrid line was derived from the genetically-created Rho0 line as described previously (73). MCF7 and MDA-MB-231 cells were grown in DMEM supplemented with 10% fetal bovine serum (Mediatech, Inc., Manassas, VA) and 0.1% penicillin/streptomycin (Mediatech). The generation and culture conditions of the MCF7 cells with tetracycline-inducible dominant negative POLG D1135A were described previously (74). POLG D1135A was induced by culturing the cells in 1000 ng/ml doxycycline for 10 days. All 143B-derived lines were maintained in DMEM (Mediatech) with 10% fetal bovine serum (Mediatech) and 0.1% penicillin/streptomycin supplemented with 50 μg/ml uridine (Sigma) and 100 μg/ml pyruvate (Sigma). Cells were maintained at 37 °C, 95% humidity, and 5% carbon dioxide.
Chemical treatment
5-aza-2′-deoxycytidine, rotenone, antimycin A, and oligomycin were delivered in DMSO. Malonate and potassium cyanide were delivered in water. All chemicals were purchased from Sigma.
Antibodies
Mouse monoclonal antibodies, NDUFB8 (ab110242), UQCRC2 (ab14745), COXII (ab110258), and OXPHOS mixture (ab110413) were obtained from MitoSciences and used at dilutions of 1:1000 for all.
miRNA microarray
One RNA sample from parental, Rho0, and cybrid (repleted mtDNA) cells was analyzed by deCODE Genetics (Reykjavik, Iceland) for expression of 661 miRNAs using the miRCURY LNATM array (v.9.2) (Exiqon A/S, Vedbaek, Denmark). After RNA passed quality control by the Bioanalyzer 2100 and NanoDrop, it was labeled using the miRCURY Hy3TM/Hy5TM power labeling kit and hybridized on the array. The quantified signals for the background correction were normalized using the global LOWESS (locally weighted scatterplot smoothing) regression algorithm. Three array slides were used; each one hybridized the Rho0 cDNA to the green channel (Hy3) and hybridized cDNA from either the parental or the cybrid line to the red channel (Hy5). Expression of miRNAs was compared between samples by comparing Hy3 and Hy5 signals.
Semi-quantitative RT-PCR and real-time PCR
RNA was isolated using TRI Reagent (Molecular Research Center, Cincinnati, OH) according to manufacturer's specifications. RNA was DNase treated (Promega, Madison, WI) and reverse transcribed using iScriptTM cDNA synthesis kit from Bio-Rad according to manufacturer's specifications. Semi-quantitative RT-PCR was conducted using Promega's GoTaq® Green Master Mix. Unless otherwise stated, reactions were carried out for 32 cycles. Real-time PCR was conducted with Bio-Rad IQTM SYBR Green Supermix and analyzed using the Roche LightCycler 480. Data were derived from three independent RNA samples. Semi-quantitative data are representative of two replicates.
Methylation-specific PCR, bisulfite genomic sequencing, and COBRA analysis
Genomic DNA was isolated using TRI Reagent and bisulfite converted using the EpiTect Bisulfite Kit (Qiagen, Hilden, Germany). Methylation-specific PCR and bisulfite sequencing of miR-663 were conducted as described previously (50). Sequencing was done on the Applied Biosystems 3730 DNA Analyzer and analyzed with BiQ Analyzer (http://biq-analyzer.bioinf.mpi-inf.mpg.de/)3. For COBRA analyses, 10 ng of bisulfite-converted DNA was amplified using the bisulfite sequencing primers to capture the BstUI cut site. The PCR product was then digested with BstUI for 1 h at 60 °C and run on a 1% agarose gel.
TCGA data collection
Publically available data for primary breast tumors were obtained from the TCGA publications (https://tcga-data.nci.nih.gov/docs/publications/brca_2012/)3. mRNA expression data were obtained for 522 breast tumors and normal samples. miRNA data normalized as reads per million mapped (RPM) were obtained for 780 breast tumors and normal samples. Methylation data for nine miR-663–associated CpGs were obtained for 552 breast tumors and normal samples.
