

Abstract
Quiescence (G0) is defined as an assortment of cell cycle arrested states that exhibit distinct properties. Leukemias harbor a subpopulation of G0 cells that can be enriched by growth factor deprivation or serum starvation. Target site reporters with shortened poly(A) tails show translation activation by microRNAs, via a noncanonical mechanism, when introduced into the nucleus of G0 cells. This is because recruitment by the activation causing FXR1a-microRNA-protein complex (FXR1a-microRNP) is nuclear and requires shortened poly(A) tails to avoid repressive factors and canonical translation. When introduced into the cytoplasm, target mRNAs and microRNAs are directed toward repression rather than translation activation. Leukemic cell lines are difficult to transfect but can be routinely nucleofected—where in vitro transcribed mRNA reporters and microRNAs are introduced into the nucleus of G0 leukemic cells. Nucleofection of a microRNA target reporter and either cognate, targeting microRNA, or control microRNA, into the nucleus of G0 cells, enables analysis of translation activation by microRNAs in G0. We discuss a modified protocol that we developed for transfection of mRNAs along with microRNAs to test translation regulation by microRNAs in G0 leukemic cells.
Keywords: MicroRNA, Noncanonical translation, Nucleofection, THP1 acute monocytic leukemia cell line, Quiescence, G0
1 Introduction
Quiescent (G0) cells are reversibly arrested cells, which are found in the body and in cancers [1–7]. Such cells show distinct properties [8], including resistance to harsh conditions [1, 2, 4, 5, 9–24]. Cells when subjected to specific stress conditions such as growth factor deprivation enter G0 transiently [3, 9]. G0 cells alter gene expression [14, 25–28]; in particular, at the translation level [29–31] where cells decrease canonical translation or protein synthesis [32, 33] and promote alternative, noncanonical modes of translation of specific genes that could enable G0 cell survival [29, 30]. We identified a noncanonical translation mechanism in G0 leukemic cells, which is mediated by microRNAs [29, 34–36]. MicroRNAs generally degrade mRNAs and repress their translation in proliferating cells, by base-pairing with specific sequences in mRNA 3′untranslated regions (UTRs) and by recruiting repressive factors to such mRNAs [37–42]. However, in G0 cells, microRNAs can activate translation of specific mRNAs by a noncanonical translation mechanism [29]. In G0 cells, microRNAs form a complex (microRNA-protein complex or microRNP) with RNA-binding proteins, FXR1a and AGO2, in the nucleus [34, 36, 42–45]. This specialized microRNP is recruited to the 3′ UTRs of specific target mRNAs that are unadenylated or possess a short poly (A) tail [29]. Short poly (A) tails avoid binding Poly (A) binding protein (PABP) that is involved in microRNA-mediated repression [37, 40], and in canonical translation that is decreased in G0 [32, 33]. Deadenylation of such target mRNAs in G0 cells is mediated by a cap-binding deadenylase protein called poly(A) ribonuclease (PARN) [29, 46, 47]. PARN becomes active in G0 cells and its binding to the 5′ cap is increased under these conditions [29, 46, 47]. PARN associates with FXR1a-microRNP that also interacts with p97/DAP5, an eIF4G paralog that brings in the 40S ribosomal subunit and mediates cap-dependent noncanonical translation of specific mRNAs in G0 cells, where canonical translation is reduced [29, 42, 48–51].
To study microRNA-mediated translation activation in G0 leukemic cells, we used luciferase reporter mRNAs bearing either a synthetic 3′UTR that possesses binding sites for a synthetic microRNA, or a specific gene 3′UTR bearing natural, endogenous microRNA-binding sites [52, 53]. Luciferase reporter mRNAs are synthesized by in vitro transcription, using T7 ultra mMESSAGE mMACHINE kit (Invitrogen™ Ambion™) with our modified protocol. The reporter mRNAs possessing gene-specific or synthetic microRNA target site 3′UTRs were generated with a 5′ Anti-reverse cap analog (ARCA) 7-methyl guanosine cap [54]. The reporter mRNAs were produced without a poly(A) tail to mimic the endogenous targets of activation that shorten their poly (A) tails to avoid PABP binding that can promote the repressive microRNP complex. The 3′ ends of the reporter mRNAs were protected by adding cordycepin (3′-deoxyadenosine analog of adenosine) that prevents transcript elongation and mRNA degradation [55].
Transfer of exogenous DNA or mRNA reporters allows us to study translation regulation in proliferating cells and upon their induction to G0. Many commercial methods have been developed to deliver exogenous DNA or RNA molecules into cultured cells. Nucleofection from Amaxa (now Lonza), an electroporation-based technology, allows for sufficient delivery of exogenous DNA or RNA molecules directly into the nucleus of a cell [56, 57]. Nucleofection [58] uses distinct sets of electrical parameters and buffers for each cell type to obtain high efficiency of transfection with low toxicity [57, 59]. Purified, in vitro transcribed reporter mRNAs along with their corresponding, targeting microRNAs can be nucleofected into the nucleus of cells of THP1 acute monocytic leukemic cell line.
