

Abstract
The ligand-binding βI and αI domains of integrin are the best-studied von Willebrand factor A domains undergoing significant conformational changes for affinity regulation. In both βI and αI domains, the α1- and α7-helixes work in concert to shift the metal-ion–dependent adhesion site between the resting and active states. An absolutely conserved Gly in the middle of the α1-helix of βI helps maintain the resting βI conformation, whereas the homologous position in the αI α1-helix contains a conserved Phe. A functional role of this Phe is structurally unpredictable. Using αLβ2 integrin as a model, we found that the residue volume at the Phe position in the α1-helix is critical for αLβ2 activation because trimming the Phe by small amino acid substitutions abolished αLβ2 binding with soluble and immobilized intercellular cell adhesion molecule 1. Similar results were obtained for αMβ2 integrin. Our experimental and molecular dynamics simulation data suggested that the bulky Phe acts as a pawl that stabilizes the downward ratchet-like movement of β6-α7 loop and α7-helix, required for high-affinity ligand binding. This mechanism may apply to other von Willebrand factor A domains undergoing large conformational changes. We further demonstrated that the conformational cross-talk between αL αI and β2 βI could be uncoupled because the β2 extension and headpiece opening could occur independently of the αI activation. Reciprocally, the αI activation does not inevitably lead to the conformational changes of the β2 subunit. Such loose linkage between the αI and βI is attributed to the αI flexibility and could accommodate the αLβ2-mediated rolling adhesion of leukocytes.
Keywords: cell adhesion, cell migration, integrin, leukocyte, von Willebrand factor, VWA domain
Introduction
Integrins are cell adhesion molecules that transmit both the mechanical and chemical signals into and out of the cells and thus allow the cells to communicate with their surroundings (1–4). The different combinations of 18 α and 8 β subunits lead to 24 integrin α/β heterodimers that play important roles in diverse physiological and pathological conditions such as hemostasis, development, immune responses, thrombosis, inflammation, and cancer (1). All the β integrin subunits consist of 10 subdomains, ranking from the N terminus to the C terminus are PSI (plexin, semaphorin, and integrin), hybrid, I (inserted), integrin epidermal growth factor (I-EGF)2 1–4, β-tail, transmembrane, and cytoplasmic tail domains (Fig. 1, A–F). All the α integrin subunits are composed of seven subdomains, namely β-propeller, thigh, genu, calf-1, calf-2, transmembrane, and cytoplasmic tail (Fig. 1, A–F). In addition, half of the α subunits including the leukocyte-specific integrins αL and αM contain an extra αI domain that is inserted into the β-propeller domain (Fig. 1, D–F). For integrins without the αI domain, the β-propeller and the βI domain form the ligand-binding site at the top of their interface (Fig. 1, A–C), whereas the αI domain is solely responsible for binding ligands when it is present in an α subunit (Fig. 1, D–F). It has been demonstrated that the conserved Glu residue at the αI C terminus acts as an internal ligand for the βI domain (Fig. 1F) (5). EM, crystallography, small-angle X-ray scattering, and mutagenesis studies have revealed at least three conformational states of integrin: bent (Fig. 1, A and D), extended with closed headpiece (Fig. 1, B and E), and extended with open headpiece (Fig. 1, C and F), which have been proposed to represent the inactive, intermediate, and active integrin conformations, respectively (5). Such a bent-to-extended global conformational transition can be triggered from the ligand-binding head region and transmit in the outside-in direction or propagated from the cytoplasmic tail and transmit in the inside-out direction. How the wave of conformational change is relayed mechanically across the tandem multiple domains of integrin has been an active area of research in the last decades (6–10).
Figure 1.

Integrin domain organization and the allosteric signal relay in the global and local conformational changes. A–C, the three major conformational states of integrins without the αI domain. D–F, the three major conformational states of integrins with an αI domain inserted into the β-propeller domain of α subunit. The local conformational changes of the α1-helix, α7-helix, and the β subunit hybrid domain were indicated by arrows. Potential intermediate conformations of β subunit are shown as dashed lines. G, sequence alignment of the α1-helixes of the αI domains. The α1/α1′-helix sequence of the β3 integrin βI domain was shown for comparison. H, structure comparison of the β3 βI domain in the closed (PDB code 3T3P) and open conformations (PDB code 2VDR). I, structure comparison of the αM αI domain in the closed (PDB code 1JLM) and open conformations (PDB code 1IDO). The closed and open conformations of α1/α1′-helix and α7-helix are shown in green and blue, respectively. Metal ions of the MIDAS and ADMIDAS are shown as spheres. β3-Gly-135 is shown as a Cα sphere. The αM-Phe-156, equivalent to αL-Phe-153, is shown as sticks. J, structure-based sequence alignment of the α1-helix from the selected VWA domains. The DXSXS motifs of the MIDAS are boxed. Residues that are equivalent to αL-Phe-153 are shown in red.
Integrin global conformational rearrangements are triggered by the local structural changes (11), which have been very well-defined for the αI and βI domains by the crystal structures (12–17). The αI and βI domains (also known as A domains) are structural homologs that belong to the large von Willebrand factor A (VWA) domain superfamily, which is widespread in the eukaryotes, bacteria, and viruses and perhaps beyond them (18). Both αI and βI domains contain a metal ion-dependent adhesion site (MIDAS) defined by a signature Asp-Xaa-Ser-Xaa-Ser (DXSXS) motif (Fig. 1G), which is connected to the N terminus of an α1-helix and also is seen in many other VWA domains (Fig. 1J). In addition, the βI domains have two additional metal ion-binding sites flanking the MIDAS, namely adjacent to MIDAS (ADMIDAS) and synergetic metal ion-binding sites (Fig. 1H). Both αI and βI domains can shift between two conformations defined as the closed inactive and the open active states. In the active state, the β1-α1 loop moves toward the MIDAS, increasing its potential for binding ligands (Fig. 1, H and I). For βI domain, this is accompanied by a bent-to-straight structural transition, resulting in an inward movement of the α1/α1′-helix (Fig. 1H), whereas for αI domain, it is associated with only a swung-in motion of the N terminus of α1-helix (Fig. 1I). For both βI and αI domains, such conformational changes of α1/(α1′)-helix are linked with a downward displacement of β6-α7 loop and α7-helix, leading to hybrid domain swinging out (Fig. 1, C, F, and H) or the engagement of αI and βI domain via an internal ligand at the C terminus of α7-helix (Fig. 1, F and I). In our recent studies, we defined the role of an absolutely conserved Gly residue at the α1/α1′-helix (Fig. 1H), which is critical in maintaining the inactive conformation of βI domains of β1, β2, and β3 integrins (19). Interestingly, a conserved Phe is present at a homologous position of the αI α1-helix among all the αI-containing integrins (Fig. 1, G and I). The equivalent Phe is also seen in the α1-helix of many VWA domains other than integrin (Fig. 1J). We speculated that such a conserved Phe residue could be important for the activation of αI domains.
In this study, we tested our above-mentioned hypothesis on αLβ2 integrin, given that the activation assay of αLβ2 has been very well-established and the high-resolution crystal structures of the αL αI domain are available in both closed and open states (15, 19–21). The αLβ2 integrin binds ligand intercellular adhesion molecule 1 (ICAM-1) and plays important roles in leukocyte adhesion and migration during the immune responses (5). To investigate a potential structural role of the conserved Phe of the αI α1-helix in regulating the activity of αLβ2 integrin, we performed the soluble ICAM-1 binding, cell adhesion, and the conformation-specific mAb-binding assays. Our results revealed an unexpected function of the Phe residue in αLβ2 activation, which may be generalized to all the integrin αI domains or other related VWA domains that couple large-scale conformational changes to activation and ligand binding.