Statistical analysis
Statistical analyses were conducted using Stata version 11 (Statacorp LP, College Station, TX). Analysis of variance (ANOVA) was used to compare multiple means, and multiple comparisons were corrected by the Bonferroni method. Kaplan-Meier curves were compared using the log-rank test. For all tests p < 0.05 was considered significant.
Lentivirus production and transduction
Lentivirus constructs for miRNA control and miR-663 expression were purchased from Applied Biological Materials (Richmond, BC, Canada), and the anti–miR-663 construct was purchased from System Biosciences (Palo Alto, CA). Transfection of constructs was done using FuGENE HD (Promega) according to manufacturer's specifications. Constructs were co-transfected with packaging plasmids PMD2G and PSPAX2 into 293T cells. Media were filtered and virus precipitated in 10% PEG 8000, dissolved in PBS, and added to target cells in complete media with 5 μg/ml Polybrene. Puromycin (5 μg/ml) was added to select transduced cells.
Design of CRISPRs
CRISPR primers were designed using E-CRISP (http://www.e-crisp.org/E-CRISP/designcrispr.html)3. One μl of each 100-μm primer was added to 48 μl of oligo annealing buffer (Promega), boiled 10 min, and cooled to room temperature. Three microliters of annealed primers and 1 μl of BbsI digested pSpCas9(BB)-2A-GFP (5 ng/μl) plasmid were ligated using the LigaFastTM rapid ligation system (Promega). CRISPRs were transfected into MCF7 cells. Positive cells were FACS sorted on GFP and plated to grow in single colonies. Colonies were selected and grown for screening by Western blotting.
Luciferase assays
Primers were designed to incorporate the entire 3′-UTR of the gene of interest, and restriction sites for NheI (5′) and SbfI (3′) were added. 3′-UTRs were amplified from MCF7 DNA, digested, and cloned into the pmirGLO Dual-Luciferase Vector (Promega). Clones were screened by restriction digest and confirmed by sequencing. Vectors were transfected into MCF7 cells in 96-well plates (100 ng/well) in the absence or presence of a miR-663 mimic (100 nm). Luminescence was analyzed in triplicate on a BioTek Synergy H1 Multi-Mode Reader using the Dual-Glo Luciferase Assay System (Promega). Firefly activity was normalized to Renilla activity.
Blue-Native polyacrylamide gel electrophoresis
Mitochondrial isolation was carried out as described previously (75). BN-PAGE was performed on isolated mitochondria to analyze endogenous protein complexes as described previously with minor modifications (30, 76).
Heteroduplex mobility assay
Primers were designed to amplify a 150–300–bp region incorporating the CRISPR guide RNA binding site of the target gene. After amplification, HMA assay was carried out as described previously (77).
MTT assay
In a 96-well plate, 5000 MCF7 or 2000 MDA-MB-231 breast cancer cells were plated per well in 100 μl of growth media. Each cell line was plated in quadruplicate, and iterations of each line were plated for daily analysis. For 4 days, cell viability was quantified as described previously (78).
Reactive oxygen species quantification
After treatment, cells were resuspended in 10 μm CM-H2DCFDA and incubated in the dark at 37 °C for 30 min and washed twice with PBS. Fluorescence of DCF-DA was measured by FACS using a BD LSR II Analyzer. Three replicates of each cell line were analyzed.
Mouse xenografts
Xenograft experiments were carried out as described previously (78). Female athymic mice (three mice per group; two tumors per mouse) were implanted with 17β-estradiol pellets 2 days before tumor cell injection. A suspension of 1 × 108 tumor cells per milliliter was made in a 1:1 mixture of Matrigel and PBS, and 0.1 ml (1 × 107 cells) was injected subcutaneously. Tumor growth was monitored externally using calipers for 50 days.
OXPHOS enzymatic assays
Mitochondria were isolated as described above and resuspended at 1 μg/ml protein. Enzymatic activities of OXPHOS complexes were measured as described previously (79).
