

Abstract
The transient receptor potential canonical channel-1 (TRPC1) is a Ca2+-permeable channel found in key metabolic organs and tissues, including the hypothalamus, adipose tissue, and skeletal muscle. Loss of TRPC1 may alter the regulation of cellular energy metabolism resulting in insulin resistance thereby leading to diabetes. Exercise reduces insulin resistance, but it is not known whether TRPC1 is involved in exercise-induced insulin sensitivity. The role of TRPC1 in adiposity and obesity-associated metabolic diseases has not yet been determined. Our results show that TRPC1 functions as a major Ca2+ entry channel in adipocytes. We have also shown that fat mass and fasting glucose concentrations were lower in TRPC1 KO mice that were fed a high-fat (HF) (45% fat) diet and exercised as compared with WT mice fed a HF diet and exercised. Adipocyte numbers were decreased in both subcutaneous and visceral adipose tissue of TRPC1 KO mice fed a HF diet and exercised. Finally, autophagy markers were decreased and apoptosis markers increased in TRPC1 KO mice fed a HF diet and exercised. Overall, these findings suggest that TRPC1 plays an important role in the regulation of adiposity via autophagy and apoptosis and that TRPC1 inhibits the positive effect of exercise on type II diabetes risk under a HF diet-induced obesity environment.
Keywords: calcium, diabetes, exercise, obesity, transient receptor potential channels (TRP channels), SOCE, TRPC1
Introduction
Intracellular Ca2+ signaling has been suggested as playing several important roles in regulating cellular energy metabolism (1); however, the specific Ca2+ ion channels involved have not yet been identified. Transient receptor potential canonical channel-1 (TRPC1)4 functions as a Ca2+ entry channel that is found in key metabolic tissues, including the hypothalamus (2), adipose tissue (3), and skeletal muscle (4), making it a likely candidate for the regulation of cellular energy metabolism. As such, functional disturbance of TRP family channels could play a role in regulating adiposity and obesity-related conditions such as insulin resistance (5–7). However, the exact role of TRPC1 in adipose tissue mass changes, development of obesity, and obesity-associated metabolic disease risks has not yet been determined.
TRP channels contain six hydrophobic stretches and a pore loop motif intercalated between the fifth and sixth transmembrane segments (8, 9). The mammalian TRP channel family consists of subfamilies of classical TRP channels (TRPC1–TRPC7), vanilloid receptor-related TRP channels (TRPV1–TRPV6), melastatin-related TRP channels (TRPM1–TRPM8), and polycystin-related TRP channels (TRPP1–TRPP2) (10). Of these, several TRPC channels are activated by G protein-coupled receptors and receptor tyrosine kinases that are linked to phosphoinositide hydrolysis via phospholipase C activation, whereas other TRPC channels (specifically TRPC1 and TRPC4) are activated upon depletion of intracellular Ca2+ stores (9, 10).
Obesity is a hallmark of metabolic syndrome and a key feature of obesity is the disruption of metabolic homeostasis leading to excess adipose accumulation (11–14), thus therapeutic targeting of proteins involved in these pathways could be essential for slowing or preventing the development of obesity and obesity-related health problems, including insulin resistance and type II diabetes. TRPC1 gene expression is induced in differentiated adipocytes (3), yet no data are currently available on whether TRPC1 has a role in adipocyte energy metabolism regulation by altering mitochondrial energy oxidation, adipocyte lipid storage or size, and adipose tissue weight.
One way that TRP channels may control energy metabolism and adiposity is by acting as sensors for chemical factors necessary in adipocyte biology (3). Dietary saturated fat intake promotes obesity and type II diabetes (15) whereas n-3 polyunsaturated fatty acids (PUFAs) mainly found in fish oil produce opposite effects (16, 17). Treatment of human embryonic kidney cells (HEK 293) with n-3 PUFAs such as linolenic, docosahexaenoic, and eicosapentaenoic acids inhibit Ca2+ entry via TRPC5 homomeric and TRPC1–TRPC5 heteromeric channels (3). Interestingly, the PUFA concentrations used in this study were within the physiologically achievable range of the human diet (3). Whether high dietary saturated fat intake modulates adipocyte energy metabolism via TRPC1-mediated signaling is not yet known.
Moreover, experimental evidence indicates that several TRP channels play an important role in the onset of diabetes (5–7) or diet-induced obesity (18); however, the role of TRPC1 in these circumstances is not yet established. Exercise regulates body energy stores and insulin resistance by reducing adipocyte size and lipid content (19, 20) and by regulating serum glucose homeostasis through inducing glucose transporter type 4 (GLUT4) protein expression (21). Interestingly, treadmill running prevents Ca2+ dysregulation and diabetic dyslipidemia in HF fed swine (22). TRPC1 knock-out (KO) mice with attenuated Ca2+ entry (23) experienced reduced muscular endurance due in part to reduced force production and a greater rate of muscle fatigue (4). However, whether a HF diet could exacerbate reduced exercise tolerance in TRPC1 KO mice or contribute to mitochondrial energy metabolism dysfunction is also not yet known. Currently, no other studies have investigated the effects of dietary HF and exercise on adipocyte energy metabolism alteration via TRPC1 protein regulation of intracellular Ca2+ homeostasis.
The present study investigated the involvement of TRPC1 in diet-induced obesity and type II diabetes. Additionally, regulation of adipocyte formation under normal-fat (NF) (21) or HF diet and control cage or voluntary exercise conditions was also evaluated to determine how optimal dietary treatments and exercise promote a healthy body weight. Our data indicate that TRPC1 KO mice fed a HF diet and exercised are protected from diet-induced obesity and type II diabetes risk indicative of an underlying mechanism resulting from loss of Ca2+ influx through TRPC1 that mediates a reduction in adiposity and insulin resistance when HF diet and exercise are combined.