Results
A bulky residue in the mid position of α1-helix of αL αI domain is critical for ligand binding
A hallmark structural change of αI domain in the transition from the low to the high-affinity state is the inward movement of the N terminus of α1-helix and the piston-like movement of the entire α7-helix (Fig. 1, E, F, and I). Structural superimposition of the active conformation onto the resting conformation of the αL αI domain shows steric clashes between the N-terminal interfacial residues of α1-helix and α7-helix (Fig. 2A), indicating that these helixes should mechanically work in concert to avoid the collisions. Interestingly, the invariant phenylalanine residue αL-Phe-153 at the middle of α1-helix in the active position has no van der Waals overlaps with the α7-helix in the resting position (Fig. 2A). To test whether the conserved αL-Phe-153 is important for the activation of αL integrin, we mutated it to Gly, Ala, Val, Leu, Met, Tyr, and Trp, respectively, ranging from the slimmest Gly to the bulkiest Trp (Fig. 2B). The activation of αLβ2 integrin was assessed by the ICAM-1-binding assay in the presence of the universal integrin activator Mn2+. Remarkably, the αL-F153G and αL-F153A mutations almost completely abrogated Mn2+-induced ICAM-1 binding to HEK293FT cells expressing αLβ2 integrin (Fig. 2C). Remarkably, the level of ICAM-1 binding correlated well with the side chain volumes of the substituted amino acids for αL-Phe-153, i.e. Gly = Ala < Val < Met < Leu = Phe = Tyr < Trp (Fig. 2C). As a comparison, the αL-Glu-146 and αL-Lys-149 were mutated to Ala. These residues show steric clashes with the α7-helix Phe-292 when overlapping their high-affinity conformation with the low-affinity conformation of α7-helix (Fig. 2A). As expected, both αL-E146A and αL-K149A mutations greatly dampened Mn2+-induced ICAM-1 binding, but to a lesser extent compared with the αL-F153A mutation (Fig. 2D). Interestingly, the αL-F147A and αL-L151A mutations, located at the opposite side of the α1- and α7-helix interface (Fig. 2A), also decreased ICAM-1 binding (Fig. 2D). By contrast, the αL-K160A mutation at the bottom of α1-helix had no effect on Mn2+-induced ICAM-1 binding of αLβ2 integrin (Fig. 2, A and D). Most of the mutations had little effect on the αLβ2 cell surface expression except the Leu substitution decreased the expression by ∼50% (Fig. 2, C and D). In addition, the αL-F153I mutation completely abolished αLβ2 cell surface expression probably because the position of αL-Phe-153 cannot accommodate an Ile residue because all the Ile rotamers show severe steric clashes with the surrounding residues when mutated in silico (data not shown). These data demonstrated that a bulky amino acid in the mid α1-helix is critical for the high-affinity ligand binding of αI domain. In addition, the N-terminal residues of α1-helix also contribute to the high-affinity conformation of α1-helix.
Figure 2.

Effect of mutations in the α1-helix of αI domain on αLβ2 ligand binding. A, conformations of the α1-helix and α7-helix of αL αI domain. The crystal structures of the αL αI domain in the closed (PDB code 1ZOP) and the open (PDB code 1MQA) conformations are superimposed, and the conformations of α1-helix in the open state (in green) and α7-helix in the closed state (in cyan) are shown. The residues at the interface of α1-helix and α7-helix are shown as sticks with dotted surfaces. The van der Waals overlaps (steric clashes) between the interfacial residues are indicated as red disks. B, structural comparison of the amino acids used for the substitution of αL-Phe-153. The αL-Phe-153 was mutated in silico using PyMOL. The rotamers that have minimal or no steric clashes with their surroundings were selected. The volumes (Å3) of amino acid side chains occupying in protein interiors are shown in parentheses (66). C and D, ICAM-1 binding of HEK293FT cells transfected with β2 WT and αL containing indicated α1-helix mutations. The ICAM-1 binding was measured by flow cytometry in the presence of 1 mm Ca2+/Mg2+ (Ca/Mg) or 0.2 mm Ca2+ plus 2 mm Mn2+ (Ca/Mn) and presented as the MFI normalized to integrin expression. The data are means ± S.D. (n ≥ 3). Two-tailed t tests were used to compare the wild type and the mutants in the same conditions. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, p > 0.05. Only the mean values are shown for the integrin expression.
Mutations that lock the high-affinity conformation of α7-helix of αI domain counteract the inactivating effect of the α1-helix Phe-153 mutation
It has been shown that the downward movement of the α7-helix is important for the activation of αI domain (15, 22, 23). We speculated that the inactivating effect of the αL-F153G or F153A mutation could be due to the destabilization of the downward movement of α7-helix. In line with this possibility, enforcing the downward movement of α7-helix, for example by mutations, may rescue the inactivating effect of the αL-F153 mutations. This hypothesis was tested using four activating mutations of αLβ2 integrin. The β2-G128A/G129T mutation (Fig. 3A) renders αLβ2 activation by facilitating the active conformation of βI domain α1-helix (19). The αL-F265S mutation induces the downward movement of αI α7-helix by facilitating the movement of the β5-α6 loop (Fig. 3, A and B) (24), whereas the αL-K287C/K294C mutation directly stabilizes the downward movement of the β6-α7 loop and α7-helix of αI domain by forming a disulfide bond (Fig. 3, A and C) (15). The αL-GFFKR/GAAKR (FFAA) mutation renders αLβ2 constitutively active by disrupting the α-β cytoplasmic association, which mimics the inside-out activation of integrin (Fig. 3A) (25). As shown in Fig. 3D, the presence of the β2-G128A/G129T mutation did not rescue the inactivating effect of αL-F153G and αL-F153A. However, compared with the β2 wild type (Fig. 2C), the requirement of residue size at the position of αL-153 for Mn2+-induced ICAM-1 binding was weakened by the β2-G128A/G129T mutation (Fig. 3D). With the β2-G128A/G129T mutation, even the αL-F153V rendered the same level of ICAM-1 binding as the αL wild type in the presence of Mn2+ but not Ca2+/Mg2+ (Fig. 3D). Consistently, the bulkiest amino acid Trp further enhanced the ICAM-1 binding at both metal ion conditions (Fig. 3D). There was ∼20% decrease of the αLβ2 cell surface expression because of these mutations (Fig. 3D). In sharp contrast, both the αL-F265S and αL-K287C/K294C mutations completely rescued the inactivating effect of αL-F153A (Fig. 3E). Interestingly, the αL-F153A mutation even enhanced the activating effect of αL-K287C/K294C (Fig. 3E). Although the αL-FFAA mutation induced the similar level of αLβ2 activation as the αL-F265S and αL-K287C/K294C mutations, it failed to rescue the inactivating effect of αL-F153A (Fig. 3E). All of these mutations clearly decreased the cell surface expression of αLβ2 integrin (Fig. 3E). These data suggest that the bulky αL-Phe-153 of α1-helix is critical for stabilizing the downward displacement of α7-helix, required for the high affinity ligand binding of αLβ2 integrin.
Figure 3.

The combined effect of the αL-Phe-153 mutations and the active αL or β2 mutations on αLβ2 ligand binding. A, model of αLβ2 integrin at the extended open headpiece conformation. The locations of the indicated mutations and the epitopes of mAbs m24 and KIM127 are shown. B, crystal structure of the αL αI domain with the active F265S mutation (PDB code 3TCX). C, crystal structure of the αL αI domain with the K287C and K294C mutations (PDB code 1T0P). The MIDAS Mg2+ ions are shown as orange spheres. The side chains of selected residues are shown as sticks. D, ICAM-1 binding of the HEK293FT cells transfected with the indicated αL-Phe-153 mutants and the active β2-G128A/G129T mutant. E, ICAM-1 binding of the HEK293FT cells transfected with β2 wild type and the αL subunits having the αL-F153A mutation combined with the active αL-F265S, αL-K287C/K294C or αL-FFAA mutation. The ICAM-1 binding was measured by flow cytometry in the presence of 1 mm Ca2+/Mg2+ (Ca/Mg) or 0.2 mm Ca2+ plus 2 mm Mn2+ (Ca/Mn) and presented as the MFI normalized to integrin expression. The data are means ± S.D. (n ≥ 3; for αl-F153G, n = 2). Two-tailed t tests were used to compare the wild type and the mutants in the same conditions or as indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, p > 0.05. Only the mean values are shown for the integrin expression.