Matrigel
A cellular invasion was measured using Corning BioCoat Matrigel Invasion Chambers (Bedford, MA) according to the manufacturer's instructions. Invading cells were stained using the Kwik-Diff Kit (Thermo Scientific) according to the manufacturer's protocol. Experiments were done in triplicate.
DNA methyltransferase activity assay
Whole-cell DNA methyltransferase activity was assessed by DNA Methyltransferase Activity Kit (EpiGentek, Farmingdale, NY). The manufacturer's protocol was used to analyze 20 μg of whole cell lysate. Analyses were done in triplicate.
Author contributions
T. C. collected and analyzed data and drafted the manuscript. B. S. assisted in conducting xenografts studies. V. M. assisted in conducting Blue Native gels. P. B. assisted in analyzing OXPHOS activity. K. K. S. conceived, planned, and directed the research project and assisted in writing and editing the manuscript.
Supplementary Material
Acknowledgments
We thank Mariola Kulawiec for her early input in this project.
This study was supported by U.S. Department of Veterans Affairs Grant 1I0IBX001716 and National Institutes of Health Grant R01 CA204430. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or reflect the position or policy of the Department of Veterans Affairs or the United States government.
This article contains supplemental Table S1 and supplemental Figs. S1–S4.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- miRNAs
- microRNAs
- UTR
- untranslated region
- ROS
- reactive oxygen species
- mtDNA
- mitochondrial DNA
- OXPHOS
- oxidative phosphorylation
- MSP
- methylation-specific PCR
- CM-H2DCFDA
- 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester
- TCGA
- The Cancer Genome Atlas
- BN-PAGE
- Blue Native-polyacrylamide gel electrophoresis
- CRISPR
- clustered regularly interspaced short palindromic repeats
- HMA
- heteroduplex mobility assay
- COBRA
- combined bisulfite restriction analysis.
References
- 1. Quirós P. M., Mottis A., and Auwerx J. (2016) Mitonuclear communication in homeostasis and stress. Nature reviews. Molecular cell biology 17, 213–226 [DOI] [PubMed] [Google Scholar]
- 2. Kulawiec M., Safina A., Desouki M. M., Still I., Matsui S., Bakin A., and Singh K. K. (2008) Tumorigenic transformation of human breast epithelial cells induced by mitochondrial DNA depletion. Cancer Biol. Ther. 7, 1732–1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chowdhury A. R., Long A., Fuchs S. Y., Rustgi A., and Avadhani N. G. (2017) Mitochondrial stress-induced p53 attenuates HIF-1α activity by physical association and enhanced ubiquitination. Oncogene 36, 397–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kulawiec M., Arnouk H., Desouki M. M., Kazim L., Still I., and Singh K. K. (2006) Proteomic analysis of mitochondria-to-nucleus retrograde response in human cancer. Cancer Biol. Ther. 5, 967–975 [DOI] [PubMed] [Google Scholar]
- 5. Delsite R., Kachhap S., Anbazhagan R., Gabrielson E., and Singh K. K. (2002) Nuclear genes involved in mitochondria-to-nucleus communication in breast cancer cells. Mol. Cancer 1, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kulawiec M., Owens K. M., and Singh K. K. (2009) Cancer cell mitochondria confer apoptosis resistance and promote metastasis. Cancer Biol. Ther. 8, 1378–1385 [DOI] [PubMed] [Google Scholar]
- 7. Singh K. K. (2004) Mitochondrial dysfunction is a common phenotype in aging and cancer. Ann. N.Y. Acad. Sci. 1019, 260–264 [DOI] [PubMed] [Google Scholar]
