

Abstract
Background
Autosomal dominant anhidrotic ectodermal dysplasia with immune deficiency (AD EDA-ID) is caused by heterozygous point mutations at or close to S32 and S36 or N-terminal truncations in IκBα that impair its phosphorylation and degradation, and thus activation of the canonical NF-κB pathway. The outcome of hematopoietic stem cell transplantation is poor in AD EDA-ID despite achievement of chimerism. Mice heterozygous for the S32I mutation in IκBα have impaired non-canonical NF-κB activity and defective lymphorganogenesis.
Objective
To establish genotype-phenotype correlation in AD EDA-ID.
Methods
A disease severity scoring system was devised. Stability of IκBα mutants was examined in transfected cells. Immunological, biochemical, and gene expression analyses were performed to evaluate canonical and non-canonical NF-κB signaling in skin-derived fibroblasts.
Results
Disease severity was greater in patients with IκBα point mutations than in patients with truncation mutations. IκBα point mutants were expressed at significantly higher levels in transfectants compared to truncation mutants. Canonical NF-κB-dependent IL-6 secretion and upregulation of the NF-κB2/p100 and RelB components of the non-canonical NF-κB pathway were diminished significantly more in patients with point mutations compared to those with truncations. Non-canonical NF-κB-driven generation of the transcriptionally active p100 cleavage product p52, and upregulation of CCL20, ICAM1 and VCAM1, important for lymphorganogenesis, were diminished significantly more in LPS+α-LTβR-stimulated fibroblasts from patients with point mutations compared to those with truncations.
Conclusions
IκBα point mutants accumulate at higher levels compared to truncation mutants and are associated with more severe disease and greater impairment of canonical and non-canonical NF-κB activity in AD EDA-ID.
Keywords: ectodermal dysplasia with immune deficiency, IκBα, canonical NF-κB pathway, non-canonical NF-κB pathway, lymphorganogenesis, hematopoietic stem cell transplantation
Graphical abstract
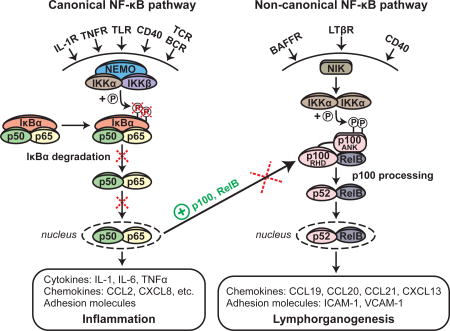
ANK, Ankyrin repeat domain; BAFFR, B cell-activating factor receptor; BCR, B cell receptor IL-1R, Interleukin-1 receptor; LTβR, Lymphotoxin beta receptor; RHD, Rel homology domain TCR, T cell receptor; TLR, Toll-like receptor; TNFR, Tumor necrosis factor receptor
INTRODUCTION
Primary immunodeficiency diseases (PID) are often monogenic with more than 300 genes implicated to date.1, 2 There is a wide phenotypic heterogeneity in the presentation, age of onset, and outcome of individual PIDs. This heterogeneity reflects in part the influence of the genotype. Genotype-phenotype correlations have been important in predicting the severity, outcome, and custom tailoring of the treatment of a number of PIDs.3–7 The inhibitor of κB (IκB) proteins inhibit the transcription factor NF-κB (nuclear factor kappa light chain enhancer in activated B cells).8 Heterozygous mutations in the IκB family member IκBα cause autosomal dominant anhidrotic ectodermal dysplasia with immune deficiency (AD EDA-ID), characterized by sparse hair, conical teeth, reduced number of sweat glands, and susceptibility to severe infections.9 The literature indicates a spectrum of disease in AD EDA-ID with variable severity and patient outcome after hematopoietic stem cell transplantation (HSCT), but a genotype-phenotype correlation has not been established.
The NF-κB family of transcription factors includes RelA (p65), RelB, c-Rel, NF-κB1 (p50/p105) and NF-κB2 (p52/p100) and plays a critical role in innate and adaptive immunity.8, 10, 11 In resting cells, NF-κB is retained in the cytoplasm through association with members of the IκB family of proteins. Two distinct pathways leading to NF-κB activation and nuclear translocation have been identified: a canonical and a non-canonical NF-κB pathway.12 The canonical, or classical, NF-κB pathway is used by a broad range of cell surface receptors, including toll-like receptors (TLRs), interleukin-1 (IL-1) receptor, tumor necrosis factor alpha (TNFα) receptor, antigen receptors, and others.8, 11 Receptor ligation causes activation of the IκB kinase (IKK) complex, which consists of the catalytic subunits IKKα and IKKβ and the scaffolding protein IKKγ/NEMO. This results in the phosphorylation of IκBα at serines 32 (S32) and 36 (S36), priming it for ubiquitination and proteasomal degradation. The released p50:p65 complex translocates into the nucleus where it induces the transcription of genes that mediate the inflammatory response.13, 14 The non-canonical NF-κB pathway is used by a limited range of cell-surface receptors, including lymphotoxin beta (LTβ) receptor, B cell activating factor (BAFF) receptor, and CD40. Receptor ligation drives NF-κB inducing kinase (NIK) to phosphorylate IKKα.15 IKKα subsequently phosphorylates NF-κB2/p100, which is in complex with RelB and contains an IκB-like domain that prevents nuclear translocation of the p100:RelB complex in resting cells. Phosphorylated p100 undergoes polyubiquitination and proteasomal processing to p52. The p52:RelB dimers translocate to the nucleus and activate transcription of genes that include chemokines and adhesion molecules involved in lymphorganogenesis.15, 16 There is crosstalk between the two NF-κB pathways: NIK-dependent activation of IKKα has been reported to cause IκBα phosphorylation and degradation.17 Importantly, transcription of p100 and RelB, as well as IκBα, is regulated by p50:p65.18–21
Two types of human IκBα mutations have been described in AD EDA-ID: mutations that result in a single amino acid substitution at or adjacent to the S32 and S36 phosphorylation sites (‘point mutations’), and mutations that introduce a premature stop codon and give rise to N-terminally truncated IκBα proteins through re-initiation of translation at downstream ATG sites (‘truncation mutations’). Both types of mutations impair signal-induced phosphorylation of IκBα and protect the mutant protein from subsequent ubiquitination and proteasomal degradation. By sequestering the NF-κB transcription factors p50 and p65 in the cytoplasm, the non-degradable IκBα mutant protein interferes with activation of the canonical NF-κB pathway. Eight AD EDA-ID patients carrying six IκBα mutations have been reported to date.22–29 and more cases are known to exist or have been submitted for publication.30 TLR-driven cytokine production is impaired in these patients. Circulating T and B cell numbers are high, but the percentages of memory T and B cells are decreased. In vitro T cell function varies from decreased to intact. In transfection studies, all IκBα mutants, except W11X, inhibited NF-κB activation more than WT IκBα, and thus have been demonstrated to function as dominant negative (DN). A majority of AD EDA-ID cases reported have undergone HSCT. However, despite good donor cell engraftment, the outcome of HSCT has been poor, with persistent recurrent and chronic bacterial and viral infections, poor antibody responses to immunizations and need for immunoglobulin (Ig) replacement, suggesting a contribution from non-hematopoietic cells to residual disease after transplantation.30–32
A knock-in mouse heterozygous for the S32I mutation in IκBα shows ectodermal dysplasia, impaired phosphorylation and degradation of the mutant IκBα protein, and diminished response to TLR ligation.33 Strikingly, the S32I mutant completely lacks lymph nodes (LNs), Peyer’s patches, marginal zone B cells and follicular dendritic cells, and fails to form germinal centers, features typical of defective non-canonical NF-κB signaling that have remained largely unaddressed in AD EDA-ID patients.17, 34–45 WT->S32I IκBα chimeras failed to form proper lymphoid organs and to reconstitute immune function despite excellent donor cell engraftment, paralleling the failure of HSCT to cure AD EDA-ID patients. In contrast, S32I IκBα->Rag2−/−bone marrow (BM) chimeras developed proper lymphoid organs. These results suggest that defective stromal cell function may contribute to the immunodeficiency in AD EDA-ID, and may explain the poor outcome of HSCT in this disease.