In this chapter, we will outline nucleofection of reporter mRNAs along with their cognate or control microRNAs, to analyze microRNA-mediated translation activation in G0 leukemic THP1 cells. As an example, we show microRNA-dependent translation activation of in vitro transcribed CX-Firefly luciferase reporter mRNA in G0 THP1 cells (Fig. 1). CX-Firefly luciferase reporter mRNA is 5′ capped and unadenylated with four binding sites for the synthetic microRNA, miRcxcr4 [34, 53, 60]. CX-Firefly luciferase reporter mRNA, along with a control microRNA (miR30a), that does not bind the CX reporter 3′ UTRor a cognate microRNA (miRcxcr4) that can bind the 3′ UTR of the reporter mRNA, as well as Renilla luciferase reporter plasmid, were co-transfected by nucleofection. Renilla luciferase serves as a transfection and normalization control. Cells are allowed to grow in medium supplemented with serum for 24 h before being shifted to a medium without serum (G0 medium) for 42–48 h [29]. G0 cells are harvested for luciferase assay and translation efficiency is determined as the ratio of firefly luciferase activity normalized to renilla luciferase activity, and then further normalized for their RNAlevels [29, 34]. Furthermore, to monitor the efficiency of transfection, THP1 cells are nucleofected with a GFP-expressing plasmid using the nucleofection protocol described here (Fig. 2). This protocol can also be used for nucleofecting siRNAs, LNAinhibitors, plasmid DNA that overexpress a gene of interest, or an shRNA to knock down a specific gene, and then monitor the effect on microRNA-mediated translation activation in G0 cells.
Fig. 1.
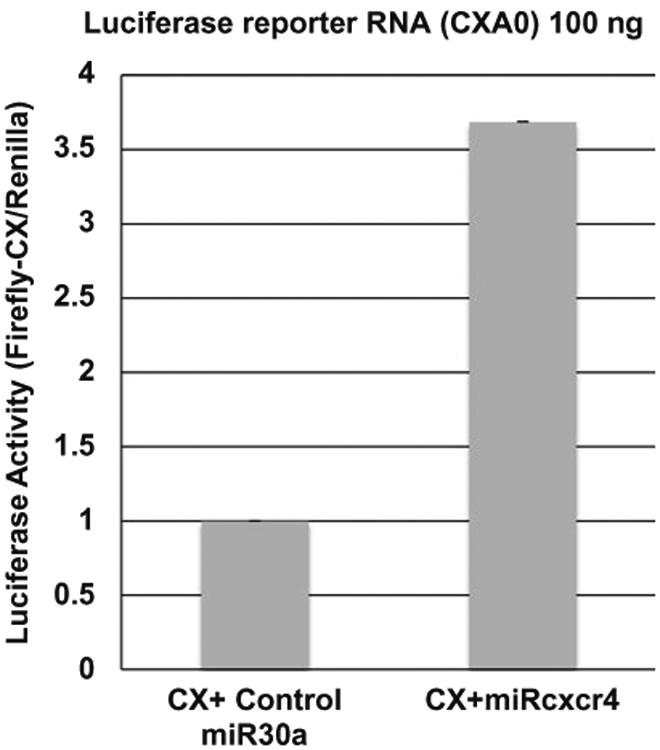
100 ng of in vitro transcribed capped CX-Firefly luciferase mRNA—without a poly(A) tail (CXA0) and with cordycepin added at the 3′ end—was nucleofected along with 20 ng of renilla plasmid, and 500 pmol of miRcxcr4 or control miR30a, into 1 × 106 THP1 cells. After nucleofection, cells were grown in RPMI medium supplemented with 10% fetal bovine serum (FBS) for 24 h, and then shifted to G0 medium (RPMI medium without FBS) for 42 h before analysis of luciferase activity. More than threefold increase in the translation of CX-firefly luciferase is observed in the presence of cognate microRNA miRcxcr4 compared to control microRNA miR30a
Fig. 2.
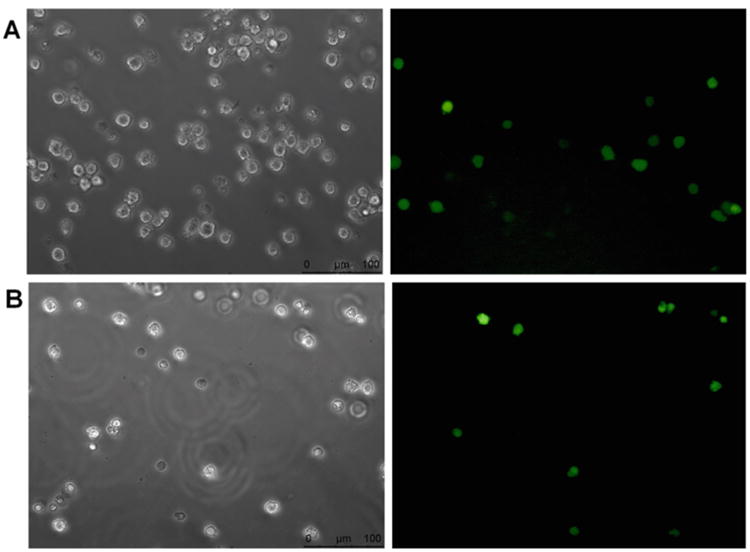
1 × 106 THP1 cells were nucleofected with 2.0 μg of pmax-GFP, using Kit V and nucleofector device II from Lonza. Left and right panels show 20× phase-contrast image and green fluorescent protein (GFP) positive THP1 cells respectively (images captured using a Leica DMI-4000B fluorescence inverted microscope). (A) Nucleofection program U-001 for high cell viability was used and transfection efficiency of about 24% was observed. (B) Program V-001 for high nucleofection efficiency was used and transfection efficiency of about 40% was achieved
2 Materials
All reagents and solutions to be used for nucleofection should be warmed to room temperature (unless indicated otherwise) prior to use. RNAs stored at −80 °C or the frozen plasmid DNA to be used for nucleofection should be thawed on ice prior to use. Nuclease-free water should be used at all times.