The αL-F153A mutation has little effect on the overall conformational change of β2 integrin
Integrin activation is associated with the long-range conformational rearrangements that are relayed among the connecting domains (5). Such conformational changes on the cell surface can be measured by the exposure of epitopes that are masked in the bent inactive conformation (26). Next, we asked whether the inactivating αL-F153A mutation could affect the global conformational change of αLβ2 integrin. Two conformation-specific mAbs were used to report the conformational states of αLβ2 integrin. The mAb m24 binds to the β2 βI domain and is specific to the open headpiece conformation (27) (Fig. 3A). The mAb KIM127 binds to the I-EGF2 domain of β2 leg and is specific to the extended conformation of β2 (28, 29) (Fig. 3A). Mn2+ induced the binding of both m24 and KIM127 to the αLβ2 integrin expressed in HEK293FT cells (Fig. 4, A and B), indicating the headpiece opening and leg extension. The inactivating mutations αL-K149A and αL-F153A decreased ICAM-1 binding by more than 90% (Fig. 2B). By contrast, these mutations had no effect on Mn2+-induced binding of m24 (Fig. 4A) or KIM127 (Fig. 4B) binding to the β2 wild type. The αL-F153W mutation had no obvious effect on the binding of both mAbs compared with the wild type (Fig. 4, A and B). The activating αL-FFAA mutation, which mimics the integrin inside-out activation, induced spontaneous binding of m24 and KIM127, which was further enhanced by Mn2+ (Fig. 4, A and B). Although the αL-F153A mutation significantly reduced the binding of both mAbs when combined with the αL-FFAA mutation, it only decreased the mAb binding by 14 or 18% (Fig. 4, A and B), which is in sharp contrast with the almost 100% decrease of ICAM-1 binding (Fig. 3E). In line with these findings, neither αL-K149A nor αL-F153A affected the binding of m24 and KIM127 in the presence of the activating mutant β2-G128A/G129T that stabilizes the active conformation of βI α1-helix (Fig. 4, A and B). As seen in our previous study on β1 and β3 integrins (19), the β2-G128A/G129T mutation increased the binding of both m24 and KIM127 compared with the wild type (Fig. 4). These data demonstrated that the inactivating mutations at the αI α1-helix had little effect on the headpiece opening and extension induced by the signals of outside-in or inside-out activation. This is consistent with the previous study showing that locking the αI domain in a high-affinity conformation by the αL-K287C/K294C mutation did not induce the binding of m24 or KIM127 (22). Thus, the conformational changes between the αI and the β2 subunit can be uncoupled during the transmission of conformational changes.
Figure 4.
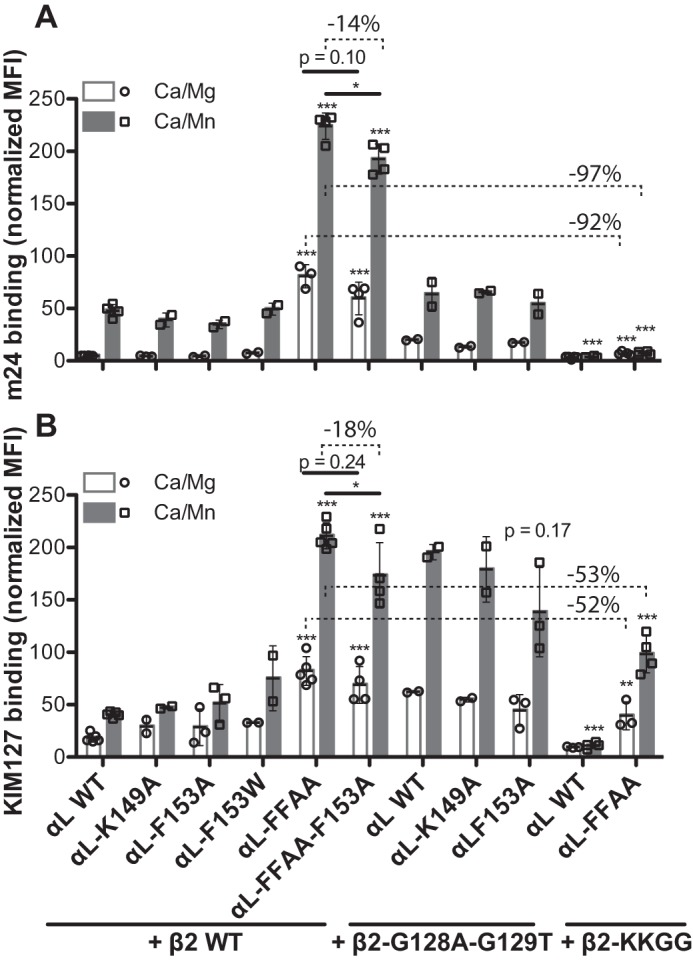
Effect of the αI α1-helix mutations on αLβ2 conformational change. HEK293FT cells were transfected with the indicated combination of αL and β2 constructs. The cells were incubated with mAb m24 (A) that reports αLβ2 headpiece opening or KIM127 (B) that reports αLβ2 extension in the presence of 1 mm Ca2+/Mg2+ (Ca/Mg) or 0.2 mm Ca2+ plus 2 mm Mn2+ (Ca/Mn). The mAb binding was measured by flow cytometry and presented as the MFI normalized to αLβ2 expression measured by TS2/4 binding. The data are means ± S.D. (n ≥ 3; except for αl-K149A and αl-F153W, n = 2). Two-tailed t tests were used to compare the wild type and the mutants in the same conditions or as indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001. The numbers of percentages indicate the decreased levels of mAb binding.
To further define the structure requirement for β2 headpiece opening and extension, we introduced a β2-KKGG mutation at the α1-helix of βI domain (Fig. 3A). This mutation has been shown in our previous study to dramatically reduced ICAM-1 binding to αLβ2 integrin by stabilizing the inactive conformation of α1-helix (19). In opposition to the β2-G128A/G129T mutation of α1-helix, the β2-KKGG mutation greatly impaired the binding of m24 and KIM127 when co-expressed with the αL wild type (Fig. 4). Moreover, it reduced the m24 binding by more than 90% (Fig. 4A), and the KIM127 binding by more than 50% (Fig. 4B), even when combined with the activating αL-FFAA mutation. These data clearly demonstrated that the active conformation of βI α1-helix; i.e. the inward movement toward the MIDAS (Fig. 1, F and H), is essential for β2 headpiece opening and extension.
The αL-F153A mutation abolishes αLβ2-mediated cell adhesion on immobilized ICAM-1
Having established the critical role of αL-F153 in the binding of αLβ2 integrin to soluble ICAM-1, we asked whether it is also important in αLβ2-mediated cell adhesion and spreading on immobilized ICAM-1, given that the receptor-ligand binding kinetics could be different in solution and in the solid phase. When seeded onto the ICAM-1-coated dish, the HEK293FT cells transfected with wild-type αLβ2 or αL-F153Y/β2 integrin spontaneously adhered and spread on the surface (Fig. 5A). However, the αL-F153A mutation completely abolished cell adhesion under the tested coating concentration of ICAM-1 (Fig. 5A), despite its comparable cell surface expression with the wild type (Fig. 5B). This is in agreement with the pivotal role of the bulky residue of αL-F153 in the high-affinity ligand binding of αLβ2.
Figure 5.

Effect of αL-Phe-153 mutations on αLβ2-mediated cell adhesion and spreading. A, HEK293FT cells transfected with the indicated integrin αLβ2 constructs were seeded onto the plates coated with human ICAM-1-Fc (5 μg/ml coating concentration) at 37 °C for 1 h. The cells were fixed and immunostained with anti-αL mAb TS2/4 shown in green. The nuclei were stained with DAPI. Scale bar, 200 μm. B, flow cytometry plots showing integrin αLβ2 expression reported by mAb TS2/4 in the corresponding HEK293FT transfectants used in A. One representative experiment of more than three repeats is shown.