The mechanism by which mutations in IκBα also affect the non-canonical NF-κB pathway in patients with AD EDA-ID is not well understood. In this study, we analyzed seven published and two unreported AD EDA-ID cases.22–28 We demonstrate that IκBα point mutants are expressed at higher levels and impair the canonical and non-canonical NF-κB pathway more severely than IκBα truncation mutants, and thus result in more severe disease. Our study establishes a genotype-phenotype correlation in AD EDA-ID, demonstrates a defect in the non-canonical NF-κB signaling in patients with AD EDA-ID, and has important implications for the management of these patients.
MATERIALS AND METHODS
Cell lines
Skin fibroblast cell lines were established from skin punch biopsies obtained from patients and three adult healthy donors after securing informed consent using protocols approved by the Ethics Committees of the centers participating in the studies, and in compliance with the Helsinki declaration. Fibroblast lines were cultured in DMEM (Life Technologies, Carlsbad, CA), supplemented with 15% FBS (Gemini Bio-Products, Broderick, CA), 10 mM HEPES buffer (Sigma, St. Louis, MO), 100 U/ml Potassium Penicillin and 100 µg/ml Streptomycin Sulfate (Lonza, Walkersville, MD). HEK293T cell line (ATCC CRL-3216) was obtained from the American Type Culture collection and cultured in DMEM, supplemented with 10% FBS, 10 mM HEPES buffer, 100 U/ml Potassium Penicillin and 100 µg/ml Streptomycin Sulfate. Before and during transfection experiments, HEK293T cells were cultured in antibiotics-free medium.
Plasmids and targeted mutagenesis
An expression vector containing cDNA encoding C-terminally FLAG-tagged human IκBα (pCMV3-NFKBIA-C-FLAG; HG12045-CF) was obtained from Sino Biological Inc. (Beijing, China). Targeted mutagenesis was performed using Quik Change II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s instructions together with custom mutagenic primers: Q9X_F: 5’-CGGCCGAGCGCCCCTAGGAGTGGGCCATGGAGG-3’; Q9X_R: 5’-CCTCCATGGCCCACTCCTAGGGGCGCTCGGCCG-3’; W11X_F: 5’-CCG-AGCGCCCCCAGGAGTAGGCCATGGAGGGCC-3’; W11X_R: 5’-GGCCCTCCATGG-CCTACTCCTGGGGGCGCTCGG-3’; E14X_F: 5’-CCCAGGAGTGGGCCATGTAGG-GCCCCCGCGACGG-3’; E14X_R: 5’-CCGTCGCGGGGGCCCTACATGGCCCAC-TCCTGGG-3’; S32I_F: 5’-GGACGACCGCCACGACATCGGCCTGGACTCC-3’; S32I_R: 5’-GGAGTCCAGGCCGATGTCGTGGCGGTCGTCC-3’; G33V_ F: 5’-CGACCGCCA-CGACAGCGTCCTGGACTCCATGAAAG-3’; G33V_R: 5’-CTTTCATGGAGTCCAGGA-CGCTGTCGTGGCGGTCG-3’; S36Y_F: 5’-CGACAGCGGCCTGGACTACATGAA-AGACGAGG-3’; S36Y_R: 5’-CCTCGTCTTTCATGTAGTCCAGGCCGCTGTCG-3’; M37K_F: 5’-GCGGCCTGGACTCCAAGAAAGACGAGGAGTACG-3’; M37K_R: 5’-CGT-ACTCCTCGTCTTTCTTGGAGTCCAGGCCGC-3’; resulting in the following changes in IκBα (RefSeq NM_020529) on the nucleotide and protein levels, respectively: c.25C>T (p.Q9X), c.32G>A (p.W11X), c.40G>T (p.E14X), c.95G>T (p.S32I), c.98G>T (p.G33V), c.107C>A (p.S36Y), c.110T>A (p.M37K). Successful mutagenesis was confirmed by Sanger sequencing of plasmid DNA.
Transient transfection of HEK293T cells
The day before transfection, 4.5×105 HEK293T cells were seeded per well of a 6-well plate in antibiotics-free medium. Transfections were carried out using FuGENE 6 transfection reagent (Promega, Madison, WI) at a 3:1 FuGENE:DNA ratio. Three µg of WT or mutant pCMV3-NFKBIA-C-FLAG were transfected per well. Cells were harvested 24 hours post-transfection and used in immunoblotting as described below.
Semi-quantitative RT-PCR from transfectants
Total RNA of HEK293T cells transiently transfected with WT or mutant pCMV3-NFKBIA-C-FLAG was extracted using TRIzol reagent (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. For each sample, 500 ng of RNA were reverse transcribed using iScript cDNA synthesis kit (BioRad, Hercules, CA). The following primers were used for RT-PCR: IκBα_F: 5’-CAATGCTCAGGAGCCCTGTAA-3’; FLAG_R: 5’-CTTATCGTCGT CATCCTTGTAATC-3’; GAPDH_F: 5’-TCGACAGTCAGCCGCATCTT-3’; GAPDH_R: 5’-CAAATGAGCCCCAGCCTTCTC-3’. To specifically amplify ectopically expressed IκBα transcripts, the reverse primer was designed to bind to the FLAG tag sequence (GATTACAAGGATGACGACGATAAG). Thirty-five PCR cycles were performed with an annealing temperature of 60°C. For analysis, PCR products were loaded on a 1% agarose gel.
Fibroblast stimulation
Primary skin fibroblasts from healthy controls and AD EDA-ID patients were grown to about 80% confluency. Cells were harvested by trypsinization, re-seeded at a density of 100,000 cells per well (6-well format) and incubated overnight. Fibroblasts were treated with media, 1 µg/ml LPS (Invivogen, San Diego, CA) or 1 µg/ml LPS + 5 µg/ml α-LTβR agonistic mAb (clone 31G4D8, BioLegend, San Diego, CA) for 48 hours. IL-6 levels in the supernatants of LPS-stimulated cells were determined 24 hours post stimulation by ELISA (eBioscience).
Immunoblotting
Samples were lysed in Western blot buffer (50 mM Tris pH 7.5, 100 mM NaCl, 1% Triton X-100) containing Protease Inhibitor cocktail (Roche, Indianapolis, IN) and lysates were clarified by spinning for 15 minutes at 20,000×g. Total protein was quantified using the BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer’s instructions. Lysates containing equal amounts of protein were resolved by SDS-PAGE, transferred to nitrocellulose membranes and probed with antibodies against the FLAG-tag (F3165, Sigma-Aldrich, St. Louis, MO); IκBα (sc-371, Santa Cruz Biotechnology, Dallas, TX); p100/p52 (#4882, Cell Signaling Technology, Danvers, MA), GAPDH (#2118, Cell Signaling Technology, Danvers, MA) and RelB (sc-226, Santa Cruz Biotechnology, Dallas, TX) following standard protocols. Band intensity was quantified using ImageJ software (National Institutes of Health, Bethesda, MD).
RNA-Seq analysis of LPS+α-LTβR-stimulated fibroblasts
Fibroblasts from healthy controls and AD EDA-ID patients were treated with media or 1 µg/ml LPS + 5 µg/ml α-LTβR agonistic mAb for 48 hours. RNA was isolated using TRIzol Plus RNA Purification Kit (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. On-column DNAse digestion was performed using PureLink DNAse Set (Invitrogen, Carlsbad, CA). Libraries were prepared using Illumina TruSeq Stranded mRNA sample preparation kits from 500 ng of purified total RNA according to the manufacturer’s protocol. The finished dsDNA libraries were quantified by Qubit fluorometer, Agilent TapeStation 2200, and RT-qPCR using the Kapa Biosystems library quantification kit according to manufacturer’s protocols. Uniquely indexed libraries were pooled in equimolar ratios and sequenced on an Illumina NextSeq500 with single-end 75bp reads by the Dana-Farber Cancer Institute Molecular Biology Core Facilities. Sequence reads were aligned to the hg19 reference genome assembly and gene counts were quantified using STAR (v2.5.1b) and differential expression testing was performed by DESeq2 (v1.10.1) as part of the VIPER analysis pipeline (https://bitbucket.org/cfce/viper/). Normalized gene counts were expressed as fragments per kilobase of exon per million reads (FPKM). For further analysis, we used genes significantly (adjusted p-value<0.05) up or downregulated by at least two folds in LPS+α-LTβR stimulated fibroblasts from healthy donors compared to unstimulated cells. FPKM values were log2-transformed and Z-scores were calculated. Signaling components and molecular targets of NF-κB were identified by comparing genes significantly up or downregulated by two folds in LPS+α-LTβR stimulated fibroblasts from healthy donors with online databases of NF-κB target genes (https://www.bu.edu/nf-kb/gene-resources/target-genes/) as well as genes included in commercially available NF-κB-specific gene arrays (TaqMan Array Human NFKB Pathway, Cat. 4414095, Thermo Fisher, Waltham, MA; RT2 Profiler PCR Array Human NF-κB Signaling Pathway and Signaling Targets, Cat. PAHS-025Z and PAHS-225Z, Qiagen, Germantown, MD). Heatmaps representing Z-scores of log2-transformed FPKM values were done in R (https://www.r-project.org) using the pheatmap and RColorBrewer packages.