2.1 Materials for In Vitro Transcription of Capped mRNA
T7 ultramMESSAGE mMACHINE transcription system kit from Invitrogen™ Ambion™ is used to in vitro transcribe RNA molecules from a linearized template DNA of interest.
Components of the kit used in our reaction are nuclease-free water, T7 Enzyme mix, 10× T7 reaction buffer containing salts, buffer, dithiothreitol, T7 2× NTP/ARCA comprising of 15 mM ATP, 15 mM CTP, 15 mM UTP, 3 mM GTP and 12 mM ARCA, and Turbo DNase I (2 U/μl).
CX-Firefly luciferase plasmid DNA described in [34, 35]—containing a T7 RNA polymerase promoter site upstream of the luciferase reporter sequence for in vitro transcription—at a concentration of 0.5 μg/μl in nuclease-free water.
ApaI restriction enzyme for linearizing the CX-Firefly luciferase reporter plasmid containing a T7 RNA polymerase promoter and corresponding buffer (New England Biolabs).
1 mM Cordycepin (from Sigma).
3 M sodium acetate, or 5 M ammonium acetate for precipitation.
5× E-PAP buffer and enzyme (From Kit).
25 mM MnCl2 (From Kit).
Phenol:Chloroform (Prepared in the lab by mixing equal volume of saturated phenol with chloroform).
Ethanol, 100% (cold, −20 °C) and 70% (room temperature).
Glycogen.
Agarose gel.
10× TAE electrophoresis buffer.
Ethidium bromide staining solution.
Nuclease-free water.
2.2 Materials for Nucleofection
Nucleofector II device from Lonza (Amaxa).
Nucleofector solution Kit V (tested and validated for usage for THP1 cells by Lonza).
Certified cuvettes supplied with the kit.
Plastic bulb pipettes supplied with the kit.
pmaxGFP plasmid supplied with the kit, in vitro transcribed firefly-luciferase reporter mRNA, renilla plasmid, and control or cognate microRNA duplex.
A 10 cm cell culture plate or a 6-well cell culture plate.
Pre-warmed RPMI1640 culture medium containing 2 mM glutamine, 100 μg/ml streptomycin, 100 U/ml penicillin, and 10% fetal bovine serum (Gibco/Invitrogen).
Appropriate number of THP1 cells (1 × 106 cells per nucleofection reaction).
2.3 Materials for Transfection and Translation Analysis
pmaxGFP plasmid (supplied with nucleofection kit).
Dual-Luciferase® Reporter 1000 Assay System (Promega). Components of this kit are: Luciferase Assay Substrate (lyophilized), luciferase Assay Buffer II, Stop & Glo® Substrate (50×), Stop & Glo® Buffer, and Passive Lysis Buffer (5×).
Fluorescence microscope (Leica DMI-4000B).
Spectrophotometer—Nanodrop (ND1000 spectrophotometer).
TD 20/20 Luminometer (Turner Biosystems).
TRIzol® Reagent(Ambion by Life Technologies).
SuperScript™ III Reverse Transcriptase kit for cDNA synthesis (Invitrogen).
iTaq ™ Universal SYBR Green Supermix (BIO-RAD).
Real-time qPCR machine (Roche 480).
3 Methods
3.1 Plasmid DNA Template Linearization for In Vitro Transcription
Digest 5–10 μg of CX-Firefly luciferase reporter plasmid DNA described in [34] with ApaI restriction enzyme overnight at 25 °C in a 50–100 μl reaction volume. ApaI cuts downstream of the T7 promoter and the luciferase reporter sequence to be transcribed.
Extract the linearized plasmid with phenol:chloroform and precipitate the DNA with 3 M sodium acetate (1/10th of the reaction volume) and 100% (cold −20 °C) ethanol (2.5 times the reaction volume).
Mix well and incubate at −80 °C for minimum 1 hour and then centrifuge the tubes at maximum speed (12,000 × g) for 30 min at 4 °C. Remove the supernatant, wash the pellet with 70% ethanol at room temperature, and briefly spin the tubes again to collect and remove the residual fluid with a 0.2–10 μl pipette.