The αM-F156A mutation reduces αMβ2-mediated cell adhesion on immobilized fibrinogen
We next tested whether the conserved Phe residue plays the same role in αM (Mac-1) integrin that binds multiple ligands including fibrinogen. The αM-Phe-156 was mutated to the small amino acid Ala and the bulky amino acid Trp. The αMβ2-mediated cell adhesion on immobilized human fibrinogen was induced by Mn2+ ions. The αM-F156A mutation significantly reduced the αMβ2-mediated cell adhesion (Fig. 6A). By contrast, the bulky αM-F156W mutation significantly enhanced the cell adhesion (Fig. 6A). This is not due to the differences in the cell surface expression of αMβ2 because both the αM-F156A and αM-F156W mutations decreased the αMβ2 expression to a similar extent compared with the wild type (Fig. 6B). In addition, the cell adhesion could be blocked by the anti-αM inhibitory mAb ICRF44 (data not shown). These data demonstrated that a bulky residue is also required for the α1-helix of the αM αI domain to support ligand binding.
Figure 6.
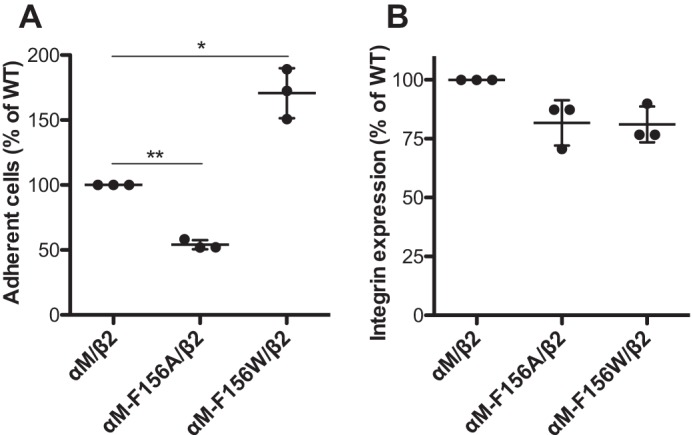
Effect of the αM-Phe-156 mutations on αMβ2-mediated cell adhesion. A, cell adhesion on immobilized human fibrinogen. HEK293FT cells transfected with indicated αMβ2 constructs and EGFP were washed with HEPES buffer and then seeded onto the 12-well cell culture plate that was precoated with 3 mg/ml human fibrinogen. The cells were incubated in the presence of 0.2 mm Ca2+ plus 2 mm Mn2+ at 37 °C for 1 h before washing and fixation. More than 20 images were randomly taken for each sample. The number of EGFP-positive adherent cells was counted for each image and averaged. The cell numbers were normalized to the wild-type level for each independent experiment. B, cell surface expression of αMβ2 in the same HEK293FT transfectants used for the cell adhesion assay in A. The cells were stained with APC-labeled anti-αM mAb ICRF44, and the mAb binding was measured by flow cytometry. The EGFP-positive cells were acquired for calculating the MFI of APC-ICRF44. The data are presented as percentages of the MFI of αMβ2 wild type. The data are means ± S.D. (n = 3). Two-tailed t tests were used to compare the wild type and the mutants. *, p < 0.05; **, p < 0.01.
Molecular dynamics simulations suggest a structural role of the bulky Phe residue of the αI α1-helix in maintaining the high affinity conformation
Our experimental data demonstrated an important role of the conserved Phe in the α1-helix of αL αI domain in high-affinity ligand binding. To gain a structural insight into how the Phe affects the conformation of αI domain, we performed the all-atom molecular dynamics (MD) simulation for wild-type and mutant αI domains in water solvent. We used the crystal structure of αL αI domain in the open conformation for the MD simulation in line with our hypothesis that the bulky Phe may stabilize the active conformation, whereas a small amino acid substitution like Ala would have the opposite effect. A 60-ns MD simulation was performed for both αL wild type and αL-F153A mutant using exactly the same parameters. To analyze the overall structural similarity of the recorded snapshots to the starting structure, the root mean square deviations (RMSDs) of backbone atoms were calculated. The αL wild type showed stable RMSD values over the 60-ns simulation. However, the RMSD values of the αL-F153A increased substantially after the first 30-ns simulation, indicating the structure instability (data not shown). To evaluate the conformational dynamics for each residue, we calculated the root mean square fluctuation (RMSF, i.e. standard deviation) of Cα positions in the snapshots relative to the starting structure. As shown in Fig. 7A, most of the residues of the wild-type αL αI domain are relatively stable over the 60-ns simulation. Remarkably, in the presence of F153A mutation, the regions of β5-α6 and β6-α7 loops showed significant fluctuations (Fig. 7A). These differences became more obvious when the RMSF values were converted to B-factors and represented as cartoon models (Fig. 7B). We next performed the same MD simulations for the open-conformation crystal structure of αM αI domain. The simulation time was extended to 100 ns. Similar to αL-F153A, the αM-F156A mutation rendered the β5-α6 and β6-α7 loops more flexible than the wild type (Fig. 7, C and D), although to a lesser extent compared with the αL αI domain (Fig. 7, A–D). These data clearly demonstrated that the bulky Phe residue of α1-helix contributes to the high affinity state of αI domain by stabilizing the active conformation of β5-α6 and β6-α7 loops.
Figure 7.

Molecular dynamics simulations of the αL and αM αI domains. A, RMSF of the Cα positions in a 60-ns MD simulation of the αI domain of αL WT or αL-F153A mutant in the open conformation. B, cartoon models of the open αL αI domains with Ala-153 and Phe-153. C, RMSF of the Cα positions in a 100-ns MD simulation of the αI domain of αM WT or αM-F156A mutant in the open conformation. D, cartoon models of the open αM αI domains with Ala-156 and Phe-156. To indicate the fluctuations of the Cα atoms, the RMSF values in the plots were converted to B-factor values and shown in the cartoon models with the Cα color and cartoon putty scaled based on the B-factors. The regions of interest are circled in the cartoons and indicated in the RMSF plots. The residues of interest are shown as green sticks with the dots representing van der Waals surface.
Discussion
Integrin is an excellent example of protein machinery in which local conformational changes in one site are propagated to a distal site, altering the ligand-binding affinity and function. Structure-based mutagenesis studies have determined certain residues that are critical for the ligand-binding activity of βI and αI domains (19, 22, 24, 30, 31). Most of the studies focused on the residues that either directly participate in the coordination with the metal ion and/or ligand or that are involved in the movements of the loops or α-helixes surrounding the active site. The importance of these residues is obvious according to the structural changes because the gain-of-function mutations are predicted to shift the conformation to the open state, whereas the loss-of-function mutations are predicted to shift it to the closed state (15, 19, 24). However, a functional role of the conserved Phe of αI α1-helix is unpredictable because this residue seems not directly participate in the movement of either α1-helix or α7-helix based on the structural comparison between the active and inactive conformations. Thus, our findings of the loss-of-function mutation of αL-Phe-153 are unexpected. Our current study further advanced our understanding of the conformational requirement for integrin ligand binding.
How does the αL-Phe-153 contribute to the ligand binding of αL αI domain? It has been indicated that the downward movement of α7-helix is required for the high affinity ligand binding of αI domain because mutations constraining it at the upward position blocked, whereas mutations facilitating the downward movement enhanced ligand binding (22, 23). In addition, small molecule inhibitors bound to the cavity under the α7-helix allosterically block ligand binding of αLβ2 by restraining the downward movement of α7-helix (32–37). Our mutagenesis study showed that the function of a residue at the position of αL-Phe-153 is determined by its size (or volume). A bulky residue is specifically required to maintain the ligand-binding capability of αI domain. At the downward position, the β6-α7 loop moves close to the middle of α1-helix (Figs. 1I and 3, B and C), where a hydrophobic and bulky side chain would act as a pawl to anchor the downward position of β6-α7 loop and α7-helix during their ratchet-like movement (15). This was indicated by the MD simulation data showing that the αL-F153A or αM-F156A mutation rendered the β5-α6 and β6-α7 loops less stable in the active conformation (Fig. 7). The bulky αL-Phe-153 is solely required for the downward movement of β6-α7 loop and α7-helix but dispensable for the inward movement of α1-helix because once the β6-α7 loop is enforced to the downward position, the trimmed α1-helix by the αL-F153A mutation still supports ligand binding. Crystal structures revealed the flexibility of α7-helix C terminus both in the isolated form of αI domain and in the context of integrin ectodomain (38–41), indicating that the α7-helix and the connecting β6-α7 loop can sample rapidly between the closed and the open conformational states. We did not observe the β6-α7 loop and α7-helix moving upward to the closed conformation in our MD simulation even in the presence of αL-F153A mutation. This is very likely due to the short time scale used for the simulation. Such a large conformational change may require much longer time of simulation to be observed. Indeed, the hydrogen-deuterium exchange kinetics measured via NMR revealed a large conformational fluctuation of α7- but not α1-helix in the resting state of the α1 integrin αI domain (42). The conserved bulky residue in the middle of α1-helix is critical to regulate the shifting between the two states of α7-helix by facilitating the transition of the αI domain to the high affinity conformation.