Quantitative PCR (qPCR) analysis of CCL20, ICAM1, and VCAM1 mRNA levels
RNA was isolated from fibroblasts treated with media or 1 µg/ml LPS + 5 µg/ml α-LTβR agonistic mAb for 48 hours. For each sample, 100 ng of RNA were reverse transcribed using iScript cDNA synthesis kit (BioRad, Hercules, CA). TaqMan primer/probes for human CCL20, ICAM1, VCAM1 and GAPDH were purchased from Thermo Fisher Scientific (Waltham, MA). Experiments were run on an Applied Biosystems 7300 Real-Time PCR system. Data was analyzed using the 2−ΔCt method.
Statistical analysis
Statistical analyses were performed using Prism 7 software (GraphPad, La Jolla, CA). Significance was calculated by using two-tailed t tests.
RESULTS
AD EDA-ID patients with point mutations in IκBα have more severe disease than patients with truncation mutations and a poor outcome after HSCT
We analyzed nine AD EDA-ID patients, including seven of the eight published cases and two unreported cases (Table I). Six patients have four different heterozygous point mutations at or adjacent to the S32 and S36 phosphorylation sites, leading to single amino acid substitutions: three with p.S32I and one each with p.G33V, p.S36Y and p.M37K (Table I). Three patients have each a heterozygous N-terminal nonsense mutation, p.Q9X, p.W11X, and p.E14X, expected to give rise to N-terminally truncated IκBα proteins that use alternative methionine start sites, presumably M13 and M37 for Q9X and W11X, and M37 for E14X24–26.
TABLE I.
Clinical characteristics.
| Patient | IκBα mutation |
Disease score† |
Secondary lymphoid organs | HSCT (age) |
Outcome | Reference | |
|---|---|---|---|---|---|---|---|
| Tonsils | Palpable lymph nodes |
||||||
| 1 | S32I | 15/15 | Absent | Absent | Yes (1 year) | Recurrent infections, on IVIG; Alive 17 years post HSCT | Courtois et al., 2003 |
| 2 | S32I | 14/15 | Absent | Absent | Yes (7 years) | Recurrent infections, on IVIG; Alive 3 years post HSCT | unpublished |
| 3 | S32I | 15/15 | Absent | Absent | Yes (2.5 years) | Recurrent infections, on IVIG; Deceased 4 months post HSCT | Janssen et al., 2004 |
| 4 | G33V | 14/15 | Absent | Absent | Yes (2 years) | Recurrent infections, on IVIG; Alive 1 year post HSCT | unpublished |
| 5 | S36Y | 11/15 | Very small | Very small | Yes (6 years) | GvHD and autoimmune thrombocytopenia, Deceased 7 months post HSCT with sepsis | Yoshioka et al., 2013 |
| 6 | M37K | 12/15 | Absent | Absent | Yes (4.8 years) | Recurrent infections, on SCIG; Deceased 18 months post HSCT with sepsis | Schimke et al., 2013 |
| 7 | Q9X | 6/12 | Yes | Yes | Yes (15 months) | Healthy 5 years post HSCT, off IVIG | Ohnishi et al., 2012 |
| 8 | W11X | 4/15 | Yes | Yes | No | Healthy, on IVIG | McDonald et al., 2007 |
| 9 | E14X | 4/12 | Yes | Yes | Yes (8 months) | Deceased day 31 post HSCT with sepsis | Lopez-Granados et al., 2008 |
HSCT, hematopoietic stem cell transplantation; GvHD, graft versus host disease; IVIG, intravenous immunoglobulin replacement therapy; SCIG, subcutaneous immunoglobulin replacement therapy.
Disease scoring is based on five clinical and laboratory criteria, each ranging in score from 0 to 3, according to the severity of the abnormality (Table II). For patients 7 and 9, data was not available on one of the criteria, resulting in a maximum disease score of 12.
To assess the severity of AD EDA-ID, we developed a disease scoring system in consultation with three experts in primary immunodeficiency who were not participants in the study. Scoring is based on five clinical and laboratory criteria, each of which was assigned a score from 0 to 3 according to the severity of the abnormality (Table II). The criteria included type of infections (mucosal, visceral, blood borne, and opportunistic), number of systems exhibiting features of EDA, spectrum of the antibody deficiency (deficiency of response to polysaccharide antigens, protein antigens, or both), magnitude of the elevation in the circulating lymphocyte count, and severity of the reduction in CD3+CD45RO+ memory T cells and/or CD19+CD27+ B cells. The maximum total score is 15. In two patients (Patients 7 and 9 in Table I) only four of the five criteria were evaluated, resulting in a maximal score of 12. Because not all criteria were evaluated in all patients, a relative disease severity score was calculated as the percentage of the maximum achievable score in each patient. Disease severity score was significantly higher in the six patients with IκBα point mutations compared to the three patients with IκBα truncation mutations (Table 1 and Fig. 1, A). The breakdown of the individual scores for each of the two groups and for each of the patients are shown in Fig. 1, B and Supplemental Table E1.
Table II.
Disease severity scoring system for AD EDA-ID.
| Score | Infections | Ectodermal dysplasia: systems affected† |
Antibody responses‡ | Lymphocyte count/µl |
Memory T and/or B cells* |
|---|---|---|---|---|---|
| 0 | None | None | Normal | Normal | Normal |
| 1 | Mucosal | Dental only | Polysaccharide (PS)-deficient | 10,000–20,000 | Modest decrease |
| 2 | Visceral and/or blood borne | Two | Protein antigen-deficient | 20,000–30,000 | Severe decrease |
| 3 | Opportunistic | Three or more | PS and protein antigen-deficient | >30,000 | Absent |
Ectodermal systems evaluated include hair, teeth, nails and sweat glands.
Deficient antibody response is defined as absent or unprotective antibody titers and/or a rise in antibody titers of less than four-fold post-immunization.
Modest decrease: Values between 2 and 3 standard deviations (SD) below the mean for age. Severe decrease: Values more than 3 SD below the mean for age but above zero.
Figure 1. Relative disease severity score in AD EDA-ID patients.

A. Cumulative disease severity score expressed as a percentage of the maximal score based on the evaluable disease criteria listed in Table 2. B. Individual disease category scores for patients with IκBα point mutations and patients with IκBα truncations. *, p<0.05; **, p<0.01; ***, p<0.001; ns, not significant; na, not applicable.
Five of the six patients with IκBα point mutations lacked visible tonsils and had no palpable LNs. Small tonsils and LNs were reported present in only one of the six patients with IκBα point mutations. In contrast, tonsils and LNs were present in all three patients with IκBα truncation mutations (Table I). All six patients with IκBα point mutations had undergone HSCT because of severe disease (Table I). Three of the six died within 18 months post HSCT and had continued to suffer until their death from recurrent infections and required Ig replacement therapy. All three transplanted patients with IκBα point mutations who are currently alive continue to suffer from recurrent infections and require Ig replacement. All six patients achieved excellent donor cell engraftment (>90%) in T cell, B cell, and myeloid cell lineages in the peripheral blood. Thus, HSCT failed to result in clinical improvement in six of six patients with IκBα point mutations despite excellent donor cell engraftment. Of the three patients with truncation mutations, the patient with the Q9X mutation was transplanted at 15 months of age and is healthy more than five years post-HSCT. The patient with the E14X mutation was transplanted at seven months of age, but died from sepsis in the immediate post-transplant period, precluding evaluation of the outcome of HSCT. The patient with the W11X mutation, now 22 years of age, is doing well on Ig replacement therapy and has not undergone HSCT.