Air-dry the pellet on ice for 5 min and then resuspend the DNA to 1 μg/μl in nuclease-free water.
Run an aliquot (∼0.5 μg) of the resuspended DNA on a 1% agarose gel to check the linearization of the plasmid.
3.2 Synthesis of In Vitro Transcribed Capped mRNA, Protected at the 3′ End by Cordycepin
Take out the frozen components of mMESSAGE mMA-CHINE T7 kit and thaw at room temperature. The RNA polymerase enzyme mix (that is stored in glycerol and not frozen) should be placed directly on ice.
After thawing the components, vortex the 10× T7 reaction buffer and the T7 2× NTP/ARCA to ensure thorough mixing.
Briefly centrifuge the tubes to collect the reagents at the bottom of the tube before opening to prevent loss or contamination of the reagents that may be present around the cap or rim of the tube.
- Assemble the transcription reaction at room temperature and mix the reagents in the following order and amounts, for a 20 μl reaction volume:
- 4 μl of nuclease-free water.
- 10 μl of T7 2} NTP/ARCA.
- 2 μl of 10} T7 Reaction Buffer.
- 1 μg of 0.5 μg/μl linear template DNA.
- 2 μl T7 enzyme mix.
Mix the reaction mixture thoroughly and then briefly centrifuge the tubes.
Incubate the reaction mixture at 37 °C for 2–4 h to achieve maximum yield.
To the reaction mixture, add 1 μl of TURBO DNase, mix by pipetting up and down gently, and then incubate at 37 °C for 15 min (see Note 1).
To 20 μl of mMESSAGE mMACHINE T7 ultra reaction, add 1.5 μl of cordycepin (1 mM final concentration), 20 μl of 5× E-PAP buffer, 10 μl of 25 mM MnCl2, 43.5 μl of nuclease-free water, and 4 μl of E-PAP enzyme. Mix gently and incubate for 2 h at 37 °C.
After cordycepin addition, purify the in vitro transcribed RNA using phenol:chloroform. Add equal volume of saturated phenol:chloroform and vortex briefly (see Note 2).
Centrifuge the tubes at 12,000 × g for 15 min at4 °C for phase separation.
Transfer the upper aqueous layer to a new tube (see Note 3).
Add 80 μl of 5 M ammonium acetate, 1 μl of glycogen as well 2.5 times the volume of the supernatant of 100% cold ethanol (−20 °C). Mix properly and incubate at −80 °C for 1 hour.
Centrifuge the tubes at maximum speed for 40 min.
Remove the supernatant and wash pellet with 1 ml of 70% ethanol (room temperature).
Briefly centrifuge the tubes to collect the residual ethanol and remove using a 10 μl pipette (see Note 3).
Air-dry the pellet on ice for 5 min and then resuspend the pellet in appropriate volume of nuclease-free water.
Run RNA on an agarose gel to check integrity and size of the RNA (see Note 4).
Measure the concentration of the RNA solution on a nanodrop (ND1000 spectrophotometer) at absorbance 260 nm.
Freeze and store the in vitro transcribed mRNA at −80 °C for further use.
3.2.1 Synthesis of MicroRNAs
Synthetic microRNA (miRcxcr4) and control microRNA (miR30a) or a scrambled microRNA can be ordered as anti-sense (targeting microRNA) and sense RNA oligos that are modified with a 5′ phosphate. These RNA oligos are designed to base-pair with each other, with a bulge in the middle of the RNA oligos (at nucleotides (nt) 9–11 of the 19 nt oligos) [34, 61]. The anti-sense strand binds the target mRNA with imperfect base pairing.
Each of the individually synthesized sense and anti-sense microRNA strands is resuspended at 200 pmol/μl concentration in annealing buffer (10 mM potassium acetate and 1 mM EDTA), as described in [34, 61].
Measure resuspended anti-sense and sense microRNA strands and then mix in equimolar proportions.
Anneal mixed RNA oligos by heating the mixture to 95 °C for 5 min and allow to cool down slowly to room temperature for annealing of the sense and anti-sense strands.
Annealed microRNAs are nucleofected as duplexes as described in [3, 34, 53, 60].
3.3 Nucleofection of Plasmid DNA or In Vitro Transcribed mRNA along with MicroRNAs into THP1 Cells
3.3.1 Culturing THP1 Cells [29]
THP1 cells from ATCC are thawed in RPMI-1640 culture medium, with 2 mM Glutamine, 100 μg/ml streptomycin, 100 U/ml penicillin, and 10% fetal bovine serum (FBS) added.
Cells should be allowed to recover after thawing for 2–3 days by incubating in a 37 °C humidified incubator supplied with 5% CO2.
Passage cells at least two times prior to starting the experiment. Allow cells to reach an optimal density of 1 × 106 cells/ml in a T75 flask (30 ml per flask).
Replace the medium two to three times a week and maintain the cell density of 3–4 × 105 cells/ml every time the medium is changed.
Seed out 2 × 105 cells/ml and subculture for 2–3 days before nucleofection.