- 8. Singh K. K., Kulawiec M., Still I., Desouki M. M., Geradts J., and Matsui S. (2005) Inter-genomic cross talk between mitochondria and the nucleus plays an important role in tumorigenesis. Gene 354, 140–146 [DOI] [PubMed] [Google Scholar]
- 9. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: The next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 10. Kaipparettu B. A., Ma Y., Park J. H., Lee T. L., Zhang Y., Yotnda P., Creighton C. J., Chan W. Y., and Wong L. J. (2013) Crosstalk from non-cancerous mitochondria can inhibit tumor properties of metastatic cells by suppressing oncogenic pathways. PLoS One 8, e61747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cruz-Bermúdez A., Vallejo C. G., Vicente-Blanco R. J., Gallardo M. E., Fernández-Moreno M. Á., Quintanilla M., and Garesse R. (2015) Enhanced tumorigenicity by mitochondrial DNA mild mutations. Oncotarget 6, 13628–13643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lin S., and Gregory R. I. (2015) MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 15, 321–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kinoshita T., Yip K. W., Spence T., and Liu F. F. (2017) MicroRNAs in extracellular vesicles: Potential cancer biomarkers. J. Hum. Genet. 62, 67–74 [DOI] [PubMed] [Google Scholar]
- 14. Hayes J., Peruzzi P. P., and Lawler S. (2014) MicroRNAs in cancer: biomarkers, functions, and therapy. Trends Mol. Med. 20, 460–469 [DOI] [PubMed] [Google Scholar]
- 15. Das S., Bedja D., Campbell N., Dunkerly B., Chenna V., Maitra A., and Steenbergen C. (2014) miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS One 9, e96820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang X., Zuo X., Yang B., Li Z., Xue Y., Zhou Y., Huang J., Zhao X., Zhou J., Yan Y., Zhang H., Guo P., Sun H., Guo L., Zhang Y., and Fu X. D. (2014) MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell 158, 607–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bucha S., Mukhopadhyay D., and Bhattacharyya N. P. (2015) Regulation of mitochondrial morphology and cell cycle by microRNA-214 targeting Mitofusin2. Biochem. Biophys. Res. Commun. 465, 797–802 [DOI] [PubMed] [Google Scholar]
- 18. Chaudhuri A. D., Choi D. C., Kabaria S., Tran A., and Junn E. (2016) MicroRNA-7 regulates the function of mitochondrial permeability transition pore by targeting VDAC1 expression. J. Biol. Chem. 291, 6483–6493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smiraglia D. J., Kulawiec M., Bistulfi G. L., Gupta S. G., and Singh K. K. (2008) A novel role for mitochondria in regulating epigenetic modification in the nucleus. Cancer Biol. Ther. 7, 1182–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Desouki M. M., Kulawiec M., Bansal S., Das G. M., and Singh K. K. (2005) Cross talk between mitochondria and superoxide generating NADPH oxidase in breast and ovarian tumors. Cancer Biol. Ther. 4, 1367–1373 [DOI] [PubMed] [Google Scholar]
- 21. Indo H. P., Davidson M., Yen H. C., Suenaga S., Tomita K., Nishii T., Higuchi M., Koga Y., Ozawa T., and Majima H. J. (2007) Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 7, 106–118 [DOI] [PubMed] [Google Scholar]
- 22. Sullivan L. B., and Chandel N. S. (2014) Mitochondrial reactive oxygen species, and cancer. Cancer Metab. 2, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hong Q., Yu S., Geng X., Duan L., Zheng W., Fan M., Chen X., and Wu D. (2015) High concentrations of uric acid inhibit endothelial cell migration via miR-663 which regulates phosphatase and tensin homolog by targeting transforming growth factor-β1. Microcirculation 22, 306–314 [DOI] [PubMed] [Google Scholar]