Results
Expression and characterization of calcium channels in subcutaneous adipose tissue
We first examined TRPC1 transcripts in subcutaneous adipose tissue from WT and TRPC1 KO mice. Adipose tissues were obtained from both WT and TRPC1 KO mice and mRNA was isolated, after which RT-PCR confirmed that full-length TRPC1 expression is lost in KO mice (Fig. 1A). We next investigated Ca2+ entry upon store depletion using primary adipocyte cells. Endoplasmic reticulum (ER) Ca2+ stores were depleted by the addition of thapsigargin (Tg) (2 μm), a SERCA (sarcoplasmic/endoplasmic reticulum Ca2+-ATPase) pump blocker, which activates store-mediated Ca2+ entry. In the absence of extracellular Ca2+, the increase in intracellular Ca2+ ([Ca2+]i) evoked by thapsigargin (first peak) was unaltered in TRPC1 KO cells when compared with WT control cells (Fig. 1, B and C). Subsequently, addition of external Ca2+ (1 mm), which initiates store-mediated Ca2+ entry, was significantly decreased in adipocytes obtained from TRPC1 KO mice (Fig. 1, B and C and supplemental Fig. 1A). Similarly, we also depleted internal stores through angiotensin II, which stimulates endogenous G protein–coupled receptors, resulting in decreased Ca2+ entry in TRPC1 KO mice indicating that TRPC1 is the functional store/receptor-operated Ca2+ entry (S/ROCE) channel in these cells (supplemental Fig. 1, B and C). Importantly, basal Ca2+ (no store depletion) was unaltered in adipocytes from WT or TRPC1 KO mice (supplemental Fig. 1, D and E). To establish the molecular identity of the Ca2+ entry channel, electrophysiological recordings were performed. Addition of thapsigargin induced an inward current which was nonselective and reversed between 0 and −5 mV (Fig. 1, D–F). Importantly, Ca2+ entry currents were significantly decreased in TRPC1 KO mice and the channel properties were similar to those previously observed with TRPC1 channels (23), suggesting that TRPC1 contributes to the endogenous store-mediated Ca2+ entry channel in adipocyte cells. Furthermore, we evaluated expression of other Ca2+ entry channels in adipocytes and found that along with TRPC1, TRPC5, STIM1, and Orai1 were also expressed in these cells (Fig. 1, G and H), although the properties of the store/receptor-operated Ca2+ entry in adipocytes was not inward rectifying as observed with Orai1-mediated ICRAC channels (24, 25) suggesting that TRPC1 is the major Ca2+ channel in adipocytes. Together, these results suggest that TRPC1 is important for Ca2+ entry in adipocyte cells.
Figure 1.

TRPC1 expression and Ca2+ signaling are decreased in subcutaneous adipose tissue of TRPC1 KO mice. A, RT-PCR expression of TRPC1 from subcutaneous adipose tissue of WT and TRPC1 KO mice following 12 weeks of diet and exercise. B, analog plots of the fluorescence ratio (340/380) from an average of 30–40 cells in each condition. C, quantification (mean ± S.D.) of 340/380 ratio. *** indicates significance (p < 0.001) versus control. D, thapsigargin (Tg)-induced currents were evaluated in adipocytes obtained from WT and TRPC1 KO mice. The holding potential for current recordings was −80 mV. E and F, I/V curves (mean current) under these conditions and the average (8–10 recordings) current intensity under various conditions is shown in F. *** indicates values (mean ± S.D.) that are significantly different from control (p < 0.001). G, subcutaneous adipose tissue from WT and TRPC1 KO mice were resolved and analyzed by Western blotting using antibodies labeled in the figure with β-actin as a loading control. H, quantification of each protein. Data are presented as mean ± S.D., n = 4. **, p < 0.01; ***, p < 0.001.
Body fat mass is decreased in TRPC1 KO mice fed a HF diet and exercised
TRPC1 KO mice had lower body weight (Fig. 2A) and body fat mass (Fig. 2D) at the start of the study and after 12 weeks of diet and exercise (Fig. 2, B and E) when compared with WT mice (p < 0.0001). However, when calculated as a -fold change, there was no change in body weight when comparing WT to TRPC1 KO mice (Fig. 2C), but body fat mass was significantly decreased (p < 0.05) in TRPC1 KO mice fed a HF diet and exercised compared with WT mice fed a HF diet and exercised (Fig. 2F). Furthermore, TRPC1 KO mice fed a HF diet and exercised had less body fat mass (p < 0.0001) than TRPC1 KO mice fed a HF diet and subjected to sedentary cage activity (Fig. 2F). Although food intake variation was influenced by the type of mouse (p < 0.01) and an exercise–diet interaction (p < 0.05), altered body composition was not a result of group differences in food consumption (p > 0.05) or exercise (p > 0.05) (supplemental Fig. 2, A and B). The data thus far show that TRPC1 is the major Ca2+ entry channel in adipocytes and that loss of TRPC1 decreases obesity risk in HF fed mice that exercise.
Figure 2.

TRPC1 KO mice fed a HF diet and exercised have decreased body fat mass. A and B, body weight was measured at week 0 (A) and week 12 (B). C, body weight change was calculated by dividing the body weight at week 12 by the body weight at week 0. D and E, body fat mass was measured by EchoMRI at week 0 (D) and week 12 (E). F, body fat change was calculated by dividing the fat weight at week 12 by the fat weight at week 0. Data are presented as mean ± S.D., n = 6–8. Significant (p < 0.05) effects from 3-way ANOVA are indicated by + (mouse type), × (diet), and # (exercise). A significant interaction was further analyzed using post hoc Tukey to perform pairwise comparisons. *, p < 0.05; **, p < 0.01; ****, p < 0.0001.