3.3.2 Nucleofecting THP1 Cells
Prepare the nucleofector mixed solution by mixing Nucleofector™ solution V and Supplement 1 solution, as instructed by Lonza. We prepare nucleofector mixed solution in a sterile 1.5 ml eppendorf tube by mixing 82 μl of Nucleofector™ solution V with 18 μl of Supplement 1 solution in a 4.5:1 ratio to make a total of 100 μl solution for a single nucleofection reaction (see Note 5).
Prepare a 10 cm plate with 10 ml of pre-warmed culture medium for every single reaction.
Count an aliquot of cells to determine the cell density.
For a single nucleofection, centrifuge 1 × 106 THP1 cells at 1000 rpm (230 × g) in a microcentrifuge for 3–5 min. Remove the supernatant completely without disturbing the cell pellet (see Note 6).
Resuspend the cell pellet gently in the 100 μl prepared nucleofection solution at room temperature, following the instructions from Lonza (see Note 7).
Mix the resuspended cell suspension with 100–200 ng of in vitro transcribed CX-Firefly luciferase reporter mRNA, 500 pmoles of control microRNA or miRcxcr4, and 20 ng of renilla plasmid (see Note 8).
Transfer cell/nucleic acid suspension into the bottom of a certified cuvette (Lonza) using a plastic bulb pipette (Lonza). Close the cuvette with the cap (see Note 9).
Select the appropriate nucleofector program in the Nucleofector device II—for nucleofection of THP1 cells V-001 for high efficiency or U-001 for high viability, as recommended by Lonza (see Note 10).
Place the cuvette containing cells and nucleic acid suspension into the Nucleofector cuvette holder and apply the selected program, following the instructions from Lonza.
Take the cuvette out of the holder once the display shows OK, following the instructions from Lonza.
Add 200–300 μl of pre-warmed culture medium to the cuvette and carefully transfer the cell suspension into the prepared 10 cm culture plate using the plastic bulb pipette, as recommended by Lonza.
Incubate the nucleofected cells at 37 °C in a humidified incubator supplied with 5% CO2 for 24–30 h, post nucleofection (see Note 11).
Change the medium to serum-free medium (G0 medium) and incubate the cells at 37 °C in a humidified incubator supplied with 5% CO2 for 42–48 h until analysis.
For luciferase reporter assay, G0 cells should be harvested by centrifuging at 1500 rpm (530 × g) for 10 min at 4 °C. Remove the supernatant completely and resuspend cells in 50 μl of 1× passive lysis buffer (1 × PLB buffer) (see Note 12).
Freeze the cells resuspended in 1× PLB buffer at —80 °C overnight and ensure complete lysis of the cells.
Thaw the frozen samples and take out 10 μl of the sample for luciferase assay.
Measure firefly and renilla luciferase activity sequentially, as per the manufacturer's guidelines on a TD 20/20 luminometer, using an appropriate dual luciferase measurement program. First, add 100 μl of luciferase assay substrate to 10 μl of the sample, mix by pipetting and measure firefly luciferase activity using the dual luciferase program on the TD 20/20 luminometer. Firefly Luciferase activity is then quenched by adding 100 μl of Stop & Glo solution and renilla luciferase activity is measured.
Translation efficiency is calculated by normalizing the ratio of firefly luciferase activity to renilla luciferase and then further normalized to their RNA levels that were measured by qPCR as described in [29] (see Note 13).
4 Notes
Make sure to treat the transcription mixture with DNase at this step, as any DNA contamination will interfere with downstream steps.
Cordycepin is an adenosine analog and should be completely removed, as it inhibits polyadenylation and translation [55]. Remove all free nucleotides and ARCA completely, as unincorporated ARCA is also an inhibitor of translation.
The upper aqueous layer should be carefully transferred to new eppendorfs without any phenol contamination upon phenol: chloroform extraction. Remove all residual ethanol and dry the RNA pellet on ice. Residual ethanol or phenol contamination will impact the nanodrop readouts while measuring the RNA concentration and will affect the nucleofection efficiency as well.
Run the in vitro transcribed mRNA on a 1% TAE agarose gel to check RNA integrity and size. CX-Firefly luciferase mRNA will run around 2 kb (1.8 kb luciferase with an additional 150 nucleotides for miRcxcr4-binding sites).
The mixture of Nucleofector™ Solution V and Supplement 1 solution from the nucleofection kit (nucleofector mixed solution), is stable only for 3 months at 4 °C, according to the instructions from Lonza.
Cell numbers lower than the minimal recommended number could lead to an increase in cell death; cell numbers more than the recommended number could influence nucleofection efficiency, as recommended by Lonza [57].
Cells should not be left in the nucleofector solution for more than 15 min, as this may affect the nucleofection efficiency and decrease cell viability, as recommended by Lonza [57].
For a 100 μl reaction containing 1 × 106 cells, increasing nucleic acid concentrations for nucleofection more than recommended may lead to increased cell death, according to the instructions from Lonza [57].
Carefully transfer the cell suspension with nucleic acid to the bottom of the cuvette and avoid air bubbles that may decrease gene transfer efficiency, as instructed by Lonza. Cap of the cuvette should be closed every time to prevent spills or contamination, as instructed by Lonza [57].