- 24. Hu W., Xu S., Yao B., Hong M., Wu X., Pei H., Chang L., Ding N., Gao X., Ye C., Wang J., Hei T. K., and Zhou G. (2014) MiR-663 inhibits radiation-induced bystander effects by targeting TGFB1 in a feedback mode. RNA Biol. 11, 1189–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Z., Zhang H., Zhang P., Dong W., and He L. (2016) MicroRNA-663 suppresses cell invasion and migration by targeting transforming growth factor β1 in papillary thyroid carcinoma. Tumour Biol. 37, 7633–7644 [DOI] [PubMed] [Google Scholar]
- 26. Li Q., Cheng Q., Chen Z., Peng R., Chen R., Ma Z., Wan X., Liu J., Meng M., Peng Z., and Jiang B. (2016) MicroRNA-663 inhibits the proliferation, migration and invasion of glioblastoma cells via targeting TGF-β1. Oncol. Rep. 35, 1125–1134 [DOI] [PubMed] [Google Scholar]
- 27. Tili E., Michaille J. J., Adair B., Alder H., Limagne E., Taccioli C., Ferracin M., Delmas D., Latruffe N., and Croce C. M. (2010) Resveratrol decreases the levels of miR-155 by up-regulating miR-663, a microRNA targeting JunB and JunD. Carcinogenesis 31, 1561–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li P., Zhu N., Yi B., Wang N., Chen M., You X., Zhao X., Solomides C. C., Qin Y., and Sun J. (2013) MicroRNA-663 regulates human vascular smooth muscle cell phenotypic switch and vascular neointimal formation. Circ. Res. 113, 1117–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ghezzi D., and Zeviani M. (2012) Assembly factors of human mitochondrial respiratory chain complexes: Physiology and pathophysiology. Adv. Exp. Med. Biol. 748, 65–106 [DOI] [PubMed] [Google Scholar]
- 30. Stroud D. A., Surgenor E. E., Formosa L. E., Reljic B., Frazier A. E., Dibley M. G., Osellame L. D., Stait T., Beilharz T. H., Thorburn D. R., Salim A., and Ryan M. T. (2016) Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 538, 123–126 [DOI] [PubMed] [Google Scholar]
- 31. Lenaz G., and Genova M. L. (2012) Supramolecular organisation of the mitochondrial respiratory chain: A new challenge for the mechanism and control of oxidative phosphorylation. Adv. Exp. Med. Biol. 748, 107–144 [DOI] [PubMed] [Google Scholar]
- 32. Vartak R., Porras C. A., and Bai Y. (2013) Respiratory supercomplexes: Structure, function and assembly. Protein Cell 4, 582–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mimaki M., Wang X., McKenzie M., Thorburn D. R., and Ryan M. T. (2012) Understanding mitochondrial complex I assembly in health and disease. Biochim. Biophys. Acta 1817, 851–862 [DOI] [PubMed] [Google Scholar]
- 34. Genova M. L., and Lenaz G. (2014) Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta 1837, 427–443 [DOI] [PubMed] [Google Scholar]
- 35. Modica-Napolitano J. S., Kulawiec M., and Singh K. K. (2007) Mitochondria and human cancer. Curr. Mol. Med. 7, 121–131 [DOI] [PubMed] [Google Scholar]
- 36. Compton S., Kim C., Griner N. B., Potluri P., Scheffler I. E., Sen S., Jerry D. J., Schneider S., and Yadava N. (2011) Mitochondrial dysfunction impairs tumor suppressor p53 expression/function. J. Biol. Chem. 286, 20297–20312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xiong S., Mu T., Wang G., and Jiang X. (2014) Mitochondria-mediated apoptosis in mammals. Protein Cell 5, 737–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shapovalov Y., Hoffman D., Zuch D., de Mesy Bentley K. L., and Eliseev R. A. (2011) Mitochondrial dysfunction in cancer cells due to aberrant mitochondrial replication. J. Biol. Chem. 286, 22331–22338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Das S., Ferlito M., Kent O. A., Fox-Talbot K., Wang R., Liu D., Raghavachari N., Yang Y., Wheelan S. J., Murphy