TRPC1 KO mice fed a HF diet and exercised are protected from type II diabetes risk
The data provided above show an important role for TRPC1 in the onset of metabolic syndrome. Thus, glucose concentrations were next evaluated under these conditions. Maximum blood glucose concentrations occurred 15–30 min after intraperitoneal injection of glucose in all groups (Fig. 3A). However, blood glucose concentrations were decreased (p < 0.0001) in TRPC1 KO mice fed a HF diet and exercised when compared with WT mice fed a HF diet and exercised (Fig. 3B). Similarly, serum insulin concentrations were decreased (p < 0.05) in TRPC1 KO mice fed a HF diet and exercised compared with WT mice fed a HF diet and exercised (Fig. 3C). Using a homeostatic model assessment of insulin resistance (HOMA IR), we found that, once more, TRPC1 KO mice fed a HF diet and exercised were less insulin resistant (p < 0.01) than WT mice fed a HF diet and exercised (Fig. 3D) although this difference was not because of altered expression of GLUT4 in the subcutaneous adipose tissue (Fig. 4A) or skeletal muscle (Fig. 4B). These studies suggest that loss of TRPC1 decreases insulin resistance and risk of diabetes in HF fed mice that exercise thereby inhibiting metabolic syndrome.
Figure 3.

TRPC1 KO mice fed a HF diet and exercised have reduced insulin resistance. A–D, blood glucose (A and B), plasma insulin (C), and calculated homeostatic model assessment of insulin resistance (D) were measured from WT and TRPC1 KO mice fasted overnight after 12 weeks of diet and exercise. Data are presented as mean ± S.E. (A) or S.D. (B–D), n = 7–8. Significant (p < 0.05) effects from 3-way ANOVA are indicated by + (mouse type), × (diet), and # (exercise). A significant interaction was further analyzed using post hoc Tukey to perform pairwise comparisons. *, p < 0.05; **, p < 0.01; ****, p < 0.0001.
Figure 4.

GLUT4 expression is unaltered in subcutaneous adipose tissue and skeletal muscle. A and B, GLUT4 expression in subcutaneous adipose tissue (A) and hind leg biceps femoris skeletal muscle (B) was measured from WT and TRPC1 KO mice following 12 weeks of diet and exercise. Data are presented as mean ± S.D., n = 7–8. No significant (p > 0.05) effects from 3-way ANOVA were identified. n.s., no significance.
Adipocyte numbers are decreased in TRPC1 KO mice fed a HF diet and exercised
To establish if adipocyte number or size is varied under these conditions, we counted the number of adipocytes present in adipose tissue depots and determined the adipocyte size. In the subcutaneous and visceral adipose tissue depots, adipocytes with size ranges of 80–160 μm were decreased (p < 0.05) in TRPC1 KO mice fed a HF diet and exercised compared with WT mice fed a HF diet and exercised (Fig. 5). In addition, TRPC1 KO mice fed a HF diet and subjected to sedentary cage activity had decreased (p < 0.05) adipocytes from 160–200 μm when compared with WT mice fed a HF diet and subjected to sedentary cage activity (Fig. 5), suggesting that loss of TRPC1 decreases the number of larger adipocytes, which could result in the decreased fat mass observed in Fig. 2.
Figure 5.
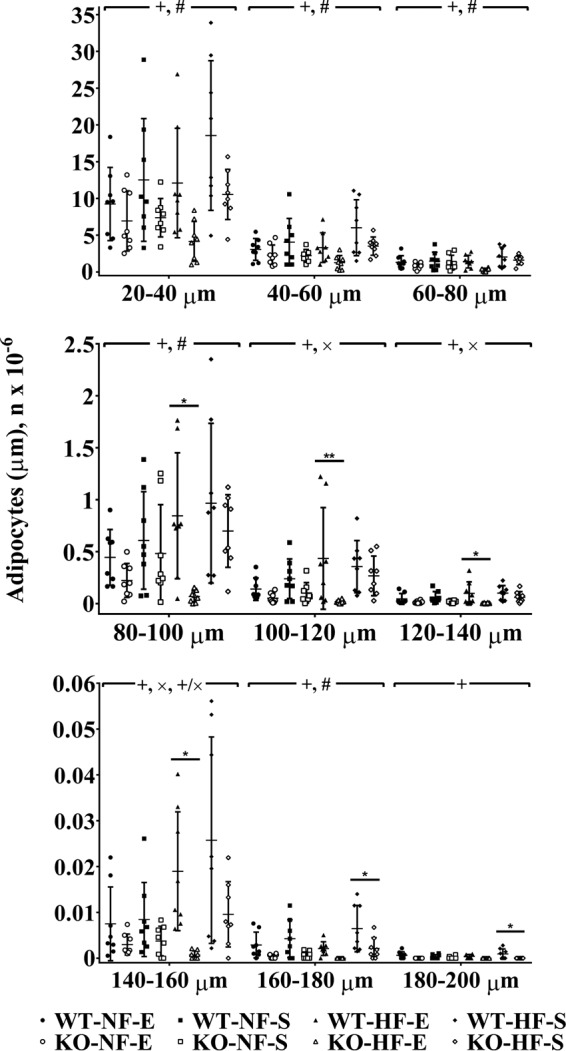
TRPC1 KO mice fed a HF diet and exercised have fewer adipocytes. Adipose tissue harvested from WT and TRPC1 KO mice following 12 weeks of diet and exercise was measured by a Multisizer. Data are presented as mean ± S.D., n = 6–8. Significant (p < 0.05) effects from 3-way ANOVA are indicated by + (mouse type), × (diet), and # (exercise). A significant interaction was further analyzed using post hoc Tukey to perform pairwise comparisons. *, p < 0.05; **, p < 0.01.