Appropriate program should be selected based on the downstream requirement of the experiment. For high efficiency of gene transfer program V-001 should be selected and if more viable cells are required the program U-001 should be used, as recommended by Lonza.
After nucleofection, cells should be allowed to grow in medium supplemented with serum for more than 24 h to alleviate the stress induced by nucleofection [29].
1× Passive lysis buffer (PLB) should be made fresh and if the cell lysate is to be used for Western blot analysis, appropriate amount of protease inhibitors should be added [29].
To measure firefly and renilla luciferase RNA levels, isolate RNA from the nucleofected samples dissolved in 1× PLB using TRIzol reagent. Prepare cDNA using random hexamer primers with superscript III kit following the manufacturer's instructions. Firefly luciferase mRNA levels can be measured by qPCR using Roche 480 real-time qPCR machine, following the manufacturer's instructions using primers; FF-F3: 5′-TTCCATCTTCCAGGGATACG-3′ and FF-R3: 5′-ATCCA-GATCCACAACCTTCG-3′ and normalized to tRNA-Lys using primers; tRNA-Lys Forward: 5′-GCCCGGATAGCTCAGTCGGTAGAG-3′ and tRNA-Lys Reverse: 5′-CGCCCGAACAGGGACTTGAACCC-3′. Renilla mRNA levels are measured using primers; Renilla (Ren1) Forward; 5′-CCATGATAATGTTGGACGAC-3′ and Renilla (Ren2) Reverse; 5′-GGCACCTCCAACAATAGCATTG-3′ and normalized to tRNA-Lys. Normalized firefly and renilla mRNA levels were used to further normalize the firefly luciferase activity and renilla activity to obtain the translation efficiency [29, 34].
References
- 1.Pardee AB. A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci U S A. 1974;71(4):1286–1290. doi: 10.1073/pnas.71.4.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aragon AD, Rodriguez AL, Meirelles O, Roy S, Davidson GS, Tapia PH, Allen C, Joe R, Benn D, Werner-Washburne M. Characterization of differentiated quiescent and nonquiescent cells in yeast stationary-phase cultures. Mol Biol Cell. 2008;19(3):1271–1280. doi: 10.1091/mbc.E07-07-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coller HA, Sang L, Roberts JM. A new description of cellular quiescence. PLoS Biol. 2006;4(3):e83. doi: 10.1371/journal.pbio.0040083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, Arruda A, Popescu A, Gupta V, Schimmer AD, Schuh AC, Yee KW, Bullinger L, Herold T, Gorlich D, Buchner T, Hiddemann W, Berdel WE, Wormann B, Cheok M, Preudhomme C, Dombret H, Metzeler K, Buske C, Lowenberg B, Valk PJ, Zandstra PW, Minden MD, Dick JE, Wang JC. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540(7633):433–437. doi: 10.1038/nature20598. [DOI] [PubMed] [Google Scholar]
- 5.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 6.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501(7467):328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crews LA, Jamieson CH. Selective elimination of leukemia stem cells: hitting a moving target. Cancer Lett. 2013;338(1):15–22. doi: 10.1016/j.canlet.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 8.Bhola PD, Mar BG, Lindsley RC, Ryan JA, Hogdal LJ, Vo TT, DeAngelo DJ, Galinsky I, Ebert BL, Letai A. Functionally identifiable apoptosis-insensitive subpopulations determine chemoresistance in acute myeloid leukemia. J Clin Investig. 2016;126(10):3827–3836. doi: 10.1172/JCI82908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sang L, Coller HA, Roberts JM. Control of the reversibility of cellular quiescence by the transcriptional repressor HES1. Science. 2008;321(5892):1095–1100. doi: 10.1126/science.1155998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tavaluc RT, Hart LS, Dicker DT, El-Deiry WS. Effects of low confluency, serum starvation and hypoxia on the side population of cancer cell lines. Cell Cycle. 2007;6(20):2554–2562. doi: 10.4161/cc.6.20.4911. [DOI] [PubMed] [Google Scholar]
- 11.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138(4):645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemons JM, Feng XJ, Bennett BD, Legesse-Miller A, Johnson EL, Raitman I, Pollina EA, Rabitz HA, Rabinowitz JD, Coller HA. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol. 2010;8(10):e1000514. doi: 10.1371/journal.pbio.1000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindeman GJ, Visvader JE. Insights into the cell of origin in breast cancer and breast cancer stem cells. Asia Pac J Clin Oncol. 2010;6(2):89–97. doi: 10.1111/j.1743-7563.2010.01279.x. [DOI] [PubMed] [Google Scholar]