E., and Steenbergen C. (2012) Nuclear miRNA regulates the mitochondrial genome in the heart. Circ. Res. 110, 1596–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li J., Donath S., Li Y., Qin D., Prabhakar B. S., and Li P. (2010) miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 6, e1000795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kren B. T., Wong P. Y., Sarver A., Zhang X., Zeng Y., and Steer C. J. (2009) MicroRNAs identified in highly purified liver-derived mitochondria may play a role in apoptosis. RNA Biol. 6, 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bandiera S., Rüberg S., Girard M., Cagnard N., Hanein S., Chrétien D., Munnich A., Lyonnet S., and Henrion-Caude A. (2011) Nuclear outsourcing of RNA interference components to human mitochondria. PLoS One 6, e20746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barrey E., Saint-Auret G., Bonnamy B., Damas D., Boyer O., and Gidrol X. (2011) Pre-microRNA and mature microRNA in human mitochondria. PLoS One 6, e20220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shi Y., Chen C., Zhang X., Liu Q., Xu J. L., Zhang H. R., Yao X. H., Jiang T., He Z. C., Ren Y., Cui W., Xu C., Liu L., Cui Y. H., Yu S. Z., Ping Y. F., and Bian X. W. (2014) Primate-specific miR-663 functions as a tumor suppressor by targeting PIK3CD and predicts the prognosis of human glioblastoma. Clin. Cancer Res. 20, 1803–1813 [DOI] [PubMed] [Google Scholar]
- 45. Shi Y., Chen C., Yu S. Z., Liu Q., Rao J., Zhang H. R., Xiao H. L., Fu T. W., Long H., He Z. C., Zhou K., Yao X. H., Cui Y. H., Zhang X., Ping Y. F., and Bian X. W. (2015) miR-663 suppresses oncogenic function of CXCR4 in glioblastoma. Clin. Cancer Res. 21, 4004–4013 [DOI] [PubMed] [Google Scholar]
- 46. Zang W., Wang Y., Wang T., Du Y., Chen X., Li M., and Zhao G. (2015) miR-663 attenuates tumor growth and invasiveness by targeting eEF1A2 in pancreatic cancer. Mol. Cancer 14, 37. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47. Yi C., Wang Q., Wang L., Huang Y., Li L., Liu L., Zhou X., Xie G., Kang T., Wang H., Zeng M., Ma J., Zeng Y., and Yun J. P. (2012) MiR-663, a microRNA targeting p21WAF1/CIP1, promotes the proliferation and tumorigenesis of nasopharyngeal carcinoma. Oncogene 31, 4421–4433 [DOI] [PubMed] [Google Scholar]
- 48. Liu Z. Y., Zhang G. L., Wang M. M., Xiong Y. N., and Cui H. Q. (2011) MicroRNA-663 targets TGFB1 and regulates lung cancer proliferation. Asian Pac. J. Cancer Prev. 12, 2819–2823 [PubMed] [Google Scholar]
- 49. Jiao L., Deng Z., Xu C., Yu Y., Li Y., Yang C., Chen J., Liu Z., Huang G., Li L. C., and Sun Y. (2014) miR-663 induces castration-resistant prostate cancer transformation and predicts clinical recurrence. J. Cell. Physiol. 229, 834–844 [DOI] [PubMed] [Google Scholar]
- 50. Yan-Fang T., Jian N., Jun L., Na W., Pei-Fang X., Wen-Li Z., Dong W., Li P., Jian W., Xing F., and Jian P. (2013) The promoter of miR-663 is hypermethylated in Chinese pediatric acute myeloid leukemia (AML). BMC Med. Genet. 14, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hu H., Li S., Cui X., Lv X., Jiao Y., Yu F., Yao H., Song E., Chen Y., Wang M., and Lin L. (2013) The overexpression of hypomethylated miR-663 induces chemotherapy resistance in human breast cancer cells by targeting heparin sulfate proteoglycan 2 (HSPG2). J. Biol. Chem. 288, 10973–10985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang Y., Wang L. L., Wang H. X., Guo Z. K., Gao X. F., Cen J., Li Y. H., Dou L. P., and Yu L. (2013) The epigenetically regulated miR-663 targets H-ras in K-562 cells. FEBS J. 280, 5109–5117 [DOI] [PubMed] [Google Scholar]