IκBα point mutants are expressed at higher levels compared to IκBα N-terminally truncated mutants
Degradation-resistant IκBα mutant proteins sequester p50:p65 in the cytoplasm impairing NF-κB activation. Thus, the level of degradation-resistant IκBα mutant protein in the cell is expected to determine the extent of impairment of NF-κB activation. We hypothesized that the levels of IκBα point mutants, which are associated with significantly more severe disease, would be higher than those of IκBα truncation mutants. Immunoblotting for IκBα was performed using an antibody directed to the C-terminus of IκBα in lysates from skin-derived fibroblasts. Fibroblasts were available from four patients with point mutations (two S32I, one G33V and one M37K) and two patients with truncations (Q9X, W11X). All patients’ fibroblasts were studied simultaneously except those from the patient with the W11X mutation, because of delay in our ability to grow these cells from frozen aliquots. As previously reported,24, 26 fibroblasts from the two patients with the Q9X and W11X IκBα truncation mutations demonstrated an IκBα band of normal size that represents the product of the WT allele (Fig. 2, A). In addition, two fainter lower molecular weight IκBα bands were detected in these fibroblasts. According to their molecular weight, the translation start sites of these products are likely to be M13 and M37, respectively. The mean ratio of the sum of the truncated IκBα products to WT IκBα in the two patients was 0.4±0.2 (n=3). As previously reported,22, 27, 28 only one IκBα band was observed in fibroblast lysates from patients with point mutations, as would be expected for mutant proteins with a single amino acid substitution. The intensity of this band was similar to the band in normal controls. Since κBα is under control of canonical NF-κB signaling46, 47, it is likely that the contribution of the product of the WT IκBα allele to the total IκBαlevel is less that of the degradation resistant product of the mutant IκBα allele.
Figure 2. Expression of IκBα in fibroblasts and transfectants.

A. Representative immunoblot of IκBα expression in cultured skin-derived fibroblasts from healthy controls (HC) and AD EDA-ID patients (P). P1 S32I and P2 S32I are patients 1 and 3 in Table I. B, C. Representative immunoblot (B) and quantitative analysis (C) of the expression of Flag-tagged WT and mutant IκBα in transfected HEK293T cells. Results in the left panel of C are expressed as the mean ratio of IκBα to tubulin of each individual mutant analyzed, relative to the mean of WT IκBα set at 1.0. Results in the right panel of C are expressed as the mean ratio of IκBα to tubulin of pooled IκBα truncation mutants and pooled IκBα point mutants relative to the mean of WT IκBα set at 1.0. D. RT-PCR analysis of mRNA encoding FLAG-tagged WT and mutant IκBα. The amounts of cDNA used were in the linear range determined for mRNA encoding FLAG-tagged WT IκBα. Data is representative of three independent experiments. Columns and bars represent the mean±SD of at least three independent experiments. **, p< 0.01; ***, p<0.001; ****, p<0.0001; ns, not significant.
Measurement of IκBα levels in cultured fibroblasts might not reflect the intrinsic stability of the mutant proteins because of the genetic heterogeneity of the fibroblast donors, the effect on IκBα expression of chronic stimulation of the fibroblast line by potential NF-κB activators in fetal calf serum, and the inability to directly determine the level of WT and mutant IκBα in fibroblasts from patients with IκBα point mutations. To assess the stability of IκBα mutant proteins, HEK293T cells were transiently transfected with expression vectors encoding C-terminally FLAG-tagged WT or mutant IκBα. Twenty-four hours post-transfection, cells were harvested and lysates were immunoblotted with an antibody to the FLAG tag. In this approach, cells of a uniform genetic background are examined under conditions of short-term culture and the expression of the FLAG-tagged point mutants is directly measurable. With the exception of M37K IκBα, which was expressed at levels not significantly different from WT IκBα, all IκBα mutant proteins showed a significant decrease in expression compared to WT IκBα (Fig. 2, B and C). Importantly, the expression levels of the three IκBα truncation mutants Q9X, W11X and E14X were significantly reduced compared to those of the four point mutants S32I, G33V, S36Y and M37K. Reduced expression levels of the mutant proteins did not result from mRNA instability, as evaluated by semi-quantitative reverse transcription polymerase chain reaction (RT-PCR) of IκBα-FLAG mRNA (Fig. 2, D). These results strongly suggest that IκBα point mutants are significantly more stable than IκBα truncation mutants.
IκBα point mutants impair LPS-driven IL-6 secretion, and accumulate following LPS stimulation significantly more than IκBα truncation mutants
Impaired production of inflammatory cytokines following activation of the canonical NF-κB pathway in AD EDA-ID has been documented in individual case reports.22–28 These reports measured the production of different cytokines (IL-1 and/or IL-6 and/or TNFα) by different cell types (PBMCs, monocytes, fibroblasts) using different modes of stimulation (LPS and/or TNFα and/or IL-1), precluding useful comparison of inflammatory cytokine production in the patients. We compared the secretion of the same cytokine, IL-6, by a single cell source, skin-derived fibroblasts, following stimulation by the same agent, LPS. IL-6 secretion was robustly induced by LPS stimulation in fibroblasts from healthy controls (Fig. 3, A). In contrast, LPS-driven IL-6 production was virtually abolished in all four patients with point mutations studied (two S32I, one G33V, one M37K), but was partially preserved in the two patients with truncation mutations studied (Q9X, W11X). These results suggest that IκBα point mutants cause a significantly greater impairment of canonical NF-κB activation compared to IκBα truncation mutants.
Figure 3. IκBα point mutants exert a stronger dominant negative effect on IL-6 secretion and accumulate to a greater extent following LPS stimulation than IκBα truncation mutants.

A. LPS-driven IL-6 secretion by fibroblasts. Left panel: mean of three determinations on each of two healthy controls (HC), two patients with S32I, and individual patients with the other IκBα mutations. Right panel: Pooled results showing the mean IL-6 secretion by fibroblasts from HC (n=2), patients with IκBα truncation mutations (n=2) and patients with IκBα point mutations (n=4). B, C. Effect of LPS stimulation on IκBα levels: Representative (B) and quantitative (C) immunoblot analysis of IκBα expression in fibroblasts from two HC and AD EDA-ID patients before and after stimulation with LPS. Results from only one patient with S32I mutation are shown in B because of the limited number of lanes in the gel, but results from both patients with S32I mutation studied are included in the quantitative analysis, which includes 2 controls, 2 patients with IκBα truncation mutations and 4 patients with IκBα point mutations. Results in C are expressed as the mean ratio of IκBα to GAPDH relative to the mean of unstimulated HC set at 1.0. D. Mean ratio of mutant to WT IκBα in unstimulated and LPS-stimulated fibroblasts from AD EDA-ID patients with truncating mutations in IκBα. Columns and bars represent the mean±SD of three independent experiments. **, p<0.01; ***, p<0.001; ****, p<0.0001.
Canonical NF-κB activation results in rapid degradation of IκBα followed by IκBα resynthesis.46, 47 The marked reduction of LPS-driven IL-6 secretion in fibroblasts from patients with IκBα truncation mutations, in which the level of the mutant was ~ half that of WT protein prior to LPS stimulation, prompted us to examine whether LPS activation alters the mutant to WT IκBα ratio, given the resistance of the mutants to stimulus-induced degradation. LPS stimulation for 48 hours caused a significant decrease in WT IκBα in normal fibroblasts (Fig. 3, B and C). Similarly, WT IκBα significantly decreased in LPS-stimulated fibroblasts from patients with Q9X and W11X IκBα truncation mutations, whereas, in contrast, the level of truncated mutant IκBα significantly increased (Fig. 3, B and C). This resulted in a significant change of the truncation mutant to WT IκBα ratio from ~0.4 in unstimulated cells to ~4 (n=3) in stimulated cells (Fig. 3, D). These results, therefore, provide a rationale for how IκBα truncation mutants can function as DN despite their lower expression compared to WT IκBα. The two fainter bands representing the IκBα truncated products in unstimulated fibroblasts detected in Fig. 2, A are poorly evident in the representative experiment shown in Fig. 3, B in which samples from all available patients were loaded on the same gel. However, these two bands were evident in other experiments (Supplemental Fig. S1). Of note, only the IκBα truncated product with the lower molecular weight, which likely lacks the S32 and S36 phosphorylation sites, increased following stimulation; whereas the higher molecular weight truncated product, which likely retains S32 and S36, may be subject to degradation (Supplemental Fig. S1).