- 14.Salony SX, Alves CP, Dey-Guha I, Ritsma L, Boukhali M, Lee JH, Chowdhury J, Ross KN, Haas W, Vasudevan S, Ramaswamy S. AKT inhibition promotes nonautonomous cancer cell survival. Molecular cancer therapeutics. 2016;15(1):142–153. doi: 10.1158/1535-7163.MCT-15-0414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dey-Guha I, Wolfer A, Yeh AC, GA J, Darp R, Leon E, Wulfkuhle J, Petricoin EF, 3rd, Wittner BS, Ramaswamy S. Asymmetric cancer cell division regulated by AKT. Proc Natl Acad Sci U S A. 2011;108(31):12,845–12,850. doi: 10.1073/pnas.1109632108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng X, Seshire A, Ruster B, Bug G, Beissert T, Puccetti E, Hoelzer D, Henschler R, Ruthardt M. Arsenic but not all-trans retinoic acid overcomes the aberrant stem cell capacity of PML/RARalpha-positive leukemic stem cells. Haematologica. 2007;92(3):323–331. doi: 10.3324/haematol.10541. [DOI] [PubMed] [Google Scholar]
- 17.Li L, Bhatia R. Stem cell quiescence. Clin Cancer Res: Off J Am Assoc Cancer Res. 2011;17(15):4936–4941. doi: 10.1158/1078-0432.CCR-10-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barnes DJ, Melo JV. Primitive, quiescent and difficult to kill: the role of non-proliferating stem cells in chronic myeloid leukemia. Cell Cycle. 2006;5(24):2862–2866. doi: 10.4161/cc.5.24.3573. [DOI] [PubMed] [Google Scholar]
- 19.Goldman J, Gordon M. Why do chronic myelogenous leukemia stem cells survive allogeneic stem cell transplantation or imatinib: does it really matter? Leuk Lymphoma. 2006;47(1):1–7. doi: 10.1080/10428190500407996. [DOI] [PubMed] [Google Scholar]
- 20.Reed JC. Molecular biology of chronic lymphocytic leukemia: implications for therapy. Semin Hematol. 1998;35(3 Suppl 3):3–13. [PubMed] [Google Scholar]
- 21.Giles FJ, DeAngelo DJ, Baccarani M, Deinin-ger M, Guilhot F, Hughes T, Mauro M, Radich J, Ottmann O, Cortes J. Optimizing outcomes for patients with advanced disease in chronic myelogenous leukemia. Semin Oncol. 2008;35(1 Suppl 1):S1–17. doi: 10.1053/j.seminoncol.2007.12.002. quiz S18–20. [DOI] [PubMed] [Google Scholar]
- 22.Krause A, Luciana M, Krause F, Rego EM. Targeting the acute myeloid leukemia stem cells. Anti Cancer Agents Med Chem. 2010;10(2):104–110. doi: 10.2174/187152010790909281. [DOI] [PubMed] [Google Scholar]
- 23.Besancon R, Valsesia-Wittmann S, Puisieux A, Caron de Fromentel C, Maguer-Satta V. Cancer stem cells: the emerging challenge of drug targeting. Curr Med Chem. 2009;16(4):394–416. doi: 10.2174/092986709787315531. [DOI] [PubMed] [Google Scholar]
- 24.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 25.Le Tonqueze O, Kollu S, Lee S, Al-Salah M, Truesdell SS, Vasudevan S. Regulation of monocyte induced cell migration by the RNA binding protein, FXR1. Cell Cycle. 2016;15(14):1874–1882. doi: 10.1080/15384101.2016.1189040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 30 untranslated regions and fewer microRNA target sites. Science. 2008;320(5883):1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138(4):673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol. 2013;14(6):329–340. doi: 10.1038/nrm3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bukhari SI, Truesdell SS, Lee S, Kollu S, Classon A, Boukhali M, Jain E, Mortensen RD, Yanagiya A, Sadreyev RI, Haas W, Vasudevan S. A Specialized Mechanism of Translation Mediated by FXR1a-Associated MicroRNP in Cellular Quiescence. Mol Cell. 2016;61(5):760–773. doi: 10.1016/j.molcel.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee S, Truesdell SS, Bukhari SI, Lee JH, LeTonqueze O, Vasudevan S. Upregulation of eIF5B controls cell-cycle arrest and specific developmental stages. Proc Natl Acad Sci U S A. 2014;111(41):E4315–E4322. doi: 10.1073/pnas.1320477111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loayza-Puch F, Drost J, Rooijers K, Lopes R, Elkon R, Agami R. p53 induces transcriptional and translational programs to suppress cell proliferation and growth. Genome Biol. 2013;14(4):R32. doi: 10.1186/gb-2013-14-4-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136(4):731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318(5858):1931–1934. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- 35.Mortensen RD, Serra M, Steitz JA, Vasudevan S. Posttranscriptional activation of gene expression in Xenopus laevis oocytes by microRNA-protein complexes (microRNPs) Proc Natl Acad Sci U S A. 2011;108(20):8281–8286. doi: 10.1073/pnas.1105401108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Truesdell SS, Mortensen RD, Seo M, Schroeder JC, Lee JH, LeTonqueze O, Vasudevan S. MicroRNA-mediated mRNA translation activation in quiescent cells and oocytes involves recruitment of a nuclear microRNP. Sci Rep. 2012;2:842. doi: 10.1038/srep00842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16(7):421–433. doi: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- 38.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5(7):522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 39.Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. 2013;14(8):475–488. doi: 10.1038/nrm3611. [DOI] [PubMed] [Google Scholar]