Autophagy marker expression is decreased whereas apoptosis marker expression is increased in TRPC1 KO mice fed a HF diet and exercised
To determine whether reduced adipocyte numbers in adipose depots of TRPC1 KO mice fed a HF diet and exercised (Fig. 5) were because of apoptosis or reduced differentiation into adipocytes, we measured mRNA of key markers for adipogenesis (PPARγ (peroxisome proliferator–activated receptor γ)), beiging (FGF21 (fibroblast growth factor 21)), hypoxia (HIF1α (hypoxia-inducible factor 1-α)), and autophagy (MAP1LC3A (microtubule-associated proteins 1A/1B light chain 3A), BECN1 (beclin 1)). Although there was no altered mRNA expression of PPARγ, FGF21, HIF1α, or BECN1 in subcutaneous adipose tissue (supplemental Fig. 3, A–D), expression of the autophagy marker MAP1LC3A was decreased (as indicated by an increased threshold cycle (Ct) value) (p < 0.05) in TRPC1 KO mice fed a HF diet and exercised compared with WT mice fed a HF diet and exercised (Fig. 6A). To confirm that our mRNA expression was replicated on the protein level, we examined protein expression of autophagy (LC3A, p62) and apoptosis (Bax, Bcl-xl) regulating proteins in WT and KO mice fed a HF diet and exercised. LC3A expression was decreased along with an increase in p62 expression in samples from TRPC1 KO mice that were fed a HF diet and exercised when compared with WT mice fed a HF diet and exercised (Fig. 6, B and C). Similarly, increased expression of Bax and an increased Bax to Bcl-xl ratio was observed in the TRPC1 KO mice fed a HF diet and exercised compared with WT mice fed a HF diet and exercised (Fig. 6, B–D). Together, these results suggest that loss of TRPC1 decreases autophagy, a survival mechanism, and increases apoptosis, which could promote loss of larger adipocytes. In addition, loss of TRPC1 significantly decreased phosphorylation of ERK2, whereas no change in the phosphorylation of ERK1 and AMPK was observed (Fig. 6, E and F). These data further indicate that loss of TRPC1 inhibits ERK2 phosphorylation, which has been shown to interact with ATG proteins and thus could modulate autophagy in these cells.
Figure 6.

Autophagy marker expression is decreased whereas apoptosis marker expression is increased in TRPC1 KO mice fed a HF diet and exercised. A, MAP1LC3A mRNA expression was measured from subcutaneous adipose tissue taken from WT and TRPC1 KO mice following 12 weeks of diet and exercise. Data are presented as mean ± S.D., n = 6–8. Significant (p < 0.05) effects from 3-way ANOVA are indicated by + (mouse type). A significant interaction was further analyzed using post hoc Tukey to perform pairwise comparisons. *, p < 0.05. B, subcutaneous adipose tissue from WT and TRPC1 KO mice fed a HF diet and exercised was resolved and analyzed by Western blotting using antibodies labeled in the figure with β-actin as a loading control. Quantification of each protein is shown in C. Data are presented as mean ± S.D., n = 7–11. *, p < 0.05. D, ratio of Bax/Bcl-xl is presented as a -fold increase of TRPC1 KO over WT values. Data are presented as mean ± S.D., n = 7–10. *, p < 0.05. E, subcutaneous adipose tissue isolated from WT and TRPC1 KO mice fed a HF diet and exercised was resolved on SDS-PAGE gels and analyzed using different antibodies as labeled in the figure. F, quantification of the phosphorylated to non-phosphorylated form of each protein. Data are presented as mean ± S.D., n = 5. *, p < 0.05.
Discussion
This study is the first to show that TRPC1 KO mice that exercise are protected from HF diet-induced obesity and type II diabetes risk because of decreased adipose tissue mass and adipocyte number as a result of reduced autophagy and increased apoptosis. Thus, in combination, exercise, HF diet, and loss of TRPC1 reduce adiposity through a yet undefined mechanism. Given that TRPC1 is involved in Ca2+ entry following depletion of internal Ca2+ stores in the ER, TRPC1 KO results in decreased Ca2+ entry in a variety of cell types including adipocytes (3), skeletal muscle (4), neuronal (26), intestinal epithelial cells (27), and salivary glands (23, 28). Thus, based on our data, it is probable that reduced Ca2+ entry due to TRPC1 KO is influenced further by HF diet and exercise, suggestive of a relationship between Ca2+ entry, diet, and exercise. Although expression of Orai1 and STIM1 was observed in adipocytes, the properties of the endogenous channel were similar to those observed with TRPC1-mediated ISOC (23, 28) and not as observed with ICRAC channels (24, 25). Furthermore, TRPC1 KO mice showed exercise-mediated inhibition of adiposity and decreased insulin resistance in the absence of TRPC1 suggesting that TRPC1 might be the dominant Ca2+ channel in these cells. However, additional studies will be needed to determine the role of Orai1 channels in exercise-mediated regulation of metabolic syndrome.