- 53. Beukers W., Hercegovac A., and Zwarthoff E. C. (2014) HRAS mutations in bladder cancer at an early age and the possible association with the Costello Syndrome. Eur. J. Hum. Genet. 22, 837–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hoenerhoff M. J., Chu I., Barkan D., Liu Z. Y., Datta S., Dimri G. P., and Green J. E. (2009) BMI1 cooperates with H-RAS to induce an aggressive breast cancer phenotype with brain metastases. Oncogene 28, 3022–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Elenbaas B., Spirio L., Koerner F., Fleming M. D., Zimonjic D. B., Donaher J. L., Popescu N. C., Hahn W. C., and Weinberg R. A. (2001) Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 15, 50–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tzenaki N., and Papakonstanti E. A. (2013) p110δ PI3 kinase pathway: emerging roles in cancer. Front. Oncol. 3, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dong T., Liu Z., Zhao S., Hu C., Liu Y., Ma W., and Zhang Q. (2015) The expression of CD9 and PIK3CD is associated with prognosis of follicular lymphoma. J. Cancer 6, 1222–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sawyer C., Sturge J., Bennett D. C., O'Hare M. J., Allen W. E., Bain J., Jones G. E., and Vanhaesebroeck B. (2003) Regulation of breast cancer cell chemotaxis by the phosphoinositide 3-kinase p110δ. Cancer Res. 63, 1667–1675 [PubMed] [Google Scholar]
- 59. Kim J. S., Park M. G., Lee S. A., Park S. Y., Kim H. J., Yu S. K., Kim C. S., Kim S. G., Oh J. S., You J. S., Kim J. S., Seo Y. S., Chun H. S., Park J. C., and Kim D. K. (2014) Downregulation of adenomatous polyposis coli by microRNA-663 promotes odontogenic differentiation through activation of Wnt/beta-catenin signaling. Biochem. Biophys. Res. Commun. 446, 894–900 [DOI] [PubMed] [Google Scholar]
- 60. Miao C. G., Shi W. J., Xiong Y. Y., Yu H., Zhang X. L., Qin M. S., Du C. L., Song T. W., Zhang B., and Li J. (2015) MicroRNA-663 activates the canonical Wnt signaling through the adenomatous polyposis coli suppression. Immunol. Lett. 166, 45–54 [DOI] [PubMed] [Google Scholar]
- 61. Mukherjee N., Bhattacharya N., Alam N., Roy A., Roychoudhury S., and Panda C. K. (2012) Subtype-specific alterations of the Wnt signaling pathway in breast cancer: Clinical and prognostic significance. Cancer Sci. 103, 210–220 [DOI] [PubMed] [Google Scholar]
- 62. Prasad C. P., Mirza S., Sharma G., Prashad R., DattaGupta S., Rath G., and Ralhan R. (2008) Epigenetic alterations of CDH1 and APC genes: Relationship with activation of Wnt/beta-catenin pathway in invasive ductal carcinoma of breast. Life Sci. 83, 318–325 [DOI] [PubMed] [Google Scholar]
- 63. Huang Y., Liu J., Fan L., Wang F., Yu H., Wei W., and Sun G. (2016) miR-663 overexpression induced by endoplasmic reticulum stress modulates hepatocellular carcinoma cell apoptosis via transforming growth factor beta 1. Onco Targets Ther. 9, 1623–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Waaijer M. E., Wieser M., Grillari-Voglauer R., van Heemst D., Grillari J., and Maier A. B. (2014) MicroRNA-663 induction upon oxidative stress in cultured human fibroblasts depends on the chronological age of the donor. Biogerontology 15, 269–278 [DOI] [PubMed] [Google Scholar]