- 40.Fabian MR, Sundermeier TR, Sonenberg N. Understanding how miRNAs post-transcriptionally regulate gene expression. Prog Mol Subcell Biol. 2010;50:1–20. doi: 10.1007/978-3-642-03103-8_1. [DOI] [PubMed] [Google Scholar]
- 41.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bukhari SI, Vasudevan S. FXR1a-associated microRNP: A driver of specialized non-canonical translation in quiescent conditions. RNA Biol. 2017;14(2):137–145. doi: 10.1080/15476286.2016.1265197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vasudevan S, Steitz JA. AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell. 2007;128(6):1105–1118. doi: 10.1016/j.cell.2007.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dube M, Huot ME, Khandjian EW. Muscle specific fragile X related protein 1 isoforms are sequestered in the nucleus of undifferentiated myoblast. BMC Genet. 2000;1:4. doi: 10.1186/1471-2156-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siomi MC, Zhang Y, Siomi H, Dreyfuss G. Specific sequences in the fragile X syndrome protein FMR1 and the FXR proteins mediate their binding to 60S ribosomal subu-nits and the interactions among them. Mol CellBiol. 1996;16(7):3825–3832. doi: 10.1128/mcb.16.7.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dehlin E, Wormington M, Korner CG, Wahle E. Cap-dependent deadenylation of mRNA. EMBO J. 2000;19(5):1079–1086. doi: 10.1093/emboj/19.5.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korner CG, Wormington M, Muckenthaler M, Schneider S, Dehlin E, Wahle E. The deadenylating nuclease (DAN) is involved in poly(A) tail removal during the meiotic maturation of Xenopus oocytes. EMBO J. 1998;17(18):5427–5437. doi: 10.1093/emboj/17.18.5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levy-Strumpf N, Deiss LP, Berissi H, Kimchi A. DAP-5, a novel homolog of eukaryotic translation initiation factor 4G isolated as a putative modulator of gamma interferon-induced programmed cell death. Mol Cell Biol. 1997;17(3):1615–1625. doi: 10.1128/mcb.17.3.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gradi A, Imataka H, Svitkin YV, Rom E, Raught B, Morino S, Sonenberg N. A novel functional human eukaryotic translation initiation factor 4G. Mol Cell Biol. 1998;18(1):334–342. doi: 10.1128/mcb.18.1.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamanaka S, Zhang XY, Maeda M, Miura K, Wang S, Farese RV, Jr, Iwao H, Innerarity TL. Essential role of NAT1/p97/DAP5 in embryonic differentiation and the retinoic acid pathway. EMBO J. 2000;19(20):5533–5541. doi: 10.1093/emboj/19.20.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sugiyama H, Takahashi K, Yamamoto T, Iwasaki M, Narita M, Nakamura M, Rand TA, Nakagawa M, Watanabe A, Yamanaka S. Nat1 promotes translation of specific proteins that induce differentiation of mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2017;114(2):340–345. doi: 10.1073/pnas.1617234114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu L, Fan J, Belasco JG. MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci U S A. 2006;103(11):4034–4039. doi: 10.1073/pnas.0510928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18(5):504–511. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pasquinelli AE, Dahlberg JE, Lund E. Reverse 5′ caps in RNAs made in vitro by phage RNA polymerases. RNA. 1995;1(9):957–967. [PMC free article] [PubMed] [Google Scholar]
- 55.Penman S, Rosbash M, Penman M. Messenger and heterogeneous nuclear RNA in HeLa cells: differential inhibition by cordycepin. Proc Natl Acad Sci U S A. 1970;67(4):1878–1885. doi: 10.1073/pnas.67.4.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hohenstein KA, Pyle AD, Chern JY, Lock LF, Donovan PJ. Nucleofection mediates high-efficiency stable gene knockdown and transgene expression in human embryonic stem cells. Stem Cells. 2008;26(6):1436–1443. doi: 10.1634/stemcells.2007-0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maess MB, Wittig B, Lorkowski S. Highly efficient transfection of human THP-1 macrophages by nucleofection. J Vis Exp. 2014;91:e51960. doi: 10.3791/51960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gresch O, Engel FB, Nesic D, Tran TT, England HM, Hickman ES, Korner I, Gan L, Chen S, Castro-Obregon S, Hammermann R, Wolf J, Muller-Hartmann H, Nix M, Siebenkotten G, Kraus G, Lun K. New non-viral method for gene transfer into primary cells. Methods. 2004;33(2):151–163. doi: 10.1016/j.ymeth.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 59.Marchenko S, Flanagan L. Transfecting human neural stem cells with the Amaxa Nucleofector. J Vis Exp. 2007;6:240. doi: 10.3791/240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Genes Dev. 2003;17(4):438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vasudevan S. Functional validation of microRNA-target RNA interactions. Methods. 2012;58(2):126–134. doi: 10.1016/j.ymeth.2012.08.002. [DOI] [PubMed] [Google Scholar]