The present study demonstrated that fat mass was reduced in TRPC1 KO mice compared with WT mice following 12 weeks of HF diet and exercise. Similarly, previous studies have shown that TRPV4 KO mice (another Ca2+ entry channel) are also protected from obesity and metabolic dysfunction with exposure to HF diet (18) suggesting that Ca2+ channels negatively regulate obesity. This is in contrast to the expectations that HF-fed mice develop obesity and glucose intolerance (29) because TRPC1 KO mice fed a HF-diet and exercised were less insulin resistant than their WT counterparts, indicative of protection from type II diabetes risk, yet GLUT4 expression was unaltered in hind leg biceps femoris skeletal muscle or subcutaneous adipose tissue. Furthermore, the number and size of subcutaneous and visceral adipocytes are decreased in TRPC1 KO mice compared with WT mice when fed a HF diet and exercised. Because TRPC1 plays a key role in cell survival and apoptosis (30–32), it was hypothesized that TRPC1 KO mice would alter expression patterns of key markers for adipogenesis, apoptosis, or autophagy in subcutaneous adipose tissue. TRPC1 KO mice fed a HF diet and exercised had decreased expression of the autophagy marker MAP1LC3A along with an increase in apoptosis markers (particularly the ratio of Bax/Bcl-xl), which is in agreement with our previous findings that silencing of TRPC1 decreased autophagy and increased cell death (33). Loss of TRPC1 also decreased phosphorylation of ERK2, which is consistent with previous studies where activation of Ca2+ channels in adipocytes increased ERK2 phosphorylation (18). In addition, loss of TRPC1 decreased the number of larger adipocytes. These findings suggest that elimination of TRPC1-mediated Ca2+ entry in TRPC1 KO mice promotes suppression of autophagy in HF diet-fed and exercised mice resulting in increased adipocyte cell death. These results are consistent with previous studies where patients with metabolic syndrome also have higher serum Ca2+ levels (34, 35), which could be because of the loss of TRPC1 or other Ca2+ channels that mediate Ca2+ entry in adipocyte cells, thereby increasing serum Ca2+ levels.
Interestingly, in skeletal muscle, even though contraction does not depend on extracellular Ca2+ (36), Ca2+ entry through TRPC1 is essential for maintaining force during sustained repeated contractions as TRPC1 KO mice experience muscle fatigue during endurance exercise though spontaneous wheel running activity is unchanged (4). Our data are in agreement as we showed no alteration in voluntary exercise. However, a reduction in endurance exercise might be expected because loss of TRPC1 could impact mitochondrial respiration by altering Ca2+ homeostasis, because of an increase in total mitochondrial protein stimulated by exercise training (37, 38), and Ca2+ is needed for proper functioning of mitochondria (39). In addition, ER stress resulting from reduced Ca2+ entry could increase translocation of apoptotic factors into mitochondria thus permeabilizing the membrane, causing release of cytochrome c and activation of caspases, leading to mitochondria-mediated cell death (30, 40–42). These findings demonstrate that loss of TRPC1 disrupts Ca2+ homeostasis potentially resulting in mitochondria-mediated cell death of adipocytes. Although a previous study has shown that knock down of TRPC1 only attenuated nonstimulated Ca2+ influx in breast cancer cells (43), our results using adipocytes did not show any decrease in basal Ca2+ entry. These results suggest that although in breast cancer cells other Ca2+ influx channels (Orai1) might be more important for SOCE, TRPC1 is essential for adipocyte function, especially in blocking the effects of exercise in HF diet-induced obesity. The mechanism by which TRPC1 KO mice fed a HF diet and exercised are protected from obesity and type II diabetes risk needs further investigation. However, our study and another published study (18) indicate that loss of Ca2+ might be the main factor that inhibits the formation of metabolic syndrome.
Experimental procedures
Study design and animals
Four-month-old male B6129SF2/J (WT) or TRPC1 knock-out (KO) mice (The Jackson Laboratory, Bar Harbor, ME) were fed diets containing either 16% (normal-fat (NF)) or 45% fat (high-fat (HF)) for 12 weeks and subjected to voluntary wheel running exercise (exercise (E)) or sedentary cage activity (sedentary (S)). Experimental groups were labeled according to diet and exercise conditions yielding eight groups: WT-NF-E, WT-NF-S, WT-HF-E, WT-HF-S, KO-NF-E, KO-NF-S, KO-HF-E, and KO-HF-S. Food intake, body weight, and body composition were measured biweekly on alternating weeks during the experimental feeding period. After 12 weeks, mice were injected with xylazine (Akorn Inc., Decatur, IL) and ketamine (Zoetis Inc., Kalamazoo, MI) and then killed by exsanguination according to the animal use and care protocol approved by the USDA Agricultural Research Service Animal Care and Use Committee.
Calcium measurements and electrophysiology
Primary adipocyte cells were incubated with 2 μm fura-2 (Molecular Probes) for 45 min and then washed twice with Ca2+-free SES buffer as described in Liu et al.(27). For patch clamp experiments, coverslips with cells were transferred to the recording chamber and perfused with an external Ringer's solution of the following composition (mm): NaCl, 145; KCl, 5; MgCl2, 1; CaCl2, 1; Hepes, 10; Glucose, 10; pH 7.4 (NaOH). The patch pipette had resistances between 3 and 5 megaohms after filling with the standard intracellular solution that contained the following (mm): cesium methane sulfonate, 145; NaCl, 8; MgCl2, 10; Hepes, 10; EGTA, 10; pH 7.2 (CsOH). The maximum peak currents were calculated at a holding potential of −80 mV. The current-voltage (I/V) curves were made using a ramp protocol where current density was evaluated at various membrane potentials and plotted. For the tabulation of statistics, peak currents were used.
EchoMRI measurements of body composition
Whole body composition, including fat mass and lean mass, was determined biweekly during the 12-week period without sedation using nuclear magnetic resonance technology with the EchoMRI700™ instrument (Echo Medical Systems, Houston, TX).
Glucose tolerance test
At the end of 12 weeks of feeding, mice were fasted overnight and then injected with 2 g/kg body weight of 20% d-glucose (Sigma-Aldrich) intraperitoneally. Approximately 5 μl of tail blood was used to measure the blood glucose concentrations as described previously (14, 44) using the Accu-Chek Aviva glucometer at baseline and then 15, 30, 60, and 120 min post glucose injection.