It should be noted that although Leu and Met have very close side chain volumes, the αL-F153L mutation exerted a similar level of ICAM-1 binding as the αL WT, whereas the αL-F153M mutation greatly reduced ICAM-1 binding (Fig. 2C). This is consistent with the numbers of rotamers that Leu and Met have. Leu can only sample four rotamer conformations, which is likely to stabilize the downward position of β6-α7 loop and α7-helix. In contrast, Met is more flexible by having more than ten rotamer conformations, one of which may adapt to the free movements of β6-α7 loop and α7-helix and thus lack the activation supporting function.
The βI and αI domains are the best-studied VWA domains that undergo conformational changes for affinity regulation. Although their β6-α7 loops and α7-helixes move in a similar fashion to accommodate for the active position of MIDAS loop and to communicate with the neighboring domains, their α1-helixes change in a different fashion (Fig. 1, H and I). This is in accordance with a conserved Gly and a conserved Phe at the homologous position of α1-helix, which play opposite roles in the βI and αI domains. As we found for a conserved negative role of the Gly in the βI α1-helix (19), the positive role of the Phe in the αI α1-helix could also be generalized to all the αI-containing integrins, given that the same conformational changes of α1- and α7-helix have been observed in the crystal structures of the αI domains of αL, αM, αX, α1, and α2 integrins (12–15, 40, 43, 44). This was demonstrated in the current study on αL and αM integrins. Interestingly, an equivalent Phe but not Gly is also present in the α1-helix of many VWA domains other than integrins (Fig. 1J). Moreover, most of the VWA domains have only one or lack an intact MIDAS, whereas the integrin βI domains have three metal ion-binding sites that regulate ligand binding. Given the different structural features and conformational regulations of βI domains, it is tempting to speculate that the βI domains might be evolved differently from a typical VWA domain and could be classified as VWA variants.
Although a conserved Phe residue at the equivalent position of αI α1-helix is found in many non-integrin VWA domains, generalizations for the similar function of this residue as seen in the αI domains can only be made with caution. For example, the VWF A1, A2, and A3 domains and the collagen VI α3N5 domain all have an equivalent Phe at the α1-helix, but they lack an intact MIDAS (Fig. 1J), and no conformational changes were observed in their α1- and α7-helixes (45–49). Similarly, the presence of an intact MIDAS does not seem to correlate with the presence of the equivalent Phe in the α1-helix. Several examples are found in the complement components C2a and factor B, in the thrombospondin repeat anonymous proteins (TRAPs) of the Plasmodium vivax and Plasmodium falciparum, and in the pilus-related adhesion RrgA of Streptococcus pneumoniae, in which a non-Phe residue is present at the equivalent position of α1-helix (Fig. 1J). We compared the crystal structures of several representative VWA domains that have an intact MIDAS, which has been suggested or confirmed to participate in ligand binding (Fig. 8). Crystal structures of the VWA domain of human anthrax toxin receptor 2 (ANTR2) in complex with anthrax toxin or pseudo-ligands highly resemble the active open conformation of integrin αI domain (Fig. 8A) (50). Although a closed conformation has yet to be seen for the ANTR2 VWA domain, mutagenesis studies suggested that it might undergo conformational changes for affinity regulation and signaling (51). Other VWA domains that have similar conformational regulations as integrin αI domains are from the adhesins of the parasites Toxoplasma gondii micronemal protein 2 (TgMIC2) and the P. vivax TRAP or P. falciparum TRAP as mentioned above. The TRAP VWA domain has been seen in both the closed and open conformations (Fig. 8B) (52). The MIC2 VWA domain has only been crystalized in the closed conformation so far (Fig. 8C), but the transition between the closed and the open states is highly predictable (53). A recent crystal structure of the Blue Mussels adhesion protein, the PTMP1 (proximal thread matrix protein 1) revealed two tandem VWA domains both in the closed conformation with the A1 domain MIDAS occupied by Zn2+ (Fig. 8D) (54). It was suggested that the MIDAS of PTMP1 might participate in collagen binding (54), which resembles the interaction of the α2 integrin αI domain and collagen. All the above-mentioned VWA domains contain the equivalent Phe at the α1-helix except that some TRAP proteins have a Leu or Met (Figs. 1J and 8, A–D). However, our data showed that a Met or Leu residue at the equivalent position of α1-helix could still support the ligand binding of αI domain (Figs. 2C and 3D). Therefore, it is tempting to speculate that these VWA domains may follow the activating mechanism of integrin αI domains.
Figure 8.

Representative VWA structures. A–F, the crystal structures of VWA domains (in green) of human anthrax toxin receptor 2 bound with anthrax toxin (PDB code 1T6B) (A); P. vivax thrombospondin repeat anonymous protein (PDB code 4HQL) (B); T. gondii micronemal protein 2 (PDB code 4OKR) (C); blue mussel proximal thread matrix protein 1 (PDB code 4CN9) (D); S. pneumoniae pilus-related adhesion, RrgA (PDB code 2WW8) (E); and human complement C3b bound with factor B and factor D (PDB code 2XWB) (F). The DXSXS motif and metal ions of MIDAS are shown in orange. The residues of α1-helix that are equivalent to the Phe of integrin αI α1-helix are shown in magenta.
We presented two examples of VWA domains from the bacteria adhesin RrgA (Fig. 8E) (55) and the complement factor B (Fig. 8F) (56), which have a small amino acid at the equivalent position of α1-helix. The crystal structure of RrgA VWA domain shows a closed conformation of MIDAS and α7-helix and an Ala at the Phe-equivalent position of α1-helix (Fig. 8E). Notably, the loop connecting the MIDAS motif and the α1-helix in RrgA is much longer than that of αL αI domain (Figs. 1J and 8E). This longer MIDAS loop may move freely toward the metal ion to accommodate with the high affinity ligand binding even without the inward movement of the α1-helix and the downward movement of the α7-helix. As such, a bulky residue in the mid α1-helix is not essential. The VWA domain of complement factor B was crystallized in both the closed and open conformations (56, 57). It has a Cys at the equivalent position of α1-helix (Figs. 1J and 8F). Although there is a swung-in motion at the N terminus of α1-helix, the scale of the downward movement of α7-helix is very small in the open conformation (Fig. 8F). The movement of α7-helix seems to be induced by the binding of factor D (56). In addition, the factor B VWA domain has an extra αl-helix, which interacts with the α7-helix and influences its conformation (Fig. 8F). All these differences may minimize the requirement of a bulky residue at the α1-helix. In summary, our study revealed a unique feature of integrin αI α1-helix in regulating ligand binding, which may be applicable to certain VWA domains of other proteins that undergo large conformational changes for signal transduction.
Another important finding in the current study is that integrin conformational transmission can be uncoupled between the αI and the βI domains. We found that although the conserved Phe of α1-helix is critical for ICAM-1 binding to αI domain, it is dispensable for β2 extension and headpiece opening. The downward displacement of α7-helix that requires the Phe of α1-helix is not essential for the conformational changes of β2 induced by Mn2+ or by inside-out activation. In addition, previous studies also showed that locking the downward conformation of α7-helix of αI domain did not result in β2 extension or headpiece opening (22). Interestingly, the internal ligand αL-Glu-310 at the C terminus of α7-helix is required for the headpiece opening of β2 subunit (58). Recent crystal structures of αXβ2 and αLβ2 suggested the intrinsic flexibility of αI domain on the platform formed by the β-propeller and βI domains (39–41). This is consistent with the loose conformational linkage between the βI and αI domains. The αLβ2 integrin mediates both the rolling adhesion and migration of leukocytes by binding the ICAM-1 expressed on the endothelial cells (59). Under the rolling condition, the loose linkage between the αI and βI enables the cells to quickly bind and release ICAM-1. When the cell signals for firm adhesion and migration are turned on, the tight engagement of the αI and βI domains could be enforced by the mechanical forces generated by the actin polymerization and ICAM-1 binding at each end of integrin (60).