LPS stimulation caused a significant (1.5±0.3 fold, n=3) increase in the level of immunoreactive IκBα protein in fibroblasts from patients with IκBα point mutations. Because of the inability to differentiate WT IκBα from point mutant IκBα, we were unable to determine the effect of LPS stimulation on the point mutant:WT IκBα ratio. The WT κBα protein is expected to be expressed at a lower level than the IκBα point mutant at baseline due to impaired NF-κB canonical activity, resulting in an expected point mutant:WT IκBα ratio of >1 at baseline. Following LPS stimulation, the WT but not the mutant protein would be degraded resulting in a further increase in the point mutant:WT κBα ratio. This ratio would be expected to be higher than that of the truncation mutant: WT ratio due to the higher stability of the point mutants, providing a likely explanation for the more severe impairment of NF-κB activation in patients with IκBα point mutations.
IκBα point mutants impair non-canonical NF-κB activation to a greater extent than do IκBα truncation mutants
Defective development of tonsils and peripheral LNs in some patients with AD EDA-ID suggests a defect in non-canonical NF-κB signaling. This hypothesis is supported by our previous observation that non-canonical NF-κB signaling is defective in mouse embryonic fibroblasts derived from mutant mice heterozygous for the S32I mutation.33 Furthermore, genetic deletion of many signaling components of the non-canonical NF-κB pathway in mice results in defective lymphorganogenesis and/or disrupted architecture of secondary lymphoid organs (SLO).17, 34–45 Non-canonical NF-κB signaling in stromal lymphoid tissue organizer (LTo) cells is critical for lymphorganogenesis and is activated upon binding of lymphotoxin beta (LTβ) expressed by hematopoietic lymphoid tissue inducer (LTi) cells to the LTβ receptor (LTβR) on LTo cells.45 Non-canonical NF-κB signaling depends on IKKα-driven phosphorylation of the cytoplasmically located NF-κB2 (p100) and its cleavage to generate the transcriptionally active p52 product, which, in complex with the NF-κB family member RelB, translocates into the nucleus and activates gene transcription.15 Importantly, both p100 and RelB are critical for the development of SLOs44, 48–51 and their expression is under the transcriptional control of the canonical NF-κB-pathway, which thus regulates the amount of p100 and RelB available for signaling through the non-canonical NF-κB-pathway.19, 20 We examined the upregulation of p100 and RelB following stimulation of the canonical NF-κB-pathway by LPS in fibroblasts, used as surrogates for stromal cells. As expected, LPS stimulation upregulated p100 and RelB expression in normal fibroblasts (Fig. 4, A). LPS-driven upregulation of p100 and RelB expression was significantly reduced in fibroblasts from the five AD EDA-ID patients studied compared to normal fibroblasts (Fig. 4, A–C). Importantly, upregulation of p100 and RelB expression was reduced significantly more in the three patients with the point mutations S32I, G33V and M37K compared to the two patients with the truncation mutations Q9X and W11X (Fig. 4, A–C). These results demonstrate that upregulation of the non-canonical NF-κB pathway family members p100 and RelB following canonical NF-κB signaling is impaired in fibroblasts from AD EDA-ID patients, and that residual canonical NF-κB activity in patients with truncation mutations correlates with their ability to upregulate expression of these two proteins.
Figure 4. IκBα point mutants impair more severely LPS-driven expression of NF-κB2/p100 and RelB, and activation of the non-canonical NF-κB pathway than IκBα truncation mutants.

A. Representative immunoblot analysis of p100 and RelB levels in fibroblasts from two healthy controls (HC) and AD EDA-ID patients (P) before and after stimulation with LPS for 48 hours. B, C. Quantitative analysis of expression of p100 (B) and RelB (C) in LPS-stimulated fibroblasts from two HC and individual patients (left panels), and pooled results representing the mean p100 and RelB expression levels in fibroblasts from patients with IκBα truncation mutations (n=2) and IκBα point mutations (n=4) (right panels). Results are expressed as the mean ratio of p100 and RelB to GAPDH relative to the mean of LPS-stimulated healthy controls set at 1.0. D. Representative immunoblot analysis of p100 and p52 levels in fibroblasts from two HC and AD EDA-ID patients before and after stimulation with LPS+anti-LTβR mAb for 48 hours. E, F. Quantitative analysis of expression of p52 (E) and p100 (F) in LPS+anti-LTβR mAb stimulated fibroblasts from healthy controls (HC) and individual patients (left panels) and pooled results representing the mean p52 and p100 expression levels in fibroblasts from HC (n=2), patients with IκBα truncation mutations (n=2) and IκBα point mutations (n=4) (right panels). Results are expressed as the ratio of p52 and p100 to GAPDH relative to the mean of LPS+anti-LTβR mAb stimulated (E) or unstimulated (F) HC set at 1.0. Columns and bars represent the mean±SD of at least three independent experiments. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
The generation of p52 was measured by stimulating fibroblasts with LPS and an agonistic monoclonal antibody (mAb) to the LTβR for 48h, then immunoblotting cellular lysates for p52 and p100. Negligible amounts of p52 were detected in unstimulated fibroblasts from patients and controls (Fig. 4, D). Stimulation with LPS and anti-LTβR mAb resulted in the appearance of a strong p52 band in healthy controls, indicating LTβR-driven, non-canonical NF-κB-mediated cleavage of p100. Fibroblasts from patients with the Q9X and W11X truncation mutations showed a partial, but significant, decrease in p52 accumulation following stimulation compared to healthy controls (Fig. 4, D–F). p52 accumulation was diminished significantly more in patients with IκBα point mutations (~80%) than in those with truncations (~50%). Of note, following LPS+anti-LTβR mAb stimulation, p100 levels robustly increased in normal fibroblasts, as observed with LPS stimulation alone, but increased significantly less in fibroblasts from patients with IκBα truncation mutations, and became virtually undetectable in fibroblasts from patients with κBα point mutations (Fig. 4, D–F). These results suggest that the patient cells can cleave p100, but fail to produce it and replenish it normally to provide sufficient substrates for p52 generation.
Induction of genes encoding chemokines and adhesion molecules important for lymphorganogenesis is impaired more severely in AD EDA-ID patients with IκBα point mutations than IκBα truncation mutations
During the development of SLOs, both the canonical and non-canonical NF-κB pathways synergize in stromal LTo cells that receive an LTβ signal to induce the expression of chemokines and adhesion molecules important for lymphorganogenesis.34, 36–38, 45 These include the chemokines CCL19, CCL20, CCL21, CXCL12 and CXCL13, and the adhesion molecules ICAM-1 and VCAM-1.45, 52 Expression of these molecules is either fully or partially dependent on the non-canonical NF-κB-pathway.16, 53–55 RNA-Seq analysis of normal fibroblasts (n=3) revealed that LPS+anti-LTβR mAb stimulation upregulated the expression of 619 genes, and downregulated the expression of 289 genes at a fold change >2, and FDR< 0.05 (Fig. 5, A). The numbers of upregulated and downregulated genes were substantially lower in LPS+anti-LTβR mAb stimulated fibroblasts from patients with IκBα truncation mutations (Q9X and W11X, n=2) compared to controls (n=3), and were drastically reduced in patients with IκBα point mutations (S32I and G33V, n=2), in whom only 4 genes were significantly upregulated and no gene was significantly downregulated (Fig. 5, A). A heat map of known targets of NF-κB the expression of which was significantly altered by LPS+anti-LTβR stimulation is shown in Fig. 5, B. The corresponding heat map including all genes the expression of which was significantly altered by LPS+anti-LTβR stimulation is shown in Supplemental Fig. S2, A.