Measurement of plasma insulin
Mice were fasted overnight and then plasma was obtained to analyze insulin concentrations (Insulin ELISA kit: EXRMI-13K, EMD Millipore, St. Charles, MO) as previously described (12) using the Bio-Rad Luminex system according to manufacturer's protocols.
Stromal vascular fraction (SVF) and primary adipocyte isolation
Subcutaneous and visceral adipose tissue were weighed and digested as described previously (12–14). Briefly, following digestion with collagenase type I (Gibco Thermo Fisher Scientific) at 37 °C for 1 h, adipose tissue cells were filtered using 100-μm nylon cell strainers (Corning Life Sciences) followed by centrifugation (1000 rpm, 10 min, 4 °C) to separate floating primary adipocytes (supernatant) from adipose SVF (cell pellet). The SVF cell pellet was treated with RBC lysis buffer (Sigma Aldrich) then quenched with DMEM + 10% FBS and the supernatant was washed and resuspended in 0.9% NaCl for adipose cell size and number determination using a Beckman Coulter Multisizer 4 with a 400-μm aperture. The instrument was set to count 6000 particles and the cell suspension was diluted so that coincident counting was <10%. After collection of pulse sizes, the data were expressed as cell numbers per particle diameter.
PCR analysis
Total RNA was extracted using the RNeasy Lipid Tissue Mini kit and Qiacube (Qiagen, Valencia, CA) from flash-frozen hind leg biceps femoris skeletal muscle or subcutaneous adipose tissue. cDNA was synthesized using the Quantitect Reverse Transcriptase kit (Qiagen, Valencia, CA) and then used to measure expression of GLUT4, HIF1α, FGF21, PPARγ, MAP1LC3A, and BECN1 by qPCR (ABI Prism 7500 PCR System, Applied Biosystems, Foster City, CA). FastStart Universal Probe Master (Rox) mix assay reagents were purchased from Roche. Primers were purchased from Integrated DNA Technology (IDT) (Coralville, IA). The endogenous control (18S rRNA) was purchased from Applied Biosystems. RT-PCR analysis for TRPC1 transcripts was done with primers from the eighth and ninth exons (forward, 5′-GCAACCTTTGCCCTCAAAGTG and reverse, 5′-GGAGGAACATTCCCAGAAATTTCC) after the EcoRI site (Eurofins MWG Operon, Huntsville, AL).
Protein extraction and immunoblotting
Protein was extracted from subcutaneous adipose tissue of WT and KO mice fed a HF diet and exercised, as described previously (13). 40 μg of proteins were resolved on NuPAGE Novex 4–12% Bis-Tris gels, transferred to nitrocellulose membranes, and probed with respective antibodies (all from Cell Signaling Technology). Respective peroxidase-conjugated secondary antibodies were used to label the proteins, which were then detected by an enhanced chemiluminescence detection kit (SuperSignal West Pico, Pierce). Densitometric analysis was performed using ImageJ (National Institutes of Health).
Statistical analysis
Data are reported as mean ± S.D. or mean ± S.E. The effects of TRPC1 KO, diet, or exercise were assessed by three-way analysis of variance (ANOVA) using GraphPad Prism 7. Statistical significance is denoted as + (mouse type), × (diet), and # (exercise) for main ANOVA interactions. When an interaction was significant (p < 0.05), Tukey contrasts were used to perform pairwise comparisons, which are reported as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Author contributions
B. B. S., J. N. R., and K. J. C.-L. designed the studies that were performed by D. K., A. S., Y. S., and P. S. The paper was written by D. K., A. S., B. B. S., J. N. R., and K. J. C.-L. All authors reviewed the results and approved the final manuscript.
Supplementary Material
Acknowledgments
We thank Amy N. Bundy and Mitch Rusten for providing technical assistance and data organization.
This work was supported by National Institutes of Health Grants R01DE017102, R01DE022765, R21DE024300, and P20GM113123 (to B. B. S.) and by the U.S. Department of Agriculture, Agricultural Research Service Project 3062–51000-052–00D (to K. J. C.-L.). The authors declare that they have no conflicts of interest with the contents of this article.The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or reflect the position or policy of the United States government.
This article contains supplemental Figs. 1–3.
- TRPC1
- transient receptor potential canonical channel-1
- TRPV
- vanilloid receptor-related TRP channels
- HF
- high fat
- ER
- endoplasmic reticulum
- NF
- normal-fat.