Experimental procedures
DNA constructs
DNA constructs of human αL and β2 integrins were as described previously (19). The full-length cDNA of human αM integrin was cloned into the pcDNA3.1 vector using the 5-′ KpnI and 3-′ XbaI sites. Mutations were introduced by site-directed mutagenesis with the QuikChange kit (Agilent Technologies).
Antibodies and protein ligands
TS2/4 (BioLegend) is a non-functional anti-αL mAb (61). KIM127 (binds to I-EGF2 domain) and mAb 24 (m24, binds to βI domain) are anti-β2 conformation-specific mAbs that report β2 integrin extension and headpiece opening, respectively (27, 29, 62, 63). ICRF44 (BioLegend) is an inhibitory anti-αM mAb. Human ICAM-1 (with a C-terminal tag of human IgG1 Fc, ICAM-1-Fc) was purchased from Sino Biological. Biotin-labeled mouse anti-human IgG1 Fc, Alexa Fluor 488-conjugated goat anti-mouse IgG, and Alexa Fluor 647-conjugated streptavidin were from ThermoFisher Scientific.
Soluble ligand-binding assay
HEK293FT cells (ThermoFisher Scientific) were cultured in DMEM plus 10% FBS at 37 °C with 5% CO2. ICAM-1 binding of HEK293FT cells was as described before (19, 21). The cells were co-transfected with αL and β2 integrin constructs with Lipofectamine 2000 (ThermoFisher Scientific) for at least 24 h. Transfected cells were incubated in HBSGB buffer (20 mm HEPES, pH 7.4, 150 mm NaCl, 5.5 mm glucose, and 1% BSA) with 15 μg/ml each of ICAM-1-Fc and biotin-labeled mouse anti-human IgG1 Fc in the presence of 5 mm EDTA or 1 mm Ca2+/Mg2+ or 0.2 mm Ca2+ plus 2 mm Mn2+ at 25 °C for 30 min. The cells were then washed and incubated on ice for 30 min with 10 μg/ml FITC-labeled TS2/4 and 10 μg/ml Alexa Fluor 647-labeled streptavidin. TS2/4-positive cells (expressing αLβ2 integrin) were acquired for calculating the mean fluorescence intensity (MFI) by flow cytometry using BD AccuriTM C6 flow cytometer. ICAM-1 binding was presented as normalized MFI, i.e. ICAM-1 MFI (after subtracting the ICAM-1 MFI in the EDTA condition) as a percentage of TS2/4 MFI (integrin expression).
Conformation-specific antibody binding
Binding of the active conformation-specific anti-β2 mAbs KIM127 and m24 to the HEK293FT transfectants was performed as described before (19, 21). In brief, the cells were first incubated with 10 μg/ml biotin-labeled KIM127 or m24 in HBSGB buffer containing 1 mm Ca2+/Mg2+ or 0.2 mm Ca2+ plus 2 mm Mn2+ at 25 °C for 30 min and then washed and incubated with 10 μg/ml FITC-labeled TS2/4 and Alexa Fluor 647-labeled streptavidin on ice for 30 min. TS2/4-positive cells were acquired for calculating the MFI by flow cytometry. The KIM127 or m24 binding was presented as normalized MFI, i.e. streptavidin MFI as a percentage of TS2/4 MFI.
Cell adhesion assay and fluorescence microscopy
For αLβ2-mediated cell adhesion assay, the Delta T dishes (Bioptechs) or 24-well cell culture plate (BD Falcon) were first coated with 5 μg/ml mouse anti-human IgG in pH 7.4, PBS, at 4 °C for 12 h, and then blocked with 1% BSA at 37 °C for 1 h before finally coated with 5 μg/ml ICAM-1-Fc at 37 °C for another 1 h. Transfected HEK293FT cells suspended in DMEM without FBS were allowed to adhere on the ICAM-1-Fc-coated Delta T dishes at 37 °C for 1 h. The unattached cells were washed off by DMEM, and the attached cells were fixed with DMEM containing 3.7% formaldehyde, blocked with 5% nonfat dry milk in PBS, and stained with 10 μg/ml mAb TS2/4 and Alexa Fluor 488-labeled goat anti-mouse IgG. The nuclei were stained with 5 μg/ml DAPI. The stained cells were fixed with 3.7% formaldehyde in PBS and imaged with an EVOS digital inverted fluorescence microscope with a 20× objective. The level of integrin expression of the transfected cells was accessed by the mAb TS2/4 staining, followed by flow cytometry.
For αMβ2-mediated cell adhesion assay, the 12-well cell culture plate was coated with 3 mg/ml human fibrinogen in PBS at 37 °C for 1 h. HEK293FT cells transfected with αMβ2 plus EGFP were seeded onto the fibrinogen-coated plate in HEPES buffer for 10 min before adding 0.2 mm Ca2+ plus 2 mm Mn2+. After incubation at 37 °C for 1 h, the plate was washed with PBS buffer for three times and fixed with 3.7% formaldehyde in PBS. The adhered cells were imaged with an EVOS digital inverted fluorescence microscope with a 10× objective. At least 20 images were randomly taken for each sample, and the EGFP-positive cells were counted for each image. The data were presented as the averaged cell numbers that were normalized to the WT level. The cell surface expression of αMβ2 was measured by flow cytometry after staining with the allophycocyanin (APC)-labeled anti-αM mAb ICRF44 (BioLegend).
Molecular dynamics simulation
The crystal structures of the αL αI (PDB code 1T0P) and the αM αI domains (PDB code 1IDO) in the open conformation were used for MD simulation. αL-Phe-153 and αM-Phe-156 were mutated to Ala in silico using PyMOL. For the αL αI structure (PDB code 1T0P), the two cysteine mutations, K287C and K294C, were mutated back to their native Lys residue by PyMOL to remove the engineered disulfide-bond that was used to stabilize the open conformation of β6-α7 loop and α7-helix. GROMACS version 2016.3 was used to prepare the structures and perform the production MD simulation using the OPLS-AA/L all-atom force field (64, 65). The protein was centered in a cubic box with a minimum distance to any wall of 1.2 nm. The box was filled with TIP3 water model and 100 mm NaCl. Energy minimization and equilibrations of temperature and pressure were performed before conducting the unrestrained 60- or 100-ns production MD simulation. The RMSDs and RMSFs of the backbone or Cα atoms were calculated for the recorded trajectories to evaluate the stability of the conformations.
Statistical analysis
The two-tailed Student's t test was performed using the GraphPad Prism software to calculate the p values for comparing the data between two experimental groups.
Author contributions
Z. W. and A. M. M. T. performed the experiments and analyzed the data. J. Z. designed the study, analyzed data, and wrote the manuscript. All authors contributed to the manuscript preparation.
Acknowledgments
We thank Drs. Chafen Lu and Timothy Springer for providing antibodies.
This work was supported by Grants HL122985 and HL131836 (to J. Zhu) from the NHLBI, National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- I-EGF
- integrin epidermal growth factor
- VWA
- von Willebrand factor A
- MIDAS
- metal ion-dependent adhesion site
- ICAM-1
- intercellular adhesion molecular 1
- ADMIDAS
- adjacent to MIDAS
- APC
- allophycocyanin
- MD
- molecular dynamics
- RMSD
- root mean square deviation
- RMSF
- root mean square fluctuation
- TRAP
- thrombospondin repeat anonymous protein
- PDB
- Protein Data Bank.