Figure 5. IκBα point mutants impair the induction of genes important for lymphorganogenesis more severely than IκBα truncation mutants.

A. Number of genes significantly (p<0.05) upregulated or downregulated by two folds or more in LPS+anti-LTβR mAb stimulated fibroblasts from healthy controls (n=3) and patients with IκBα truncation (n=2) and point mutations (n=2). B. Heat map analysis of NF-κB regulated genes the expression of which was significantly altered by LPS+anti-LTβR stimulation of normal fibroblasts. C. RNA-Seq analysis of the expression of CCL20, ICAM1 and VCAM1 mRNA in unstimulated and LPS+anti-LTβR mAb stimulated fibroblasts from healthy controls (n=3) and patients with IκBα truncation (n=2) and point mutations (n=2). Results are expressed as Fragments per Kilobase of Exon per Million (FPKM). Columns and bars represent the mean±SD for each of the three groups. D. q-PCR analysis of the expression of CCL20, ICAM1 and VCAM1 mRNA in unstimulated and LPS+anti-LTβR mAb stimulated fibroblasts from healthy controls (n=3) and patients with IκBα truncation (n=2) and point mutations (n=3). Results for ICAM1 and VCAM1 are expressed as the ratio of the gene of interest to GAPDH as calculated by the 2−ΔCt method relative to that of unstimulated healthy controls set at 1.0. Because CCL20 was undetectable in unstimulated fibroblasts, results are expressed as the ratio of CCL20 to GAPDH as calculated by the 2−ΔCt method. Columns and bars in D represent the mean±SD of three independent experiments. *, p<0.05; **, p<0.01; ***, p<0.001; ns, not significant.
Of the genes known to be implicated in lymphorganogenesis, CCL20, ICAM1 and VCAM1 were upregulated more than two fold in LPS+anti-LTβR mAb stimulated normal fibroblasts (Fig. 5, B and C). Upregulation of all three genes was partially decreased in fibroblasts from patients with IκBα truncation mutations and virtually nil in patients with IκBα point mutations (Fig. 5, B and C). Importantly, upregulation of CCL20, ICAM1 and VCAM1 was more severely impaired in patients with IκBα point mutations than in patients with IκBα truncation mutations (Fig. 5, B and C). To validate these results, we performed qPCR analysis of the mRNA (Fig. 5, D). Upregulation of CCL20, ICAM1 and VCAM1 mRNA levels was significantly impaired in patients with IκBα point mutations (n=3) compared to healthy controls (n=3), confirming the RNA-Seq results. The expression levels of CCL20, ICAM1 and VCAM1 mRNA in LPS+anti-LTβR mAb stimulated fibroblasts from patients with IκBα truncation mutations (n=2) were reduced compared to healthy controls (n=3) but the difference was not significant. These results demonstrate that expression of chemokines and adhesion molecules important for lymphorganogenesis is more severely impaired in AD EDA-ID patients with IκBα point mutations than in patients with IκBα truncation mutations, thus providing a mechanistic understanding of the absence of lymph nodes and apparently poor response to HSCT seen in patients with IκBα point mutations. As expected, LPS+anti-LTβR mAb stimulation strongly upregulated the expression of NFKBIA, NFKB2 and RELB in normal fibroblasts, but substantially less in fibroblasts from AD EDA-ID patients (Supplemental Fig. S2, B).
DISCUSSION
We establish a robust genotype-phenotype correlation in AD EDA-ID and demonstrate that IκBα point mutants are expressed at higher levels, impair more severely canonical and non-canonical NF-κB signaling, and result in more severe disease than IκBα truncation mutants. Non-canonical NF-κB dependent genes important for lymphorganogenesis were inducible in patients with IκBα truncation mutations, but absent in patients with IκBα point mutations, providing a potential explanation for their virtual lack of tonsils and LNs, and their poor immune reconstitution following HSCT.
Disease severity score was significantly higher in the six patients with IκBα point mutations compared to the three patients with IκBα N-terminal truncation mutations, demonstrating that IκBα point mutations are associated with more severe disease compared to IκBα truncation mutations in patients with AD EDA-ID. The outcome of HSCT was poor in all six patients with point mutations in IκBα despite excellent donor cell chimerism, suggesting that a defect in stromal cells may be at play.
Using fibroblasts from the patients, we were able to demonstrate lower expression of the truncated IκBα compared to WT IκBα. However, we were not able to determine the contribution of WT IκBα versus point mutant IκBα. Using HEK293T cells transfected with IκBα mutants, we observed that truncation and point mutants were expressed at significantly lower levels than WT IκBα, suggesting that both types of mutants, and more so the truncation mutants, are intrinsically unstable. We cannot, however, rule out a contribution of inefficient translation from downstream ATG sites to the decreased expression of the truncation mutants. In addition, expression of the truncated IκBα mutants compared to WT IκBα was relatively lower in transfected cells than in fibroblasts from the patients.
Consistent with their higher expression levels, IκBα point mutants were associated with more significant reduction in LPS-driven IL-6 secretion and upregulation of p100 in fibroblasts, which are dependent on the canonical NF-κB pathway. Whereas pathways other than the canonical NF-κB pathway can be activated by LPS, LPS induction of IL-6 and p100 are strictly dependent on NF-κB.56, 57 The impairment of IL-6 secretion exceeded what would have been expected from the baseline ratio of mutant to WT IκBα for both patients with point mutations and truncation mutations. The decrease in the level of WT IκBα 48 hours post LPS stimulation suggests that upregulation of WT κBα is outweighed by its degradation. The marked increase in the truncation mutant to WT IκBα ratio in LPS-stimulated cells explains the DN effect exerted by these mutants despite their lower expression compared to WT protein in unstimulated cells. We previously reported that the W11X mutant did not exert a DN effect on canonical NF-κB signaling when transiently co-transfected with WT IκBα and an NF-κB reporter into HEK293T cells, which were then stimulated with TNFα for 6 hours.24 In that study, the short stimulation time, the low expression level of the unstable mutant protein and endogenous WT IκBα may have masked the potential of the mutant to exert a DN effect.
We show for the first time that the non-canonical NF-κB pathway is impaired in patients with AD EDA-ID. We demonstrate that upregulation of the expression of p100 and RelB, two key components of this pathway known to be transcriptionally regulated by the canonical NF-κB pathway, is deficient in LPS-stimulated fibroblasts from these patients. The defect was more severe in patients with point mutations than in those with truncation mutations, paralleling the relative magnitude of the defect in LPS-driven IL-6 secretion. As expected from their lower content of the p100 substrate, generation of the transcriptionally active product p52, which is dependent on activation of the non-canonical NF-κB pathway, was deficient in LPS+anti-LTβR mAb stimulated fibroblasts from these patients. The defect was more severe in patients with IκBα point mutations than in those with IκBα truncation mutations, a reflection of the relative level of p100 in these patients. It should be noted that defect in the non-canonical NF-κB pathway in the patients is an extrinsic one due to the decreased ability to generate p100, a critical substrate for IKKα. In fact, although the level of p52 following activation with LPS+anti-LTβR is lower in the patients, the p52:p100 ratio is higher, suggesting that the intrinsic ability to activate NIK and IKKα, which is essential for p100 phosphorylation and degradation, is intact. It is likely that the higher p52:p100 ratio is a reflection of the limited amount of substrate rather than an intrinsically more active non-canonical NF-κB activation.
Defective signaling via the non-canonical NF-κB pathway in LPS+anti-LTβR mAb stimulated fibroblasts from patients with AD EDA-ID was associated with defective regulation of gene expression as demonstrated by RNA-Seq analysis. Importantly, three genes critical for lymphorganogesis, CCL20, ICAM1 and VCAM1 were significantly upregulated in LPS+anti-LTβR mAb stimulated normal fibroblasts. Several other genes in human LTos are important for lymphorganogenesis. These include CCL19, CCL21, and CXCL13.58 The failure to detect upregulation of genes other than CCL20, ICAM1 and VCAM1 important and essential for lymphorganogenesis is likely because fibroblasts are an imperfect surrogate for the stromal LTo cells. qPCR analysis confirmed that upregulation of CCL20, ICAM1 and VCAM1 was abolished in patients with IκBα point mutations. Notably, expression of these three genes in fibroblasts from patients with IκBα truncation mutations, albeit lower for VCAM1 and ICAM 1, was not significantly different from healthy controls. This finding suggests that the residual NF-κB pathway signaling in patients with IκBα truncation mutations is sufficient for the expression of these genes, and is consistent with the presence of LNs and tonsils in these patients. Taken together, our results suggest defective activation of stromal organizer cells in AD EDA-ID as a possibility.