- 65. Vislovukh A., Kratassiouk G., Porto E., Gralievska N., Beldiman C., Pinna G., El'skaya A., Harel-Bellan A., Negrutskii B., and Groisman I. (2013) Proto-oncogenic isoform A2 of eukaryotic translation elongation factor eEF1 is a target of miR-663 and miR-744. Br. J. Cancer 108, 2304–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Minocherhomji S., Tollefsbol T. O., and Singh K. K. (2012) Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics 7, 326–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Singh K. K. (2015) MIPIGENETICS and MIPIGENOMICS: Integrating mitochondria-induced mayhem contributing to mystondria. Mitochondrion 24, S6 [Google Scholar]
- 68. Xie C. H., Naito A., Mizumachi T., Evans T. T., Douglas M. G., Cooney C. A., Fan C. Y., and Higuchi M. (2007) Mitochondrial regulation of cancer-associated nuclear DNA methylation. Biochem. Biophys. Res. Commun. 364, 656–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lehmann U., Hasemeier B., Christgen M., Müller M., Römermann D., Länger F., and Kreipe H. (2008) Epigenetic inactivation of microRNA gene hsa-mir-9–1 in human breast cancer. J. Pathol. 214, 17–24 [DOI] [PubMed] [Google Scholar]
- 70. Lee S., and Vasudevan S. (2013) Post-transcriptional stimulation of gene expression by microRNAs. Adv. Exp. Med. Biol. 768, 97–126 [DOI] [PubMed] [Google Scholar]
- 71. Tucker E. J., Wanschers B. F., Szklarczyk R., Mountford H. S., Wijeyeratne X. W., van den Brand M. A., Leenders A. M., Rodenburg R. J., Reljić B., Compton A. G., Frazier A. E., Bruno D. L., Christodoulou J., Endo H., Ryan M. T., Nijtmans L. G., Huynen M. A., and Thorburn D. R. (2013) Mutations in the UQCC1-interacting protein, UQCC2, cause human complex III deficiency associated with perturbed cytochrome b protein expression. PLoS Genet. 9, e1004034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Saffran H. A., Pare J. M., Corcoran J. A., Weller S. K., and Smiley J. R. (2007) Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep. 8, 188–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kulawiec M., Owens K. M., and Singh K. K. (2009) mtDNA G10398A variant in African-American women with breast cancer provides resistance to apoptosis and promotes metastasis in mice. J. Hum. Genet. 54, 647–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Singh K. K., Ayyasamy V., Owens K. M., Koul M. S., and Vujcic M. (2009) Mutations in mitochondrial DNA polymerase-gamma promote breast tumorigenesis. J. Hum. Genet. 54, 516–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Johnston A. J., Hoogenraad J., Dougan D. A., Truscott K. N., Yano M., Mori M., Hoogenraad N. J., and Ryan M. T. (2002) Insertion and assembly of human Tom7 into the preprotein translocase complex of the outer mitochondrial membrane. J. Biol. Chem. 277, 42197–42204 [DOI] [PubMed] [Google Scholar]
- 76. Schägger H. (1995) Native electrophoresis for isolation of mitochondrial oxidative phosphorylation protein complexes. Methods Enzymol. 260, 190–202 [DOI] [PubMed] [Google Scholar]
- 77. Challa A. K., Boitet E. R., Turner A. N., Johnson L. W., Kennedy D., Downs E. R., Hymel K. M., Gross A. K., and Kesterson R. A. (2016) Novel hypomorphic alleles of the mouse tyrosinase gene induced by CRISPR-Cas9 nucleases cause non-albino pigmentation phenotypes. PLoS One 11, e0155812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ayyasamy V., Owens K. M., Desouki M. M., Liang P., Bakin A., Thangaraj K., Buchsbaum D. J., LoBuglio A. F., and Singh K. K. (2011) Cellular model of Warburg effect identifies tumor-promoting function of UCP2 in breast cancer and its suppression by genipin. PLoS One 6, e24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Owens K. M., Kulawiec M., Desouki M. M., Vanniarajan A., and Singh K. K. (2011) Impaired OXPHOS Complex III in breast cancer. PLoS One 6, e23846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.