References
- 1. Filadi R., Theurey P., and Pizzo P. (2017) The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 62, 1–15 [DOI] [PubMed] [Google Scholar]
- 2. Qiu J., Fang Y., Rønnekleiv O. K., and Kelly M. J. (2010) Leptin excites proopiomelanocortin neurons via activation of TRPC channels. J. Neurosci. 30, 1560–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sukumar P., Sedo A., Li J., Wilson L. A., O'Regan D., Lippiat J. D., Porter K. E., Kearney M. T., Ainscough J. F., and Beech D. J. (2012) Constitutively active TRPC channels of adipocytes confer a mechanism for sensing dietary fatty acids and regulating adiponectin. Circ. Res. 111, 191–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zanou N., Shapovalov G., Louis M., Tajeddine N., Gallo C., Van Schoor M., Anguish I., Cao M. L., Schakman O., Dietrich A., Lebacq J., Ruegg U., Roulet E., Birnbaumer L., and Gailly P. (2010) Role of TRPC1 channel in skeletal muscle function. Am. J. Physiol. Cell Physiol. 298, C149–C162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Graham S., Yuan J. P., and Ma R. (2012) Canonical transient receptor potential channels in diabetes. Exp. Biol. Med. (Maywood) 237, 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sabourin J., Le Gal L., Saurwein L., Haefliger J. A., Raddatz E., and Allagnat F. (2015) Store-operated Ca2+ entry mediated by Orai1 and TRPC1 participates to insulin secretion in rat β-cells. J. Biol. Chem. 290, 30530–30539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hu G., Oboukhova E. A., Kumar S., Sturek M., and Obukhov A. G. (2009) Canonical transient receptor potential channels expression is elevated in a porcine model of metabolic syndrome. Mol. Endocrinol. 23, 689–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Venkatachalam K., and Montell C. (2007) TRP channels. Annu. Rev. Biochem. 76, 387–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pani B., and Singh B. B. (2009) Lipid rafts/caveolae as microdomains of calcium signaling. Cell Calcium 45, 625–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Montell C. (2005) The TRP superfamily of cation channels. Sci. STKE re3. [DOI] [PubMed] [Google Scholar]
- 11. Blüher M. (2009) Adipose tissue dysfunction in obesity. Exp. Clin. Endocrinol. Diabetes 117, 241–250 [DOI] [PubMed] [Google Scholar]
- 12. Claycombe K. J., Uthus E. O., Roemmich J. N., Johnson L. K., and Johnson W. T. (2013) Prenatal low-protein and postnatal high-fat diets induce rapid adipose tissue growth by inducing Igf2 expression in Sprague Dawley rat offspring. J. Nutr. 143, 1533–1539 [DOI] [PubMed] [Google Scholar]
- 13. Claycombe K. J., Vomhof-DeKrey E. E., Garcia R., Johnson W. T., Uthus E., and Roemmich J. N. (2016) Decreased beige adipocyte number and mitochondrial respiration coincide with increased histone methyl transferase (G9a) and reduced FGF21 gene expression in Sprague-Dawley rats fed prenatal low protein and postnatal high-fat diets. J. Nutr. Biochem. 31, 113–121 [DOI] [PubMed] [Google Scholar]
- 14. Claycombe K. J., Vomhof-DeKrey E. E., Roemmich J. N., Rhen T., and Ghribi O. (2015) Maternal low-protein diet causes body weight loss in male, neonate Sprague-Dawley rats involving UCP-1-mediated thermogenesis. J. Nutr. Biochem. 26, 729–735 [DOI] [PubMed] [Google Scholar]
- 15. Holzer R. G., Park E. J., Li N., Tran H., Chen M., Choi C., Solinas G., and Karin M. (2011) Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 147, 173–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ouguerram K., Maugeais C., Gardette J., Magot T., and Krempf M. (2006) Effect of n-3 fatty acids on metabolism of apoB100-containing lipoprotein in type 2 diabetic subjects. Br J. Nutr. 96, 100–106 [DOI] [PubMed] [Google Scholar]
- 17. Kuda O., Brezinova M., Rombaldova M., Slavikova B., Posta M., Beier P., Janovska P., Veleba J., Kopecky J. Jr., Kudova E., Pelikanova T., and Kopecky J. (2016) Docosahexaenoic acid-derived fatty acid esters of hydroxy fatty acids (FAHFAs) with anti-inflammatory properties. Diabetes 65, 2580–2590 [DOI] [PubMed] [Google Scholar]
- 18. Ye L., Kleiner S., Wu J., Sah R., Gupta R. K., Banks A. S., Cohen P., Khandekar M. J., Boström P., Mepani R. J., Laznik D., Kamenecka T. M., Song X., Liedtke W., Mootha V. K., Puigserver P., Griffin P. R., Clapham D. E., and Spiegelman B. M. (2012) TRPV4 is a regulator of adipose oxidative metabolism, inflammation, and energy homeostasis. Cell 151, 96–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gollisch K. S., Brandauer J., Jessen N., Toyoda T., Nayer A., Hirshman M. F., and Goodyear L. J. (2009) Effects of exercise training on subcutaneous and visceral adipose tissue in normal- and high-fat diet-fed rats. Am. J. Physiol. Endocrinol. Metab. 297, E495–E504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Craig B. W., Hammons G. T., Garthwaite S. M., Jarett L., and Holloszy J. O. (1981) Adaptation of fat cells to exercise: Response of glucose uptake and oxidation to insulin. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 51, 1500–1506 [DOI] [PubMed] [Google Scholar]
- 21. Stanford K. I., Middelbeek R. J., Townsend K. L., Lee M. Y., Takahashi H., So K., Hitchcox K. M., Markan K. R., Hellbach K., Hirshman M. F., Tseng Y. H., and Goodyear L. J. (2015) A novel role for subcutaneous adipose tissue in exercise-induced improvements in glucose homeostasis. Diabetes 64, 2002–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Witczak C. A., Wamhoff B. R., and Sturek M. (2006) Exercise training prevents Ca2+ dysregulation in coronary smooth muscle from diabetic dyslipidemic yucatan swine. J. Appl. Physiol. (1985) 101, 752–762 [DOI] [PubMed] [Google Scholar]