References
- 1. Hynes R. O. (2002) Integrins: bi-directional, allosteric, signalling machines. Cell 110, 673–687 [DOI] [PubMed] [Google Scholar]
- 2. Iwamoto D. V., and Calderwood D. A. (2015) Regulation of integrin-mediated adhesions. Curr. Opin. Cell Biol. 36, 41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sun Z., Guo S. S., and Fässler R. (2016) Integrin-mediated mechanotransduction. J. Cell Biol. 215, 445–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Humphrey J. D., Dufresne E. R., and Schwartz M. A. (2014) Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 15, 802–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Springer T. A., and Dustin M. L. (2012) Integrin inside-out signaling and the immunological synapse. Curr. Opin. Cell Biol. 24, 107–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takagi J., and Springer T. A. (2002) Integrin activation and structural rearrangement. Immunol. Rev. 186, 141–163 [DOI] [PubMed] [Google Scholar]
- 7. Arnaout M. A., Mahalingam B., and Xiong J. P. (2005) Integrin structure, allostery, and bidirectional signaling. Annu. Rev. Cell Dev. Biol. 21, 381–410 [DOI] [PubMed] [Google Scholar]
- 8. Kim C., Ye F., and Ginsberg M. H. (2011) Regulation of integrin activation. Annu. Rev. Cell Dev. Biol. 27, 321–345 [DOI] [PubMed] [Google Scholar]
- 9. Shattil S. J., Kim C., and Ginsberg M. H. (2010) The final steps of integrin activation: the end game. Nat. Rev. Mol. Cell Biol. 11, 288–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liddington R. C. (2014) Structural aspects of integrins. Adv. Exp. Med. Biol. 819, 111–126 [DOI] [PubMed] [Google Scholar]
- 11. Luo B.-H., Carman C. V., and Springer T. A. (2007) Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee J.-O., Bankston L. A., Arnaout M. A., and Liddington R. C. (1995) Two conformations of the integrin A-domain (I-domain): a pathway for activation? Structure 3, 1333–1340 [DOI] [PubMed] [Google Scholar]
- 13. Lee J.-O., Rieu P., Arnaout M. A., and Liddington R. (1995) Crystal structure of the A domain from the α subunit of integrin CR3 (CD11b/CD18). Cell 80, 631–638 [DOI] [PubMed] [Google Scholar]
- 14. Emsley J., Knight C. G., Farndale R. W., Barnes M. J., and Liddington R. C. (2000) Structural basis of collagen recognition by integrin α2β1. Cell 101, 47–56 [DOI] [PubMed] [Google Scholar]
- 15. Shimaoka M., Xiao T., Liu J.-H., Yang Y., Dong Y., Jun C.-D., McCormack A., Zhang R., Joachimiak A., Takagi J., Wang J.-H., Springer T. A. (2003) Structures of the αL I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell 112, 99–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xiao T., Takagi J., Wang J.-H., Coller B. S., Springer T. A. (2004) Structural basis for allostery in integrins and binding of fibrinogen-mimetic therapeutics. Nature 432, 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhu J., Zhu J., and Springer T. A. (2013) Complete integrin headpiece opening in eight steps. J. Cell Biol. 201, 1053–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Whittaker C. A., and Hynes R. O. (2002) Essay from the genome annotation series: distribution and evolution of the von Willebrand/integrin A domain: a widely dispersed domain with roles in cell adhesion and elsewhere. Mol. Biol. Cell 13, 3369–3387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang C., Liu J., Jiang X., Haydar N., Zhang C., Shan H., and Zhu J. (2013) Modulation of integrin activation and signaling by α1/α1′-helix unbending at the junction. J. Cell Sci. 126, 5735–5747 [DOI] [PubMed] [Google Scholar]
- 20. Qu A., and Leahy D. J. (1996) The role of the divalent cation in the structure of the I domain from the CD11a/CD18 integrin. Structure 4, 931–942 [DOI] [PubMed] [Google Scholar]
- 21. Liu J., Wang Z., Thinn A. M., Ma Y. Q., and Zhu J. (2015) The dual structural roles of the membrane distal region of the α-integrin cytoplasmic tail during integrin inside-out activation. J. Cell Sci. 128, 1718–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu C., Shimaoka M., Zang Q., Takagi J., and Springer T. A. (2001) Locking in alternate conformations of the integrin αLβ2 I domain with disulfide bonds reveals functional relationships among integrin domains. Proc. Natl. Acad. Sci. U.S.A. 98, 2393–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shimaoka M., Lu C., Salas A., Xiao T., Takagi J., and Springer T. A. (2002) Stabilizing the integrin αM inserted domain in alternative conformations with a range of engineered disulfide bonds. Proc. Natl. Acad. Sci. U.S.A. 99, 16737–16741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jin M., Song G., Carman C. V., Kim Y.-S., Astrof N. S., Shimaoka M., Wittrup D. K., and Springer T. A. (2006) Directed evolution to probe protein allostery and integrin I domains of 200,000-fold higher affinity. Proc. Natl. Acad. Sci. U.S.A. 103, 5758–5763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu C. F., and Springer T. A. (1997) The α subunit cytoplasmic domain regulates the assembly and adhesiveness of integrin lymphocyte function-associated antigen-1 (LFA-1). J. Immunol. 159, 268–278 [PubMed] [Google Scholar]
- 26. Humphries M. J. (2004) Monoclonal antibodies as probes of integrin priming and activation. Biochem. Soc. Trans. 32, 407–411 [DOI] [PubMed] [Google Scholar]
- 27. Chen X., Xie C., Nishida N., Li Z., Walz T., and Springer T. A. (2010) Requirement of open headpiece conformation for activation of leukocyte integrin αXβ2. Proc. Natl. Acad. Sci. U.S.A. 107, 14727–14732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu C., Ferzly M., Takagi J., and Springer T. A. (2001) Epitope mapping of antibodies to the C-terminal region of the integrin β2 subunit reveals regions that become exposed upon receptor activation. J. Immunol. 166, 5629–5637 [DOI] [PubMed] [Google Scholar]
- 29. Nishida N., Xie C., Shimaoka M., Cheng Y., Walz T., and Springer T. A. (2006) Activation of leukocyte β2 integrins by conversion from bent to extended conformations. Immunity 25, 583–594 [DOI] [PubMed] [Google Scholar]
- 30. Shimaoka M., Shifman J. M., Jing H., Takagi J., Mayo S. L., and Springer T. A. (2000) Computational design of an integrin I domain stabilized in the open, high affinity conformation. Nat. Struct. Biol. 7, 674–678 [DOI] [PubMed] [Google Scholar]
- 31. Barton S. J., Travis M. A., Askari J. A., Buckley P. A., Craig S. E., Humphries M. J., and Mould A. P. (2004) Novel activating and inactivating mutations in the integrin β1 subunit A domain. Biochem. J. 380, 401–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kallen J., Welzenbach K., Ramage P., Geyl D., Kriwacki R., Legge G., Cottens S., Weitz-Schmidt G., and Hommel U. (1999) Structural basis for LFA-1 inhibition upon lovastatin binding to the CD11a I-domain. J. Mol. Biol. 292, 1–9 [DOI] [PubMed] [Google Scholar]
- 33. Crump M. P., Ceska T. A., Spyracopoulos L., Henry A., Archibald S. C., Alexander R., Taylor R. J., Findlow S. C., O'Connell J., Robinson M. K., and Shock A. (2004) Structure of an allosteric inhibitor of LFA-1 bound to the I-domain studied by crystallography, NMR, and calorimetry. Biochemistry 43, 2394–2404 [DOI] [PubMed] [Google Scholar]
- 34. Weitz-Schmidt G., Welzenbach K., Dawson J., and Kallen J. (2004) Improved LFA-1 inhibition by statin derivatives: Molecular basis determined by X-ray analysis and monitoring of LFA-1 conformational changes in vitro and ex vivo. J. Biol. Chem. 279, 46764–46771 [DOI] [PubMed] [Google Scholar]
- 35. Wattanasin S., Kallen J., Myers S., Guo Q., Sabio M., Ehrhardt C., Albert R., Hommel U., Weckbecker G., Welzenbach K., and Weitz-Schmidt G. (2005) 1,4-Diazepane-2,5-diones as novel inhibitors of LFA-1. Bioorg. Med. Chem. Lett. 15, 1217–1220 [DOI] [PubMed] [Google Scholar]