Severely defective activation of the non-canonical NF-κB pathway in stromal cells from patients with IκBα point mutations likely explains the absence of visible tonsils and the lack of detectable peripheral LNs in these patients, as well as their poor outcome following HSCT. Secondary lymphoid organs are critical for adaptive immunity because they provide a scaffold for the interactions of antigen-bearing dendritic cells derived from peripheral tissues, with re-circulating antigen-specific T cells, which are essential for T cell immunity. They also provide a scaffold for the interaction of B cells with T helper cells, which is important for T cell-dependent antibody responses, and with antigen-presenting cells in the splenic marginal zone, which is important for type II T cell-independent antibody responses to polysaccharide antigens.
Our study establishes a genotype-phenotype correlation in patients with AD-EDA ID that has important implications for their management. Higher levels of mutant protein in patients with IκBα point mutations may explain the more severe disease, the impaired development of secondary lymphoid organs, and the poor response of these patients to HSCT. Given the severe functional defect in the non-canonical pathway in patients with point mutations, strategies aimed at correcting the stromal cell niche essential for lymphorganogenesis need to be considered in these patients.
Supplementary Material
Key messages.
IκBα point mutations are associated with more severe disease compared to IκBα truncation mutations in patients with AD EDA-ID.
IκBα point mutants are expressed at significantly higher levels in fibroblasts and transfectants compared to IκBα truncation mutants.
IκBα point mutations are associated with greater impairment of canonical and non-canonical NF-κB activity, and severely defective expression of genes important for lymphorganogenesis, compared to IκBα truncation mutations.
Acknowledgments
The authors thank Drs J. Orange, L.D. Notarangelo and F. Bonilla for their valuable advice in devising the AD EDA-ID scoring system, and Ms. B. Cangemi for technical assistance. This work was supported by a scholarship from the DAAD Thematic Network: ‘Research for Rare Diseases and Personalized Medicine’ and a Carl Duisberg scholarship from Bayer Foundations to DP and USPHS grant 1R21AI124101-01 to RSG.
Abbreviations
- AD EDA-ID
Autosomal dominant anhidrotic ectodermal dysplasia with immune deficiency
- BAFF-R
B cell activating factor receptor
- BM
Bone marrow
- CCL
C-C motif chemokine ligand
- CXCL
C-X-C motif chemokine ligand
- DN
Dominant negative
- HSCT
Hematopoietic stem cell transplantation
- ICAM-1
Intercellular adhesion molecule 1
- Ig
Immunoglobulin
- IκBα
NF-κB inhibitor alpha
- IKK
IκB kinase
- IL-6
Interleukin-6
- LN
Lymph node
- LPS
Lipopolysaccacharide
- LTβR
Lymphotoxin beta receptor
- LTi
Lymphoid tissue inducer
- LTo
Lymphoid tissue organizer
- NEMO
NF-κB essential modulator
- NF-κB
Nuclear factor kappa light chain enhancer of activated B cells
- NF-κB2
Nuclear factor kappa B subunit 2
- NIK
NF-κB inducing kinase
- qPCR
Quantitative polymerase chain reaction
- RAG2
Recombination activating gene 2
- RelB
RELB proto-oncogene, NF-κB subunit
- RT-PCR
Reverse transcription polymerase chain reaction
- SLO
Secondary lymphoid organ
- TLR
Toll-like receptor
- TNFα
Tumor necrosis factor alpha
- VCAM-1
Vascular adhesion molecule 1
- WT
Wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have declared that no conflict of interest exists.
References
- 1.Bousfiha A, Jeddane L, Al-Herz W, Ailal F, Casanova JL, Chatila T, et al. The 2015 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol. 2015;35:727–38. doi: 10.1007/s10875-015-0198-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Picard C, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, et al. Primary Immunodeficiency Diseases: an Update on the Classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol. 2015;35:696–726. doi: 10.1007/s10875-015-0201-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee PP, Chen TX, Jiang LP, Chan KW, Yang W, Lee BW, et al. Clinical characteristics and genotype-phenotype correlation in 62 patients with X-linked agammaglobulinemia. J Clin Immunol. 2010;30:121–31. doi: 10.1007/s10875-009-9341-5. [DOI] [PubMed] [Google Scholar]
- 4.Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol. 2014;133:1099–108. doi: 10.1016/j.jaci.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moratto D, Giliani S, Notarangelo LD, Mazza C, Mazzolari E, Notarangelo LD. The Wiskott-Aldrich syndrome: from genotype-phenotype correlation to treatment. Expert Rev Clin Immunol. 2007;3:813–24. doi: 10.1586/1744666X.3.5.813. [DOI] [PubMed] [Google Scholar]
- 6.Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123:809–21. doi: 10.1182/blood-2013-07-515528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weemaes CM, van Tol MJ, Wang J, van Ostaijen-ten Dam MM, van Eggermond MC, Thijssen PE, et al. Heterogeneous clinical presentation in ICF syndrome: correlation with underlying gene defects. Eur J Hum Genet. 2013;21:1219–25. doi: 10.1038/ejhg.2013.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–34. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Picard C, Casanova JL, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clin Microbiol Rev. 2011;24:490–7. doi: 10.1128/CMR.00001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 11.Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–80. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 12.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–8. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 13.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 14.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun SC. The noncanonical NF-kappaB pathway. Immunol Rev. 2012;246:125–40. doi: 10.1111/j.1600-065X.2011.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, et al. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002;17:525–35. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 17.Matsushima A, Kaisho T, Rennert PD, Nakano H, Kurosawa K, Uchida D, et al. Essential role of nuclear factor (NF)-kappaB-inducing kinase and inhibitor of kappaB (IkappaB) kinase alpha in NF-kappaB activation through lymphotoxin beta receptor, but not through tumor necrosis factor receptor I. J Exp Med. 2001;193:631–6. doi: 10.1084/jem.193.5.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shih VF, Tsui R, Caldwell A, Hoffmann A. A single NFkappaB system for both canonical and non-canonical signaling. Cell Res. 2011;21:86–102. doi: 10.1038/cr.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bren GD, Solan NJ, Miyoshi H, Pennington KN, Pobst LJ, Paya CV. Transcription of the RelB gene is regulated by NF-kappaB. Oncogene. 2001;20:7722–33. doi: 10.1038/sj.onc.1204868. [DOI] [PubMed] [Google Scholar]
- 20.Liptay S, Schmid RM, Nabel EG, Nabel GJ. Transcriptional regulation of NF-kappa B2: evidence for kappa B-mediated positive and negative autoregulation. Mol Cell Biol. 1994;14:7695–703. doi: 10.1128/mcb.14.12.7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ito CY, Kazantsev AG, Baldwin AS., Jr Three NF-kappa B sites in the I kappa B-alpha promoter are required for induction of gene expression by TNF alpha. Nucleic Acids Res. 1994;22:3787–92. doi: 10.1093/nar/22.18.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Courtois G, Smahi A, Reichenbach J, Doffinger R, Cancrini C, Bonnet M, et al. A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest. 2003;112:1108–15. doi: 10.1172/JCI18714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janssen R, van Wengen A, Hoeve MA, ten Dam M, van der Burg M, van Dongen J, et al. The same IkappaBalpha mutation in two related individuals leads to completely different clinical syndromes. J Exp Med. 2004;200:559–68. doi: 10.1084/jem.20040773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDonald DR, Mooster JL, Reddy M, Bawle E, Secord E, Geha RS. Heterozygous N-terminal deletion of IkappaBalpha results in functional nuclear factor kappaB haploinsufficiency, ectodermal dysplasia, and immune deficiency. J Allergy Clin Immunol. 2007;120:900–7. doi: 10.1016/j.jaci.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 25.Lopez-Granados E, Keenan JE, Kinney MC, Leo H, Jain N, Ma CA, et al. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum Mutat. 2008;29:861–8. doi: 10.1002/humu.20740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohnishi H, Miyata R, Suzuki T, Nose T, Kubota K, Kato Z, et al. A rapid screening method to detect autosomal-dominant ectodermal dysplasia with immune deficiency syndrome. J Allergy Clin Immunol. 2012;129:578–80. doi: 10.1016/j.jaci.2011.09.042. [DOI] [PubMed] [Google Scholar]