- 23. Liu X., Cheng K. T., Bandyopadhyay B. C., Pani B., Dietrich A., Paria B. C., Swaim W. D., Beech D., Yildrim E., Singh B. B., Birnbaumer L., and Ambudkar I. S. (2007) Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(−/−) mice. Proc. Natl. Acad. Sci. U.S.A. 104, 17542–17547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peinelt C., Vig M., Koomoa D. L., Beck A., Nadler M. J., Koblan-Huberson M., Lis A., Fleig A., Penner R., and Kinet J. P. (2006) Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat. Cell Biol. 8, 771–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yuan J. P., Kim M. S., Zeng W., Shin D. M., Huang G., Worley P. F., and Muallem S. (2009) TRPC channels as STIM1-regulated SOCs. Channels (Austin) 3, 221–225 [DOI] [PubMed] [Google Scholar]
- 26. Fiorio Pla A., Maric D., Brazer S. C., Giacobini P., Liu X., Chang Y. H., Ambudkar I. S., and Barker J. L. (2005) Canonical transient receptor potential 1 plays a role in basic fibroblast growth factor (bFGF)/FGF receptor-1-induced Ca2+ entry and embryonic rat neural stem cell proliferation. J. Neurosci. 25, 2687–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rao J. N., Platoshyn O., Golovina V. A., Liu L., Zou T., Marasa B. S., Turner D. J., Yuan J. X., and Wang J. Y. (2006) TRPC1 functions as a store-operated Ca2+ channel in intestinal epithelial cells and regulates early mucosal restitution after wounding. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G782–G792 [DOI] [PubMed] [Google Scholar]
- 28. Liu X., Wang W., Singh B. B., Lockwich T., Jadlowiec J., O'Connell B., Wellner R., Zhu M. X., and Ambudkar I. S. (2000) Trp1, a candidate protein for the store-operated Ca2+ influx mechanism in salivary gland cells. J. Biol. Chem. 275, 3403–3411 [DOI] [PubMed] [Google Scholar]
- 29. Kalupahana N. S., Claycombe K., Newman S. J., Stewart T., Siriwardhana N., Matthan N., Lichtenstein A. H., and Moustaid-Moussa N. (2010) Eicosapentaenoic acid prevents and reverses insulin resistance in high-fat diet-induced obese mice via modulation of adipose tissue inflammation. J. Nutr. 140, 1915–1922 [DOI] [PubMed] [Google Scholar]
- 30. Selvaraj S., Watt J. A., and Singh B. B. (2009) TRPC1 inhibits apoptotic cell degeneration induced by dopaminergic neurotoxin MPTP/MPP(+). Cell Calcium 46, 209–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bollimuntha S., Ebadi M., and Singh B. B. (2006) TRPC1 protects human SH-SY5Y cells against salsolinol-induced cytotoxicity by inhibiting apoptosis. Brain Res. 1099, 141–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pani B., Cornatzer E., Cornatzer W., Shin D. M., Pittelkow M. R., Hovnanian A., Ambudkar I. S., and Singh B. B. (2006) Up-regulation of transient receptor potential canonical 1 (TRPC1) following sarco(endo)plasmic reticulum Ca2+ ATPase 2 gene silencing promotes cell survival: a potential role for TRPC1 in Darier's disease. Mol. Biol. Cell 17, 4446–4458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sukumaran P., Sun Y., Vyas M., and Singh B. B. (2015) TRPC1-mediated Ca2+ entry is essential for the regulation of hypoxia and nutrient depletion-dependent autophagy. Cell Death Dis. 6, e1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sorva A., and Tilvis R. S. (1990) Low serum ionized to total calcium ratio: Association with geriatric diabetes mellitus and with other cardiovascular risk factors? Gerontology 36, 212–216 [DOI] [PubMed] [Google Scholar]
- 35. Saltevo J., Niskanen L., Kautiainen H., Teittinen J., Oksa H., Korpi-Hyövälti E., Sundvall J., Männistö S., Peltonen M., Mäntyselkä P., and Vanhala M. (2011) Serum calcium level is associated with metabolic syndrome in the general population: FIN-D2D study. Eur. J. Endocrinol. 165, 429–434 [DOI] [PubMed] [Google Scholar]
- 36. Armstrong C. M., Bezanilla F. M., and Horowicz P. (1972) Twitches in the presence of ethylene glycol bis(β-aminoethyl ether)-N,N′-tetracetic acid. Biochim. Biophys. Acta 267, 605–608 [DOI] [PubMed] [Google Scholar]
- 37. Holloszy J. O. (1967) Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 242, 2278–2282 [PubMed] [Google Scholar]
- 38. Scalzo R. L., Peltonen G. L., Binns S. E., Shankaran M., Giordano G. R., Hartley D. A., Klochak A. L., Lonac M. C., Paris H. L., Szallar S. E., Wood L. M., Peelor F. F. 3rd, Holmes W. E., Hellerstein M. K., Bell C., Hamilton K. L., and Miller B. F. (2014) Greater muscle protein synthesis and mitochondrial biogenesis in males compared with females during sprint interval training. FASEB J. 28, 2705–2714 [DOI] [PubMed] [Google Scholar]
- 39. Chan C. S., Gertler T. S., and Surmeier D. J. (2009) Calcium homeostasis, selective vulnerability and Parkinson's disease. Trends Neurosci. 32, 249–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wei M. C., Zong W. X., Cheng E. H., Lindsten T., Panoutsakopoulou V., Ross A. J., Roth K. A., MacGregor G. R., Thompson C. B., and Korsmeyer S. J. (2001) Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Budihardjo I., Oliver H., Lutter M., Luo X., and Wang X. (1999) Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 15, 269–290 [DOI] [PubMed] [Google Scholar]
- 42. Pan Z., Bhat M. B., Nieminen A. L., and Ma J. (2001) Synergistic movements of Ca2+ and Bax in cells undergoing apoptosis. J. Biol. Chem. 276, 32257–32263 [DOI] [PubMed] [Google Scholar]
- 43. Davis F. M., Peters A. A., Grice D. M., Cabot P. J., Parat M. O., Roberts-Thomson S. J., and Monteith G. R. (2012) Non-stimulated, agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on calcium entry. PLoS One 7, e36923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Claycombe K. J., Roemmich J. N., Johnson L., Vomhof-DeKrey E. E., and Johnson W. T. (2015) Skeletal muscle Sirt3 expression and mitochondrial respiration are regulated by a prenatal low-protein diet. J. Nutr. Biochem. 26, 184–189 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.