- 36. Potin D., Launay M., Monatlik F., Malabre P., Fabreguettes M., Fouquet A., Maillet M., Nicolai E., Dorgeret L., Chevallier F., Besse D., Dufort M., Caussade F., Ahmad S. Z., Stetsko D. K., et al. (2006) Discovery and development of 5-[(5S,9R)-9-(4-cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-tria zaspiro[4.4]non-7-yl-methyl]-3-thiophenecarboxylic acid (BMS-587101): a small molecule antagonist of leukocyte function associated antigen-1. J. Med. Chem. 49, 6946–6949 [DOI] [PubMed] [Google Scholar]
- 37. Zhang H., Astrof N. S., Liu J. H., Wang J. H., and Shimaoka M. (2009) Crystal structure of isoflurane bound to integrin LFA-1 supports a unified mechanism of volatile anesthetic action in the immune and central nervous systems. FASEB J. 23, 2735–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang H., Casasnovas J. M., Jin M., Liu J. H., Gahmberg C. G., Springer T. A., and Wang J. H. (2008) An unusual allosteric mobility of the C-terminal helix of a high-affinity αL integrin I domain variant bound to ICAM-5. Mol. Cell 31, 432–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xie C., Zhu J., Chen X., Mi L., Nishida N., and Springer T. A. (2010) Structure of an integrin with an α I domain, complement receptor type 4. EMBO J. 29, 666–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sen M., Yuki K., and Springer T. A. (2013) An internal ligand-bound, metastable state of a leukocyte integrin, αXβ2. J. Cell Biol. 203, 629–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sen M., and Springer T. A. (2016) Leukocyte integrin αLβ2 headpiece structures: The αI domain, the pocket for the internal ligand, and concerted movements of its loops. Proc. Natl. Acad. Sci. U.S.A. 113, 2940–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nunes A. M., Zhu J., Jezioro J., Minetti C. A., Remeta D. P., Farndale R. W., Hamaia S. W., and Baum J. (2016) Intrinsic local destabilization of the C-terminus predisposes integrin α1 I domain to a conformational switch induced by collagen binding. Protein Sci. 25, 1672–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chin Y. K., Headey S. J., Mohanty B., Patil R., McEwan P. A., Swarbrick J. D., Mulhern T. D., Emsley J., Simpson J. S., and Scanlon M. J. (2013) The structure of integrin α1 I domain in complex with a collagen-mimetic peptide. J. Biol. Chem. 288, 36796–36809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bajic G., Yatime L., Sim R. B., Vorup-Jensen T., and Andersen G. R. (2013) Structural insight on the recognition of surface-bound opsonins by the integrin I domain of complement receptor 3. Proc. Natl. Acad. Sci. U.S.A. 110, 16426–16431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brondijk T. H., Bihan D., Farndale R. W., and Huizinga E. G. (2012) Implications for collagen I chain registry from the structure of the collagen von Willebrand factor A3 domain complex. Proc. Natl. Acad. Sci. U.S.A. 109, 5253–5258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jakobi A. J., Mashaghi A., Tans S. J., and Huizinga E. G. (2011) Calcium modulates force sensing by the von Willebrand factor A2 domain. Nat. Commun. 2, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhou M., Dong X., Baldauf C., Chen H., Zhou Y., Springer T. A., Luo X., Zhong C., Gräter F., and Ding J. (2011) A novel calcium-binding site of von Willebrand factor A2 domain regulates its cleavage by ADAMTS13. Blood 117, 4623–4631 [DOI] [PubMed] [Google Scholar]
- 48. Zhang Q., Zhou Y.-F., Zhang C.-Z., Zhang C. Z., Zhang X., Lu C., and Springer T. A. (2009) Structural specializations of A2, a force-sensing domain in the ultralarge vascular protein von Willebrand factor. Proc. Natl. Acad. Sci. U.S.A. 106, 9226–9231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huizinga E. G., Tsuji S., Romijn R. A., Schiphorst M. E., de Groot P. G., Sixma J. J., and Gros P. (2002) Structures of glycoprotein Ibα and its complex with von Willebrand factor A1 domain. Science 297, 1176–1179 [DOI] [PubMed] [Google Scholar]
- 50. Santelli E., Bankston L. A., Leppla S. H., and Liddington R. C. (2004) Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature 430, 905–908 [DOI] [PubMed] [Google Scholar]
- 51. Deuquet J., Lausch E., Superti-Furga A., and van der Goot F. G. (2012) The dark sides of capillary morphogenesis gene 2. EMBO J. 31, 3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Song G., Koksal A. C., Lu C., and Springer T. A. (2012) Shape change in the receptor for gliding motility in Plasmodium sporozoites. Proc. Natl. Acad. Sci. U.S.A. 109, 21420–21425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Song G., and Springer T. A. (2014) Structures of the Toxoplasma gliding motility adhesin. Proc. Natl. Acad. Sci. U.S.A. 111, 4862–4867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Suhre M. H., Gertz M., Steegborn C., and Scheibel T. (2014) Structural and functional features of a collagen-binding matrix protein from the mussel byssus. Nat. Commun. 5, 3392. [DOI] [PubMed] [Google Scholar]
- 55. Izoré T., Contreras-Martel C., El Mortaji L., Manzano C., Terrasse R., Vernet T., Di Guilmi A. M., and Dessen A. (2010) Structural basis of host cell recognition by the pilus adhesin from Streptococcus pneumoniae. Structure 18, 106–115 [DOI] [PubMed] [Google Scholar]
- 56. Forneris F., Ricklin D., Wu J., Tzekou A., Wallace R. S., Lambris J. D., and Gros P. (2010) Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science 330, 1816–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Milder F. J., Gomes L., Schouten A., Janssen B. J., Huizinga E. G., Romijn R. A., Hemrika W., Roos A., Daha M. R., and Gros P. (2007) Factor B structure provides insights into activation of the central protease of the complement system. Nat. Struct. Mol. Biol. 14, 224–228 [DOI] [PubMed] [Google Scholar]
- 58. Salas A., Shimaoka M., Kogan A. N., Harwood C., von Andrian U. H., and Springer T. A. (2004) Rolling adhesion through an extended conformation of integrin αLβ2 and relation to α I and β I-like domain interaction. Immunity 20, 393–406 [DOI] [PubMed] [Google Scholar]
- 59. Springer T. A. (1994) Traffic signals for lymphocyte recirculation and leukocyte emigration: the multi-step paradigm. Cell 76, 301–314 [DOI] [PubMed] [Google Scholar]
- 60. Nordenfelt P., Elliott H. L., and Springer T. A. (2016) Coordinated integrin activation by actin-dependent force during T-cell migration. Nat. Commun. 7, 13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sanchez-Madrid F., Krensky A. M., Ware C. F., Robbins E., Strominger J. L., Burakoff S. J., and Springer T. A. (1982) Three distinct antigens associated with human T lymphocyte-mediated cytolysis: LFA-1, LFA-2, and LFA-3. Proc. Natl. Acad. Sci. U.S.A. 79, 7489–7493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Robinson M. K., Andrew D., Rosen H., Brown D., Ortlepp S., Stephens P., and Butcher E. C. (1992) Antibody against the Leu-cam β-chain (CD18) promotes both LFA-1-and CR3-dependent adhesion events. J. Immunol. 148, 1080–1085 [PubMed] [Google Scholar]
- 63. Dransfield I., and Hogg N. (1989) Regulated expression of Mg2+ binding epitope on leukocyte integrin α subunits. EMBO J. 8, 3759–3765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jorgensen W. L., Maxwell D. S., and Tirado-Rives J. (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 [Google Scholar]
- 65. Kaminski G. A., Friesner R. A., Tirado-Rives J., and Jorgensen W. L. (2001) Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 105, 6474–6487 [Google Scholar]
- 66. Harpaz Y., Gerstein M., and Chothia C. (1994) Volume changes on protein folding. Structure 2, 641–649 [DOI] [PubMed] [Google Scholar]