- 27.Schimke LF, Rieber N, Rylaarsdam S, Cabral-Marques O, Hubbard N, Puel A, et al. A novel gain-of-function IKBA mutation underlies ectodermal dysplasia with immunodeficiency and polyendocrinopathy. J Clin Immunol. 2013;33:1088–99. doi: 10.1007/s10875-013-9906-1. [DOI] [PubMed] [Google Scholar]
- 28.Yoshioka T, Nishikomori R, Hara J, Okada K, Hashii Y, Okafuji I, et al. Autosomal dominant anhidrotic ectodermal dysplasia with immunodeficiency caused by a novel NFKBIA mutation, p.Ser36Tyr, presents with mild ectodermal dysplasia and non-infectious systemic inflammation. J Clin Immunol. 2013;33:1165–74. doi: 10.1007/s10875-013-9924-z. [DOI] [PubMed] [Google Scholar]
- 29.Lee AJ, Moncada-Velez M, Picard C, Llanora G, Huang CH, Abel L, et al. Severe Mycobacterial Diseases in a Patient with GOF IkappaBalpha Mutation Without EDA. J Clin Immunol. 2016;36:12–5. doi: 10.1007/s10875-015-0223-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boisson B, Puel A, Picard C, Casanova JL. Human IκBα Gain of Function: a Severe and Syndromic Immunodeficiency. J Clin Immunol. 2017 doi: 10.1007/s10875-017-0400-z. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dupuis-Girod S, Cancrini C, Le Deist F, Palma P, Bodemer C, Puel A, et al. Successful allogeneic hemopoietic stem cell transplantation in a child who had anhidrotic ectodermal dysplasia with immunodeficiency. Pediatrics. 2006;118:e205–11. doi: 10.1542/peds.2005-2661. [DOI] [PubMed] [Google Scholar]
- 32.Fish JD, Duerst RE, Gelfand EW, Orange JS, Bunin N. Challenges in the use of allogeneic hematopoietic SCT for ectodermal dysplasia with immune deficiency. Bone Marrow Transplant. 2009;43:217–21. doi: 10.1038/bmt.2008.308. [DOI] [PubMed] [Google Scholar]
- 33.Mooster JL, Le Bras S, Massaad MJ, Jabara H, Yoon J, Galand C, et al. Defective lymphoid organogenesis underlies the immune deficiency caused by a heterozygous S32I mutation in IkappaBalpha. J Exp Med. 2015;212:185–202. doi: 10.1084/jem.20140979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alimzhanov MB, Kuprash DV, Kosco-Vilbois MH, Luz A, Turetskaya RL, Tarakhovsky A, et al. Abnormal development of secondary lymphoid tissues in lymphotoxin beta-deficient mice. Proc Natl Acad Sci U S A. 1997;94:9302–7. doi: 10.1073/pnas.94.17.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banks TA, Rouse BT, Kerley MK, Blair PJ, Godfrey VL, Kuklin NA, et al. Lymphotoxin-alpha deficient mice. Effects on secondary lymphoid organ development and humoral immune responsiveness. J Immunol. 1995;155:1685–93. [PubMed] [Google Scholar]
- 36.De Togni P, Goellner J, Ruddle NH, Streeter PR, Fick A, Mariathasan S, et al. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 1994;264:703–7. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 37.Futterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. 1998;9:59–70. doi: 10.1016/s1074-7613(00)80588-9. [DOI] [PubMed] [Google Scholar]
- 38.Koni PA, Sacca R, Lawton P, Browning JL, Ruddle NH, Flavell RA. Distinct roles in lymphoid organogenesis for lymphotoxins alpha and beta revealed in lymphotoxin beta-deficient mice. Immunity. 1997;6:491–500. doi: 10.1016/s1074-7613(00)80292-7. [DOI] [PubMed] [Google Scholar]
- 39.Kuprash DV, Alimzhanov MB, Tumanov AV, Anderson AO, Pfeffer K, Nedospasov SA. TNF and lymphotoxin beta cooperate in the maintenance of secondary lymphoid tissue microarchitecture but not in the development of lymph nodes. J Immunol. 1999;163:6575–80. [PubMed] [Google Scholar]
- 40.Koike R, Nishimura T, Yasumizu R, Tanaka H, Hataba Y, Hataba Y, et al. The splenic marginal zone is absent in alymphoplastic aly mutant mice. Eur J Immunol. 1996;26:669–75. doi: 10.1002/eji.1830260324. [DOI] [PubMed] [Google Scholar]
- 41.Miyawaki S, Nakamura Y, Suzuka H, Koba M, Yasumizu R, Ikehara S, et al. A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur J Immunol. 1994;24:429–34. doi: 10.1002/eji.1830240224. [DOI] [PubMed] [Google Scholar]
- 42.Paxian S, Merkle H, Riemann M, Wilda M, Adler G, Hameister H, et al. Abnormal organogenesis of Peyer's patches in mice deficient for NF-kappaB1, NF-kappaB2, and Bcl-3. Gastroenterology. 2002;122:1853–68. doi: 10.1053/gast.2002.33651. [DOI] [PubMed] [Google Scholar]
- 43.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–9. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 44.Yilmaz ZB, Weih DS, Sivakumar V, Weih F. RelB is required for Peyer's patch development: differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J. 2003;22:121–30. doi: 10.1093/emboj/cdg004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10:664–74. doi: 10.1038/nri2832. [DOI] [PubMed] [Google Scholar]
- 46.Chiao PJ, Miyamoto S, Verma IM. Autoregulation of I kappa B alpha activity. Proc Natl Acad Sci U S A. 1994;91:28–32. doi: 10.1073/pnas.91.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scott ML, Fujita T, Liou HC, Nolan GP, Baltimore D. The p65 subunit of NF-kappa B regulates I kappa B by two distinct mechanisms. Genes Dev. 1993;7:1266–76. doi: 10.1101/gad.7.7a.1266. [DOI] [PubMed] [Google Scholar]
- 48.Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, et al. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med. 1998;187:147–59. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM, et al. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998;187:185–96. doi: 10.1084/jem.187.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, et al. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. 1995;80:331–40. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 51.Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, et al. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–6. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- 52.Williams IR. CCR6 and CCL20: partners in intestinal immunity and lymphorganogenesis. Ann N Y Acad Sci. 2006;1072:52–61. doi: 10.1196/annals.1326.036. [DOI] [PubMed] [Google Scholar]
- 53.Lo JC, Basak S, James ES, Quiambo RS, Kinsella MC, Alegre ML, et al. Coordination between NF-kappaB family members p50 and p52 is essential for mediating LTbetaR signals in the development and organization of secondary lymphoid tissues. Blood. 2006;107:1048–55. doi: 10.1182/blood-2005-06-2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cildir G, Low KC, Tergaonkar V. Noncanonical NF-kappaB Signaling in Health and Disease. Trends Mol Med. 2016;22:414–29. doi: 10.1016/j.molmed.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 55.Rumbo M, Sierro F, Debard N, Kraehenbuhl JP, Finke D. Lymphotoxin beta receptor signaling induces the chemokine CCL20 in intestinal epithelium. Gastroenterology. 2004;127:213–23. doi: 10.1053/j.gastro.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 56.Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krappmann D, Wegener E, Sunami Y, Esen M, Thiel A, Mordmuller B, et al. The IkappaB kinase complex and NF-kappaB act as master regulators of lipopolysaccharide-induced gene expression and control subordinate activation of AP-1. Mol Cell Biol. 2004;24:6488–500. doi: 10.1128/MCB.24.14.6488-6500.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoorweg K, Narang P, Li Z, Thuery A, Papazian N, Withers DR, et al. A Stromal Cell Niche for Human and Mouse Type 3 Innate Lymphoid Cells. J Immunol. 2015;195:4257–63. doi: 10.4049/jimmunol.1402584. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.